

Abstract
Platinating agents, including cisplatin, carboplatin, and oxaliplatin, have been used clinically for nearly thirty years as part of the treatment of many types of cancers, including head and neck, testicular, ovarian, cervical, lung, colorectal and relapsed lymphoma. The cytotoxic lesion of platinating agents is thought to be the platinum intrastrand crosslink that forms on DNA, although treatment activates a number of signal transduction pathways. Treatment with these agents is characterized by resistance, both acquired and intrinsic. This resistance can be caused by a number of cellular adaptations, including reduced uptake, inactivation by glutathione and other anti-oxidants, and increased levels of DNA repair or DNA tolerance. Here we investigate the pathways that treatment with platinating agents activate, the mechanisms of resistance, potential candidate genes involved in the development of resistance, and associated clinical toxicities. Although the purpose of this review is to provide an overview of cisplatin, carboplatin, and oxaliplatin, we have focused primarily on preclinical data that has clinical relevance generated over the past five years.
Keywords: cisplatin, drug resistance, DNA damage
Background
Cisplatin [cis-diammine-dichloroplatinum (II)]4 (Figure 1) is a commonly used chemotherapeutic agent that was discovered in 1970 as an inhibitor of growth in Eschericia coli [1]. The clinical benefits of cisplatin as an anti-cancer agent have been recognized for over 30 years. Cisplatin is considered to be curative treatment for testicular cancer, when combined with bleomycin and etoposide. It is closely related to its second generation analog carboplatin; the two compounds share a mechanism of action, are fully cross-resistant, and form identical lesions on DNA. Both agents are used for many other types of cancer, including ovarian, cervical, head and neck, non-small cell lung, and lymphoma. However, for many, particularly head and neck, lung, and relapsed lymphomas, cisplatin treatment is plagued by problems -including toxicities and resistance, both intrinsic and acquired [2]. Oxalate (trans-l-1,2-diamminocyclohexane) platinum (II) (oxaliplatin) is an analog which does not share the same mechanism as cisplatin, and likewise does not share in cross-resistance (reviewed in [3]).
Figure 1.

The chemical structures of cisplatin, transplatin, carboplatin, oxaliplatin, and satraplatin.
In this review, we will focus on the most commonly used platinating agents - cisplatin, carboplatin, and oxaliplatin. It is worth mentioning, however, that a fourth platinating agent, satraplatin (bis(aceto)amminedichloro(cyclohexylamine) platinum (IV)), is available, and is currently the only orally available platinating agent. Satraplatin does not share cross-resistance with cisplatin, which is thought to be due to a different detoxification mechanism, and it shows activity in advanced hormone-refractory prostate cancer [4, 5].
Mechanism of Action
DNA Lesions
Upon entering a cell, all platinating agents become aquated, losing chloride or oxalate ions, and gaining two water molecules. This positively charged molecule is then able to interact with nucleophilic molecules within the cell, including DNA, RNA, and proteins. It is generally agreed that DNA is the preferential and cytotoxic target for cisplatin and other platinating agents (reviewed in [6]).When binding to DNA, platinating agents favor the N7 atoms of the imidazole rings of guanosine and adenosine. Three different types of lesions can form on purine bases of DNA: monoadducts, intrastrand crosslinks, and interstrand crosslinks (Figure 2). Monoadducts are first formed as one molecule of water is lost from aquated platinating agents; however, greater than 90% of monoadducts then react to form crosslinks. Almost all of these crosslinks are intrastrand, with the majority being 1,2-d(GpG) crosslinks. Additional DNA lesions include interstrand crosslinks. Oxaliplatin forms fewer crosslinks than cisplatin at equimolar concentrations; however, it is equally as potent at these concentrations [7, 8] and is able to induce similar numbers of single-strand and double-strand breaks on DNA [9].
Figure 2.

Platinating agent adducts on DNA. Platinating agents are able to interact with DNA to form monoadducts, intrastrand crosslinks (1,2-d(GpG), 1,2-d(ApG), 1,3-d(GpXpGp)), interstrand crosslinks (G-G), and DNA-protein crosslinks.
All crosslinks result in contortion of the DNA (reviewed in [10]). Cisplatin and carboplatin intrastrand crosslinks bend the double helix by 32-35° toward the major groove, whereas oxaliplatin treatment bends the helix even further [11]. Both 1,2-d(GpG) and 1,2-d(ApG) intrastrand crosslinks unwind DNA by 13°, while the 1,3-d(GpXpG) intrastrand lesion unwinds DNA by 34°. Interstrand lesions induce even more steric changes in DNA, with extrusion of the cytosines at the crosslinked d(GpC)d(GpC) sites, bending of the double helix toward the minor groove by 20-40°, and extensive DNA unwinding of up to 80°. Oxaliplatin adducts are bulkier and more hydrophobic than those formed from cisplatin or carboplatin, leading to different effects in the cell (reviewed in [12]).
HMG Involvement
There are different theories as to which lesion is responsible for cytotoxicity. Some believe that the interstrand crosslink is cytotoxic because of the level of distortion in the DNA; however, most believe that the predominant 1,2-intrastrand crosslinks are the cytotoxic lesion because of comparisons with the biologically inactive trans isomer of cisplatin, trans-diamine-dichloroplatinum (II) (trans-DDP) (Figure 1). trans-DDP is unable to form 1,2-intrastrand crosslinks, but is able to form 1,3-intra- and inter-strand linkages [13]. Additionally, high mobility group (HMG) proteins are able to recognize and bind to DNA at the 1,2-d(GpG) intrastrand crosslinks. HMG domains are basic domains of 80 amino acids which contain three α-helical domains. They are intimately associated with the curvature of chromatin. Their presence is thought to be crucial for sensitivity to cisplatin and carboplatin, partly because the testis, which is exquisitely sensitive to cisplatin, expresses several HMG domain proteins. Members of the HMGB family, including HMGB1 (HMG-1), have been shown to bind to 1,2-d(GpG) crosslinks induced by cisplatin, but not to DNA treated with trans-DDP. The binding of HMGB1 to cisplatin aids in preventing replicative bypass (translesion synthesis) [14]. Additionally, HMGB proteins such as SRY, UBF, and LEF-1 have been shown to block nucleotide excision repair (NER) components from repairing the lesion via a “shielding mechanism” (reviewed in [15]). The cisplatin-DNA-HMGB1 ternary complex is also able to block transcription factors, thus preventing both transcription and replication. This block in cellular processes may be responsible for sending out DNA damage signals that result in initiation of apoptosis [16]. In support of this theory, He, et al., found that overexpression of HMGB1 caused by estrogen exposure sensitized breast tumor cells to cisplatin [17]. HMGB1 generally functions to facilitate binding steroid hormone receptors to their promoter sites on DNA. In the MCF-7 breast cancer line, priming the cells with estrogen or progesterone resulted in increased transcription of HMGB1 by approximately two-fold. This increased HMGB1 expression is associated with a concomitant increase in sensitivity to cisplatin in this cell line. HMG has a much lower affinity for oxaliplatin crosslinks on DNA than it does for cisplatin or carboplatin adducts [14]. The molecular geometry of the oxaliplatin adduct, with a narrower major groove and correspondingly wider minor groove, is thought to be responsible for this observation.
Endoplasmic Reticulum Stress
Although cisplatin is known to induce apoptosis following DNA damage, it has also been shown to cause activation of apoptotic caspases through activation of the endoplasmic reticulum (ER) stress pathway (Figure 3) [18]. The ER stress pathway is based on the cellular unfolded protein response (UPR). When the ER experiences stress such as starvation or treatment with inhibitors of N-glycosylation (e.g. tunicamycin), it cannot fold or transport proteins correctly, and the UPR is activated. The first step of the UPR is phosphorylation of eIF2α at Ser51, halting new protein synthesis, then regulatory components of the ER stress pathway, including ATF4, ATF6, XBP1, and BiP (Grp78), are upregulated [19]. In some cases, these regulatory proteins are able to restore normal ER function. In other circumstances, the UPR initiates apoptosis. This ER stress-induced apoptosis is dependent upon the activation of caspase-12 [19]. Caspase 12 is located at the cytosolic face of the ER and is cleaved by the calpain protease [20]. Inhibition of calpain by calpeptin prevents cisplatin-induced caspase-12 cleavage [18].
Figure 3.

Mechanism of cisplatin activity and mechanisms of resistance to platinating agents, as exemplified here by cisplatin. Cisplatin can act in the cell either by causing DNA damage, or by activating the ER stress pathway, both of which can lead to cellular apoptosis. In addition, many mechanisms of resistance (italics) are present, including transport, cellular antioxidants, increased DNA damage repair, and DNA tolerance.
Treatment of enucleated melanoma 224 and colon carcinoma HCT 116 cell lines with cisplatin resulted in activation of caspase 12, followed by caspase 3 activation. Grp78 (BiP) was also upregulated in 224 cells [18]. Additionally, it has been observed in a pancreatic cancer cell line that cisplatin is capable of activating ER stress pathways, including upregulation of chaperone proteins and caspase 12 cleavage [21]. The stimulation of pro-apoptotic pathways in enucleated cells by cisplatin-induced ER stress was a novel finding and one that other groups are beginning to evaluate further as a secondary mechanism of cisplatin cytotoxicity. ER stress activation has not been shown as yet for either carboplatin or oxaliplatin.
Signaling Cascades and Transcription Factors
Treatment with cisplatin or carboplatin results in the activation of complex signaling cascades in the cell [2]. Transcription factors activated by these cascades serve to vary the gene expression pattern after treatment with cisplatin or carboplatin. Oxaliplatin induces notably different cascades from either cisplatin or carboplatin. Here, we highlight some of the most important transcription factors and signaling cascades.
The transcription factor c-Fos has been implicated in cisplatin resistance. c-Fos transcription is induced upon cisplatin treatment in cisplatin-resistant human ovarian cells [22]. In fact, transfection of a sensitive ovarian carcinoma cell line with a c-fos vector generates cisplatin-resistant cells [23]. Decreasing c-Fos expression restores some cisplatin sensitivity [22]. In rat fibroblasts, c-fos expression induces resistance to cisplatin by 2-to-3 fold [24]. c-Fos is part of the transcription factor activator protein 1 (AP-1), which can interact with the Ets-1 transcription factor. In C13* cells, Ets-1 is upregulated as compared to 2008 cells, and 2008 cells designed to overexpress Ets-1 show similar or greater resistance to cisplatin than C13* cells [25]. Similarly, HT-29 colon carcinoma cells transfected with ets-1 are more resistant to cisplatin than non-transfected cells [25]. Ets-1 is a transcription factor for metallothioneins, which are involved in detoxification of cisplatin, and a microarray experiment in 2008 cells identified these, in addition to several genes involved in DNA repair, as potential targets of Ets-1 [25].
Most cisplatin-mediated cell death is through an apoptotic pathway. Inhibition of this pathway by genes such as bcl-2 can lead to drug resistance [26]. Other proteins, known as inhibitor of apoptosis proteins (IAPs), have been identified in mammals, and at least one protein, Xiap, has a potential role in cisplatin resistance in ovarian cancer [27]. Treatment of ovarian cancer lines with antisense Xiap induced apoptosis in both cisplatin-sensitive and cisplatin-resistant cell lines, with sensitive lines being more susceptible to apoptosis. In fact, cisplatin treatment itself downregulated Xiap expression in cisplatin sensitive cells, but not in resistant cells, indicating a different signaling cascade is activated in sensitive cells [27].
Oxaliplatin is involved in the inhibition of survivin, an inhibitor of apoptosis. Upon treatment with oxaliplatin, expression of both survivin and the cell cycle protein cdc2 decreased immediately after treatment [28]. In p53 mutant cells, which represents the majority of colorectal cancers, oxaliplatin also decreased the phosphorylation of Bcl-2 and Bax, thereby further promoting cellular apoptosis [28].
Using diploid deletion strains of Saccharomyces, Huang, et al., identified genes that potentially contribute to cisplatin resistance [29]. In addition to genes that have already been characterized as leading to resistance when deleted-MMR genes, CTR1, MAC1, NPR2, and SKY1, there were twenty other genes characterized [30-33]. These genes had a wide range of activities in the cell - including nucleotide metabolism, mRNA catabolism, RNA Pol II-dependent gene regulation, and transport [29]. Several of the genes encode proteins involved in transcription factor networks, including NOT3, STP1, SNF6, and the uncharacterized open reading frame (ORF) YJL175W (overlaps the SWI3 transcription factor). Further studies in human cells, including the use of siRNA, will decipher the role of these gene products in development of resistance to cisplatin.
DNA methyltransferase is involved in methylating DNA, thus repressing gene transcription. In murine neuroblastoma (MNB) cells, methyltransferase activity, specifically of DNA methyltransferase-3b (Dnmt3b), was shown to correlate with cisplatin resistance [34]. In cisplatin-sensitive MNB cells, transfection of Dnmt3a or 3b resulted in increased resistance to cisplatin. Cisplatin sensitivity could be regained by treating these Dnmt3-expressing cells with 5′-azacytidine, an inhibitor of methyltransferase [34]. DNA methylation appears to be a global effect; the role of gene specific methylation in cisplatin resistance still remains to be elucidated.
Dihydrodiol dehydrogenase was identified as a differentially expressed gene in microarray analysis between the ovarian carcinoma sensitive-resistant pair 2008 and C13*, with higher expression in the resistant C13* line [35]. This upregulation was confirmed using real-time PCR. Overexpression of dihydrodiol dehydrogenase in the sensitive 2008 cell line conferred cisplatin resistance to the cell line. This resistance did correspond to an increase in enzyme activity, indicating that expression of the dihydrodiol dehydrogenase gene is involved in resistance to cisplatin, most likely as a result of an as yet unknown signaling cascade [35]. Clearly, resistance to cisplatin is multigenic and some resistance mechanisms are likely to be tissue specific.
Repair of DNA Lesions
DNA damage recognition of cisplatin and carboplatin adducts may be due to a conformational change induced by the intra- or inter-strand crosslink on DNA. In order to address this question, synthetic oligonucleotides were generated which contained one putative intrastrand binding site for cisplatin (GTG) [36]. Nucleotides were either exposed to cisplatin or not before being incubated with histones to generate nucleosomes. DNA that had been platinated generated a very different pattern of exonuclease III cleavage than untreated DNA. It is thought that this specific cleavage pattern occurs because the platinum crosslink locks the nucleosome in place [36], similar to those observed in UV-induced thymidine dimers [37, 38].
Nucleotide Excision Repair
Platinating agent adduct repair occurs primarily through nucleotide excision repair (NER) (Figure 3, Table 1) [39]. While all three types of intrastrand crosslinks (1,2-d(ApG), 1,2-d(GpG), and 1,3-d(GpNpG)) are recognized by the NER mechanism, the 1,2 intrastrand crosslinks are repaired less efficiently than the 1,3 intrastrand crosslinks, supporting the hypothesis that the 1,2 intrastrand crosslinks are the cytotoxic lesion [39, 40]. Increased expression of several NER genes has been correlated with cisplatin resistance. In ovarian cancer, XPA and ERCC1-XPF were shown to have increased expression in tumors of patients resistant to platinum treatment [41, 42]. In primary ovarian tumors, levels of XPB transcripts were significantly higher in tumors resistant to cisplatin than in tumor samples from patients who responded well to platinum treatment [43]. Similarly, gastric cancer showed a correlation between cisplatin resistance and ERCC1-XPF mRNA levels [44]. Testicular cancer, generally very responsive to cisplatin, has low levels of XPA and ERCC1-XPF, providing further correlative evidence for the importance of NER in cisplatin resistance [45, 46]. Oxaliplatin treatment also induces expression of NER proteins, including XPA and ERCC-1 [47], and the rate and kinetics of NER are similar between cisplatin and oxaliplatin [48].
Table 1.
Molecular Mechanisms of Platinating Agent Repair and Resistance.
| Molecular Mechanism | Preclinical Evidence | Clinical Evidence |
|---|---|---|
| HMG Proteins | ▷ HMGB1 binds to 1,2-d(GpG) crosslinks induced by cisplatin [15] | ▷ Testis expresses several HMG domain proteins [15] |
| ▷ HMGB proteins block NER and transcription factors [15, 16] | ||
| ▷ In breast cancer cells, HMGB1 overexpression correlates with cisplatin sensitivity [17] | ||
| Nucleotide Excision Repair | ▷ SWI/SNF chromatin remodeling may be necessary for NER to occur around nucleosomes [36, 49, 51, 52] | ▷ Ovarian cancer patients resistant to cisplatin have higher levels of XPA, XPB, and ERCC1-XPF [41-43] |
| ▷ Testicular carcinomas have low levels of XPA and ERCC1-XPF [45, 46] | ||
| Mismatch Repair | ▷ Tumor cells deficient in MMR are resistant to cisplatin; restoration of MMR restores sensitivity [57, 58] | ▷ Correlation between cisplatin resistance and microsatellite instability in colon carcinoma, and in ovarian adeno-, serous, and clear cell carcinomas [53, 62-70] |
| Homologous Recombination | ▷ Double-strand DNA breaks not observed in NER-deficient lines, nor in small cell lung carcinoma lines [77, 78] | |
| Replicative Bypass | ▷ The error-prone polymerases β, η, ζ, and ι can bypass cisplatin lesions [79-83] | |
| ▷ Overexpression of Pol β leads to resistance [86] | ||
| ▷ Pol ζ plays a role in bypass in MMR-deficient cells [88] | ||
| ▷ Absence of pol η results in enhancement to cisplatin, carboplatin, and oxaliplatin [89] | ||
| Copper Transport | ▷ Cisplatin inhibits copper transport [133] | ▷ Eighty-two primary ovarian carcinomas were examined; those that express ATP7B (43%) had a significantly poorer prognosis [139] |
| ▷ SR2 expresses half of Ctr1 protein as SCLC, take up less cisplatin and carboplatin [134] | ▷ Primary tumors with ATP7A histochemical staining had a lower response rate [140] | |
| ▷ In ovarian tumor cell lines, high ATP7B expression correlates with resistance to cisplatin [136, 138] | ||
| ▷ ATP7A can sequester cisplatin, carboplatin, and oxaliplatin [142] | ||
| MRP2 | ▷ MRP2 levels increase upon cisplatin treatment [145] | |
| ▷ Upregulated in cisplatin-resistant melanoma cell lines, but downregulated in cisplatin-resistant liver carcinoma cell lines [146, 147] | ||
| ▷ No link between MRP2 and resistance in ovarian clear cell carcinomas [169] | ||
| Glutathione and Thioredoxin | ▷ Depletion of glutathione in bladder carcinoma cells sensitizes cells to cisplatin [151] | ▷ Head and neck and ovarian primary tumors show a correlation between GST-π and cisplatin resistance [153-156] |
| ▷ TrxR1 inhibited by cisplatin, oxaliplatin, and transplatin [161, 162] | ||
| Metallothioneins | ▷ Elevated levels of MT in cisplatin-resistant cell lines [163] | ▷ In ovarian cancer, higher MT level corresponds with shorter survival [155] |
| ▷ Bladder cancer xenograft levels of MT correlate with increased cisplatin dosage [163] | ▷ Correlation found in esophageal, transitional cell, and head and neck carcinomas [165-167] | |
| ▷ Cells transfected with MT are more resistant to cisplatin [164] | ▷ No correlation between MT and cisplatin in testicular or germ cell tumors [168] |
NER may be inhibited by the presence of nucleosomes along DNA. Previous studies have indicated that the presence of nucleosomes on DNA is able to inhibit NER in cells treated with DNA damaging agents, including cisplatin and UV rays [49-51]. In synthetically generated platinated oligonucleotides, the presence of nucleosomes disrupted NER to ∼10% of the levels observed in platinated oligonucleotides without nucleosomes [36]. Nucleosome induced NER inhibition may be overcome by the activity of the SWI/SNF chromatin remodeling complex, which is activated upon damage recognition by the NER factors XPA and SPC [52]. SWI/SNF has not been shown to be a necessary component for repair of cisplatin lesions; however, inhibition of this pathway may lead to a targeted therapy for sensitization of tumors to cisplatin and other DNA damaging agents.
Mismatch Repair
The mismatch repair pathway (MMR) has also been proposed to be involved in development of cisplatin and carboplatin resistance (Figure 3, Table 1) [53]. The presence of MMR is thought to mediate cisplatin- and carboplatin-induced apoptosis [54-56]. Tumor cells deficient in MMR are 2-3 fold more resistant to cisplatin treatment compared to cells proficient in MMR [57]. Similarly, restoration of hMLH1 expression in a resistant derivative of the A2780 ovarian cell line which lacked hMLH1 restored cisplatin sensitivity to the level of the parent line [58]. While MMR proteins appear to recognize cisplatin and carboplatin adducts on DNA, they are not involved in the repair of the damage [40]. For example, the presence of the MSH2 protein is required for sensitivity to cisplatin in S. cerevisiae; however, loss of function of either the ATP binding or ATP hydrolysis domain of MSH2 does not affect cisplatin sensitivity [59], indicating that the repair activity of MSH2 is not required to prevent cisplatin resistance. This is significantly different from repair of DNA mismatch lesions by MMR, which is dependent on the ATP processing activity of MSH2.
Alteration of the ability of MSH2 to sense DNA damage is thought to be necessary for cisplatin sensitivity, based on the results from genetic variations in yeast [60]. Identification of genetic polymorphisms, in addition to tumor-induced mutations, in MSH2 and other mismatch repair proteins may be valuable as physicians try to predict not only which patients are most likely to respond to cisplatin treatment, but also in predicting patients at risk for cisplatin-induced toxicities.
One measure of MMR deficiency has been microsatellite instability (MSI) [61]. There have been reports of a correlation between cisplatin resistance and MSI in colon carcinoma and in ovarian adenocarcinoma, serous carcinoma, and clear cell carcinoma [53, 62-70]. However, an additional study using quasimonomorphic markers did not identify significant levels of MSI in 34 ovarian carcinomas [61].
A notable difference between the mechanisms of cisplatin/carboplatin and oxaliplatin is the mismatch repair system. Deficiency of MMR does not affect cytotoxicity of oxaliplatin, and different cellular cascades are activated for the two different types of platinating agents [53, 71]. This is particularly important in the usage of oxaliplatin to treat colorectal cancer, as mismatch repair is frequently deficient in these tumors.
Homologous Recombination
Homologous recombination has been proposed to play a role in repairing double strand breaks (DSB) resulting from cisplatin-induced interstrand DNA adducts (Table 1) [72]. The NER components XPF and ERCC1 are also thought to be important for homologous recombination [73-76]. When Chinese hamster ovary (CHO) cells deficient in NER genes were examined for induction of DSB from interstrand crosslinks, CHO cells deficient in ERCC1 or XPF were extremely sensitive to cisplatin, but DSB as measured by pulsed field gel electrophoresis were not observed in these lines [77]. Similarly, in small cell lung cancer lines, cisplatin-resistant and cisplatin-sensitive lines did not differ significantly in their formation of DSB [78]. This provides anecdotal evidence supporting intrastrand crosslinks as the cytotoxic lesion, as DSBs are thought to be formed only during the repair of interstrand crosslinks.
Replicative Bypass
In some cell lines, platinum tolerance can be achieved without the need for DNA repair. In order for platinated DNA to be replicated and tolerance to form, DNA polymerase must skip the platinum adduct, which is most commonly an intrastrand lesion (Table 1) (reviewed in [6]). The classic DNA replication polymerases - α, θ, and ε - cannot bypass the lesion; however, several polymerases have been shown to bypass intrastrand crosslinks by translesion synthesis - namely, β, η, ζ, and ι [79-83]. Overexpression of DNA pol β has been shown to lead to cisplatin resistance, while downregulation using anti-sense RNA leads to sensitivity [84-87]. Pol ζ has been shown in MMR deficient cells to play a role in DNA tolerance and bypass of lesions [88].
Recent experiments with pol η null and expressing XP-variant human fibroblasts have shown that the absence of pol η results in a statistically significant enhancement in cisplatin sensitivity [89]. This enhancement is also observed when the cells were treated with carboplatin and oxaliplatin, but not with transplatin, which only forms interstrand crosslinks [89]. Pol η is a potential target for future therapy, as inhibiting it may prevent tolerance and increase sensitivity to cisplatin. Pol ζ, pol γ, and low concentrations of pol β have a preference for bypassing oxaliplatin adducts over cisplatin or carboplatin adducts, thus providing an additional clue for the difference between these mechanisms [14].
Clinical Utility of Platinating Agents
Cisplatin, carboplatin, and oxaliplatin are all commonly used intravenous platinating agents. Cisplatin is still used regularly for head and neck and germ cell tumors, while carboplatin has supplanted the use of cisplatin for most ovarian tumors and for the treatment of non-small cell lung carcinoma [90]. Oxaliplatin is currently approved for treatment in colorectal cancer, but has also been shown to have activity against breast and endometrial cancers and malignant melanoma in Phase I studies (reviewed in [12]). Additional Phase II trials show oxaliplatin to be active against non-small cell lung cancer, prostate cancer, germ-cell malignancies, ovarian carcinoma, non-Hodgkin’s lymphoma, and malignant mesothelioma; minimal or no activity was observed in head and neck carcinoma and in malignant astrocytoma (reviewed in [12]).
Toxicities
Toxicities associated with cisplatin range from mild to severe, with nephrotoxicity and peripheral neurotoxicity being the most serious (Figure 4) [91, 92]. Nephrotoxicity is primarily due to uptake by the proximal tubule cells of the nephron, with uptake by other cells having a lesser effect [92]. Nephrotoxicity has largely been controlled by diuretics and pre-hydration of patients, such that neurotoxicity has now become the dose-limiting effect. Cisplatin is thought to act on the dorsal root ganglion to generate both transient and chronic neuropathies [91].
Figure 4.
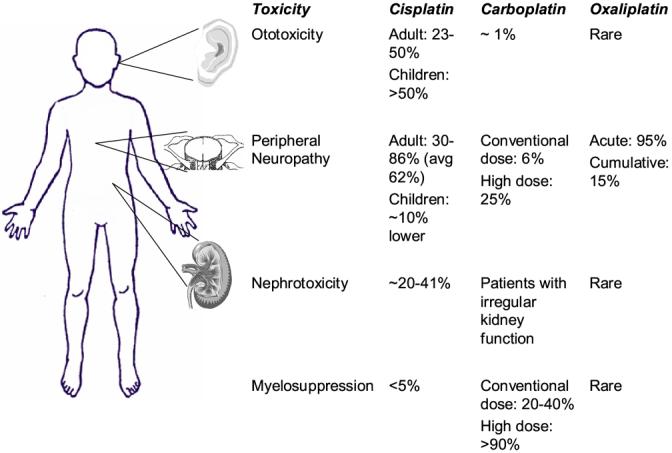
Toxicities associated with treatment with platinating agents. A. The most common side effects associated with cisplatin treatment are ototoxicity, peripheral neuropathy, myelosuppression, and nephrotoxicity. Ototoxicity is notably higher in pediatric patients, while neuropathy is relatively more common in adult patients. B. The most common toxicity associated with carboplatin is myelosuppression, with rare cases of neurotoxicity and nephrotoxicity. Oxaliplatin most commonly causes neurotoxicity.
One notable distinction between cisplatin and carboplatin is a difference in the spectrum of toxicities. Carboplatin rarely results in nephrotoxicity and peripheral neuropathy, with its major toxicity being myelosuppression [93]. The most common toxicity associated with oxaliplatin treatment is peripheral neuropathy, which ranges from acute and transient to a cumulative neuropathy. Oxaliplatin is generally free of ototoxicity and nephrotoxicity, with only moderate isolated cases of neutropenia and thrombocytopenia (reviewed in [12]).
Ototoxicity
Ototoxicity occurs in approximately 23-54% of patients receiving cisplatin treatment, and in greater than half of pediatric patients receiving cisplatin (Figure 4) [94]. Bolus higher doses of cisplatin have been shown to be more ototoxic and nephrotoxic than repeated infusions at lower doses in adults [95]. In children, however, prolonged infusions are less nephrotoxic than bolus doses but still result in considerable ototoxicity [96].
Platinum-based chemotherapeutic agents damage the outer hair cells of the cochlea (inner ear), resulting in functional deficits (reviewed in [94]). The mechanisms underlying these troublesome side effects most likely involve the production of reactive oxygen species (ROS) in the cochlea, which can trigger cell-death pathways. This is thought to be due to activation of the NADPH oxidase isoform NOX3, which is expressed only in the inner ear, as kidney cells transfected with the nox3 gene exhibit enhanced superoxide formation upon treatment with cisplatin [97]. The superoxide radical can then be transformed by cellular enzymes into hydrogen peroxide and the hydroxyl radical, which has been hypothesized to play a major role in cisplatin-induced ototoxicity [98]. In the outer hair cells of the cochlea, cell death pathways are triggered by the release of cytochrome c and activation of caspases 9 and 3 [99].
One strategy to protect the inner ear from ototoxicity is pretreatment with thiol-containing drugs that act as antioxidants, including sodium thiosulfate (STS), methionine, glutathione ester, and amifostine (reviewed in [94]). α-Tocopherol (Vitamin E) was shown to reduce cisplatin-induced ototoxicity in male rats as measured both by hearing threshold differences and cochlear morphology [100]. In Hartley albino guinea pigs, treatment with α-tocopherol alone was less effective, but when given in conjunction with the thiol-containing compound tiopronin, cisplatin-induced hearing loss was significantly slowed [101].
High serum concentrations of carboplatin have also been linked to oxotoxicity, although this phenomenon is relatively rare, occurring in ∼1% of patients. Oxaliplatin has not been linked to ototoxicity (reviewed in [90]).
Nephrotoxicity
Nephrotoxicity is associated with cisplatin treatment, but is rare with therapies involving its later generation analogs carboplatin or oxaliplatin (Figure 4) [102, 103]. Due to the renal excretion of cisplatin, the kidney accumulates a higher effective concentration of cisplatin than any other organ [104]. This accumulation preferentially affects the terminal proximal tubule and the distal nephron and can cause either apoptosis or necrosis, depending on exposure time and concentration [104]. Low, prolonged doses of cisplatin typically induce apoptosis, whereas necrosis is caused by short exposures to higher concentrations of cisplatin [105]. Similar to cisplatin-induced ototoxicity, nephrotoxicity related to cisplatin treatment is due to the production of ROS. Notably, ROS are only thought to mediate the apoptosis pathway, and are not involved in the necrotic death pathway [105].
ROS damage is thought to be mitigated by hypoxia-inducible factor 1α (HIF1α). Immortalized rat renal tubular cells expressing dominant negative HIF1α were more susceptible to apoptosis following cisplatin treatment in hypoxic conditions than cells expressing wild-type HIF1α [106]. Future studies hope to use activation of HIF-1 as a target for further protecting patients from nephrotoxicity, possibly with siRNA or gene therapy.
The human organic cation transpoter (hOCT) has been proposed to be involved in potentiating cisplatin-induced nephrotoxicity in the proximal tubule. This transporter is expressed primarily in the kidney [107]. After treatment with a concentration of cisplatin known to induce apoptosis, the hOCT2 substrate cimetidine was able to suppress cisplatin-induced apoptosis [108]. Cotreatment of cisplatin with a hOCT2 inhibitor could lead to reduction in nephrotoxicity [108]. More evidence in favor of the OCT2 transporter as a target for cisplatin-induced nephrotoxicity was uncovered using HEK293 cells transfected with the rat OCT2 transporter. In these cells, cisplatin-induced cytotoxicity was increased by the presence of the rOCT2 transporter, as a direct result of increased platinum uptake. This indicates that rOCT2 expression was a definitive marker of cisplatin-induced nephrotoxicity [109].
Many antioxidant treatments, including tiopronin, N-acetylcysteine pre-treatment and sodium thiosulfate post-treatment. STS post-treatment was time-sensitive, with a 2h delay being protective against nephrotoxicity, and up to 4 h giving otoprotection [110, 111].
Irregular kidney function can result in toxicities in rare cases of carboplatin treatment, usually in patients with renal dysfunction; in contrast, patients with a high glomerular filtration rate can have subtherapeutic systemic concentrations of carboplatin [90].
Neurotoxicity
The dorsal root ganglia of the spinal cord are the primary location of cisplatin damage in the central nervous system (Figure 4) [112]. This explains the primary sensory neuropathy commonly observed in patients treated with cisplatin [112]. Cisplatin-induced neuropathy is characterized by decreased sensory nerve conduction velocity, possibly by acting as a calcium channel blocker [[90]]. Co-treatment of rats with acetyl-L-carnitine was able to protect animals from neurotoxicity while having no effect on the anti-neoplastic activity of cisplatin [113].
Vitamin E has been shown to be decreased in patients treated with cisplatin [114], and vitamin E deficiency causes a sensory neuropathy very similar to that observed with cisplatin treatment [115]. Therefore, vitamin E was tested as a means to protect against cisplatin-induced neuropathy in a controlled clinical trial. One group of patients received vitamin E concomitantly with cisplatin, and for three months following the last cisplatin treatment; the other group received cisplatin as prescribed by dosing recommendations [116]. Neurotoxicity, as measured by a peripheral neuropathy score, was significantly decreased in patients treated with cisplatin plus vitamin E as compared with those treated with cisplatin alone.
Erythropoietin has also been associated with neuroprotection in vivo. In preclinical experiments in rats, erythropoietin was protective against cisplatin-induced neuropathy [117, Bianchi, 2006 #232]118]. A carbamylated derivative of erythropoietin was also tested, to avoid the erythropoietic effects of the parent drug, and it was also shown to be effective as a neuroprotectant for cisplatin neurotoxicity in rats [118]. Carbamylated erythropoietin is currently undergoing further experimentation for long-term side effects, with future clinical trials planned.
Carboplatin is notably less neurotoxic than cisplatin at conventional doses, with a similar sensory neuropathy occurring in approximately 6% of patients [119]. In rare cases, high doses of carboplatin have been shown to result in a sensory ataxia soon after treatment. These patients had all received cisplatin prior to carboplatin, and experienced a mild neuropathy from the first platinating agent [120]. Among gynecologic carcinoma patients treated with a combination of carboplatin and paclitaxel, 25% of patients developed peripheral neurotoxicity [121].
Oxaliplatin neuropathy has a wide spectrum, ranging from an acute sensory neuropathy immediately following treatment to a chronic, dose-limiting neuropathy that usually takes several weeks of treatment to appear. Acute neurotoxicity causes numbness and pain in the distal extremities, and worsens upon exposure to cold temperatures; this is thought to be due to inhibition of voltage-gated sodium currents by oxaliplatin, and may also be due to the presence of free oxalate ions acting as calcium chelators (reviewed in [90]). This neurotoxicity occurs in greater than 95% of patients and can be managed by treatment of calcium gluconate or magnesium sulfate preceding and following treatment [122-124].
The cumulative neuropathy caused by oxaliplatin occurs in approximately 15% of patients, and its reversible symptoms include non-cold-related numbness and pain, sensory loss, and sensory ataxia. Amifostine and glutathione have been used to reduce the severity of this neuropathy, as have the anti-epileptic agents gabapentin and carbamazepine (reviewed in [90, 125]. Chronomodulating the delivery of oxaliplatin also helps to prevent this toxicity (reviewed in [90]).
Myelosuppression
The dose-limiting side effect of carboplatin is myelosuppression, specifically neutropenia and thrombocytopenia (Figure 4). While conventional carboplatin doses result in thrombocytopenia in 20-40% of patients and severe neutropenia in less than 20%, high doses can result in life-threatening toxicity, made more manageable by addition of granulocyte colony stimulating factor (GM-CSF). In the majority of cases, neither cisplatin nor oxaliplatin is associated with severe myelosuppression.
Resistance to Platinating Agents
In ovarian cancers, greater than 70% of patients initially respond to therapy with platinating agents; however, this reprieve is short-lived, as the five-year survival rate for ovarian carcinoma is less than 25% [126]. Similarly, the relapse rate for small cell lung carcinomas after cisplatin or carboplatin treatment is 95% [16]. Head and neck cancers, for which cisplatin is first-line therapy, have only a 20-30% response rate to platinating agents [127]. Resistance can develop as a result of decreased influx or increased efflux of drug, glutathione or metallothionein conjugation, drug detoxification, DNA repair, or skipping lesions during DNA replication (see above) (Figure 3). While it is possible that only one of these mechanisms may lead to resistance to cisplatin, it is more likely that a combination of these mechanisms results in a cisplatin-resistant tumor (Table 1).
Influx/Efflux
A major mechanism of resistance to cisplatin is a decreased effective concentration of drug in the cell (Figure 3, Table 2). Reduction in cisplatin concentration of 20-70% has been observed in cell lines resistant to cisplatin (reviewed in [16]). This can be due either to decreased influx or increased efflux. It has long been presumed that cisplatin and carboplatin are taken up passively by the cell, as uptake is not saturable, nor is it inhibited by structural analogs (reviewed in [2]). Oxaliplatin uptake is most likely passive, as a correlation between hydrophobicity and uptake has been shown (reviewed in [11]). Interestingly, however, ouabain, a small molecule which inhibits the membrane sodium/potassium ATPase pump, blocks cisplatin import, indicating that cisplatin uptake may be dependent on the membrane potential of the cell [128]. Additionally, benzaldehyde and similar aldehyde molecules have been shown to decrease intracellular accumulation of cisplatin by inhibiting uptake. This is thought to occur through formation of Schiff bases with integral membrane transport proteins [129-132].
Cisplatin, at plasma concentrations, not only prevents copper from being transported by the high-affinity copper transporter Ctr1, but also downregulates protein expression of Ctr1 in human ovarian carcinoma cell lines [133]. In comparing sensitive and resistant cell line pairs, the resistant small cell lung carcinoma (SCLC) line SR2 expresses less than half of Ctr1 protein than its sensitive counterpart SCLC [134]. As expected, SR2 cells take up much less cisplatin and carboplatin than SCLC cells [134]. Expression of transfected Ctr1 protein in SR2 cells results in an increase in the uptake rate of cisplatin or copper, but neither is rescued to SCLC levels of uptake, thus indicating that Ctr1 expression alone is not enough to maintain copper (or cisplatin) homeostasis in the cell [134]. More evidence for the role of copper transport in cisplatin sensitivity is based on the observation that cisplatin and copper are competitive inhibitors for the transport of the other molecule into the cell [31, 135] Mouse embryonic fibroblasts (MEF) null for CTR1 provide 3.2-fold increased resistance as compared with transfected cells. Carboplatin resistance is also mediated by CTR1, providing 2-fold resistance in CTR1-/- cells as compared with wildtype. Oxaliplatin is thought to be dependent upon an alternate entry mechanism [136].
Two other copper transporters have also been implicated in resistance to cisplatin - ATP7A and ATP7B are responsible for the export of copper from the cell. ATP7A/B shuttle copper between the Golgi and the plasma membrane. ATP7B was first proposed to be involved in cisplatin resistance when Komatsu, et al., overexpressed this transporter in human epidermoid carcinoma cells and observed that these cells gained resistance to cisplatin as a result of ATP7B expression [137]. Cisplatin accumulation in ATP7B-transfected cells was approximately 60% that observed in cells transfected with empty vector. In human ovarian tumors, SKOV3, OMC6, and PA1 cells exhibited high ATP7B expression levels. SKOV3 cells, which exhibited the highest level of ATP7B expression, also showed the highest resistance to cisplatin [138]. In these cell lines, association of cisplatin with other known drug transporters, including MDR1, MRP1, LRP, and BCRP, was not found.
Additionally, eighty-two primary ovarian carcinomas were profiled for expression of several known resistance genes - including ATP7B, MDR1, MRP1, MRP2, LRP, and BCRP [139]. With the exception of ATP7B, none were indicators for resistance of ovarian cancer to cisplatin. Patients whose carcinomas expressed high levels of ATP7B had a significantly poorer prognosis than patients with tumors that expressed low levels of ATP7B [138]. Additionally, it has been shown that at least one additional resistant ovarian cell line, IGROV-1/CP exhibited increased expression of ATP7B [140].
ATP7A is able to sequester into vesicles not only cisplatin, but also both carboplatin and oxaliplatin. Increased expression of ATP7A in the 2008 line (2008/MNK) leads to increased resistance to all three of these agents; interestingly, overexpression leads to increased sequestration of platinating agents and not to decreased total accumulation [141]. ATP7A is overexpressed as measured by protein levels in some cisplatin-resistant ovarian carcinoma cell lines [140]. In two cell lines, A2780/CP and 2008/C13*5.25 overexpression of ATP7A was shown to be causative for resistance to cisplatin; however, these lines were generated from growth in cisplatin-containing media. Patients whose ovarian tumors exhibited increased or new ATP7A histochemical staining post-platinum treatment were found to have a lower survival rate than patients without increased ATP7A staining post-treatment [140].
Fibroblasts (cell line Me32a, established from a patient with Menkes’ disease) that express neither ATP7A nor ATP7B exhibited much higher accumulation of copper than the same fibroblasts transfected with vectors containing either ATP7A (MeMNK) or ATP7B (MeWND) [142]. Me32a cells were also more sensitive to both cisplatin and carboplatin than lines expressing either ATP7A or ATP7B. Interestingly, however, Me32a cells accumulated less total cellular platinum than either MeMNK or MeWMD cells, indicating that cisplatin in Me32a cells is likely sequestered in cellular vesicles. No difference between the transfected lines and Me32a was observed, however, in total platinum DNA adducts [142]. This suggests that sensitivity to cisplatin-induced cytotoxicity in the cell may involve pathways unrelated to DNA damage, such as ER stress induction.
In addition to the copper transporters, the multi-drug resistance protein (MRP) is thought to function as an ATP-dependent pump for many drugs, including cisplatin [143, 144]. In rat hepatocyte cell lines treated with cisplatin, MRP-2 protein and mRNA levels increased approximately 3-fold upon treatment with cisplatin [145]. Cisplatin also slightly increased the mRNA expression of MRP3, but did not affect MRP1 expression [143, 145]. Using sensitive and resistant melanoma cell lines, MRP2 expression was shown to be upregulated in the cisplatin-resistant cell lines as compared to the sensitive line [146]. The resistant cells also show a decreased level of platinum intrastrand crosslinks on DNA [146]. However, additional studies on liver carcinoma cell lines showed conflicting results. Using cell lines sensitive and resistant to cisplatin, it was observed that expression of both MRP1 and MRP2 were downregulated in the resistant cell lines [147]. While MRP expression may play a role in cisplatin resistance in some tissue types, it does not appear to be a global mechanism by which resistance to cisplatin occurs.
Increased levels of MRP2, ATP7A, and ATP7B have also been associated with decreased lysosomal cellular compartment in the cisplatin-resistant C13*5.25 ovarian carcinoma cell line [148]. This cell line also has decreased protein levels of the lysosome associated proteins (LAMP) 1 and 2. It is obvious that the transport story for platinating agents has yet to be clearly elucidated; however, some transporters may be cell type dependent, and each cell type may have multiple transporters, further complicating the situation.
Glutathione, Metallothioneins, and Anti-oxidants
In the cytoplasm, platinating agents becomes aquated, which then enables them to react with thiol-containing molecules, including glutathione (GSH) and metallothioneins. Increased concentrations of these compounds are known to induce resistance against cisplatin (Figure 3, Table 2) (reviewed in [16]). Glutathione itself acts as an antioxidant of the cell; it helps to maintain the redox environment while maintaining reduced sulfhydryl groups. Cisplatin is detoxified by glutathione through adduct formation [149]. Several ovarian cell lines known to be resistant to cisplatin showed a correlation between the degree of resistance and the levels of GSH, likely due to increased d-glutamylcysteine synthetase [150].
In bladder carcinoma cell lines that are known to be resistant to cisplatin, exposure to buthiomine sulfoximine (BSO), which significantly depleted cellular glutathione concentration, resulted in a significant enhancement in cisplatin cytotoxicity [151]. Additionally, the NSAID (non-steroidal anti-inflammatory drug) indomethacin significantly decreases cellular concentrations of GSH and sensitizes bladder carcinoma cells to cisplatin treatment [151]. However, neither of these treatments sensitizes these cells to the level of their parent sensitive strain, indicating either that glutathione levels are only one component of cisplatin resistance [151], or that the NSAID may have other effects in the cell that prevent complete sensitization.
There have not been consistent observations in attempts to correlate glutathione-S-transferase-π (GST-π) expression and resistance to cisplatin between and among cell lines and clinical tumors. Colon, lung adenocarcinoma, and glioblastoma tumor cell lines [152]; and ovarian [153-155] and head and neck clinical samples [156] do exhibit a correlation between high GST-π levels and cisplatin resistance. However, in other studies of ovarian, cervical, and lung carcinoma, no relationship was evident [157-160]. Another study has shown that ovarian cancers with high expression of GST-π typically have lower survival and a less favorable response to cisplatin. Much like the MRP data, much of the GSH/GST data is conflicting, leading to questions about its importance. While it may have some role in certain types of cancers, it does not appear to be a global indicator of cisplatin resistance.
An additional member of the antioxidant defense system is thioredoxin (Trx), which, similar to glutathione, regulates the oxidation reduction environment of the cell [161]. Thioredoxin is involved in the regulation of transcription factors, apoptosis, and DNA synthesis, among others. Trx is reduced by thioredoxin reductase (TrxR), which involves the oxidation of NADPH [162]. In clinical samples, a correlation has been observed between Trx levels and cisplatin resistance in bladder, prostate, liver, gastic, and colon cancer cells [161]. Both cisplatin and transplatin show this inhibition of TxrR1 [161], as does oxaliplatin but not carboplatin [162]. No study has shown that glutaredoxin (Grx), the analogous enzyme to TrxR in the glutathione system, is inhibited by cisplatin or oxaliplatin; however, the glutathione-cisplatin adduct (GS-Pt) is also able to inhibit both mammalian TrxR1 and Grx [161]. As transplatin is not cytotoxic to cells, inhibition of TxR1 and Grx may be evidence against the importance of Txr in cisplatin resistance.
Metallothioneins (MT) are very low molecular weight proteins comprised of several cysteine and aromatic amino acid residues [163]. Interestingly, metallothioneins are thought to be involved in controlling levels of copper and zinc, as well as protecting cells from oxidative stress and toxicities associated with heavy metals, including copper, cadmium, and zinc [163, 164]. Elevated levels of metallothionein II have been described in cisplatin-resistant cell lines, and cisplatin-resistant bladder cancer lines exhibit cross resistance to the heavy metal cadmium, further implicating MTII in cisplatin resistance [163]. Human bladder cancer xenografts [164] and esophageal [165] and transitional cell primary carcinomas [166] that express high levels of MT exhibit less of a clinical response to cisplatin. In head and neck cancers, cisplatin induces metallothionein expression [167], while in germ cell and testicular tumors, no relationship between MT and cisplatin was observed [168]. The association of MT levels with cisplatin resistance may be tissue specific and may play a minor role depending on the cellular environment.
Summary
The platinating agents remain an important class of anti-cancer agents, with cisplatin and carboplatin used extensively in treating testicular, gynecologic, head and neck, and lung carcinomas, and oxaliplatin becoming a mainstay of colorectal cancer treatment. These agents are characterized by the ability to generate platinum lesions on DNA as their proposed major mode of cytotoxicity. However, clinical problems of tumor resistance and a number of associated toxicities limit these agents from reaching their full potential. Increased DNA repair, either through activation of NER, MMR, and/or HR pathways, in addition to cytoplasmic detoxification of cisplatin are all known mechanisms by which resistance occurs. Newer observations have pointed toward transporters as a mechanism for cisplatin resistance, including the copper transporters CTR1, ATP7A, and ATP7B. Toxicities associated with cisplatin include nephrotoxicity, neurotoxicity, and ototoxicity, any or all of which can be dose-limiting. Ototoxicity is particularly prevalent among pediatric patients treated with platinating agents. Anti-oxidant compounds are being developed to prevent these toxic side effects. Carboplatin and oxaliplatin produce fewer side effects than the parent cisplatin, with myelosuppression and neurotoxicity, respectively, being dose-limiting. The ability to overcome platinating agent resistance in tumors and decrease toxic side effects in patients and/or identifying patients at risk for nonresponse or toxicity will be beneficial to the large numbers of cancer patients who receive these drugs.
4. Abbreviations
- Cisplatin
cis-platinum(II) diammine dichloride
- carboplatin
cyclobutane-1,1-dicarboxylic acid platinum (II)
- oxaliplatin
oxalate (trans-l-1,2-diamminocyclohexane) platinum (II)
- satraplatin
bis(aceto)amminedichloro(cyclohexylamine) platinum (IV)
- trans-DDP
trans-platinum (II) diammine dichloride
- HMG
high mobility group
- NER
nucleotide excision repair
- ER
endoplasmic reticulum
- UPR
unfolded protein response
- AP-1
activator protein 1
- IAP
inhibitor of apoptosis proteins
- MNB
murine neuroblastoma
- Dnmt3b
DNA methyltransferase 3b
- MMR
mismatch repair
- MSI
microsatellite instability
- DSB
double-strand breaks
- CHO
Chinese hamster ovary
- SCLC
small cell lung carcinoma
- ROS
reactive oxygen species
- STS
sodium thiosulfate
- HIF1α
hypoxia inducible factor 1α
- hOCT2
human organic cation transporter 2
- GSH
glutathione
- BSO
buthiomine sulfoximine
- GST
glutathione-S-transferase
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- Grx
glutaredoxin
- MT
metallothionein
- UCN-01
7-hydroxystaurosporine
- RAR
retinoic acid receptors
- RXR
retinoid-X receptors
- AGT
O6-alkylguanine DNA alkyltransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Some of the work in this review was supported by NIH/NCI grant CA81485 and Medical Scientist National Research Service Award Grant 5 T32 GM07281 (C.A.R.).
Correspondence and requests for reprints should be addressed to: M. Eileen Dolan, 5841 S. Maryland Ave., Box MC2115, University of Chicago, Chicago, IL 60637. Phone: (773) 702-4441; Fax: (773) 702-0963; E-mail: edolan@medicine.bsd.uchicago.edu
References
- 1.Rosenberg B, et al. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222(191):385–6. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 2.Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478(12):23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 3.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–20. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 4.Samimi G, et al. Novel mechanisms of platinum drug resistance identified in cells selected for resistance to JM118 the active metabolite of satraplatin. Cancer Chemother Pharmacol. 2006 doi: 10.1007/s00280-006-0271-0. [DOI] [PubMed] [Google Scholar]
- 5.Sternberg CN, et al. Phase III trial of satraplatin, an oral platinum plus prednisone vs. prednisone alone in patients with hormone-refractory prostate cancer. Oncology. 2005;68(1):2–9. doi: 10.1159/000084201. [DOI] [PubMed] [Google Scholar]
- 6.Zorbas H, Keppler BK. Cisplatin damage: are DNA repair proteins saviors or traitors to the cell? Chembiochem. 2005;6(7):1157–66. doi: 10.1002/cbic.200400427. [DOI] [PubMed] [Google Scholar]
- 7.Woynarowski JM, et al. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol Pharmacol. 1998;54(5):770–7. doi: 10.1124/mol.54.5.770. [DOI] [PubMed] [Google Scholar]
- 8.Woynarowski JM, et al. Oxaliplatin-induced damage of cellular DNA. Mol Pharmacol. 2000;58(5):920–7. doi: 10.1124/mol.58.5.920. [DOI] [PubMed] [Google Scholar]
- 9.Faivre S, et al. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem Pharmacol. 2003;66(2):225–37. doi: 10.1016/s0006-2952(03)00260-0. [DOI] [PubMed] [Google Scholar]
- 10.Fuertes MA, Alonso C, Perez JM. Biochemical modulation of Cisplatin mechanisms of action: enhancement of antitumor activity and circumvention of drug resistance. Chem Rev. 2003;103(3):645–62. doi: 10.1021/cr020010d. [DOI] [PubMed] [Google Scholar]
- 11.Di Francesco AM, Ruggiero A, Riccardi R. Cellular and molecular aspects of drugs of the future: oxaliplatin. Cell Mol Life Sci. 2002;59(11):1914–27. doi: 10.1007/PL00012514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misset JL, et al. Oxaliplatin clinical activity: a review. Crit Rev Oncol Hematol. 2000;35(2):75–93. doi: 10.1016/s1040-8428(00)00070-6. [DOI] [PubMed] [Google Scholar]
- 13.Eastman A, Barry MA. Interaction of trans-diamminedichloroplatinum(II) with DNA: formation of monofunctional adducts and their reaction with glutathione. Biochemistry. 1987;26(12):3303–7. doi: 10.1021/bi00386a009. [DOI] [PubMed] [Google Scholar]
- 14.Vaisman A, et al. Effect of DNA polymerases and high mobility group protein 1 on the carrier ligand specificity for translesion synthesis past platinum-DNA adducts. Biochemistry. 1999;38(34):11026–39. doi: 10.1021/bi9909187. [DOI] [PubMed] [Google Scholar]
- 15.Reeves R, Adair JE. Role of high mobility group (HMG) chromatin proteins in DNA repair. DNA Repair (Amst) 2005;4(8):926–38. doi: 10.1016/j.dnarep.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 17.He Q, Liang CH, Lippard SJ. Steroid hormones induce HMG1 overexpression and sensitize breast cancer cells to cisplatin and carboplatin. Proc Natl Acad Sci U S A. 2000;97(11):5768–72. doi: 10.1073/pnas.100108697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandic A, et al. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278(11):9100–6. doi: 10.1074/jbc.M210284200. [DOI] [PubMed] [Google Scholar]
- 19.Breckenridge DG, et al. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22(53):8608–18. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150(4):887–94. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nawrocki ST, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65(24):11658–66. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- 22.Kashani-Sabet M, Wang W, Scanlon KJ. Cyclosporin A suppresses cisplatin-induced c-fos gene expression in ovarian carcinoma cells. J Biol Chem. 1990;265(19):11285–8. [PubMed] [Google Scholar]
- 23.Scanlon KJ, et al. Ribozyme-mediated cleavage of c-fos mRNA reduces gene expression of DNA synthesis enzymes and metallothionein. Proc Natl Acad Sci U S A. 1991;88(23):10591–5. doi: 10.1073/pnas.88.23.10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moorehead RA, Singh G. Influence of the proto-oncogene c-fos on cisplatin sensitivity. Biochem Pharmacol. 2000;59(4):337–45. doi: 10.1016/s0006-2952(99)00333-0. [DOI] [PubMed] [Google Scholar]
- 25.Wilson LA, Yamamoto H, Singh G. Role of the transcription factor Ets-1 in cisplatin resistance. Mol Cancer Ther. 2004;3(7):823–32. [PubMed] [Google Scholar]
- 26.Eliopoulos AG, et al. The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and Bcl-2. Oncogene. 1995;11(7):1217–28. [PubMed] [Google Scholar]
- 27.Li J, et al. Human ovarian cancer and cisplatin resistance: possible role of inhibitor of apoptosis proteins. Endocrinology. 2001;142(1):370–80. doi: 10.1210/endo.142.1.7897. [DOI] [PubMed] [Google Scholar]
- 28.Fujie Y, et al. Oxaliplatin, a potent inhibitor of survivin, enhances paclitaxel-induced apoptosis and mitotic catastrophe in colon cancer cells. Jpn J Clin Oncol. 2005;35(8):453–63. doi: 10.1093/jjco/hyi130. [DOI] [PubMed] [Google Scholar]
- 29.Huang RY, et al. Genome-wide screen identifies genes whose inactivation confer resistance to cisplatin in Saccharomyces cerevisiae. Cancer Res. 2005;65(13):5890–7. doi: 10.1158/0008-5472.CAN-04-4093. [DOI] [PubMed] [Google Scholar]
- 30.Fox ME, Feldman BJ, Chu G. A novel role for DNA photolyase: binding to DNA damaged by drugs is associated with enhanced cytotoxicity in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14(12):8071–7. doi: 10.1128/mcb.14.12.8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishida S, et al. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A. 2002;99(22):14298–302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schenk PW, et al. SKY1 is involved in cisplatin-induced cell kill in Saccharomyces cerevisiae, and inactivation of its human homologue, SRPK1, induces cisplatin resistance in a human ovarian carcinoma cell line. Cancer Res. 2001;61(19):6982–6. [PubMed] [Google Scholar]
- 33.Schenk PW, et al. Anticancer drug resistance induced by disruption of the Saccharomyces cerevisiae NPR2 gene: a novel component involved in cisplatin- and doxorubicin-provoked cell kill. Mol Pharmacol. 2003;64(2):259–68. doi: 10.1124/mol.64.2.259. [DOI] [PubMed] [Google Scholar]
- 34.Qiu YY, Mirkin BL, Dwivedi RS. Inhibition of DNA methyltransferase reverses cisplatin induced drug resistance in murine neuroblastoma cells. Cancer Detect Prev. 2005;29(5):456–63. doi: 10.1016/j.cdp.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Deng HB, et al. Increased expression of dihydrodiol dehydrogenase induces resistance to cisplatin in human ovarian carcinoma cells. J Biol Chem. 2002;277(17):15035–43. doi: 10.1074/jbc.M112028200. [DOI] [PubMed] [Google Scholar]
- 36.Danford AJ, et al. Platinum anticancer drug damage enforces a particular rotational setting of DNA in nucleosomes. Proc Natl Acad Sci U S A. 2005;102(35):12311–6. doi: 10.1073/pnas.0506025102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosmoski JV, Smerdon MJ. Synthesis and nucleosome structure of DNA containing a UV photoproduct at a specific site. Biochemistry. 1999;38(29):9485–94. doi: 10.1021/bi990297h. [DOI] [PubMed] [Google Scholar]
- 38.Suquet C, Smerdon MJ. UV damage to DNA strongly influences its rotational setting on the histone surface of reconstituted nucleosomes. J Biol Chem. 1993;268(32):23755–7. [PubMed] [Google Scholar]
- 39.Zamble DB, et al. Repair of cisplatin--DNA adducts by the mammalian excision nuclease. Biochemistry. 1996;35(31):10004–13. doi: 10.1021/bi960453+. [DOI] [PubMed] [Google Scholar]
- 40.Moggs JG, et al. Differential human nucleotide excision repair of paired and mispaired cisplatin-DNA adducts. Nucleic Acids Res. 1997;25(3):480–91. doi: 10.1093/nar/25.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dabholkar M, et al. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J Clin Invest. 1994;94(2):703–8. doi: 10.1172/JCI117388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Q, et al. Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer Res. 2000;20(2A):645–52. [PubMed] [Google Scholar]
- 43.Dabholkar M, et al. Increased mRNA levels of xeroderma pigmentosum complementation group B (XPB) and Cockayne’s syndrome complementation group B (CSB) without increased mRNA levels of multidrug-resistance gene (MDR1) or metallothionein-II (MT-II) in platinum-resistant human ovarian cancer tissues. Biochem Pharmacol. 2000;60(11):1611–9. doi: 10.1016/s0006-2952(00)00448-2. [DOI] [PubMed] [Google Scholar]
- 44.Metzger R, et al. ERCC1 mRNA levels complement thymidylate synthase mRNA levels in predicting response and survival for gastric cancer patients receiving combination cisplatin and fluorouracil chemotherapy. J Clin Oncol. 1998;16(1):309–16. doi: 10.1200/JCO.1998.16.1.309. [DOI] [PubMed] [Google Scholar]
- 45.Koberle B, et al. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr Biol. 1999;9(5):273–6. doi: 10.1016/s0960-9822(99)80118-3. [DOI] [PubMed] [Google Scholar]
- 46.Welsh C, et al. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int J Cancer. 2004;110(3):352–61. doi: 10.1002/ijc.20134. [DOI] [PubMed] [Google Scholar]
- 47.Hector S, et al. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer Chemother Pharmacol. 2001;48(5):398–406. doi: 10.1007/s002800100363. [DOI] [PubMed] [Google Scholar]
- 48.Reardon JT, et al. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999;59(16):3968–71. [PubMed] [Google Scholar]
- 49.Hara R, Mo J, Sancar A. DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol Cell Biol. 2000;20(24):9173–81. doi: 10.1128/mcb.20.24.9173-9181.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kosmoski JV, Ackerman EJ, Smerdon MJ. DNA repair of a single UV photoproduct in a designed nucleosome. Proc Natl Acad Sci U S A. 2001;98(18):10113–8. doi: 10.1073/pnas.181073398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang D, et al. Nucleotide excision repair from site-specifically platinum-modified nucleosomes. Biochemistry. 2003;42(22):6747–53. doi: 10.1021/bi034264k. [DOI] [PubMed] [Google Scholar]
- 52.Hara R, Sancar A. The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol Cell Biol. 2002;22(19):6779–87. doi: 10.1128/MCB.22.19.6779-6787.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fink D, et al. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996;56(21):4881–6. [PubMed] [Google Scholar]
- 54.Karran P, Offman J, Bignami M. Human mismatch repair, drug-induced DNA damage, and secondary cancer. Biochimie. 2003;85(11):1149–60. doi: 10.1016/j.biochi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Obmolova G, et al. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407(6805):703–10. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 56.Aebi S, et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56(13):3087–90. [PubMed] [Google Scholar]
- 57.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst) 2004;3(89):1091–101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 58.Durant ST, et al. Dependence on RAD52 and RAD1 for anticancer drug resistance mediated by inactivation of mismatch repair genes. Curr Biol. 1999;9(1):51–4. doi: 10.1016/s0960-9822(99)80047-5. [DOI] [PubMed] [Google Scholar]
- 59.Drotschmann K, et al. Mutations in the nucleotide-binding domain of MutS homologs uncouple cell death from cell survival. DNA Repair (Amst) 2004;3(7):729–42. doi: 10.1016/j.dnarep.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 60.Clodfelter JE, M BG, Drotschmann K. MSH2 missense mutations alter cisplatin cytotoxicity and promote cisplatin-induced genome instability. Nucleic Acids Res. 2005;33(10):3323–30. doi: 10.1093/nar/gki646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mesquita B, et al. No significant role for beta tubulin mutations and mismatch repair defects in ovarian cancer resistance to paclitaxel/cisplatin. BMC Cancer. 2005;5:101. doi: 10.1186/1471-2407-5-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aebi S, et al. Resistance to cytotoxic drugs in DNA mismatch repair-deficient cells. Clin Cancer Res. 1997;3(10):1763–7. [PubMed] [Google Scholar]
- 63.Arzimanoglou II, et al. Microsatellite instability differences between familial and sporadic ovarian cancers. Carcinogenesis. 1996;17(9):1799–804. doi: 10.1093/carcin/17.9.1799. [DOI] [PubMed] [Google Scholar]
- 64.Cai KQ, et al. Microsatellite instability and alteration of the expression of hMLH1 and hMSH2 in ovarian clear cell carcinoma. Hum Pathol. 2004;35(5):552–9. doi: 10.1016/j.humpath.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 65.Fujita M, et al. Microsatellite instability and alterations in the hMSH2 gene in human ovarian cancer. Int J Cancer. 1995;64(6):361–6. doi: 10.1002/ijc.2910640602. [DOI] [PubMed] [Google Scholar]
- 66.Geisler JP, et al. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer. 2003;98(10):2199–206. doi: 10.1002/cncr.11770. [DOI] [PubMed] [Google Scholar]
- 67.Haas CJ, et al. Microsatellite analysis in serous tumors of the ovary. Int J Gynecol Pathol. 1999;18(2):158–62. doi: 10.1097/00004347-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 68.Orth K, et al. Genetic instability in human ovarian cancer cell lines. Proc Natl Acad Sci U S A. 1994;91(20):9495–9. doi: 10.1073/pnas.91.20.9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pieretti M, et al. Genetic alterations distinguish different types of ovarian tumors. Int J Cancer. 1995;64(6):434–40. doi: 10.1002/ijc.2910640614. [DOI] [PubMed] [Google Scholar]
- 70.Singer G, et al. Different types of microsatellite instability in ovarian carcinoma. Int J Cancer. 2004;112(4):643–6. doi: 10.1002/ijc.20455. [DOI] [PubMed] [Google Scholar]
- 71.Nehme A, et al. Induction of JNK and c-Abl signalling by cisplatin and oxaliplatin in mismatch repair-proficient and -deficient cells. Br J Cancer. 1999;79(78):1104–10. doi: 10.1038/sj.bjc.6690176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frankenberg-Schwager M, et al. Cisplatin-mediated DNA double-strand breaks in replicating but not in quiescent cells of the yeast Saccharomyces cerevisiae. Toxicology. 2005;212(23):175–84. doi: 10.1016/j.tox.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 73.Adair GM, et al. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. Embo J. 2000;19(20):5552–61. doi: 10.1093/emboj/19.20.5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ivanov EL, Haber JE. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15(4):2245–51. doi: 10.1128/mcb.15.4.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sargent RG, et al. Role of the nucleotide excision repair gene ERCC1 in formation of recombination-dependent rearrangements in mammalian cells. Nucleic Acids Res. 2000;28(19):3771–8. doi: 10.1093/nar/28.19.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sargent RG, et al. Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc Natl Acad Sci U S A. 1997;94(24):13122–7. doi: 10.1073/pnas.94.24.13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Silva IU, et al. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002;30(17):3848–56. doi: 10.1093/nar/gkf479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Groen HJ, et al. Carboplatin- and cisplatin-induced potentiation of moderate-dose radiation cytotoxicity in human lung cancer cell lines. Br J Cancer. 1995;72(6):1406–11. doi: 10.1038/bjc.1995.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Havener JM, et al. Translesion synthesis past platinum DNA adducts by human DNA polymerase mu. Biochemistry. 2003;42(6):1777–88. doi: 10.1021/bi0270079. [DOI] [PubMed] [Google Scholar]
- 80.Hoffmann JS, et al. DNA polymerase beta bypasses in vitro a single d(GpG)-cisplatin adduct placed on codon 13 of the HRAS gene. Proc Natl Acad Sci U S A. 1995;92(12):5356–60. doi: 10.1073/pnas.92.12.5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Masutani C, et al. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. Embo J. 2000;19(12):3100–9. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vaisman A, Chaney SG. The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase beta. J Biol Chem. 2000;275(17):13017–25. doi: 10.1074/jbc.275.17.13017. [DOI] [PubMed] [Google Scholar]
- 83.Vaisman A, et al. Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase eta. Biochemistry. 2000;39(16):4575–80. doi: 10.1021/bi000130k. [DOI] [PubMed] [Google Scholar]
- 84.Albertella MR, Lau A, O’Connor MJ. The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst) 2005;4(5):583–93. doi: 10.1016/j.dnarep.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 85.Bergoglio V, et al. Enhanced expression and activity of DNA polymerase beta in human ovarian tumor cells: impact on sensitivity towards antitumor agents. Oncogene. 2001;20(43):6181–7. doi: 10.1038/sj.onc.1204743. [DOI] [PubMed] [Google Scholar]
- 86.Canitrot Y, et al. Overexpression of DNA polymerase beta in cell results in a mutator phenotype and a decreased sensitivity to anticancer drugs. Proc Natl Acad Sci U S A. 1998;95(21):12586–90. doi: 10.1073/pnas.95.21.12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Horton JK, et al. Strategic down-regulation of DNA polymerase beta by antisense RNA sensitizes mammalian cells to specific DNA damaging agents. Nucleic Acids Res. 1995;23(19):3810–5. doi: 10.1093/nar/23.19.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin X, et al. DNA polymerase zeta accounts for the reduced cytotoxicity and enhanced mutagenicity of cisplatin in human colon carcinoma cells that have lost DNA mismatch repair. Clin Cancer Res. 2006;12(2):563–8. doi: 10.1158/1078-0432.CCR-05-1380. [DOI] [PubMed] [Google Scholar]
- 89.Albertella MR, et al. A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65(21):9799–806. doi: 10.1158/0008-5472.CAN-05-1095. [DOI] [PubMed] [Google Scholar]
- 90.Hartmann JT, Lipp HP. Toxicity of platinum compounds. Expert Opin Pharmacother. 2003;4(6):889–901. doi: 10.1517/14656566.4.6.889. [DOI] [PubMed] [Google Scholar]
- 91.Donzelli E, et al. Neurotoxicity of platinum compounds: comparison of the effects of cisplatin and oxaliplatin on the human neuroblastoma cell line SH-SY5Y. J Neurooncol. 2004;67(12):65–73. doi: 10.1023/b:neon.0000021787.70029.ce. [DOI] [PubMed] [Google Scholar]
- 92.Stachurska A, et al. Cisplatin up-regulates the in vivo biosynthesis and degradation of renal polyamines and c-Myc expression. Biochim Biophys Acta. 2004;1689(3):259–66. doi: 10.1016/j.bbadis.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 93.Wagstaff AJ, et al. Carboplatin. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the treatment of cancer. Drugs. 1989;37(2):162–90. doi: 10.2165/00003495-198937020-00005. [DOI] [PubMed] [Google Scholar]
- 94.Rybak LP, Whitworth CA. Ototoxicity: therapeutic opportunities. Drug Discov Today. 2005;10(19):1313–21. doi: 10.1016/S1359-6446(05)03552-X. [DOI] [PubMed] [Google Scholar]
- 95.Reddel RR, et al. Ototoxicity in patients receiving cisplatin: importance of dose and method of drug administration. Cancer Treat Rep. 1982;66(1):19–23. [PubMed] [Google Scholar]
- 96.Lanvers-Kaminsky C, et al. Continuous or repeated prolonged cisplatin infusions in children: A prospective study on ototoxicity, platinum concentrations, and standard serum parameters. Pediatr Blood Cancer. 2005 doi: 10.1002/pbc.20673. [DOI] [PubMed] [Google Scholar]
- 97.Banfi B, et al. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279(44):46065–72. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 98.Lee JE, et al. Role of reactive radicals in degeneration of the auditory system of mice following cisplatin treatment. Acta Otolaryngol. 2004;124(10):1131–5. doi: 10.1080/00016480410017521. [DOI] [PubMed] [Google Scholar]
- 99.Wang J, et al. Caspase inhibitors, but not c-Jun NH2-terminal kinase inhibitor treatment, prevent cisplatin-induced hearing loss. Cancer Res. 2004;64(24):9217–24. doi: 10.1158/0008-5472.CAN-04-1581. [DOI] [PubMed] [Google Scholar]
- 100.Kalkanis JG, Whitworth C, Rybak LP. Vitamin E reduces cisplatin ototoxicity. Laryngoscope. 2004;114(3):538–42. doi: 10.1097/00005537-200403000-00028. [DOI] [PubMed] [Google Scholar]
- 101.Fetoni AR, et al. Protective effects of alpha-tocopherol and tiopronin against cisplatin-induced ototoxicity. Acta Otolaryngol. 2004;124(4):421–6. doi: 10.1080/00016480410016559. [DOI] [PubMed] [Google Scholar]
- 102.Cassidy J, Misset JL. Oxaliplatin-related side effects: characteristics and management. Semin Oncol. 2002;29(5 Suppl 15):11–20. doi: 10.1053/sonc.2002.35524. [DOI] [PubMed] [Google Scholar]
- 103.Wolfgang GH, et al. Comparative nephrotoxicity of a novel platinum compound, cisplatin, and carboplatin in male Wistar rats. Fundam Appl Toxicol. 1994;22(1):73–9. doi: 10.1006/faat.1994.1010. [DOI] [PubMed] [Google Scholar]
- 104.Ikari A, et al. Sodium-dependent glucose transporter reduces peroxynitrite and cell injury caused by cisplatin in renal tubular epithelial cells. Biochim Biophys Acta. 2005;1717(2):109–17. doi: 10.1016/j.bbamem.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 105.Lieberthal W, Triaca V, Levine J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs. necrosis. Am J Physiol. 1996;270(4 Pt 2):F700–8. doi: 10.1152/ajprenal.1996.270.4.F700. [DOI] [PubMed] [Google Scholar]
- 106.Tanaka T, et al. Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2005;289(5):F1123–33. doi: 10.1152/ajprenal.00081.2005. [DOI] [PubMed] [Google Scholar]
- 107.Gorboulev V, et al. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16(7):871–81. doi: 10.1089/dna.1997.16.871. [DOI] [PubMed] [Google Scholar]
- 108.Ciarimboli G, et al. Cisplatin Nephrotoxicity Is Critically Mediated via the Human Organic Cation Transporter 2. Am J Pathol. 2005;167(6):1477–1484. doi: 10.1016/S0002-9440(10)61234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yonezawa A, et al. Association between tubular toxicity of cisplatin and expression of organic cation transporter rOCT2 (Slc22a2) in the rat. Biochem Pharmacol. 2005;70(12):1823–31. doi: 10.1016/j.bcp.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 110.Dickey DT, et al. Protection against cisplatin-induced toxicities by N-acetylcysteine and sodium thiosulfate as assessed at the molecular, cellular, and in vivo levels. J Pharmacol Exp Ther. 2005;314(3):1052–8. doi: 10.1124/jpet.105.087601. [DOI] [PubMed] [Google Scholar]
- 111.Viale M, et al. Cisplatin combined with tiopronin or sodium thiosulfate: cytotoxicity in vitro and antitumor activity in vivo. Anticancer Drugs. 1999;10(4):419–28. doi: 10.1097/00001813-199904000-00011. [DOI] [PubMed] [Google Scholar]
- 112.Meijer C, et al. Cisplatin-induced DNA-platination in experimental dorsal root ganglia neuronopathy. Neurotoxicology. 1999;20(6):883–7. [PubMed] [Google Scholar]
- 113.Pisano C, et al. Paclitaxel and Cisplatin-induced neurotoxicity: a protective role of acetyl-L-carnitine. Clin Cancer Res. 2003;9(15):5756–67. [PubMed] [Google Scholar]
- 114.Bove L, et al. A pilot study on the relation between cisplatin neuropathy and vitamin E. J Exp Clin Cancer Res. 2001;20(2):277–80. [PubMed] [Google Scholar]
- 115.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford University Press; Oxford: 1993. pp. 188–276. [Google Scholar]
- 116.Argyriou AA, et al. A randomized controlled trial evaluating the efficacy and safety of vitamin E supplementation for protection against cisplatin-induced peripheral neuropathy: final results. Support Care Cancer. 2006 doi: 10.1007/s00520-006-0072-3. [DOI] [PubMed] [Google Scholar]
- 117.Orhan B, et al. Erythropoietin against cisplatin-induced peripheral neurotoxicity in rats. Med Oncol. 2004;21(2):197–203. doi: 10.1385/MO:21:2:197. [DOI] [PubMed] [Google Scholar]
- 118.Bianchi R, et al. Protective effect of erythropoietin and its carbamylated derivative in experimental Cisplatin peripheral neurotoxicity. Clin Cancer Res. 2006;12(8):2607–12. doi: 10.1158/1078-0432.CCR-05-2177. [DOI] [PubMed] [Google Scholar]
- 119.Canetta R, Rozencweig M, Carter SK. Carboplatin: the clinical spectrum to date. Cancer Treat Rev. 1985;12(Suppl A):125–36. doi: 10.1016/0305-7372(85)90027-1. [DOI] [PubMed] [Google Scholar]
- 120.Heinzlef O, Lotz JP, Roullet E. Severe neuropathy after high dose carboplatin in three patients receiving multidrug chemotherapy. J Neurol Neurosurg Psychiatry. 1998;64(5):667–9. doi: 10.1136/jnnp.64.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Markman M, et al. Neurotoxicity associated with a regimen of carboplatin (AUC 5-6) and paclitaxel (175 mg/m2 over 3 h) employed in the treatment of gynecologic malignancies. J Cancer Res Clin Oncol. 2001;127(1):55–8. doi: 10.1007/s004320000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gamelin E, et al. Clinical aspects and molecular basis of oxaliplatin neurotoxicity: current management and development of preventive measures. Semin Oncol. 2002;29(5 Suppl 15):21–33. doi: 10.1053/sonc.2002.35525. [DOI] [PubMed] [Google Scholar]
- 123.Grolleau F, et al. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J Neurophysiol. 2001;85(5):2293–7. doi: 10.1152/jn.2001.85.5.2293. [DOI] [PubMed] [Google Scholar]
- 124.Gamelin E, Gamelin L, Delva R. Prevention of oxaliplatin peripheral sensory neuropathy by calcium + gluconate/Mg + chloride infusions: a retrospective study. Proc. Am. Soc. Clin. Oncol. 2002 [Google Scholar]
- 125.Grothey A. Clinical management of oxaliplatin-associated neurotoxicity. Clin Colorectal Cancer. 2005;5(Suppl 1):S38–46. doi: 10.3816/ccc.2005.s.006. [DOI] [PubMed] [Google Scholar]
- 126.Tewari KS, Monk BJ. Gynecologic oncology group trials of chemotherapy for metastatic and recurrent cervical cancer. Curr Oncol Rep. 2005;7(6):419–34. doi: 10.1007/s11912-005-0007-z. [DOI] [PubMed] [Google Scholar]
- 127.Jacobs C, et al. A phase III randomized study comparing cisplatin and fluorouracil as single agents and in combination for advanced squamous cell carcinoma of the head and neck. J Clin Oncol. 1992;10(2):257–63. doi: 10.1200/JCO.1992.10.2.257. [DOI] [PubMed] [Google Scholar]
- 128.Andrews PA, et al. cis-Diamminedichloroplatinum(II) accumulation in sensitive and resistant human ovarian carcinoma cells. Cancer Res. 1988;48(1):68–73. [PubMed] [Google Scholar]
- 129.Dornish JM, Melvik JE, Pettersen EO. Reduced cellular uptake of cis-dichlorodiammine-platinum by benzaldehyde. Anticancer Res. 1986;6(4):583–8. [PubMed] [Google Scholar]
- 130.Dornish JM, Pettersen EO. Protection from cis-dichlorodiammineplatinum-induced cell inactivation by aldehydes involves cell membrane amino groups. Cancer Lett. 1985;29(3):235–43. doi: 10.1016/0304-3835(85)90133-8. [DOI] [PubMed] [Google Scholar]
- 131.Dornish JM, Pettersen EO. Modulation of cis-dichlorodiammineplatinum(II)-induced cytotoxicity by benzaldehyde derivatives. Cancer Lett. 1989;46(1):63–8. doi: 10.1016/0304-3835(89)90216-4. [DOI] [PubMed] [Google Scholar]
- 132.Pettersen EO, et al. Antitumour effect of benzylidene-glucose (BG) in rats with chemically induced hepatocellular carcinoma. Anticancer Res. 1986;6(2):147–52. [PubMed] [Google Scholar]
- 133.Holzer AK, et al. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin Cancer Res. 2004;10(19):6744–9. doi: 10.1158/1078-0432.CCR-04-0748. [DOI] [PubMed] [Google Scholar]
- 134.Song IS, et al. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol Cancer Ther. 2004;3(12):1543–9. [PubMed] [Google Scholar]
- 135.Holzer AK, et al. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol Pharmacol. 2004;66(4):817–23. doi: 10.1124/mol.104.001198. [DOI] [PubMed] [Google Scholar]
- 136.Holzer AK, Manorek GH, Howell SB. The contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin and oxaliplatin. Mol Pharmacol. 2006 doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 137.Komatsu M, et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000;60(5):1312–6. [PubMed] [Google Scholar]
- 138.Nakayama K, et al. Expression and cisplatin sensitivity of copper-transporting P-type adenosine triphosphatase (ATP7B) in human solid carcinoma cell lines. Oncol Rep. 2001;8(6):1285–7. doi: 10.3892/or.8.6.1285. [DOI] [PubMed] [Google Scholar]
- 139.Nakayama K, et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) as a cisplatin based chemoresistance marker in ovarian carcinoma: comparative analysis with expression of MDR1, MRP1, MRP2, LRP and BCRP. Int J Cancer. 2002;101(5):488–95. doi: 10.1002/ijc.10608. [DOI] [PubMed] [Google Scholar]
- 140.Katano K, et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62(22):6559–65. [PubMed] [Google Scholar]
- 141.Samimi G, et al. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin Cancer Res. 2004;10(14):4661–9. doi: 10.1158/1078-0432.CCR-04-0137. [DOI] [PubMed] [Google Scholar]
- 142.Samimi G, et al. Modulation of the cellular pharmacology of cisplatin and its analogs by the copper exporters ATP7A and ATP7B. Mol Pharmacol. 2004;66(1):25–32. doi: 10.1124/mol.66.1.25. [DOI] [PubMed] [Google Scholar]
- 143.Ishikawa T, et al. Coordinated induction of MRP/GS-X pump and gamma-glutamylcysteine synthetase by heavy metals in human leukemia cells. J Biol Chem. 1996;271(25):14981–8. doi: 10.1074/jbc.271.25.14981. [DOI] [PubMed] [Google Scholar]
- 144.Lautier D, et al. Multidrug resistance mediated by the multidrug resistance protein (MRP) gene. Biochem Pharmacol. 1996;52(7):967–77. doi: 10.1016/0006-2952(96)00450-9. [DOI] [PubMed] [Google Scholar]
- 145.Schrenk D, et al. Up-regulation of transporters of the MRP family by drugs and toxins. Toxicol Lett. 2001;120(13):51–7. doi: 10.1016/s0378-4274(01)00306-x. [DOI] [PubMed] [Google Scholar]
- 146.Liedert B, et al. Overexpression of cMOAT (MRP2/ABCC2) is associated with decreased formation of platinum-DNA adducts and decreased G2-arrest in melanoma cells resistant to cisplatin. J Invest Dermatol. 2003;121(1):172–6. doi: 10.1046/j.1523-1747.2003.12313.x. [DOI] [PubMed] [Google Scholar]
- 147.Shen DW, et al. Decreased accumulation of [14C]carboplatin in human cisplatin-resistant cells results from reduced energy-dependent uptake. J Cell Physiol. 2000;183(1):108–16. doi: 10.1002/(SICI)1097-4652(200004)183:1<108::AID-JCP13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 148.Safaei R, et al. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol Cancer Ther. 2005;4(10):1595–604. doi: 10.1158/1535-7163.MCT-05-0102. [DOI] [PubMed] [Google Scholar]
- 149.Rudin CM, et al. Inhibition of glutathione synthesis reverses Bcl-2-mediated cisplatin resistance. Cancer Res. 2003;63(2):312–8. [PubMed] [Google Scholar]
- 150.Godwin AK, et al. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci U S A. 1992;89(7):3070–4. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Byun SS, et al. Augmentation of cisplatin sensitivity in cisplatin-resistant human bladder cancer cells by modulating glutathione concentrations and glutathione-related enzyme activities. BJU Int. 2005;95(7):1086–90. doi: 10.1111/j.1464-410X.2005.05472.x. [DOI] [PubMed] [Google Scholar]
- 152.Goto S, Kamada K, Soh Y, Ihara Y, Kondo T. Significance of nulcear glutathione S-transferase pi in resistance to anti-cancer drugs. Jpn J Cancer Res. 2002;93:1047–1056. doi: 10.1111/j.1349-7006.2002.tb02482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mayr D, Pannekamp U, Baretton GB, Gropp M, Meier W, Flens MJ, Scheper R, Diebold J. Immunohistochemical analysis of drug resistance-associated proteins in ovarian carcinomas. Pathol Res Pract. 2000;196:469–475. doi: 10.1016/S0344-0338(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 154.Satoh T, Nishida M, Tsunoda H, Kubo T. Expression of glutathione S-transferase pi (GST-pi) in human ovarian tumors. Eur J Obstet Gynecol Reprod Biol. 2001;96:202–208. doi: 10.1016/s0301-2115(00)00473-5. [DOI] [PubMed] [Google Scholar]
- 155.Surowiak P, Materna V, Kaplenko I, Spaczynski M, Dietel M, Lage H, Zabel M. Augemented expression of metallothionein and glutathione S-transferase pi as unfavourable prognostic factors in cisplatin-treated ovarian cancer patients. Virchows Arch. 2005;447:626–633. doi: 10.1007/s00428-005-1228-0. [DOI] [PubMed] [Google Scholar]
- 156.Nishimura T, Newkirk K, Session RB, ANdrews PA, Trock BJ, Rasmussen AA, Montgomery EA, Bischoff EK, Cullen KJ. Immunohistochemical staining for glutathione S-transferase predicts response to platinum-based chemotherapy in head and neck cancer. Clin Cancer Res. 1996;2:1859–1865. [PubMed] [Google Scholar]
- 157.Ikeda K, Sakai K, Yamamotot R, Hareyama H, Tsumura N, Watari H, Shimizus M, Minanami H, Sakuragi N. Multivariate analysis for prognostic significance of histologica subtype, GST-pi, MDR-1, and p53 in stages II-IV ovarian cancer. Int J Gynecol Cancer. 2003;13:776–786. doi: 10.1111/j.1525-1438.2003.13381.x. [DOI] [PubMed] [Google Scholar]
- 158.Van der Zee AGJ, Hollema H, Suurmeijer AJH, Krans M, Sluiter WJ, WIlemse PHB, Aalders JG, Vries EGE. Value of P-glycoprotein, glutathione-S-transferase pi, c-erbB2, and p53 as prognostic factors in ovarian carcinomas. J Clin Oncol. 1995;13:70–78. doi: 10.1200/JCO.1995.13.1.70. [DOI] [PubMed] [Google Scholar]
- 159.Konishi I, Nanbu K, Mandai M, Tsuruta Y, Kataoka N, Nagata Y. Tumorr esponse to neoadjuvant chemotherapy correlates witht he expression of P-glycoprotein and PCNA but not GST-pi in the tumor cells of cervical carcinoma. Gynecol Oncol. 1998;70:365–371. doi: 10.1006/gyno.1998.5077. [DOI] [PubMed] [Google Scholar]
- 160.Miyatake K, Gemba K, Ueoka H, Mishii K, Kiura K, Tabata M, Shibayama T, Takigawa N, Kawaraya M, Tanimoto M. Prognostic significance of mutant p53 protein, P-glycoprotein and glutathione S-transferase-pi in patients with unresectable non-small cell lung cancer. Anticancer Res. 2003;23:2829–2836. [PubMed] [Google Scholar]
- 161.Arner ESJ, Nakamura H, Sasada T, Yodoi J, HOmgren A, Spyrou G. Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase; glutaredoxin by cis-diamminidischloroplatinum (II) and its major metabolite, the glutathione-platinum complex. Free Radic. Biol. Med. 2001;31:1170–1178. doi: 10.1016/s0891-5849(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 162.Witte AB, et al. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic Biol Med. 2005;39(5):696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 163.Siegsmund MJ, Marx C, Seeman O, Schummer B, Steidler A, Toktomambetova L, Kohrmann K-U, Rassweiler J, Alken P. Cisplatin-resistant bladder carcinoma cells: enhanced expression of metallotioneins. Urol Res. 1999;27:157–163. doi: 10.1007/s002400050103. [DOI] [PubMed] [Google Scholar]
- 164.Toyoda H, Mizushima T, Satoh T, Izuka N, Nomoto A, Chiba H, Mita M, Naganuma A, Himeno S, Imura N. HeLa cell transformants overproducing mouse metallothionein show in vivo resistant to cis-platinum in nude mice. Jpn J Cancer Res. 2000;91:91–98. doi: 10.1111/j.1349-7006.2000.tb00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hishikawa Y, Koji T, Dhar DK, Kinugasa S, Yamaguchi M, Nagasue N. Metallothionein expression correlates with metastatic and proliferativ epotential in squamous cell carcinoma of the esophagus. Br J Cancer. 1999;81:712–720. doi: 10.1038/sj.bjc.6690753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Siu LL, Banerjee D, Khurana RJ, Pan X, Pflueger R, Tannock IF. The prognostic role of p53, metallothionein, P-glycoprotein, and MIB-1 in muslce-invasive urothelial transitional cell carcinoma. Clin Cancer Res. 1998;4:559–564. [PubMed] [Google Scholar]
- 167.Muramatsu Y, Yasegawa Y, Fukano H, Ogawa T, Namuba M, Mour S, Fujimoto Y, Matsuura H, Takai Y, Mori M. Metallothionein immunoreactivitiy in head and neck carcinomas - special reference to clinical behaviors and chemotherapy responses. Anticancer Res. 2000;20:257–264. [PubMed] [Google Scholar]
- 168.Meijer C, TImmer A, DeVries EG, Groten JP, Knol A, Zwart N, Sleijefer DT, Mulder NH. Role of metallothionein in cisplatin sensitivity of germ-cell tumors. Int J Cancer. 2000;85(6):777–781. doi: 10.1002/(sici)1097-0215(20000315)85:6<777::aid-ijc6>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 169.Itamochi H, et al. Mechanisms of cisplatin resistance in clear cell carcinoma of the ovary. Oncology. 2002;62(4):349–53. doi: 10.1159/000065067. [DOI] [PubMed] [Google Scholar]