

Abstract
The retinoblastoma tumor suppressor protein (pRB) plays a critical role in the control of cell proliferation and in the DNA damage checkpoints. pRB inhibits cell cycle progression through interactions with the E2F family of transcription factors. Here, we report that DNA damage induced not only the dephosphorylation of pRB at Cdk phosphorylation sites and the binding of pRB to E2F-1, but also the phosphorylation of pRB at Ser612. Phosphorylation of pRB at Ser612 enhanced the formation of a complex between pRB and E2F-1. Substitution of Ser612 with Ala decreased pRB–E2F-1 binding and the transcriptional repression activity. Until now, Ser612 of pRB has been thought to be phosphorylated by Cdk2. However, the phosphorylation of pRB at Ser612 was conducted by Chk1/2 after DNA damage, and inhibition of ATM-Chk1/2 activity suppressed the phosphorylation of Ser612 and the binding of pRB to E2F-1. These results suggest that Ser612 is phosphorylated by Chk1/2 after DNA damage, leading to the formation of pRB–E2F-1. This is the first report that pRB is phosphorylated in vivo by a kinase other than Cdk.
Keywords: Cdk, Chk1, Chk2, E2F, RB
Introduction
The DNA damage signaling pathway is a highly conserved response to genotoxic stress (Zhou and Elledge, 2000). In mammalian cells, the pathway functions to protect cells from agents that induce cellular death or transformation, participating in DNA repair and checkpoint control leading to survival or apoptosis. Ataxia-telangiectasia mutated (ATM) kinase and related kinases, which become activated after DNA damage, transduce signals to downstream targets, including p53 and the checkpoint kinases Chk1 and Chk2 (Banin et al, 1998; Canman et al, 1998; Abraham, 2001; Shiloh, 2001). In turn, checkpoint kinases phosphorylate key substrates, such as p53, E2F-1, Cdc25A, and Cdc25C, to facilitate the DNA damage response (Sanchez et al, 1997; Hirao et al, 2000; Shieh et al, 2000; Falck et al, 2001a; Stevens et al, 2003).
The retinoblastoma tumor suppressor protein (pRB) is a negative regulator of cell proliferation (Classon and Harlow, 2002). The antiproliferative activity of pRB is mediated by its ability to inhibit the transcription of genes that are required for cell cycle progression. This transcriptional regulatory function of pRB is achieved through several distinct mechanisms, which are best illustrated by its interaction with the E2F family and the inhibition of E2F-regulated gene expression. The binding of E2F to pRB requires the large pocket of pRB (amino acids 379–870).
The ability of pRB to inhibit cellular proliferation is counterbalanced by the action of Cdks (Taya, 1997; Sherr and Roberts, 1999). pRB is phosphorylated in a cell cycle-dependent manner by Cyclin-dependent kinases (Cdks). In quiescent and early G1 cells, pRB exists in a predominantly unphosphorylated state. As cells progress toward S phase, pRB becomes phosphorylated. The initial phosphorylation of pRB is most likely catalyzed by Cdk4–Cyclin D or Cdk6–Cyclin D complexes. Subsequently, Cdk2–Cyclin E and Cdk2–Cyclin A phosphorylate pRB (Ortega et al, 2002; Sherr and McCormick, 2002). During mitosis, pRB is rapidly dephosphorylated (Ludlow et al, 1993). Sixteen potential sites for Cdk-mediated phosphorylation (Ser/Thr-Pro motifs) exist in pRB, and twelve of these sites have been shown to be phosphorylated in vivo (Knudsen and Wang, 1996; Zarkowska and Mittnacht, 1997). Inactivation of pRB by phosphorylation leads to the dissociation and activation of E2F, allowing the expression of many genes required for cell cycle progression and S phase entry. We have previously shown that Cdk4–Cyclin D1, but not Cdk2–Cyclin E, specifically phosphorylated Ser780 in pRB and that pRB phosphorylated at Ser780 cannot bind to E2F-1 (Kitagawa et al, 1996).
Growth arrest induced by DNA damage in mammalian cells requires the function of pRB (Harrington et al, 1998; Brugarolas et al, 1999). It has been shown that pRB plays a role in the activation of p53-dependent (Slebos et al, 1994; Smith et al, 1994) and -independent (Dou et al, 1995) checkpoints. pRB-deficient cells are hypersensitive to DNA damage-induced apoptosis (Almasan et al, 1995; Knudsen et al, 2000). On the other hand, E2F-1 has a role distinct from other E2Fs in the regulation of apoptosis (DeGregori et al, 1997). Following DNA damage, E2F-1 is stabilized by ATM and Chk2 phosphorylation (Lin et al, 2001; Stevens et al, 2003). E2F-1 has been found to induce the expression of many apoptotic genes (Attwooll et al, 2004).
In this report, we have explored the possibility that pRB is regulated by the DNA damage signaling pathway, and report a new pathway through which ATM-Chk1/2 kinase signals to pRB and promotes the formation of a complex between pRB and E2F-1.
Results
DNA damage induces the phosphorylation of pRB at Ser612
To test whether DNA damage affects the phosphorylation of several sites on pRB, MCF7 cells that express wild-type pRB were treated with ionizing radiation (IR) and immunoprecipitates obtained with anti-pRB antibody from cell lysates were immunoblotted with phosphospecific pRB antibodies generated by us (Kitagawa et al, 1996; Adams et al, 1999; Brugarolas et al, 1999; Watanabe et al, 1999; Taya et al, 2003). After IR, the dephosphorylation of Thr356 and Ser780 was detected and E2F-1 was co-precipitated with pRB (Figure 1A). Other Cdk phosphorylation sites (e.g. Ser608, Thr821) were also dephosphorylated after IR (data not shown). Until now, Ser612 of pRB has been thought to be phosphorylated by Cdk. Interestingly, DNA damage enhanced the phosphorylation rather than dephosphorylation of pRB at Ser612. The phosphorylation of Ser612 occurs in response to 10 Gy of IR, non-apoptotic level of IR in MCF7 cells (Supplementary Figure S1A).
Figure 1.
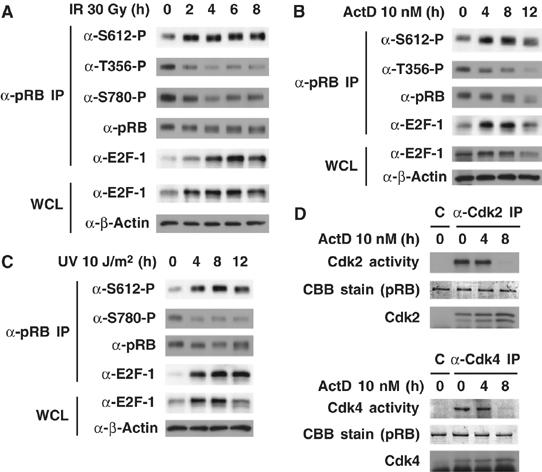
DNA damage induces the phosphorylation of pRB at Ser612. (A–C) Ser612 but not other Cdk phosphorylation sites is phosphorylated after DNA damage. MCF7 cells were exposed to 30 Gy of γ irradiation (IR) (A), 10 nM actinomycin D (ActD) (B), or 10 J/m2 of UV radiation (UV) (C) for the indicated periods. Cell lysates were immunoprecipitated with anti-pRB antibodies and Western blotting was performed. (D) Analysis of Cdk2 and Cdk4 kinase activities after DNA damage. MCF7 cells were exposed to 10 nM ActD for the indicated periods. Cell lysates were immunoprecipitated with anti-Cdk2 or anti-Cdk4 antibody, and an in vitro kinase assay was performed by using recombinant pRB as a substrate. The reaction products were analyzed by immunoblotting using anti-phospho-Thr821 (for Cdk2 activity) or anti-phospho-Ser608 (for Cdk4 activity) antibodies.
Next, we treated MCF7 cells with other DNA-damaging agents, actinomycin D (ActD) and ultraviolet radiation (UV), and investigated the phosphorylation of several sites on pRB. Both ActD and UV could induce the phosphorylation of Ser612 and dephosphorylation of other Cdk phosphorylation sites, similar to the effect of IR (Figure 1B and C). The binding of pRB to E2F-1 was enhanced after both treatments (Figure 1A–C). Similar results were also observed in HepG2 and HCT116 cells (data not shown). To investigate whether Cdks are involved in the phosphorylation of Ser612 after DNA damage, we carried out an in vitro kinase assay. Cdk2 and Cdk4 proteins were immunoprecipitated from ActD-treated MCF7 cells and their kinase activities toward pRB were analyzed (Figure 1D). Cdk2 and Cdk4 activities were sustained at 4 h and both activities were lost at 8 h after ActD treatment. However, the phosphorylation of Ser612 was enhanced at 8 h after the treatment (Figure 1B). To rule out the possibility that Cdk2 is responsible for the phosphorylation of pRB at Ser612 after DNA damage, we tested the effect of the Cdk inhibitor (GW8510) on the Ser612 phosphorylation level after DNA damage. The pretreatment of GW8510 did not affect Ser612 phosphorylation after IR (Supplementary Figure S1B). Also, it has been reported that PP1 and PP2A are candidates for pRB phosphatases (Alberts et al, 1993; Ludlow et al, 1993). We tested the effect of okadaic acid (OA), a PP1/PP2A phosphatase inhibitor, on the Ser612 phosphorylation level after DNA damage. The pretreatment of OA did not affect Ser612 phosphorylation after IR (Supplementary Figure S1C). These results showed that all of the agents, which can induce DNA damage, are probably capable of inducing phosphorylation of pRB at Ser612, and that Cdk2 and Cdk4 are unlikely to be involved in the phosphorylation of Ser612 after DNA damage.
Phosphorylation of pRB at Ser612 promotes the formation of a complex between pRB and E2F-1
We next investigated whether the site-specific phosphorylation or dephosphorylation of pRB affected the formation of a complex between pRB and E2F-1. For this purpose, whole-cell lysate prepared from MCF7 cells after treatment with adriamycin (ADR) was subjected to two rounds of immunoprecipitation (IP) with anti-E2F-1 antibody. E2F-1 was almost completely depleted in the whole-cell lysate. Subsequently, the residual cell lysate was subjected to IP with anti-pRB antibody and the pRB unbound to E2F-1 (free pRB) was collected. Immunoprecipitates were normalized for the amount of pRB, and equivalent amounts of E2F-1-bound and unbound pRB were analyzed by Western blotting with various phospho-specific antibodies for pRB. The treatment with ADR for 24 h led to dephosphorylation of pRB at Thr356 and Thr821 but not at Ser612 (Supplementary Figure S1D). However, residual phosphorylated pRB was detected by Western blotting when lyates prepared from cells after treatment with ADR were immunoprecipitated. As shown in Figure 2A, Thr356-, Ser612- and Thr821-phosphorylated pRB were present in E2F-1-bound pRB but not in free pRB. In contrast, Ser608-, Ser780-, Ser807- and Ser811-phosphorylated pRB were present in free pRB but not in E2F-1-bound pRB.
Figure 2.
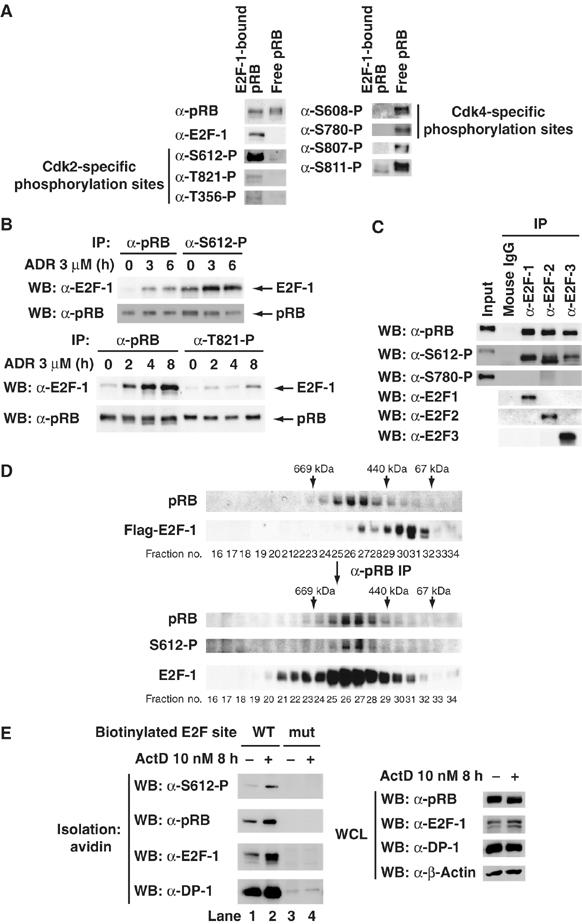
pRB phosphorylated at Ser612 prefers to bind to E2F-1. (A) Analysis of phosphorylation profiles in E2F-1-bound pRB and E2F-1 unbound pRB after DNA damage. MCF7 cells were treated with 3 μM adriamycin (ADR) for 24 h. Cell lysates were subjected to two rounds of immunoprecipitation with anti-E2F-1 antibody (E2F-1-bound pRB). Next, the residual supernatants were subjected to immunoprecipitation with anti-pRB antibody (free pRB). Immunoprecipitates normalized for the amount of precipitated pRB were subjected to a Western blot analysis with the indicated antibodies. (B) E2F-1 is effectively co-precipitated with Ser612- but not Thr821-phosphorylated pRB. MCF7 cells were treated with 3 μM ADR for the indicated periods. Cell lysates were subjected to immunoprecipitation with the indicated antibodies. Immunoprecipitates normalized for the amount of precipitated pRB were subjected to a Western blot analysis with the indicated antibodies. (C) E2F-2 and E2F-3 effectively bind to Ser612-phosphorylated pRB as well as E2F-1 does. Lysates from MOLT-4 cells were subjecied to immunoprecipitation with the indicated antibodies. Immunoprecipitates normalized for the amount of precipitated pRB were subjected to a Western blot analysis with the indicated antibodies. (D) Ser612-phosphorylated pRB forms a complex with E2F-1 in vivo. MCF7 cells were transiently transfected with Flag–E2F-1. Cells were treated with 3 μM ADR for 24 h. Cell lysate was immunoprecipitated with anti-Flag antibody-conjugated beads. Flag–E2F-1 complex was eluted from the beads with Flag peptide and further purified by gel filtration on a column of superose 6. Upper panels are an immunoblot analysis of gel filtration column fractions using antibodies. pRB in the E2F-1 complex was concentrated using anti-pRB antibody to immunoprecipitate pRB from fractions of a superose 6 gel filtration column, and Ser612-phosphorylated pRB in the E2F-1 complex was detected (lower panels). (E) Ser612-phosphorylated pRB can be recruited to E2F binding sites through its interaction with the E2F-1/DP-1 heterodimer. MCF7 cells were exposed to 10 nM ActD for 8 h, and then cell extracts were subjected to a pull-down analysis using biotinylated oligonucleotides harboring either a wild-type E2F site or a mutated site. Bound proteins were then analyzed using the indicated antibodies.
The Ser608, Ser780 and Ser795 residues on pRB are preferentially phosphorylated by Cdk4–Cyclin D, whereas Thr356, Ser612 and Thr821 are preferentially phosphorylated by Cdk2–CyclinE or Cdk2–Cyclin A (our unpublished data). Because DNA damage induced the dephosphorylation of pRB at Thr356 and Thr821 but not at Ser612, phosphorylation of Ser612 may affect the formation of a complex between pRB and E2F-1 distinct from the Cdk phosphorylation sites of pRB. We performed IP with phospho-specific antibodies for pRB and normalized co-precipitated E2F-1 for the amount of pRB (Figure 2B). E2F-1 was effectively co-precipitated with Ser612-phosphorylated pRB compared with pRB precipitated by ordinary pRB antibody. However, E2F-1 was ineffectively co-precipitated with Thr821-phosphorylated pRB. We also confirmed that anti-Ser612-phosphorylated pRB antibody could not directly immunoprecipitate and recognize E2F-1 (data not shown). These results suggest that phosphorylation of pRB at Ser612 promotes the binding of pRB to E2F-1.
pRB has been shown to interact with four members of the E2F family (E2F-1, E2F-2, E2F-3 and E2F-4) in vivo (Attwooll et al, 2004). Therefore, we investigated whether other family members were effectively co-precipitated with Ser612-phosphorylated pRB. Immunoprecipitates from lysates of proliferating MOLT-4 cells obtained with anti-E2F family antibodies were normalized for the amount of pRB and analyzed by Western blotting (Figure 2C). Co-precipitated pRB with anti-E2F family antibodies were normalized to the input. Interestingly, Ser612-phosphorylated pRB was effectively co-precipitated with E2F-1, E2F-2, and E2F-3. Ser780-phosphorylated pRB was not co-precipitated, consistent with previous reports (Kitagawa et al, 1996). This result suggests that E2F-2 and E2F-3 also effectively bind to Ser612-phosphorylated pRB.
To confirm that Ser612-phosphorylated pRB forms a complex with E2F-1 in vivo, we purified the E2F-1 complex from ADR-treated MCF7 cell lysates by superose 6 gel filtration column chromatography. As shown in Figure 2D, pRB in the E2F-1 complex was detected in fractions 24–29 (upper panels). pRB in the E2F-1 complex was concentrated using anti-pRB antibody to immunoprecipitate pRB from fractions of a superose 6 gel filtration column. As expected, Ser612-phosphorylated pRB in the E2F-1 complex was detected similar to pRB (lower panels). Thus, our data suggest that pRB phosphorylated at Ser612 prefers to bind E2F-1.
We also investigated whether Ser612-phosphorylated pRB could be recruited to E2F-binding sites through its interaction with the E2F-1/DP-1 heterodimer. We incubated ActD-treated MCF7 cell lysates with biotinylated oligonucleotides harboring either a wild-type or mutated E2F site (Nicolas et al, 2003). Proteins bound to these oligonucleotides were then collected by use of streptavidin beads and analyzed by Western blotting (Figure 2E). Ser612-phosphorylated pRB was able to bind to the wild-type oligonucleotide but not to the mutated one (compare lanes 1 and 3). This binding was enhanced after DNA damage (compare lanes 1 and 2). Taken together, these results suggest that Ser612-phosphorylated pRB can be recruited to E2F sites through interaction with E2F-1/DP-1 in response to DNA damage.
Substitution of Ser612 with Ala decreases pRB–E2F-1 binding and the transcriptional repression
To test the effect of the phosphorylation of Ser612 on the binding of pRB to E2F-1, we constructed pRB variants in which Ser612 was changed to alanine (S612A). RB-deficient SAOS2 cells were transfected with Flag–E2F-1 and myc-RB, and immunoprecipitates obtained with anti-Flag antibody from cell lysates were immunoblotted with anti-myc antibody. As expected, the S612A mutation decreased the binding of myc-pRB to Flag–E2F-1 (Figure 3A; compare lanes 2 and 3). Mutations at Ser780 and Ser608, Ser780, and Ser795 slightly enhanced the binding of myc-pRB to Flag–E2F-1 (lanes 4 and 5). Although a mutation to aspartic acid can sometimes mimic the effect of phosphorylation, we did not observe increased binding of pRB–E2F-1 on such a mutation (data not shown). This was not entirely unexpected as aspartic acid may not always mimic phosphorylation.
Figure 3.
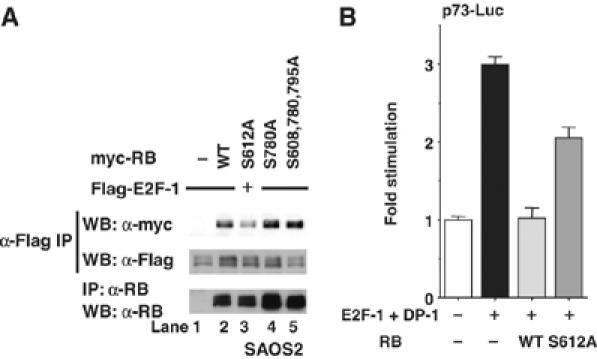
Substitution of Ser612 with Ala decreases the binding of pRB to E2F-1 and the transcriptional repression. (A) Substitution of Ser612 with Ala decreases pRB–E2F-1 binding. SAOS2 cells were transiently transfected with the indicated constructs. After 40 h, cell lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with anti-myc antibody (top) or anti-Flag antibody (middle). The expression level of pRB was assessed by the immunoblotting of immunoprecipitates with anti-pRB antibody (bottom). (B) Transcriptional repressive activity of pRB S612A is lower than that of wild-type pRB. MCF7 cells were co-transfected with the p73-Luc reporter gene, E2F-1, DP-1, and pGL4.74[hRluc/TK] in the absence or presence of pRB. After 24 h, the luciferase activity in cell lysates was measured and was normalized with the Renilla luciferase activity. The change in the mean±s.d. based on three cultures is shown.
During cell cycle progression, E2F activates genes such as Cyclin E, Dhfr, and Cdc6. In conditions that activate apoptosis, such as DNA damage, certain E2F subunits like E2F-1 are stabilized to induce apoptosis, sometimes through direct effects on target genes like p73 and Apaf1 (Attwooll et al, 2004). The pRB–E2F complex forms on the promoters of a multitude of E2F target genes to repress transcription. We next examined the ability of wild-type pRB or the S612A mutant to regulate E2F-1's transcriptional activity toward the p73 promoter. A reporter assay was performed using MCF7 cells transfected with E2F-1 and DP-1, along with a construct containing the luciferase gene under the control of the p73 promoter (Figure 3B). The p73 promoter activity was increased by E2F-1 and DP-1, but cotransfection with RB completely suppressed this activation. The transcriptional repression by pRB S612A was weaker than that by the wild-type pRB toward the activation of the p73 promoter induced by E2F-1 and DP-1, and this result was consistent with the finding that the mutation S612A decreased the binding of pRB to E2F-1. Collectively, phosphorylation of pRB at Ser612 promotes this binding (Figure 2) and inhibition of Ser612 phosphorylation decreases it (Figure 3).
Loss of Ser612 phosphorylation reduces antiapoptotic activity of pRB
Re-expression of pRB in pRB-deficient human osteosarcoma cell line SAOS2 has become a major model experimental system to investigate functions of pRB. Therefore, we utilized SAOS2-derived cell line SRB1, in which wild-type pRB expression is controlled by a tetracycline-repressable promoter (Ookawa et al, 1997, 2001; Uchida et al, 2005). We also established SRBSA1, in which pRB S612A expression is controlled by a tetracycline-repressable promoter. We first confirmed that the expression of wild-type pRB and pRB S612A was induced with similar levels after the withdrawal of tetracycline (Supplementary Figure S2A). Consistent with previous result, the mutation of S612A reduced the binding of pRB to E2F-1 at 24 h post pRB induction (Figure 4A). We next determined the effects of both isoforms of pRB on the expression levels of Cyclin A and E2F-1. These proteins are known to be repressed by pRB in SAOS2 cells (Jiang et al, 2000; Ookawa et al, 2001). As shown in Figure 4B, the expression levels of Cyclin A and E2F-1 were significantly repressed by wild-type pRB at 48 h post-induction, whereas S612A mutant had less activity to repress these target genes. On the other hand, the expression levels of E2F-4, Cdk4 and β-actin did not change in both cell lines. Thus, loss of Ser612 phosphorylation reduces the E2F repression activity of pRB.
Figure 4.
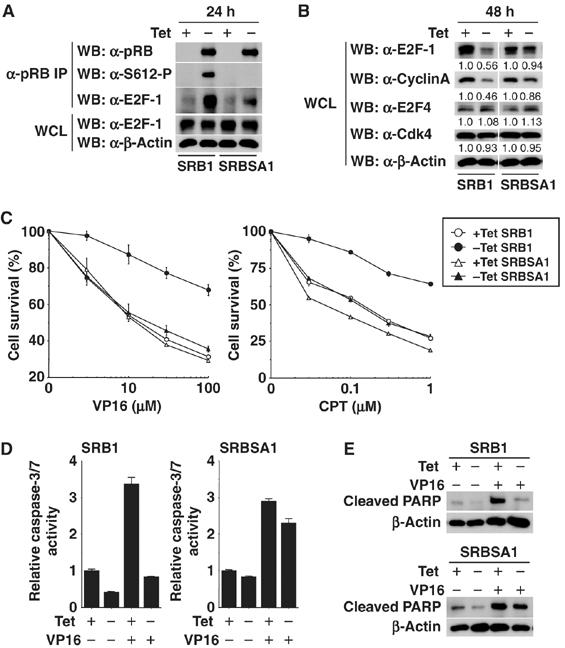
Loss of Ser612 phosphorylation reduces antiapoptotic activity of pRB. (A) Wild-type pRB (SRB1) and pRB S612A (SRBSA1) were induced by tetracycline withdrawal for 24 h in a stable, pRB-inducible SAOS2 cell line. Cell lysates were immunoprecipitated with anti-pRB antibody and Western blotting was performed. (B) pRB was induced by tetracycline withdrawal for 48 h and E2F target genes (E2F-1 and Cyclin A) were examined by Western blot analysis. (C) SRB1 cells and SRBSA1 cells were cultured for 24 h in 10% FBS–DMEM with or without 1 μg/ml of tetracycline. Cells were treated with VP16 or CPT for 48 h at the indicated doses. The percentage of survival cells was measured by crystal violet staining. (D) Caspase-3/7 activity in SRB1 cells or SRBSA1 cells treated with 100 μM of VP16 for 48 h was measured. pRB expression was induced as in (C). (E) PARP cleavage in SRB1 cells or SRBSA1 cells treated with 100 μM of VP16 for 48 h was examined by Western blot analysis. pRB expression was induced as in (C).
pRB plays a central role in DNA damage-induced cell cycle checkpoints and is required for induction of G1 and S phase arrest following DNA-damaging events (Harrington et al, 1998; Brugarolas et al, 1999). pRB-deficient cells are hypersensitive to DNA damage-induced apoptosis (Almasan et al, 1995; Knudsen et al, 2000). To evaluate the role of Ser612 phosphorylation in response to DNA damage, we compared the ability of wild-type pRB and pRB S612A to rescue the survival of SAOS2 cells in response to DNA damage. Wild-type pRB efficiently blocked VP16- and camptothecin (CPT)-induced cell death in SAOS2 cells (Figure 4C and Supplementary Figure S2B). In contrast, cells expressing pRB S612A remained hypersensitive to DNA damage. Caspase-3/7 activity and PARP cleavage were inhibited by re-expression of wild-type pRB but not pRB S612A (Figure 4D and E). These results suggest that Ser612 phosphorylation of pRB is essential for cell survival and antiapoptotic activity in response to DNA damage.
ATM is required for stimulating phosphorylation of pRB at Ser612
The primary mobilizer of the response to DNA damage is the nuclear protein kinase ATM (Abraham, 2001; Shiloh, 2001). Therefore, we tested whether ATM plays a role in regulating the phosphorylation of pRB at Ser612. MCF7 cells were treated with caffeine to inhibit ATM/ATR. As shown in Figure 5A, treatment with caffeine inhibited the phosphorylation of Ser612 induced by IR (lane 3). However, this treatment did not inhibit the dephosphorylation of pRB at Ser780. In a time course experiment, treatment with caffeine blocked the induction of phosphorylation at Ser612 and binding of pRB–E2F-1 after IR (Supplementary Figure S3A). Furthermore, pretreatment of cells with KU-55933, a specific ATM inhibitor (Hickson et al, 2004), blocked Ser612 phosphorylation of pRB induced by IR (Figure 5A; compare lanes 6 and 8). A prompt response to genotoxic stress has been reported to induce stabilization of E2F-1 through its phosphorylation by ATM and ATR (Lin et al, 2001). E2F-1 levels were increased after IR and caffeine, or KU-55933 also blocked the stabilization of E2F-1. Therefore, we speculate that the increased binding of pRB to E2F-1 is not only derived from the phosphorylation of pRB at Ser612, but also derived from the stabilization of E2F-1 in response to DNA damage.
Figure 5.
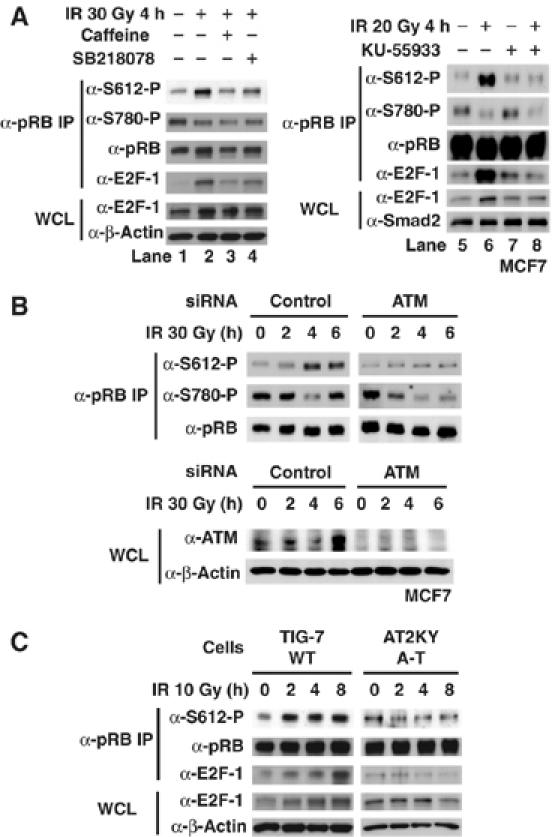
ATM is required for stimulating the phosphorylation of pRB at Ser612. (A) Effect of the ATM/ATR inhibitor and Chk1 inhibitor on the phosphorylation of pRB at Ser612 after DNA damage. MCF7 cells were pretreated with caffeine (5 mM), SB218078 (1 μM), or KU-55933 (10 μM) for 1 h and subsequently exposed to IR for 4 h. Cell lysates were immunoprecipitated with anti-pRB antibody and a Western blot analysis was performed. (B) The reduction of ATM by siRNA completely abolished the phosphorylation at Ser612, but had no effect on the dephosphorylation of pRB at Ser780. MCF7 cells were transfected with control siRNA or ATM siRNA for 48 h, and exposed to 30 Gy of IR for the indicated periods. Cell lysates were immunoprecipitated with anti-pRB antibody and a Western blot analysis was performed. (C) IR-induced Ser612 phosphorylation of pRB is abolished in ATM-null cells. Cells were exposed to 10 Gy of IR for the indicated periods. Cell lysates were immunoprecipitated with anti-pRB antibody and Western blotting was performed.
To further test the role of ATM, its expression in MCF7 cells was suppressed by siRNA. This reduction in the expression of ATM completely abolished the phosphorylation at Ser612, but had no effect on the dephosphorylation of pRB at Ser780 (Figure 5B). Furthermore, the phosphorylation of pRB at Ser612 and pRB–E2F-1 complex formation were also seen in human diploid fibroblasts TIG-7 cells but not in A-T fibroblasts AT2KY cells subjected to IR treatment (Figure 5C), even though AT2KY cells were irradiated at high dose (Supplementary Figure S3B). Although ATM is primarily activated by DNA double-strand breaks (DSBs) caused by IR or radiomimeric drugs, ATR responds to replicative stress and UV irradiation. The phosphorylation of pRB at Ser612 and pRB–E2F-1 complex formation were seen after UV treatment in AT2KY cells (Supplementary Figure S3C). Together, these results suggest that ATM plays an important role in the phosphorylation of pRB at Ser612 and the binding of pRB to E2F-1 after DNA DSBs.
Chk1 and Chk2 phosphorylate Ser612 of pRB
ATM phosphorylates its substrates at serine or threonine residues, followed by glutamine residues (SQ/TQ motif) (Abraham, 2001; Shiloh, 2001). Because Ser612 of pRB is followed by a proline residue, we speculated that Ser612 is not directly phosphorylated by ATM. Activation of ATM by DNA damage leads to the phosphorylation and activation of downstream kinases Chk1, and Chk2. Chk2 is activated after DNA damage and is thought to function as a signal amplifier for ATM. To investigate whether Chk2 can phosphorylate pRB, we performed an in vitro kinase assay. An in vitro kinase assay was established to monitor the phosphorylation of recombinant pRB by purified recombinant Chk2. As a positive control, GST-BRCA1 was used as substrate of Chk2 (Lee et al, 2000). Recombinant Chk2 was able to phosphorylate pRB and GST-BRCA1 when [γ-32P]ATP was added to the kinase reaction (Figure 6A and Supplementary Figure S4A). Approximately 1.5 moles of phosphate per mole of protein were incorporated into the stoichiometric kinase reaction (Supplementary Figure S4B). Chk2 is a Ser/Thr-specific kinase for which a small number of physiological substrates have been identified (Sanchez et al, 1997; Hirao et al, 2000; Lee et al, 2000; Shieh et al, 2000; Falck et al, 2001a; Stevens et al, 2003). A consensus sequence motif for phosphorylation has been identified (Figure 6B) (O'Neill et al, 2002). The amino-acid sequence surrounding Ser612 of pRB closely resembles this consensus phosphorylaton site, and pRB's site is found to be more similar to the site in BRCA1 than that in Cdc25A, Cdc25C or E2F-1 (Figure 6B). Therefore, we investigated using a different method whether Chk2 can phosphorylate pRB at Ser612 in vitro. As a positive control, purified recombinant Cdk2-Cyclin A or p38α was used. Thr356, Ser612 and Thr821 residues on pRB are preferentially phosphorylated by Cdk2-Cyclin A in vitro (our unpublished data). Also, p38 phosphorylates its substrates at serine or threonine residues, followed by proline residues (SP/TP motif) in vitro. Following in vitro phosphorylation, pRB was probed with phospho-specific antibodies against the sites Thr356-P, Thr373-P, Ser608-P, Ser612-P, Ser780-P, Ser788-P, Ser795-P, Ser807-P, Ser811-P, Thr821-P, and Thr826-P (Figure 6C and Supplementary Figure S5A). As mentioned above, all sites of pRB were phosphorylated by p38α. Chk2 was able to phosphorylate pRB at Ser612 as efficiently as Cdk2–Cyclin A or p38α. However, Chk2 could hardly phosphorylate other phosphorylation sites of pRB. This result suggests that Chk2 is able to selectively phosphorylate Ser612 of pRB in vitro.
Figure 6.
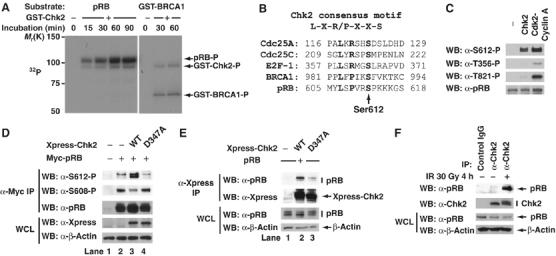
Chk1 and Chk2 phosphorylate Ser612 of pRB. (A) In vitro phosphorylation of pRB by Chk2. Recombinant pRB or GST-BRCA1 were incubated with Chk2 in the presence of [γ-32P]ATP. The reaction products were resolved by SDS–PAGE and detected by autoradiography. (B) Comparison of the Chk2 phosphorylation site in Cdc25A, Cdc25C, E2F-1, and BRCA1 with that in pRB. Residues in bold fit the consensus phosphorylation motif. (C) In vitro phosphorylation of Ser612 by Chk2. Recombinant pRB was incubated with Chk2 or Cdk2–Cyclin A in the presence of ATP. The reaction products were analyzed by immunoblotting using the indicated antibodies. (D) Coexpression of wild-type Chk2 and pRB in MCF7 cells stimulated phosphorylation of pRB at Ser612. MCF7 cells were transiently transfected with the indicated constructs. After 40 h, cell lysates were immunoprecipitated with anti-myc antibody and immunoblotted with the indicated antibodies. (E) Chk2 binds to pRB in vivo. MCF7 cells were transiently transfected with the indicated constructs. After 40 h, cell lysates were immunoprecipitated with anti-Xpress antibody and immunoblotted with the indicated antibodies. (F) Endogenous pRB and Chk2 interact in MCF7 cells after DNA damage. MCF7 cells were exposed to 30 Gy of IR for 4 h. Cell lysates were immunoprecipitated with normal rabbit IgG or anti-Chk2 antibody and a Western blot analysis was performed.
Chk1 and Chk2 have a similar substrate specificity but distinct functions (Ahn et al, 2004). Therefore, we next tested whether Chk1 can phosphorylate Ser612 of pRB in vitro. Chk1 phosphorylated GST-E2F-1 at Ser364 and GST-BRCA1 at Ser988 as efficiently as Chk2 (Supplementary Figure S5B). Although Chk1 was able to phosphorylate Ser612 of pRB in vitro, its effect was weaker than that of Chk2.
Coexpression of wild-type Chk2 and pRB in MCF7 cells stimulated the phosphorylation of pRB at Ser612 (Figure 6D, lane 3). However, phosphorylation at Ser608 was decreased by the expression of wild-type Chk2. Chk2 D347A, a catalytically inactive mutant (Shieh et al, 2000), had little effect on the phosphorylation of pRB at Ser608 and Ser612. These results suggest that Chk2 and Chk1 contribute to the phosphorylation of pRB at Ser612 after DNA damage.
Chk2 interacts with pRB in vitro and in vivo, and Chk2–pRB interaction is enhanced in response to DNA damage
To investigate whether Chk2 interacts with pRB, we performed a GST pull-down assay. Recombinant pRB was able to bind to GST-Chk2 but not to GST (Supplementary Figure S5C). pRB also interacted with GST-Chk1 in vitro (Supplementary Figure S5D). pRB LP (large pocket; amino acids 379–928) interacted sufficiently with Chk2 in vitro (Supplementary Figure S5E). These results suggest that Chk1 and Chk2 directly interact with pRB in vitro.
We next examined by using co-IP whether Chk2 and pRB interact in vivo. myc-tagged pRB and Xpress-tagged wild-type Chk2 interacted in MCF7 cells (Figure 6E, lane 2). Although Chk2 D347A also interacted with pRB, this binding was much weaker than that of wild-type Chk2 (compare lanes 2 and 3). Endogenous pRB and Chk2 hardly interacted in MCF7 cells before DNA damage, but DNA damage enhanced the binding of these two proteins (Figure 6F). Collectively, Chk2 directly interacts with pRB in vitro and in vivo, and Chk2–pRB interaction is enhanced after DNA damage.
Silencing of Chk2 decreases phosphorylation at Ser612 and binding of pRB to E2F-1 after DNA damage
DNA damage leads to phosphorylation and activation of Chk1 and Chk2 (Figure 7A). To demonstrate that Chk2 is responsible for the phosphorylation of Ser612 after DNA damage, we stably knocked down cellular Chk2 using RNA interference in MCF7 cells. The phosphorylation of pRB at Ser612 and pRB–E2F-1 complex formation were seen in cells expressing control short-hairpin RNA (shRNA) after IR. In contrast, Ser612 phosphorylation and E2F-1 binding severely decreased in Chk2-depleted cells irradiated by γ-ray (Figure 7B). Reduction of Chk2 by siRNA also decreased but did not abolish the phosphorylation at Ser612, but had no effect on the dephosphorylation of pRB at Ser780 (Supplementary Figure S6A). The binding of pRB to E2F-1 in response to DNA damage decreased after the depletion of Chk2 by siRNA (Supplementary Figure S6B). These results indicate that Chk2 is a responsible kinase for Ser612 phosphorylation to promote the binding affinity of pRB to E2F-1.
Figure 7.
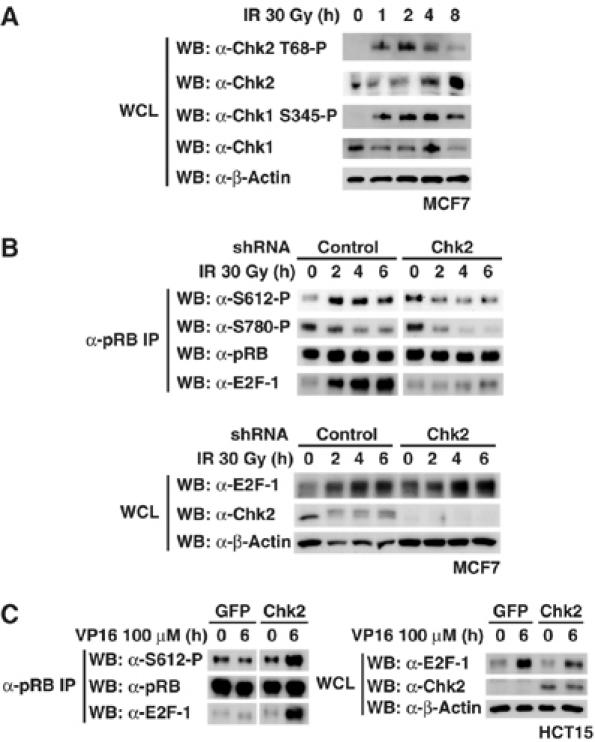
Silencing of Chk2 decreases the phosphorylation at Ser612 and binding of pRB to E2F-1 after DNA damage. (A) Kinetics of Chk1 and Chk2 activation after DNA damage. MCF7 cells were exposed to 30 Gy of IR and cell lysates were immunoblotted as indicated. (B) Stable knockdown of Chk2 diminishes the phosphorylation at Ser612 after DNA damage. The shcontrol-MCF7 and shChk2-MCF7 cells were exposed to 30 Gy of IR for the indicated periods. Cell lysates were immunoprecipitated with anti-pRB antibody and Western blotting was performed. (C) HCT15 cells were nucleofected with the indicated constructs. After 24 h, cells were exposed to 100 μM of VP16 for 6 h. Immunoprecipitation and Western blotting were performed as in (B).
HCT15 colon carcinoma cells carry Chk2 bearing a loss-of-function mutation (R145W) (Lee et al, 2001; Falck et al, 2001b). As shown in Figure 7C, re-expression of wild-type Chk2 in HCT15 cells recovered the phosphorylation of pRB at Ser612 and the binding of pRB to E2F-1 in response to VP16 treatment. In contrast, expression of GFP in HCT15 cells did not recover Ser612 phosphorylation and the pRB–E2F-1 binding in response to VP16 treatment. These experiments, together with previous results, support the idea that Ser612 of pRB is phosphorylated by the ATM-Chk1/2 pathway in response to DNA damage, leading to the formation of the complex pRB–E2F-1.
Discussion
In this paper, we report a new pathway through which ATM-Chk1/2 kinase signals to pRB and promotes the formation of a complex between pRB and E2F-1. DNA damage induces not only the dephosphorylation of pRB at Cdk-phosphorylation sites and the binding of pRB to E2F-1, but also the phosphorylation of pRB at Ser612. The phosphorylation at Ser612 is induced by ATM-Chk1/2 kinase after DNA damage, leading to enhancement of the formation of a complex between pRB and E2F-1. Although it is widely assumed that phosphorylation of pRB converts it into an inactive form, we demonstrated that the phosphorylation of pRB at Ser612 in response to DNA damage represented an activating event.
In addition to its tumor suppressor activity, pRB is a potent antiapoptotic protein. Loss of pRB in normal fibroblasts confers sensitivity to DNA-damaging agents (Almasan et al, 1995; Knudsen et al, 2000), and reintroduction of pRB into pRB-null tumor cells confers resistant to these agents (Haas-Kogan et al, 1995). As shown in Figure 4C–E, SAOS2 expressing wild-type pRB exhibited increased viability and decreased apoptosis following treatment of VP16 or CPT. However, re-expression of pRB S612A did not protect SAOS2 cells from apoptosis. These results suggest that Ser612 phosphorylation of pRB is essential for cell survival and antiapoptotic activity in response to DNA damage.
Based on our results, we propose the following model. In asynchronously growing cells, pRB exists in a predominantly phosphorylated state, which is dissociated from E2F, allowing E2F-dependent transcription. DNA damage induces dephosphorylation of pRB at Cdk phosphorylation sites, leading to the complex pRB–E2F-1 being formed. DNA damage also activates ATM kinase and transduces signals to the checkpoint kinases Chk1 and Chk2. Activated Chk1 and Chk2 phosphorylate Ser612 of pRB and this phosphorylation enhances the formation of a complex between pRB and E2F-1, leading to repression of the transcriptional activity of E2F-1. Consequently, pRB-dependent cell cycle arrest and repression of apoptosis occur.
We showed in this study that Chk1 and Chk2 are directly involved in the phosphorylation of pRB at Ser612 after DNA damage. Because the interaction of Chk2 with pRB is disrupted by Cdk2–Cyclin A-mediated phosphorylation of pRB in vitro (our unpublished data), we speculate that Chk2 cannot bind to pRB and phosphorylate pRB at Ser612 in growing cells. Therefore, we think that Chk2 (probably also Chk1) is specifically involved in the phosphorylation of pRB at Ser612 after DNA damage.
On the other hand, E2F-1 has been shown to be phosphorylated by ATM and Chk2 after DNA damage, resulting in the protein's stabilization (Lin et al, 2001; Stevens et al, 2003). Therefore, ATM and Chk2 help to upregulate the activity of E2F-1. Our results show that ATM and Chk2 promote the repressive activity of pRB. We propose the following model to explain the paradoxical situation whereby E2F-1 as well as pRB are targeted for phosphorylation by Chk2. In the early stages of DNA damage, pRB becomes active and represses E2F-1 activity, which, in turn provides a signal for cell cycle arrest. However, if DNA damage is severe, pRB is cleaved by caspase (Tan et al, 1997) and becomes inactive, leading to activation of E2F-1. Activated E2F-1 induces apoptosis through direct effects on target genes like p73, Arf, Apaf1, and caspases (Attwooll et al, 2004).
The mechanism by which the phosphorylation of pRB at Ser612 promotes the formation of a complex between pRB and E2F-1 after DNA damage remains to be further investigated. Ser612 is located in the spacer region (amino acids 573–645) between the A pocket (amino acids 379–572) and B pocket (amino acids 646–772). According to Lee et al (2002), E2F binds to the conserved groove at the interface. The phosphorylation at Ser612 may induce a conformational change in pRB, leading to promotion of the formation of a complex between pRB and E2F.
In summary, we have shown that DNA damage induces the phosphorylation of pRB at Ser612 through an ATM- and Chk1/2-dependent pathway, resulting in promotion of the formation of a complex between pRB and E2F-1. Repression of E2F-1 activity contributes to cell cycle arrest. This is the first report that pRB is phosphorylated in vivo by a kinase other than Cdk, and that this novel phosphorylation plays an important role in response to DNA damage.
Materials and methods
Cell lines and plasmids and transfections
MOLT-4 cells and HCT15 cells were cultured in RPMI1640 medium containing 10% fetal bovine serum (FBS). MCF7 cells and SAOS2 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS. Normal human fibroblast TIG-7 and fibroblast cell line from A-T patient AT2KY (a gift from Dr S Mizutani, Tokyo Medical and Dental University, Tokyo, Japan) were grown in DMEM containing 10% FBS.
The SRB1 cell line, a wild-type pRB-inducible clone of SAOS2 cells regulated by tetracycline (Tet-Off system), and STA-e clone, a tet repressor herpes simplex virus transactivator protein VP16 fusion protein expression clone of SAOS2, were previously reported (Ookawa et al, 1997, 2001; Uchida et al, 2005). The SRBSA1 cell, a stable transfectant with pT2RB(S612A)neo (Ookawa et al, 1997), was established from STA-e by screening with G418. SRB1 cells and SRBSA1 cells were grown in DMEM containing 10% FBS, 0.5 mg/ml of G418, 0.3 mg/ml of hygromycin and 1 μg/ml of tetracycline.
pFLAGCMV2-E2F-1, pCMV5-myc-RB, pCDNA4-HisMax-Chk2, pGEX6P3-E2F-1, and pGEX6P3-BRCA1 (amino acids 758–1064) were amplified by PCR using mRNA derived from MCF7 cells. p73-Luc was generated by ligating the human p73 promoter region (−705 to +537) (Stiewe and Putzer, 2000) with pGL4.10. All constructs were verified by sequencing. MCF7 cells and SAOS2 cells were transfected by the lipofection method using Lipofectamine 2000 reagent (Invitrogen) as recommended by the manufacturer. HCT15 cells were transfected by the nucleofection method. Two million HCT15 cells were resuspended in buffer provided in Nucleofector kit V (Amaxa), mixed with plasmid DNA (1 μg), and electroporated with T-16 program of nucleofector (Amaxa) as suggested by the manufacturer. Transfection efficiency is ∼80%.
Antibodies
Phospho-specific monoclonal (Thr356-P and Ser807-P) and polyclonal (Thr373-P, Ser788-P and Thr826-P) antibodies were raised against chemically synthesized, KLH-conjugated phosphopeptides as previously described (Kitagawa et al, 1996; Shieh et al, 1997; Taya et al, 2003). The sequences of phosphopeptides are; T356: SFETQRpTPRKSNL, T373: NVIPPHpTPVRTVM, S788: IPHIPRpSPYKFPS, S807: GGNIYIpSPLKSPYKI, and T826: PTPTKMpTPRSPRIL. Monoclonal and polyclonal antibodies were further purified as described previously (Taya et al, 2003). Phospho-specific monoclonal (Ser612-P and Ser780-P) and polyclonal (Ser608-P, Ser780-P, Ser795-P, Ser811-P, and Thr821-P) antibodies have been described (Kitagawa et al, 1996; Adams et al, 1999; Brugarolas et al, 1999; Watanabe et al, 1999; Taya et al, 2003).
Anti-E2F1 (C-20), anti-E2F2 (C-20), anti-E2F3 (C-18), anti-Cdk2 (M2), anti-Cdk4 (C-22), anti-Chk1 (FL-476), anti-Chk2 (H-300), anti-pBRCA1 (Ser988), Omni-probe (M-21), and anti-GST (B-14) were obtained from Santa Cruz; anti-Flag (M2) and anti-β-actin (AC-15) were from Sigma; anti-myc (9E10) was from Roche; anti-pChk1 (Ser345), anti-pChk2 (Thr68), and anti-Cleaved PARP were from Cell Signaling; anti-Cyclin A and anti-Smad2 were from BD Biosciences; and anti-ATM (2C1) was from GeneTex.
Reporter gene assay and IP and Western blot analysis
See Supplementary data for details.
In vitro kinase assays
Recombinant E2F-1 and BRCA (amino acids 758–1064) proteins fused with GST were produced in BL21. Purified Chk1 (recombinant protein expressed in Sf21), Chk2 (recombinant protein expressed in Escherichia coli), and p38α/SAPK2α (recombinant protein expressed in E. coli) were from Upstate. Recombinant Cdk2–cyclin A was from NEB. Recombinant full-length pRB was from QED Bioscience. One microgram of substrate was incubated with purified Chk1 or Chk2 at 30°C for 30 min in kinase buffer (50 mM HEPES, pH 7.5, 10 mM MgCl2, 1 mM DTT, 2.5 mM EGTA, and 200 μM ATP) in a final volume of 20 μl. The samples were boiled, fractionated by SDS–PAGE, and subjected to Western blotting using phospho-specific antibodies.
RNA interference
Double-stranded RNA duplexes corresponding to human ATM (5′-GCGCCTGATTCGAGATCCT-3′) (Yoshida et al, 2003) and control siRNA (5′-TTCTCCGAACGTGTCACGT-3′) were purchased from Qiagen. SMARTpool ATM siRNA, SMARTpool Chk2 siRNA, and negative control siRNA were purchased from Dharmacon. Cells were transfected with Lipofectamine 2000 according to the manufacturer's directions in the presence of siRNAs.
For stable knockdown in MCF7 cells, shRNA against Chk2 (5′-gatccccGAACCTGAGGACCAAGAACttcaaga gaGTTCTTGGTCCTCAGGTTCtttttggaaa-3′) (Ahn et al, 2003) was expressed in the retroviral vector pSUPERretro (Oligoengine). The sequence provided is the primer sequences as cloned into the pSUPERretro vector, with the uppercase letters representing the sequences complementary to the target gene. Next, the pSUPERlenti-Chk2 construct was generated by subcloning a ClaI/SpeI insert of pSUPERretro-Chk2 into ClaI/XbaI of pLenti6/V5-DEST (Invitrogen). MCF7 cells were infected with the viral particles according to standards protocols and selected with 8 μg/ml blasticidin.
Gel filtration
E2F-1 complex purified using anti-Flag M2 beads (Sigma) was further purified by gel filtration on a column of superose 6 (1 by 30 cm; Amersham Pharmacia Biotech) that had been equilibrated with IP buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 0.1% Tween-20 and 10% glycerol). Elution was performed at 0.3 ml/min, and 0.5-ml fractions were collected. The fractions were immunoprecipitated with anti-pRB antibody.
Cell survival and apoptosis assays
Cells were washed with PBS. Crystal violet was added to stain cells and the stained cells were lysed in 1% SDS and absorbance was measured at 595 nm. Caspase-3/7 activity was measured with Caspase-Glo 3/7 Assay (Promega) according to the instructions.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary data
Acknowledgments
We thank Shuki Mizutani (Tokyo Medical and Dental University) for AT2KY cells and Chiharu Uchida (Hamamatsu University School of Medicine), Koji Okamoto, and Masato Enari for discussions and suggestions. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, a Grant-in-Aid for Third Term Comprehensive Control Research for Cancer from the Ministry of Health, Labor and Welfare, Japan, a Grant-in-Aid from Tokyo Biocheminal Research Foundation, and Research Grants from the Princess Takamatsu Cancer Research Fund and Takeda Science Foundation to YT. YI is an Awardee of a Research Resident Fellowship from the Foundation for the Promotion of Cancer Research (Japan) for the 3rd Term Comprehensive 10-Year-Strategy for Cancer Control.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Adams PD, Li X, Sellers WR, Baker KB, Leng X, Harper JW, Taya Y, Kaelin WG Jr (1999) Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol Cell Biol 19: 1068–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Urist M, Prives C (2003) Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J Biol Chem 278: 20480–20489 [DOI] [PubMed] [Google Scholar]
- Ahn J, Urist M, Prives C (2004) The Chk2 protein kinase. DNA Repair 3: 1039–1047 [DOI] [PubMed] [Google Scholar]
- Alberts AS, Thorburn AM, Shenolikar S, Mumby MC, Feramisco JR (1993) Regulation of cell cycle progression and nuclear affinity of the retinoblastoma protein by protein phosphatases. Proc Natl Acad Sci USA 90: 388–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasan A, Yin Y, Kelly RE, Lee EY, Bradley A, Li W, Bertino JR, Wahl GM (1995) Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci USA 92: 5436–5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwooll C, Lazzerini Denchi E, Helin K (2004) The E2F family: specific functions and overlapping interests. EMBO J 23: 4709–4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin C, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677 [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Moberg K, Boyd S, Taya Y, Jacks T, Lees J (1999) Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc Natl Acad Sci USA 96: 1002–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281: 1677–1679 [DOI] [PubMed] [Google Scholar]
- Classon M, Harlow E (2002) The retinoblastoma tumor suppressor in development and cancer. Nat Rev Cancer 2: 910–917 [DOI] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR (1997) Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci USA 94: 7245–7250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou QP, An B, Will PL (1995) Induction of a retinoblastoma phosphatase activity by anticancer drugs accompanies p53-independent G1 arrest and apoptosis. Proc Natl Acad Sci USA 92: 9019–9023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Lukas C, Protopopova M, Lukas J, Selivanova G, Bartek J (2001b) Functional impact of concomitant versus alternative defects in the Chk2-p53 tumor suppressor pathway. Oncogene 20: 5503–5510 [DOI] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J (2001a) The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410: 842–847 [DOI] [PubMed] [Google Scholar]
- Haas-Kogan DA, Kogan SC, Levi D, Dazin P, T'Ang A, Fung YK, Israel MA (1995) Inhibition of apoptosis by the retinoblastoma gene product. EMBO J 14: 461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington EA, Bruce JL, Harlow E, Dyson N (1998) pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci USA 95: 11945–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64: 9152–9159 [DOI] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287: 1824–1827 [DOI] [PubMed] [Google Scholar]
- Jiang H, Karnezis AN, Tao M, Guida PM, Zhu L (2000) pRB and p107 have distinct effects when expressed in pRB-deficient tumor cells at physiologically relevant levels. Oncogene 19: 3878–3887 [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y (1996) The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J 15: 7060–7069 [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Wang JY (1996) Differential regulation of retinoblastoma protein function by specific cdk phosphorylation sites. J Biol Chem 271: 8313–8320 [DOI] [PubMed] [Google Scholar]
- Knudsen KE, Booth D, Naderi S, Sever-Chroneos Z, Fribourg AF, Hunton IC, Feramisco JR, Wang JY, Knudsen ES (2000) RB-dependent S-phase response to DNA damage. Mol Cell Biol 20: 7751–7763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Chang JH, Lee HS, Cho Y (2002) Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev 16: 3199–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Collins KM, Brown AL, Lee CH, Chung JH (2000) hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 404: 201–204 [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim SH, Bell DW, Wahrer DC, Schiripo TA, Jorczak MM, Sgroi DC, Garber JE, Li FP, Nichols KE, Varley JM, Godwin AK, Shannon KM, Harlow E, Haber DA (2001) Destabilization of CHK2 by a missense mutation associated with Li-Fraumeni syndrome. Cancer Res 61: 8062–8067 [PubMed] [Google Scholar]
- Lin WC, Lin FT, Nevins JR (2001) Selective induction of E2F-1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev 15: 1833–1844 [PMC free article] [PubMed] [Google Scholar]
- Ludlow JW, Glendening CL, Livingston DM, DeCaprio JA (1993) Specific enzymatic dephosphorylation of the retinoblastoma protein. Mol Cell Biol 13: 367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas E, Roumillac C, Trouche D (2003) Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Mol Cell Biol 23: 1614–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill T, Giarratani L, Chen P, Iyer L, Lee CH, Bobiak M, Kanai F, Zhou BB, Chung JH, Rathbun GA (2002) Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J Biol Chem 277: 16102–16115 [DOI] [PubMed] [Google Scholar]
- Ookawa K, Tsuchida S, Adachi J, Yokota J (1997) Differentiation induced by RB expression and apoptosis induced by p53 expression in an osteosarcoma cell line. Oncogene 14: 1389–1396 [DOI] [PubMed] [Google Scholar]
- Ookawa K, Tsuchida S, Kohno T, Yokota J (2001) Alterations in expression of E2F-1 and E2F-responsive genes by RB, p53 and p21(Sdi1/WAF1/Cip1) expression. FEBS Lett 500: 25–30 [DOI] [PubMed] [Google Scholar]
- Ortega S, Malumbres M, Barbacid M (2002) Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochem Biophys Acta 1602: 73–87 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501–1512 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F (2002) The RB and p53 pathways in cancer. Cancer Cell 2: 103–112 [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C (2000) The human homologues of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 14: 289–300 [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2001) ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 11: 71–77 [DOI] [PubMed] [Google Scholar]
- Slebos RJ, Lee MH, Plunkett BS, Kessis TD, Williams BO, Jacks T, Hedrick L, Kastan MB, Cho K R (1994) p53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proc Natl Acad Sci USA 91: 5320–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Zhan Q, Bae I, Fornance AJ Jr (1994) Role of retinoblastoma gene product in p53-mediated DNA damage response. Exp Cell Res 215: 386–389 [DOI] [PubMed] [Google Scholar]
- Stevens C, Smith L, La Thangue NB (2003) Checkpoint kinase activates E2F-1 in response to DNA damage. Nat Cell Biol 5: 401–409 [DOI] [PubMed] [Google Scholar]
- Stiewe T, Putzer BM (2000) Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet 26: 464–469 [DOI] [PubMed] [Google Scholar]
- Tan X, Martin SJ, Green DR, Wang JY (1997) Degradation of retinoblastoma protein in tumor necrosis factor- and CD95-induced cell death. J Biol Chem 272: 9613–9616 [DOI] [PubMed] [Google Scholar]
- Taya Y (1997) RB kinases and RB-binding proteins: new points of view. Trends Biochem Sci 22: 14–17 [DOI] [PubMed] [Google Scholar]
- Taya Y, Nakajima K, Yoshizawa-Kumagaye K, Tamai K (2003) Generation and application of phospho-specific antibodies for p53 and pRB. Methods Mol Biol 223: 17–26 [DOI] [PubMed] [Google Scholar]
- Uchida C, Miwa S, Kitagawa K, Hattori T, Isobe T, Otani S, Oda T, Sugimura H, Kamijo T, Ookawa K, Yasuda H, Kitagawa M (2005) Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J 24: 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Watanabe T, Kitagawa M, Taya Y, Nakayama KI, Motoyama N (1999) pRb phosphorylation is regulated differentially by cyclin-dependent kinase (Cdk) 2 and Cdk4 in retinoic acid-induced neuronal differentiation of P19 cells. Brain Res 842: 342–350 [DOI] [PubMed] [Google Scholar]
- Yoshida K, Wang HG, Miki Y, Kufe D (2003) Protein kinase Cδ is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J 22: 1431–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkowska T, Mittnacht S (1997) Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem 272: 12738–12746 [DOI] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary data