

Abstract
Genetic variation in CC chemokine receptor 5 (CCR5), the major HIV-1 coreceptor, has been shown to influence HIV-1 transmission and disease progression. However, it is generally assumed that the same CCR5 genotype (or haplotype) has similar phenotypic effects in different populations. To test this assumption, we used an evolutionary-based classification of CCR5 haplotypes to determine their associated HIV-1 disease-modifying effects in a large well-characterized racially mixed cohort of HIV-1-seropositive individuals. We demonstrate that the spectrum of CCR5 haplotypes associated with disease acceleration or retardation differs between African Americans and Caucasians. Also, we show that there is a strong interactive effect between CCR5 haplotypes with different evolutionary histories. The striking population-specific phenotypic effects associated with CCR5 haplotypes emphasize the importance of understanding the evolutionary context in which disease susceptibility genes are expressed.
Human populations have varied evolutionary histories and, more importantly, have coevolved with different combinations of microbes. Hence, the repertoire of alleles that afford resistance or susceptibility to pathogens (e.g., malaria) may vary in different populations (1). Evolutionary forces may have had similar effects on the genes encoding proteins that affect susceptibility to HIV-1, especially in African populations where cross-species transmission of HIV-like retroviruses likely first occurred (2).
CC chemokine receptor 5 (CCR5) serves as the major portal of entry for HIV-1, and it has been hypothesized that polymorphisms in the coding and/or cis-regulatory regions may influence cell surface expression and consequently could influence an individual’s susceptibility to HIV-1 (3, 4). Thus, significant attention has been focused on understanding the HIV-1 disease-modifying effects of CCR5 polymorphisms (5–14). For example, the CCR5-Δ32 allele and a CCR5 allele in linkage disequilibrium with the CCR2-64I polymorphism have been associated with disease retardation. These associations were found in cohorts composed of predominantly homosexual Caucasian men. Whether the results of these association studies can be generalized to other ethnic/population groups is unclear.
In the U.S., AIDS is evolving from a disease that once predominantly affected homosexual Caucasian men to one that now largely strikes minority groups (15). This changing epidemiology of HIV-1 makes stratification for population-specific disease-modifying genetic determinants in cohorts used to evaluate HIV-1 intervention strategies compelling. We tested the hypothesis that variability in HIV-1 disease progression differs according to CCR5 haplotype and ethnicity in a large well- characterized racially mixed cohort of HIV-1-seropositive individuals. This cohort has several epidemiologic features that make it ideally suited for dissecting the population-specific genetic determinants of HIV-1 infection (16). In this cohort, we found previously that the CCR2-64I allele was associated with a delay in disease progression in African Americans but not in Caucasians (16). To determine whether the population-specific risk of HIV-1 infection varied according to CCR5 haplotype, we compared the genotype of 1,151 individuals from this cohort to that of 1,199 uninfected individuals representing ethnic groups living in Africa, Asia, and Europe.
Methods
Subjects.
Patients with HIV-1 participating in the U.S. Air Force portion of the Tri-Service HIV Natural History Study contributed samples for this study. Wilford Hall Medical Center (WHMC) is the referral hospital for all Air Force personnel who develop infection with HIV. The voluntary fully informed consent of the subjects used in this research was obtained as required by Air Force Regulation 169–9. A total of 1,151 patients were evaluated and included 528 seroincident and 623 seroprevalent cases. The demographic background of this cohort was 54% Caucasian, 37% African American, 6% Hispanic, and 3% “other.” The median age at the time of diagnosis was 28 yr (range, 18 to 70 yr), and 94% of the subjects were male. The median followup time was 5.9 yr for the entire cohort. It was 6.3 yr for the seroconvertors using as the initial time point the estimated seroconversion date (the midpoint between the last negative and first positive HIV test). The median time from the last negative HIV test to estimated seroconversion was 10.4 mo. Thirty-eight percent of this cohort progressed to AIDS (1987 criteria), and 34% died during the study period, which ended October 1998. Additional epidemiological features of the cohort and the different ethnic populations analyzed are described in the supplemental material (see www.pnas.org).
Genotype Analysis.
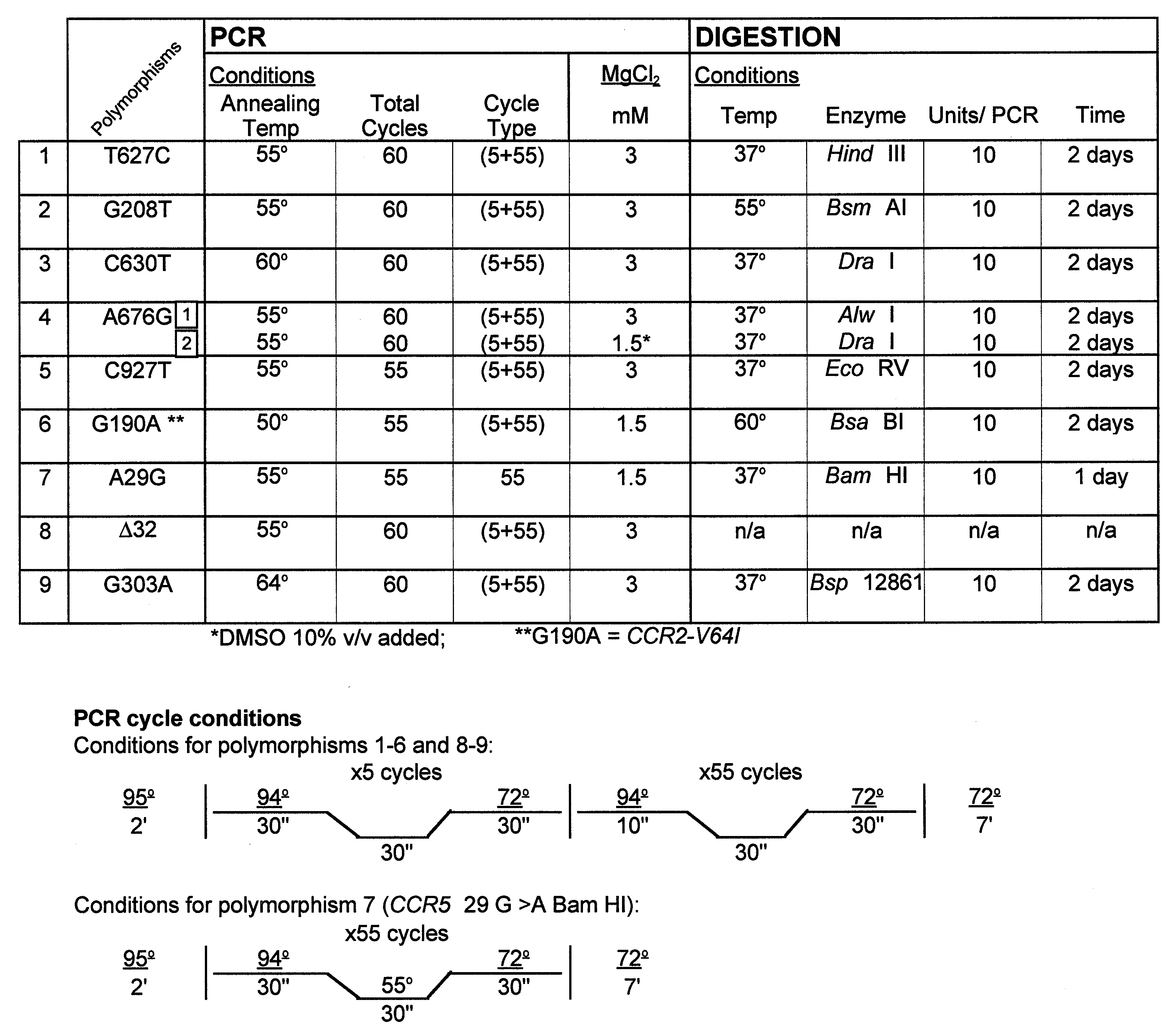
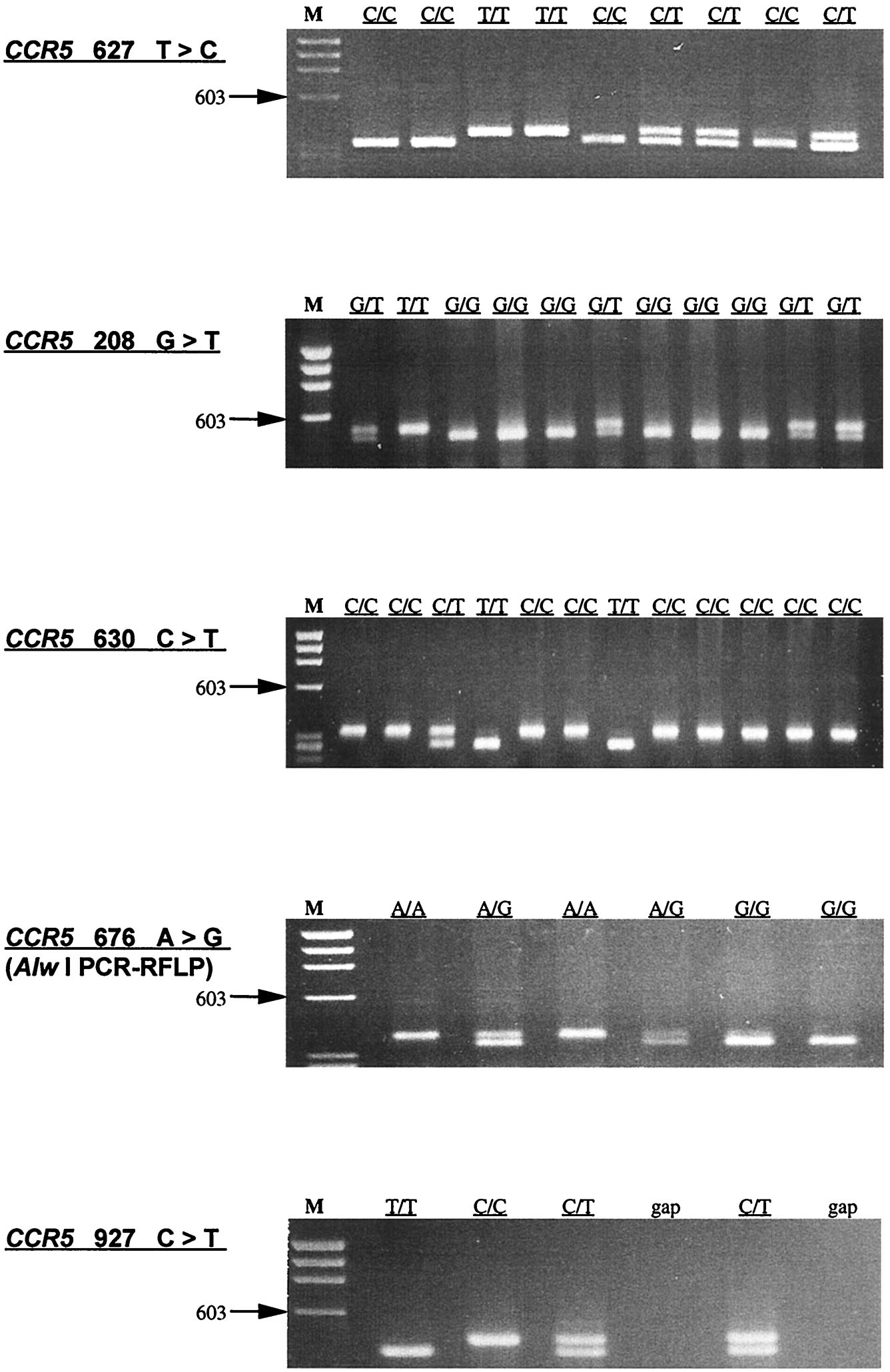
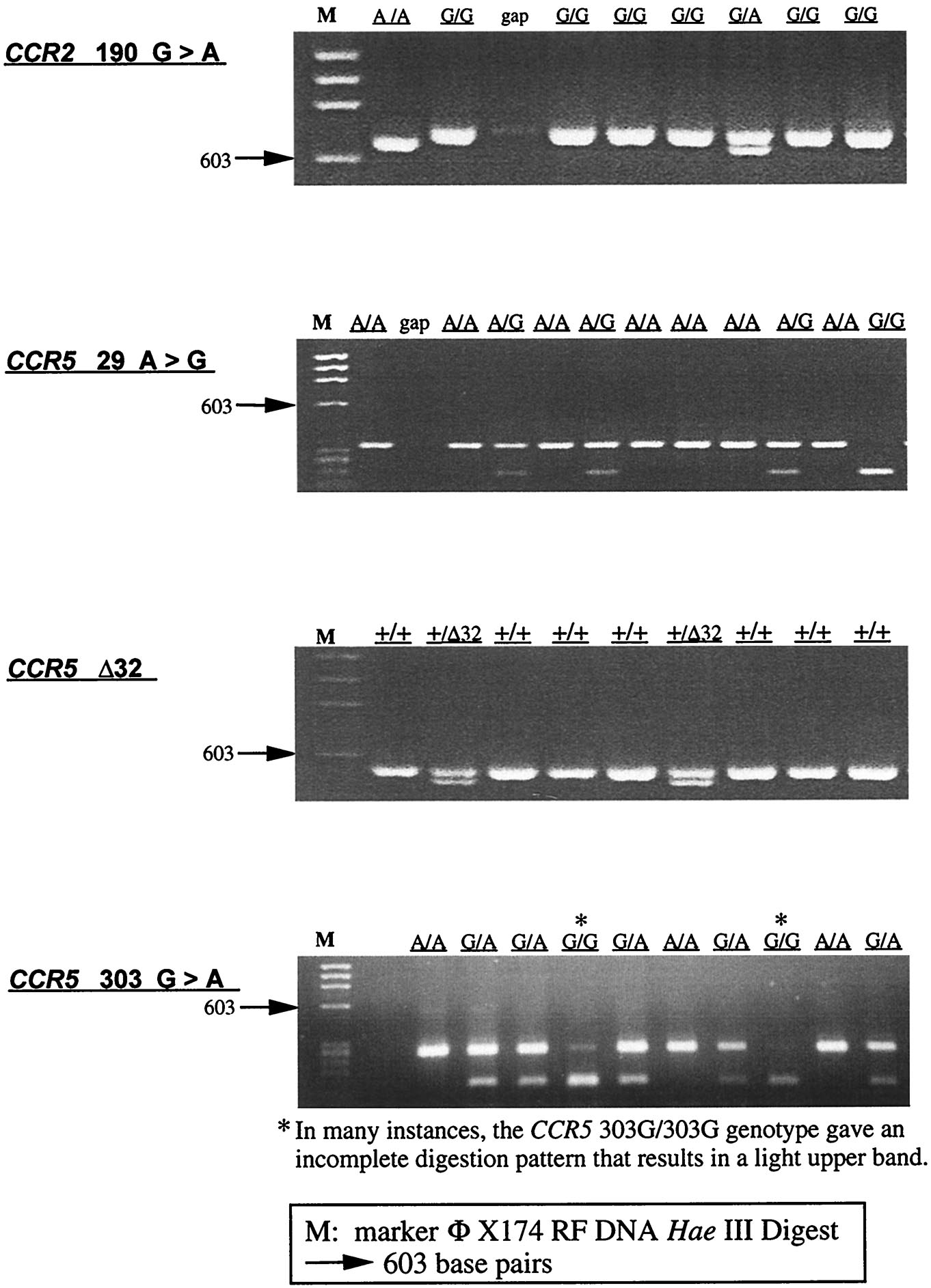
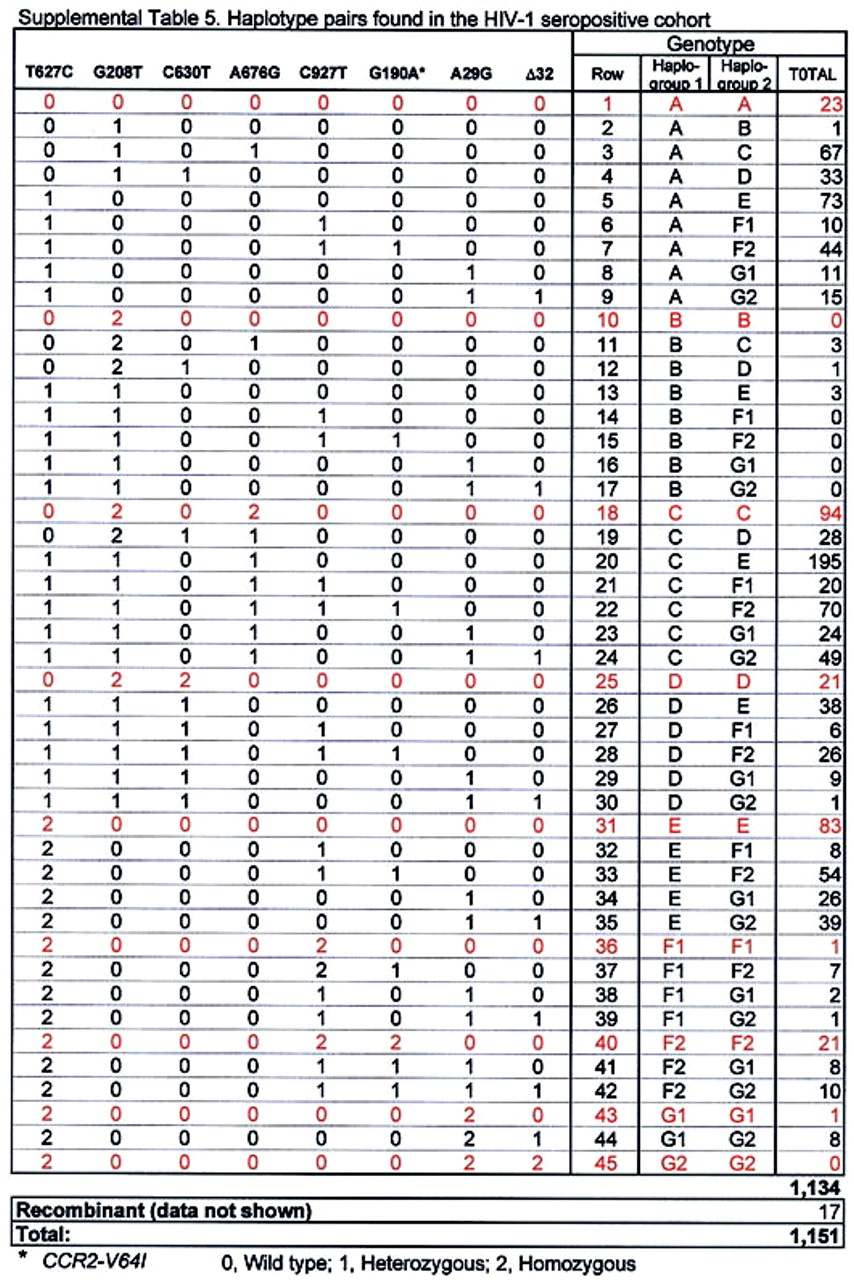
PCR–restriction fragment length polymorphism (RFLP)-based assays were used to genotype the WHMC cohort and ethnic populations at a single nucleotide polymorphism (SNP) in the CCR2 coding region (G190A; CCR2-V64I), the SNPs in a CCR5 cis-regulatory region [A29G, G208T, G303A (only WHMC cohort), T627C, C630T, A676G, C927T (16, 17)]. The CCR5-Δ32 mutation was genotyped as described previously (16). Molecular beacon-based genotyping methods were used to confirm the genotype at CCR5-627 and CCR5-676 in the WHMC cohort. Detailed protocols are provided in supplemental Figs. 6–9 (see www.pnas.org). The relationship between CCR5-C927T and CCR2-V64I, CCR5-G303A and CCR5-T627C, and CCR5-A29G and CCR5-Δ32 is described in supplemental Tables 2–4. In 1,138 individuals from the WHMC cohort, CCR5-303G and CCR5-303A were found to be in nearly complete linkage disequilibrium with CCR5-627T and CCR5-627C, respectively (supplemental Table 3). For this reason, the haplotype reported was restricted to the genotype analysis of SNPs at CCR2 190, CCR5 29, 208, 627, 630, 676, 927, and the CCR5-Δ32 mutation. Methods for CCR5 haplotype assignment and the frequency of the different haplotype pairs found in the WHMC cohort are shown in supplemental Table 5.
Statistical Analysis.
Time curves for progression to AIDS (1987 criteria) and survival were prepared by the Kaplan–Meier (KM) method by using sas, version 6.12 (SAS Institute, Cary, NC). Between-group analyses were completed by using the log-rank test. Relative hazards were calculated by using univariate and multivariate Cox-proportional hazard models. The number of individuals in each group whose KM curves are shown in Figs. 2–5 is shown in supplemental Fig. 10 (see www.pnas.org). The reference group for each of the analyses is indicated in the figure legends. In 17 individuals, one CCR5 allele appeared to be the product of a recombination event, and these patients were excluded from analysis. CI indicates 95% confidence interval limits. Because of the disease-modifying effects associated with HHF*2 (CCR2-64I) and HHG*2 (CCR5-Δ32) (16), adjustments were made for their protective effects in African Americans and Caucasians, respectively; in survival analysis for the entire cohort, adjustments were made for these two haplotypes.
Figure 2.

Disease-modifying effects of CCR5 haplotypes in Caucasians. (a and b) CCR5-HHG*1 and -HHG*2 haplotypes are associated with different HIV-1 disease-modifying effects in Caucasians. The KM curves of the development of AIDS (1987 criteria) (a) or death (b) for Caucasians who possessed at least one HHG*1 (green line) or HHG*2 (brown line) allele. The reference group for the survival analyses was Caucasians that did not possess either of these two alleles (purple line; −HHG*1/−HHG*2). For statistical analysis comparing HHG*2 to non-HHG-bearing patients, individuals who were homozygous for HHG*1 and also had a CCR5-Δ32 mutation (HHG*2) on one of these alleles were considered as part of HHG*2. They were excluded from the comparison of HHG*1 and HHG*2. P and RH indicate the significance value by log-rank test and the relative hazard with respect to the reference group, respectively. The data shown are for the combination of the seroconverting and seroprevalent Caucasians. (c and d) KM curves comparing the clinical course of Caucasians lacking an HHE haplotype (0, purple line) or possessing one (1, brown line) or two HHE haplotypes (2, green line). The reference group for the survival analyses is Caucasians who do not possess HHE haplotypes (purple line). The unadjusted P and RH values are followed in parentheses by values adjusted for the protective effects of HHG*2. (e and f) KM curves comparing the clinical course of Caucasians who possess (nonpurple colored lines) or lack (purple line) the haplotype pairs listed in f. (g and h) The KM curves of the development of AIDS or death in Caucasians with the following haplotype pairs [presence (+) and absence (−)]: +HHC/+HHG*2 (brown line); −HHC/+HHG*2 (green line); −HHG*2/−HHG*2 (purple line). The reference group for the statistical analyses was Caucasians who are −HHG*2/−HHG*2. (i) CCR5 haplotypes in Caucasians associated with different outcomes of HIV-1 disease. The haplotype pairs associated with no statistically significant disease-modifying effects are designated as being neutral and are not shown.
Figure 5.

HHC-associated allele–allele interactions in African Americans and the disease-modifying role of HHD haplotypes. (a–f) KM curves comparing the clinical course of African Americans who possess (nonpurple colored lines) or lack (purple line) the haplotype pairs listed. The reference group for the statistical analyses is the purple-colored KM curve in a–f. The data shown are for the combination of the seroconverting and seroprevalent populations. (g) HHF*2-unadjusted and -adjusted relative risk of AIDS and death associated with three HHC-containing haplotype pairs in African Americans. The reference groups for the log-rank test are for African Americans who lack these haplotype pairs, and they are represented by the purple-colored KM curve in a and b. (h) CCR5 haplotypes in African Americans that are associated with different HIV-1 disease progression rates.
Results
Spectrum of CCR5 Haplotypes in World-Wide Populations.
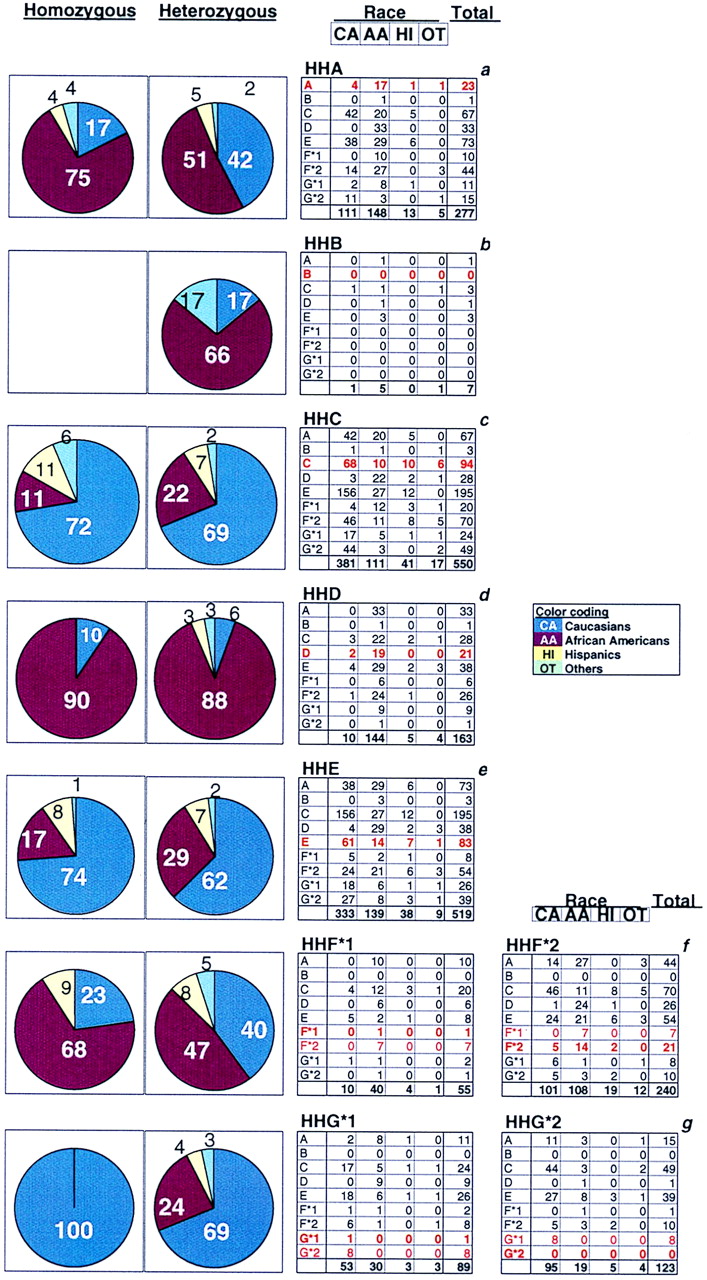
We grouped CCR5 haplotypes into seven phylogenetically distinct clusters that we designate CCR5 human haplogroups (HH)-A, -B, -C, -D, -E, -F, and -G, with HHA representing the ancestral CCR5 haplogroup (Fig. 1a; S.M., M.B., M.J.D., and S.K.A., unpublished work). HHA haplotypes were defined as ancestral to all other haplotypes by comparison to the CCR5 alleles of Great Apes and Old and New World monkeys. CCR5 haplogroup frequencies were similar between HIV-infected and -uninfected Caucasians and African Americans (Table 1). Among uninfected populations, CCR5 haplogroup frequencies varied substantially among races and ethnic groups (Table 1). Overall, haplotype diversity was highest in Africans, and only a subset of these haplotypes was found in non-African populations.
Figure 1.

(a) CCR5 human haplogroups and distribution of CCR5 haplogroup/haplotype pairs. The phylogenetic tree shown is a highly abbreviated version adapted from S.M., M.B., M.J.D., and S.K.A. (unpublished work). Detailed methods of the phylogenetic analysis and rationale for the CCR5 haplogroup classification will be reported by S.M., M.B., M.J.D., and S.K.A. elsewhere. Briefly, a CCR5 haplogroup is an aggregate of several distinct haplotypes that share a common ancestry. Therefore, each haplotype within a haplogroup is characterized by the constellation of invariant CCR5/CCR2 polymorphisms indicated in a, but may differ from each other by unique but rare SNPs. The sequences common to those found in the ancestral CCR5 haplotype, designated as HHA, are colored in red. HHF*2 and HHG*2 designate the subset of haplotypes within HHF and HHG that are in linkage disequilibrium with the CCR2-64I and CCR5-Δ32 polymorphisms, respectively. (b) Histogram illustrating the heterogeneity in the frequency of CCR5 haplogroup pairs (%) in Caucasians and African Americans (note differences in scale). The frequencies for the haplotype pairs associated with HHB were very low and are not shown.
Table 1.
CCR5 haplogroup frequencies in different racial and ethnic groups
| Haplogroup | African
|
African American
|
Asian
|
Caucasian
|
Hispanic American
|
|||
|---|---|---|---|---|---|---|---|---|
| Pygmies | Non-pygmies | Uninfected | HIV-1 infected | Uninfected | Uninfected | HIV-1 infected | HIV-1 infected | |
| HHA | 70.6 (34) | 26.5 (49) | 22 (209) | 20.1 (410) | 16.8 (158) | 10.7 (248) | 9.3 (618) | 9.5 (74) |
| HHC | 2.0 (25) | 10.6 (71) | 15.6 (212) | 14.8 (410) | 36.5 (163) | 37.1 (206) | 36.3 (618) | 34.5 (74) |
| HHD | 0 (37) | 20.1 (82) | 18.4 (212) | 20.1 (410) | 4.4 (34) | 0 (429) | 1.0 (618) | 3.4 (74) |
| HHE | 11.8 (38) | 20.7 (58) | 18.4 (193) | 18.7 (410) | 25 (376) | 31.8 (140) | 31.9 (618) | 30.4 (74) |
| HHF*1 | 6.3 (40) | 11.8 (68) | 4.1 (195) | 5.0 (410) | 1.6 (478) | 2.0 (154) | 0.8 (618) | 2.7 (74) |
| HHF*2 | 6.3 (40) | 14.7 (68) | 14.1 (195) | 14.9 (410) | 12.8 (478) | 5.5 (154) | 8.6 (618) | 14.2 (74) |
| HHG*1 | 2.5 (40) | 0.7 (71) | 4.5 (210) | 3.7 (410) | 0.8 (518) | 3.3 (151) | 4.4 (618) | 2.0 (74) |
| HHG*2 | 0 (40) | 0 (71) | 2.6 (210) | 2.3 (410) | 0.1 (518) | 5.6 (151) | 7.7 (618) | 3.4 (74) |
The number in parentheses denotes the number of individuals from whom the haplotype frequency (%) was derived. HHB haplotypes are rare, and their frequencies are not shown. Also, because of failure to amplify by PCR all CCR5 polymorphisms and/or limited DNA quantities, the number of noninfected individuals for whom complete haplotype frequency data are available varies. Hence, for these two reasons the frequencies approximate but do not total to 100%. Individuals in whom a CCR5 haplotype appeared to be a product of a recombination event were excluded from analysis.
The distribution of haplotype pairs between African Americans and Caucasians was also different [Fig. 1b; supplemental Fig. 10 (see www.pnas.org)]. Fifty-two different haplotype pairs were found in the HIV-1-positive cohort, and 99% of individuals in the cohort had one of 39 of these pairs (Fig. 1b, supplemental Table 5). In Caucasians, most individuals had one of only a few different haplotype pairs, and the three most common haplotype pairs were HHC/HHE (25%), HHC/HHC (≈11%), and HHE/HHE (≈10%; Fig. 1b; supplemental Fig. 10). In contrast, no single haplotype pair was common in African Americans, and the prevalence of each haplotype pair was less than 10% (Fig. 1b; supplemental Fig. 10). This heterogeneous distribution of haplotype pairs suggested that the spectrum of CCR5 haplotype pairs associated with differences in HIV-1 disease progression might differ between Caucasians and African Americans.
Varied Disease-Modifying Effects of CCR5 Haplotypes.
There was a delay in progression to AIDS and death in Caucasians for those with the HHG*2 haplotypes (CCR5-Δ32) compared with those without it (Fig. 2 a and b). Although both HHG*1 (CCR5-29G without CCR5-Δ32) and HHG*2 were found on a haplotype background with CCR5-29G (Fig. 1a and supplemental Table 4), only haplotypes with the CCR5-Δ32 mutation were associated with disease retardation in comparison to the population not possessing any HHG haplotypes (Fig. 2 a and b). The disease-modifying effects of the HHG*1 and HHG*2 haplotypes differed with respect to each other for both progression to AIDS (P = 0.07) and death (P = 0.02).
The two haplotypes that contain the SNP CCR5-927T (Fig. 1a) were associated with different disease-modifying effects. HHF*2 haplotypes [combining homozygotes (+/+) and heterozygotes (+/−)] were associated with a delay in progression to AIDS [P = 0.01; relative hazard (RH) = 0.58; CI = 0.38–0.88] and death (P = 0.005; RH = 0.50; CI = 0.31–0.81) in African Americans but not in Caucasians [(for AIDS, P = 0.77; RH = 0.95; CI = 0.68–1.33) (for death, P = 0.84; RH = 1.04; CI = 0.74–1.46); KM curves not shown]. In contrast, HHF*1 haplotypes (+/+ and +/−) were associated with an acceleration to AIDS in the entire cohort (P = 0.05; RH = 1.47; CI = 1.0–2.16) and in African Americans (P = 0.04; RH = 1.64; CI = 1.01–2.66).
In the entire cohort, HHA haplotypes (combining +/+ and +/−) were associated with a delay in progression to AIDS (adjusted for HHF*2 and HHG*2, P = 0.04; RH = 0.77; CI = 0.60–0.99) and death (adjusted P = 0.04; RH = 0.79; CI = 0.62–0.99; KM curves not shown). This association was demonstrable in African Americans (Fig. 3 a and b) but not Caucasians (for AIDS, adjusted for HHG*2, P = 0.71; for death, adjusted P = 0.94). These findings suggested that HHA haplotypes in African Americans were associated with disease retardation, and that this association was independent of the effect of HHF*2. However, they did not exclude the possibility of an additive and/or interactive effect between HHA and HHF*2 haplotypes. Thus, we stratified the African-American and Caucasian patients into four groups, with each group composed of a different pairwise haplotype combination (Fig. 3 c and d). For African Americans, the three groups that contain an HHA and/or HHF*2 haplotype were each associated with a delay in progression to AIDS and death, with the combination of HHA and HHF*2 providing the greatest advantage. In Caucasians, there are no demonstrable differences between various combinations of these two haplotypes (Fig. 3 c and d Inset).
Figure 3.

(a and b) The KM curves (adjusted for the effects of HHF*2) of the development of AIDS or death in African Americans that possess (brown line) or lack (purple line) an HHA allele. The P and RH (95% CI) values have been adjusted for the disease-retarding effects of HHF*2 haplotypes. (c–d) The KM curves of the development of AIDS or death in African Americans with the following haplotype pairs [presence (+) and absence (−)]: +HHA/−HHF*2 (brown line); −HHA/+HHF*2 (green line); +HHA/+HHF*2 (black line). The reference groups are individuals who are −HHA/−HHF*2 (purple line). P, RH, and CI determined by log-rank test. The KM curves for Caucasians with the identical haplotype pairs are shown as Inset in c and d. The data shown are for the combination of the seroconverting and seroprevalent African Americans.
In the overall cohort, there was no difference in clinical outcomes for groups possessing zero, one, or two HHC haplotypes (data not shown). If the cohort was stratified by race, the effect of HHC haplotypes on HIV-1 disease differed between African Americans, Caucasians, and Hispanics (Fig. 4). In Caucasians and Hispanics, HHC haplotypes were associated with disease retardation, particularly a delayed progression to death (Fig. 4 e–j). In contrast, for African Americans, possession of HHC haplotypes was associated with disease acceleration (Fig. 4 a–d).
Figure 4.

The KM curves for the development of AIDS or death in African Americans (a–d), Caucasians (e–h), and Hispanic Americans (i and j) lacking (0, purple line) or possessing one (1, brown line) or two (2, green line) HHC alleles. The unadjusted P and RH values shown are for the combined analysis of individuals who are homozygous or heterozygous for HHC vs. the reference group that is composed of individuals who lack an HHC allele (purple line). The unadjusted P and RH values are followed in parentheses by values adjusted for HHF*2 (in African Americans) or HHG*2 (in Caucasians). The values for Hispanic Americans are unadjusted. ALL refers to the combination of the seroconverting and seroprevalent population; SCT indicates the seroconverting population.
HHE homozygosity was associated with acceleration to AIDS (adjusted for both HHC and HHF*2, P = 0.02; RH = 1.55; CI = 1.09- 2.20) and death (adjusted P = 0.003; RH = 1.72; CI = 1.20- 2.46; KM curves not shown) in the entire cohort, whereas HHE heterozygotes had similar outcomes to non-HHE-bearing individuals. For Caucasians, HHE homozygosity (but not HHE heterozygosity) was associated with disease acceleration, particularly an accelerated progression to death (Fig. 2 c and d). HHE homozygosity was not associated with disease-modifying effects in the African Americans (data not shown).
CCR5 Haplotype Interactions in African Americans.
Because we know that the distribution of haplotypes differs between Caucasians and African Americans, the potential partner allele for a single HHC allele also differs. We therefore next looked at the effect of HHC allele pairs on disease progression (Fig. 5). For African Americans, the pairing of an HHC haplotype with an HHD or HHE haplotype was associated with accelerated disease (Fig. 5 a, b, and g). This phenotype was similar to that observed in HHC homozygotes (Figs. 4 a–d and 5 a, b, and g). For African Americans who possessed one of the haplotype pairs HHC/HHC, HHC/HHD, or HHC/HHE, the combined median time to AIDS and death was 5.21 and 6.34 yr, respectively. In contrast, the median time to AIDS was 9.37 yr in African Americans lacking an HHC haplotype. The median time to death had not been reached in African Americans lacking an HHC haplotype, but a calculated estimate was greater than 12 yr. A disease-accelerating effect was also observed for the haplotype pair, HHC/HHF*1 (Fig. 5 c and d). In contrast, if an HHC haplotype was paired with one of the haplotypes that was associated with protection in African Americans [HHA or HHF*2 (CCR2-64I)], the disease-accelerating effects of the HHC haplotype were negated (Fig. 5 c and d).
To test the disease-modifying effects of the HHD haplotype independent of its association with HHC, we stratified African Americans into four groups of haplotype pairs (Fig. 5 e and f). The disease course of individuals who possess both an HHD and HHC haplotype was significantly more rapid than in those who have an HHD haplotype paired with a non-HHC haplotype (for AIDS, P = 0.005; for death, P = 0.02; for comparison of green and black colored KM curves, see Fig. 5 e and f). These findings suggest that in African Americans, the detrimental phenotypic effect associated with the HHC haplotype was evident when combined with HHD or HHE, but not with HHA or HHF*2 haplotypes. Collectively, these findings (Figs. 3, 4 a–d, and 5) permitted us to identify CCR5 haplotype pairs that were associated with a broad spectrum of effects on HIV-1 disease in African Americans (Fig. 5h). Notably, HHC/HHC and HHC/HHD, the haplotype pairs associated with maximal disease progression in African Americans, represent individuals who are homozygous for the CCR5-208T SNP (Fig. 1a).
CCR5 Haplotype Interactions in Caucasians.
In Caucasians, the KM curves for haplotype pairs that contained at least one HHC haplotype were above or superimposed on the KM curve of haplotype pairs that did not contain a HHC haplotype (Fig. 2 e and f). Together, these haplotype pairs accounted for ≈50% of all Caucasians. HHC/HHC and HHC/HHE accounted for nearly 34% of Caucasian haplotype pairs, but they represented only a small proportion of African-American haplotype pairs (Fig. 1b). Yet, there were sharply contrasting disease-modifying effects between African Americans and Caucasians for HHC/HHC and HHC/HHE (compare Figs. 2 e and f and 5 a and b). Furthermore, after adjustment for the protective effects of HHG*2, the haplotype pair HHC/HHE was associated with a delay in time to death in Caucasians (adjusted P = 0.04; RH = 0.70; CI = 0.50–0.98; Fig. 2 e and f) in contrast to the accelerated progression seen in HHE/HHE homozygotes.
The haplotype pair HHC/HHG*2 was also associated with a trend towards a delay in progression to AIDS (P = 0.08, RH = 0.59; CI = 0.34–1.05; Fig. 2e) and death (P = 0.08; RH = 0.59; CI = 0.32–1.06; Fig. 2f). Because the strength of this association was similar to that for all HHG*2 alleles (Fig. 2 a and b), we compared the effects of HHC/HHG*2 vs. all haplotype pairs that contained an HHG*2 (CCR5-Δ32) haplotype and a non-HHC haplotype. Although an HHG*2 haplotype was most commonly found in association with an HHA, HHC, or HHE haplotype (Fig. 1b), the pairing of HHG*2 with HHC accounts for most of HHG*2’s beneficial effect (Fig. 2, compare a and g and b and h). These findings suggest that the phenotypic effects associated with the CCR5-Δ32-bearing haplotype depend, in large part, on the identity of its partner allele.
Discussion
Population-Specific Effects of CCR5 Haplotypes.
Collectively, our findings indicate that the CCR5 haplotypes associated with altered rates of HIV-1 disease progression in Caucasians were different from those in African Americans (compare Figs. 2i and 5h). Our studies also highlight the importance of understanding the interactions between CCR5 haplotypes and emphasize that analysis of a single mutation or haplotype in isolation may obscure the complexity underlying CCR5 genotype-phenotype relationships. HHA and HHF*2 haplotypes have significantly higher frequencies in African Americans than in Caucasians, and in the WHMC cohort their effect was dominant (i.e., even a single allele confers disease retardation). However, this phenotypic effect was demonstrable only in African Americans, not Caucasians. Conversely, HHC haplotypes have significantly higher frequencies in Caucasians than in African Americans. In African Americans, HHC haplotypes were associated with a detrimental effect that was mitigated when paired with haplotypes associated with protective effects (i.e., HHA or HHF*2). These population-specific CCR5 haplotype-pair associations may be the consequence of the evolution of different combinations of alleles encoding mediators of the immune response in Africans vs. Caucasians. Such combinations of alleles may have offered selective advantages to ancestral Caucasian and African populations that were exposed to different spectrums of pathogens. These findings also suggest that disruption of combinations of alleles that may have been previously favored by selection might result in deleterious effects in very specific circumstances.
The heterogeneous distribution of CCR5 haplotypes in African Americans and Caucasians may influence the results of genotype-phenotype association studies. For example, among all Caucasians who possess a CCR5-Δ32-bearing haplotype (HHG*2), the haplotype pair, HHC/HHG*2, affords the strongest protective effects (Fig. 2 g and h). Thus, the frequencies of HHC and HHG*2 haplotypes in Caucasians will determine the frequency of HHC/HHG*2 haplotype pairs, and therefore the likelihood of associating a CCR5-Δ32-bearing haplotype with a protective phenotype. Varying frequencies of both HHC and HHG*2 haplotypes in cohorts could therefore explain some of the intercohort outcome differences reported for the CCR5-Δ32 mutation (reviewed in ref. 18). This suggests that it may be more appropriate to estimate whether haplotype pairs, rather than individual haplotypes, are associated with particular disease-modifying phenotypes.
It is noteworthy that despite presumably intimate contact with a SIVcpz/HIV-1 reservoir for thousands of years, the frequency of zoonotic transmission of SIVcpz/HIV-1 to pygmies appears to be very low (2, 19–22). Yet among these secluded ethnic populations there is a high prevalence of other blood-borne infections such as hepatitis B virus, hepatitis C virus, and human T-lymphotrophic virus (HTLV)-1 (19, 20). The very close relationships among some simian T-lymphotrophic virus-1 strains from chimpanzees and HTLV-1 subtype B strains present in pygmies (23, 24) reinforce the possibility of zoonotic transmission of other retroviruses such as SIVcpz from chimpanzees to this ethnic group. Our results indicate that HHA haplotypes are associated with a delay in disease progression in individuals of African descent, although there is no evidence that HHA haplotypes are associated with a reduction in transmission risk. Nonetheless, the highest prevalence of ancestral HHA haplotypes was in individuals of African descent (≥0.22), reaching its maximum in Mbuti and Biaka pygmies (0.71; Table 1). Whether protection against HIV-1 infection in pygmies could have been afforded, in part, by HHA haplotypes is unclear.
To lessen the potential of conflating protective and nonprotective CCR5 haplotypes, we organized the complex patterns of human CCR5 SNPs/polymorphisms into evolutionarily meaningful relationships (Fig. 1a; S.M., M.B., M.J.D., and S.K.A., unpublished work) that provided the framework necessary for defining the effects of interactions between CCR5 haplotypes. This organization/classification of CCR5 haplotypes differs from that reported recently (13). On the basis of genotypic data from a region of CCR5 spanning +208 to +811, 10 CCR5 promoter alleles have been described (i.e., P1–P10). These CCR5 alleles represent only a subset of the haplotypes that we observed in world-wide populations (Fig. 1a; S.M., M.B., M.J.D., and S.K.A., unpublished work). P2, P3, and P4 correspond to HHA, HHD, and HHC, respectively. The additional alleles defined by P5–P10 likely are members of haplogroup A, B, C, or D. In this study, we found possession of HHD alleles to be restricted primarily to individuals of African descent, whereas the previously reported allelic frequency was 0.14 for this allele in Caucasians (13). The basis for this discrepancy is not readily apparent.
Homozygosity for the P1 (13) or 303A (referred to as the 59029A allele in ref. 14) allele has been associated with disease acceleration. However, Fig. 1a shows that the P1/303A allele is a composite of at least three haplogroups that share 303A and 627C (HHE, HHF*1, and HHG*1). The reason for this is that although the CCR2-64I allele is in nearly complete linkage disequilibrium with CCR5-927T, the converse is not true. In the WHMC cohort, 18% of CCR5-927T alleles were linked to CCR2-64V (HHG*1 allele; supplemental Table 2). Similarly, although the CCR5-Δ32 mutation is in nearly complete disequilibrium with CCR5-29G, 42% of CCR5-29G alleles are not linked to the CCR5-Δ32 mutation (HHG*1 allele; supplemental Table 4). Thus, HHE is composed of P1/303A alleles lacking CCR5-29G and CCR5-927T (Fig. 1a). Inclusion of HHG*1 (neutral phenotype) and HHF*1 (disease-accelerating phenotype) haplotypes into HHE in the WHMC cohort would have increased the number of HHE homozygotes by 43%, and this would have altered the significance of the phenotypic effects of this genotype. Thus, the P1 (13) or 303A (59029A; ref. 14) allele is a conflation of at least three alleles with different evolutionary histories and HIV-1 disease-modifying phenotypic effects.
The mechanistic basis for the HIV-1 disease-modifying effects of genetic variation in CCR5 is unclear and may in part be attributable to differences in haplotype-specific transcriptional efficiency and/or differential nuclear factor binding to polymorphic CCR5 cis-regulatory sites (S.M. and S.K.A., unpublished work). However, the translation of in vitro data on differences in transcriptional efficiency and/or DNA–protein interactions to differences in CCR5 surface expression, much less differences in disease progression, will be challenging.
In summary, the findings of this study support our hypothesis that CCR5 haplotypes are associated with powerful population-specific HIV-1 disease-modifying effects. Our findings also highlight the importance of understanding the evolutionary context in which disease-associated haplotypes are found and underscore the potential impact of allele–allele interactions, especially between alleles with different evolutionary histories. Given that defining the phenotypic effects of variation in the human genome is one of the next major challenges in genetic medicine, our studies illustrate the complexity of the relationships that are certain to be encountered.
Supplementary Material
Acknowledgments
We thank S. Hilsenbeck, G. Crawford, K. Phelps, F. R. Kramer, and L. Jorde for advice; B. Kramer, N. Nicholls, A. Bock, V. Kostecki, and V. Telles for technical assistance, and A. S. Ahuja for forbearance. The H. M. Jackson Foundation and the Military HIV Program, Walter Reed Army Institute of Research contributed support for the WHMC patient cohort as part of the Tri-Service HIV Program. S.S.A and S.K.A. are indebted to the R. J. Kleberg, Jr. and H. C. Kleberg Foundation for their strong support. This work was also supported by a Veterans’ Administration Merit Award to S.K.A., National Institutes of Health grants (AI43279 and AI46326) to S.K.A and F. R. Kramer (HL43521), and by grant support from the National Science Foundation (SBR-9514733 and SBR-9700729) for M.B. E.G. is supported by a National Institutes of Health Minority Supplement Grant (AI 43279). Because of space constraints, we regret our inability to refer to the other excellent papers that have also examined the HIV disease-modifying effects of CCR5 polymorphisms.
Abbreviations
- CCR
CC chemokine receptor
- WHMC
Wilford Hall Medical Center
- HH
human haplogroups
- CI
confidence interval limits
- RH
relative hazard
- KM
Kaplan–Meier method
- SNP
single nucleotide polymorphism
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hill A V. Annu Rev Immunol. 1998;16:593–617. doi: 10.1146/annurev.immunol.16.1.593. [DOI] [PubMed] [Google Scholar]
- 2.Gao F, Bailes E, Robertson D L, Chen Y, Rodenburg C M, Michael S F, Cummins L B, Arthur L O, Peeters M, Shaw G M, et al. Nature (London) 1999;397:436–441. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- 3.Moore J P, Trkola A, Dragic T. Curr Opin Immunol. 1997;9:551–562. doi: 10.1016/s0952-7915(97)80110-0. [DOI] [PubMed] [Google Scholar]
- 4.Cohen O J, Kinter A, Fauci A S. Immunol Rev. 1997;159:31–48. doi: 10.1111/j.1600-065x.1997.tb01005.x. [DOI] [PubMed] [Google Scholar]
- 5.Dean M, Carrington M, Winkler C, Huttley G A, Smith M W, Allikmets R, Goedert J J, Buchbinder S P, Vittinghoff E, Gomperts E, et al. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 6.Huang Y, Paxton W A, Wolinsky S M, Neumann A U, Zhang L, He T, Kang S, Ceradini D, Jin Z, Yazdanbakhsh K, et al. Nat Med. 1996;2:1240–1243. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- 7.Michael N L, Chang G, Louie L G, Mascola J R, Dondero D, Birx D L, Sheppard H W. Nat Med. 1997;3:338–340. doi: 10.1038/nm0397-338. [DOI] [PubMed] [Google Scholar]
- 8.Smith M W, Dean M, Carrington M, Winkler C, Huttley G A, Lomb D A, Goedert J J, O’Brien T R, Jacobson L P, Kaslow R, et al. Science. 1997;277:959–965. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- 9.Zimmerman P A, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, et al. Mol Med. 1997;3:23–36. [PMC free article] [PubMed] [Google Scholar]
- 10.van Rij R P, de Roda Husman A M, Brouwer M, Goudsmit J, Coutinho R A, Schuitemaker H. J Infect Dis. 1998;178:1806–1811. doi: 10.1086/314522. [DOI] [PubMed] [Google Scholar]
- 11.Kostrikis L G, Huang Y, Moore J P, Wolinsky S M, Zhang L, Guo Y, Deutsch L, Phair J, Neumann A U, Ho D D. Nat Med. 1998;4:350–353. doi: 10.1038/nm0398-350. [DOI] [PubMed] [Google Scholar]
- 12.Rizzardi G P, Morawetz R A, Vicenzi E, Ghezzi S, Poli G, Lazzarin A, Pantaleo G. Nat Med. 1998;4:252–253. doi: 10.1038/nm0398-252. [DOI] [PubMed] [Google Scholar]
- 13.Martin M P, Dean M, Smith M W, Winkler C, Gerrard B, Michael N L, Lee B, Doms R W, Margolick J, Buchbinder S, et al. Science. 1998;282:1907–1911. doi: 10.1126/science.282.5395.1907. [DOI] [PubMed] [Google Scholar]
- 14.McDermott D H, Zimmerman P A, Guignard F, Kleeberger C A, Leitman S F, Murphy P M. Lancet. 1998;352:866–870. doi: 10.1016/s0140-6736(98)04158-0. [DOI] [PubMed] [Google Scholar]
- 15.Centers for Disease Control and Prevention. HIV/AIDS Surveillance Report. 1998;10:1–40. [Google Scholar]
- 16.Mummidi S, Ahuja S S, Gonzalez E, Anderson S A, Santiago E N, Stephan K T, Craig F E, O’Connell P, Tryon V, Clark R A, et al. Nat Med. 1998;4:786–793. doi: 10.1038/nm0798-786. [DOI] [PubMed] [Google Scholar]
- 17.Mummidi S, Ahuja S B, McDaniel B L, Ahuja S K. J Biol Chem. 1997;272:30662–30671. doi: 10.1074/jbc.272.49.30662. [DOI] [PubMed] [Google Scholar]
- 18.Garred P. Lancet. 1998;351:2–3. doi: 10.1016/S0140-6736(98)22001-0. [DOI] [PubMed] [Google Scholar]
- 19.Kowo M P, Goubau P, Ndam E C, Njoya O, Sasaki S, Seghers V, Kesteloot H. Trans R Soc Trop Med Hyg. 1995;89:484–486. doi: 10.1016/0035-9203(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 20.Ndumbe P M, Atchou G, Biwole M, Lobe V, Ayuk-Takem J. Med Microbiol Immunol. 1993;182:281–284. doi: 10.1007/BF00191943. [DOI] [PubMed] [Google Scholar]
- 21.Brun-Vezinet F, Jaeger G, Rouzioux C, Rey M A, Dazza M C, Chamaret S, Montagnier L, Charmot G. Lancet. 1986;1:854. doi: 10.1016/s0140-6736(86)90960-8. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez J P, Georges-Courbot M C, Martin P M, Mathiot C C, Salaun D, Georges A J. Lancet. 1987;1:1499. doi: 10.1016/s0140-6736(87)92257-4. [DOI] [PubMed] [Google Scholar]
- 23.Koralnik I J, Boeri E, Saxinger W C, Monico A L, Fullen J, Gessain A, Guo H G, Gallo R C, Markham P, Kalyanaraman V, et al. J Virol. 1994;68:2693–2707. doi: 10.1128/jvi.68.4.2693-2707.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saksena N K, Herve V, Durand J P, Leguenno B, Diop O M, Digouette J P, Mathiot C, Muller M C, Love J L, Dube S, et al. Virology. 1994;198:297–310. doi: 10.1006/viro.1994.1033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}