

Abstract
Background & Aims
Each cholangiocte in the biliary tree has a primary cilium extending from the apical plasma membrane into the ductal lumen. While the physiological significance of cholangiocyte cilia is unknown, studies in renal epithelia suggest that primary cilia possess sensory functions. Here, we tested the hypothesis that cholangiocyte cilia are sensory organelles that detect and transmit luminal bile flow stimuli into intracellular Ca2+ ([Ca2+]i) and cAMP signaling.
Methods
Scanning, transmission electron and immunofluorescent confocal microscopy of rat isolated intrahepatic bile duct units (IBDUs) were used to detect and characterize cholangiocyte cilia. The fluid flow-induced changes in Ca2+ and cAMP levels in cholangiocytes of microperfused IBDUs were detected by epifluorescence microscopy and a fluorescence assay, respectively.
Results
In intrahepatic bile ducts with a luminal diameter of approximately 100 μm, cilia are 6.68±1.67 μm in length and 0.23±0.02 μm in diameter. In microperfused IBDUs, luminal fluid flow induces a 37.5±3.1% increase in [Ca2+]i and causes suppression of the forskolin-induced cAMP increase. The fluid flow-induced changes in [Ca2+]i and cAMP levels were significantly reduced or abolished when cilia were removed by chloral hydrate or when ciliary-associated proteins polycystin-1 (PC-1), a mechanoreceptor, polycystin-2 (PC-2), a Ca2+ channel, and the Ca2+-inhibitable adenylyl cyclase isoform 6 (AC6), were individually down-regulated by siRNAs.
Conclusions
Cholangiocyte cilia are sensory organelles containing PC-1, PC-2 and AC6 through which luminal fluid flow affects both [Ca2+]i and cAMP signaling in the cell. The data suggest a new model for regulation of ductal bile secretion involving cholangiocyte cilia.
Introduction
The primary cilium is a common, solitary, non-motile, long, tubular organelle extending from the plasma membrane of the cell. With few exceptions (e.g., nucleated blood cells, adipocytes, hepatocytes), primary cilia are ubiquitous cell organelles in vertebrates.1-3 While the existence of primary cilia has been known for over a century, interest of biomedical scientists in these organelles has increased only recently triggered by three critical observations; first, that primary cilia in the node of gastrulation-stage embryos are essential for the determination of left-right asymmetry of the body;4, 5 second, that the two most frequent lethal genetic disorders (i.e., autosomal dominant polycystic kidney disease [ADPKD] and autosomal recessive polycystic kidney disease [ARPKD]), both characterized by progressive cyst development in kidney, liver and pancreas, are cilia-related diseases;6-13 and third, that Bardet-Biedl syndrome (patients with this syndrome have kidney failure, lose their eyesight, are obese and develop diabetes) is a result of mutations in genes, which determine ciliary structure and function, i.e., is a cilia-related disease.14-15 Thus, it has become evident that primary cilia are functionally important organelles that are involved in both normal developmental and pathological processes.
It has also been recently discovered that ADPKD is a result of mutations in PKD1 or PKD2, genes encoding polycystin-1 (PC-1), a cell surface receptor, and polycystin-2 (PC-2), a Ca2+ channel, two integral membrane proteins normally localized to primary cilia. ARPKD is a result of mutations in PKHD1, the gene encoding fibrocystin, an integral membrane protein with unknown functions, which is normally also localized to primary cilia. When these genes are mutated, the absence of their protein products results in ciliary dysfunction and cyst formation.6-11
Development of cysts in the liver is the most frequent extra-renal manifestation in both ADPKD and ARPKD.10-13 Although in ADPKD the liver generally functions normally, the progressive increase in size of the polycystic liver causes abdominal pain, early postprandial fullness and/or shortness of breath, and thus significantly affects quality of life.12 In ARPKD, which belongs to a group of congenital hepatorenal fibrocystic syndromes, approximately 30% of affected neonates die because of polycystic kidneys. In survivors, hepatic lesions become progressively more severe with age and polycystic liver then becomes the major cause of morbidity and mortality.12-13 Currently, there are no effective medical therapies for polycystic liver for at least two reasons: (i) very little is known about the mechanisms of hepatic cyst development and progression; and (ii) nothing is known about the physiological and pathophysiological functions of primary cilia in cholangiocytes, the cells from which the liver cysts originate as a potential result of an increased cholangiocyte proliferation and abnormal fluid secretion.
In contrast, in last four years, considerable new information has been generated regarding the physiological functions of primary cilia in renal epithelia.2, 16-21 As shown in MDCK cells, a cultured cell line derived from the collecting duct of canine kidney, bending of a single cilium by a micropipette or by alterations in perfusate flow rates increases [Ca2+]i while pharmacological removal of cilia abolishes the flow-induced [Ca2+]i increase.16, 17 Similar results (i.e., interruption of a flow-induced [Ca2+]i signaling response in the renal epithelial cells) were obtained in mice with mutated Pkd1 and Pkd2, genes encoding PC-1 and PC-2, respectively, and in wild type renal epithelial cells preincubated with antibodies raised against extracellular but not intracellular loops of PC-1 and PC-2.18, 19 Finally, an acute increase in tubular fluid flow rates in microperfused cortical collecting ducts of the rabbit and mouse nephrons also led to an increase in [Ca2+]i with the involvement of primary cilia.20, 21 Thus, the conclusion was made that one of the physiological functions of primary cilia in kidney is to detect fluid flow over the surface of the tubular epithelial cells and transmit mechanostimuli into an [Ca2+]i signaling response.2
Based on these observations and given the importance of both [Ca2+]i and cAMP signaling in biliary epithelia, we hypothesized that cholangiocyte cilia may also act as mechanosensors that monitor and transmit luminal bile flow stimuli into integrated intracellular Ca2+ and cAMP signaling. The mechanosensory function of cholangiocyte cilia was addressed directly by using a microperfused rat intrahepatic bile duct unit model (IBDU) that allows controlled manipulation of luminal fluid flow rates, detection of both [Ca2+]i and cAMP levels, and effective utilization of gene silencing by siRNAs.22-24
Material and Methods
Materials
All chemicals were of highest purity commercially available and were purchased from Sigma Chemical Co. (St. Louis, MO) unless otherwise indicated.
Animals
Male Fisher 344 rats (225-250 g) were obtained from Harlan Sprague Dawley Inc. (Indianapolis, IN), housed in temperature-controlled room (22° C) with 12 hour light-dark cycles and maintained on a standard diet with free access to water. All experimental procedures were approved by the Animal Use and Care Committee of the Mayo Foundation.
Solutions
The composition of standard Ringer-HCO3 buffer (KRB) (in mM) was: 120.0 NaCl, 5.9 KCl, 1.2 Na2HPO4, 1.0 MgSO4, 25.0 NaHCO3, 1.25 CaCl2, 5.0 d-glucose, pH 7.4. In Ca2+-free KRB, CaCl2 was removed and 1 mM EGTA added, pH 7.4.
Isolated intrahepatic bile duct units (IBDUs)
IBDUs ranging in luminal diameter from 100-125 μm and in length from 1.0-1.5 mm, were isolated from normal rat liver as we previously described.22
Scanning (SEM) and Transmission (TEM) Electron Microscopy
For SEM and TEM of isolated IBDUs, samples were prepared as we previously described.10 SEM observation was performed with a Hitachi S-4700 microscope (Hitachi Inc., Pleasanton, CA); TEM observation was performed with a Jeol 1200 electron microscope (Jeol USA Inc., Peabody, MA). The length of cilia was measured by NIH Images Program ImageJ. To test directly whether luminal fluid flow affects cholangiocyte cilia, IBDUs were fixed immediately after perfusion (i.e., flow was stopped, IBDUs were removed from the perfusion apparatus and put in 2.5% phosphate-buffered glutaraldehyde for 1 hour, rinsed with phosphate buffer three times, post-fixed in 1% osmium tetroxide for 1 hour on ice), and then prepared for SEM as we previously described.10 The position and the angle of bent cilia then were measured blindly on SEM images of 23 cilia in 5 control (i.e., no fluid flow) IBDUs and 21 cilia in 5 IBDUs exposed to luminal fluid flow. Depending on magnification, SEM images contained from 3 to 15 cilia; all of cilia present on SEM images were analyzed.
Immunofluorescence confocal microscopy
Immunofluorescence microscopy was performed with a confocal microscope Zeiss LSM 510 (Carl Zeiss Inc., Thornwood, NY) with a 100x Pan-Apochromat 1.4 nm oil objective and epifluorescent Nikon Eclipse TE300 microscope as we previously described.10 Samples were incubated with antibodies against acetylated α-tubulin (1:200) (Sigma Chemical Co., St. Louis, MO), PC-1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), PC-2 (Zymed Laboratories Inc., South San Francisco, CA) and AC5 and AC6 (FabGennix Inc. International, Frisco, TX) for 2 hours followed by incubation for 1 hour with secondary antibodies (1:200) (Molecular Probes, Eugene, OR). Nuclei were stained with 4’, 6-diamidino-2-phenylindole (DAPI). Corresponding negative controls (i.e., no primary antibodies) were performed in all experiments and showed no positive labeling of proteins of interest.
Gene silencing by siRNAs against Pkd1, Pkd2, Adcy5 and Adcy6
The sequence of the full-length rat Pkd1 was assembled from available rat genomic sequences (contig XM340765) by BLAT analysis with mouse Pkd1 cDNA sequence. The rat Pkd2 mRNA sequence was found by performing a BLAT search on the UCSC Genomic server using the mouse Pkd2 mRNA (accession # NM 008861). The rat Pkd2 sequence shares 93% homology with the mouse Pkd2 mRNA sequence. The rat Adcy5 sequence (accession # NM_022600) and rat Adcy6 sequence (accession # NM_012821) were extracted from the NCBI database. A target site within the gene was chosen and searched with NCBI BLASTN to confirm specificity to Pkd1, Pkd2, Adcy5 and Adcy6. The siRNAs were prepared by a transcription-based method (Silencer siRNA construction kit, Ambion, Austin, TX). Both 29-mer sense and antisense DNA oligonucleotide templates were synthesized by the Mayo Molecular Core Facility. Scrambled siRNAs to Pkd1, Pkd2, Adcy5 and Adcy6 contain the same percent of base pairs but in random sequence. Isolated IBDUs were cultured for 24 hours in NRC medium with 20 nM of functional siRNA or corresponded scrambled siRNA by procedure that we previously described.24 For quantitation of Pkd1, Pkd2, Adcy5 and Adcy6 message after siRNAs, a standard curve was generated by amplifying Pkd1, Pkd2, Adcy5 and Adcy6 from freshly isolated cholangiocyte cDNA with rat specific primers: (Pkd1, sense 5’-ATCACCTTTTTCCCATACCCC-3’ and antisense 5’-TGACTCCACAGAAGCATTGAC-3’), (Pkd2, sense 5’-CAAAACATGACCTGCATTACC-3’ and antisense 5’-CCTTCCATGAACTTCCAGAAC-3’), (Adcy5, sense 5’-ATCGAGCTCATCTACGTGC-3’ and antisense 5’-AGCATGCAGATACAGAGCC-3’), (Adcy6, sense 5’-CTGCTTGTGTTCATCTCTG-3’ and antisense 5’-GACGCTAAGCAGTAGATCA-3’). The 282-bp, 241-bp, 201-bp and 172-bp amplicons were eletrophoresed on a 1.5 % agarose gel, visualized with ethidium bromide, gel-extracted and used as a template for the Real time-PCR for Pkd1, Pkd2, Adcy5 and Adcy6, recpectively, and normalized with 18S ribosome (Ambion, Austin, TX) using Lightcycler (Roche Diagnostic, Germany).
Measurement of [Ca2+]i in cholangiocytes of microperfused IBDUs
Changes in cytosolic Ca2+ [Ca2+]i in cholangiocytes of microperfused IBDUs in response to luminal flow of 1 to 3 μl/sec applied for 60 seconds, were detected using the cell-permeant Ca2+-sensitive fluorescent probe, Fluo-4 AM (Molecular Probes, Eugene, OR), as we previously described in detail.23 To address the effect of chloral hydrate on [Ca2+]i signaling in cholangiocytes, IBDUs were perfused through their lumen with 100 μM ATP and changes in [Ca2+]i were detected as we previously described.23
Measurement of cAMP in cholangiocytes of microperfused IBDUs
After equilibration of an IBDU in the perfusion chamber for 30 min, we stimulated them with forskolin (FSK) by adding 100 μM FSK into the bathing KRB containing 0.5 mM IBMX, an inhibitor of phosphodiesterases, and simultaneously perfused the lumen of an IBDU with standard KRB with a flow rate of 3μl/sec for 60 seconds. For the next 14 minutes, IBDUs remained in the bathing KRB containing forskolin and IBMX; then we removed them from the microperfusion system and measured the cAMP level. The FSK-stimulated increase in cAMP levels in IBDUs treated with IBMX was approximately three times higher than in FSK-stimulated IBDUs without IBMX, suggesting that treatment of IBDUs with IBMX prevented degradation of cAMP in cholangiocytes The concentrations of cAMP were determined by the Bridge-It™ cAMP designer cAMP assay (Mediomics, LLC, St. Louis, MO). Results were expressed in pmol per? g DNA. The amount of DNA in a single IBDU was determined by employing the DNeasy Tissue Kit (QIAGEN Inc., Valencia, CA).
Statistical analysis
All values are expressed as mean ± SE. Statistical analysis was performed by the Student’s t-text, and results were considered statistically different at P<0.05.
Results
Primary cilia in intrahepatic bile ducts exposed to luminal fluid flow
Normally, cholangiocytes have a single primary cilium extending from the apical plasma membrane into the ductal lumen (Figure 1A,B). In intrahepatic bile ducts with a luminal diameter of approximately 100 μm, cilia were 6.68±1.67 μm in length and 0.23±0.02 μm in diameter being longer than in renal tubular cells (i.e., 2-3 μm)19 and comparable to primary cilia in MDCK cells (i.e., ∼8 μm)16 which had previously been used as models for testing the sensory function of primary cilia. In microperfused IBDUs, luminal fluid flow affects cilia causing the bending of these organelles through an angle of 72±7° and 2.7±0.2 μm above the cell surface (Figure 1C). The length of cilia exposed to luminal fluid flow varied from 5.5 μm to 7.5 μm (i.e., on average 6.65±0.29 μm). The distance above the cell surface at which ciliary bending occurred varied from 1.75 μm to 3.5 μm. The number of cilia bent by luminal flow varied from 63% to 100% (i.e., on average, 72.6±8.6% vs 4.8±3.0% in control).
Figure 1.

Primary cilia in rat intrahepatic bile ducts. TEM (A) and SEM (B, C) images of the primary cilium extending from the cholangiocyte apical plasma membrane. (A and B) A completely straight cilium projects into the lumen of an intrahepatic bile duct in the absence of fluid flow. (A) Structurally, the primary cilium consists of a microtubule-based axoneme covered by a specialized plasma membrane, and a basal body, which is derived from a mature mother centriole. The axoneme of a primary cilium has ‘9+0’ microtubule pattern (A, inset). (C) A SEM image of ciliary bending by luminal fluid flow moving in microperfused IBDUs from right to left. A cilium bent through an angle of approximately 60° (dashed lines) with respect to the axoneme axis and at position of 2.7 μm above the cell surface. Scale bar, 1 μm.
Luminal flow induces ciliary-dependent changes in [Ca2+]i and cAMP in cholangiocytes of microperfused IBDUs
In IBDUs exposed to alterations in luminal fluid flow rates from 1 μl/sec to 3 μl/sec, relative Fluo-4 fluorescence (a reflection of [Ca2+]i in cholangiocytes) increased (Figures 2A and 2B). The amplitude of the fluid flow-induced Ca2+ signaling response was dependent on the 5perfusion rate: cholangiocytes develop a maximal increase in Fluo-4 fluorescence of 37.5 ± 3.1% at perfusion rate of 3 μl/sec (Figure 2B). The [Ca2+]i peaked within 10-15 seconds from the initial increase of luminal perfusion rate and remained elevated above baseline for 30-40 seconds after the peak was reached (Figure 2A), which is in agreement with a previously published observation on the fluid flow-induced ciliary-mediated changes in [Ca2+]i in renal epithelial cells.18
Figure 2.

Luminal flow induces changes in [Ca2+]i in cholangiocytes of microperfused IBDUs. (A) A representative tracing of Fluo-4 fluorescence in response to luminal flow. (B) The maximal increase in Fluo-4 fluorescence was observed when IBDUs were exposed to an acute increase of luminal flow of 3 μl/sec. (C) The flow-induced increase in Fluo-4 fluorescence was suppressed in IBDUs perfused with Ca2+-free solutions or pretreated with thapsigargin. (n=6-8 IBDUs in each group, *, P<0.05).
Data in Figure 2C show that the fluid flow-induced increase in [Ca2+]i was abolished when IBDUs were perfused with Ca2+-free perfusate and bath solutions (i.e., 1.7±1.0% compared to 31.1±3.3% in control), and was reduced by 2.6-fold (i.e., to 12.1±3.3% compared to 31.1±3.3% in control) when intracellular IP3-sensitive Ca2+ stores were depleted by thapsigargin. Taken together, these data suggest that mechanostimuli generated by luminal fluid flow are transmitted into cholangiocyte Ca2+ signaling responses and that both extracellular and intracellular Ca2+ sources are involved in this process.
To determine whether the luminal fluid flow affects cAMP signaling, studies were performed on microperfused IBDUs exposed to a perfusion rate that induced a maximal increase in [Ca2+]i (i.e., 3 μl/sec). Initially, we measured cAMP in cholangiocytes of isolated nonperfused IBDUs (i.e., basal cAMP level), and in nonperfused IBDUs exposed to forskolin (FSK, 100 μM), a cell permeable diterpenoid isolated from Coleus forskohlii, which has the unique ability to stimulate most isoforms of adenylyl cyclase resulting in increased intracellular cAMP (i.e., FSK-stimulated cAMP level). The data in Figure 3 show that the basal level of cAMP in cholangiocytes was 2.89±0.35 pmol/μg DNA whereas in FSK-stimulated cholangiocytes the concentration of cAMP increased five fold (i.e., to 14.41±1.52 pmol/μg DNA), suggesting that cAMP signaling in isolated IBDUs could be regulated by exogenous stimuli.
Figure 3.

Luminal flow induces changes in cAMP in cholangiocytes of microperfused IBDUs. In the basal state (No FSK), cholangiocytes did not respond to luminal flow with changes in cAMP. In contrast, in forskolin-stimulated cholangiocytes (FSK) exposed to luminal fluid flow of 3 μl/sec, cAMP decreased to the basal levels (n=5-7 IBDUs in each group; *, P<0.001)
Luminal fluid flow did not affect the cAMP level in IBDUs unexposed to FSK (i.e., 2.62±0.36 pmol/μg DNA compared to 2.89±0.35 pmol/μg DNA in control, Figure 3). However, when IBDUs were exposed to both stimuli, i.e., FSK and luminal fluid flow, no forskolin-stimulated cAMP increase was observed (i.e., 2.07±0.63 pmol/μg DNA compared to 14.41±1.52 pmol/μg DNA in IBDUs exposed to FSK alone, P<0.001) (Figure 3). Collectively, these data indicate that luminal fluid flow suppresses or prevents a forskolin-stimulated cAMP increase in cholangiocytes, i.e., affects cAMP signaling if this pathway is initially activated by a specific agonist. Based on these observations, subsequent studies on the effects of luminal fluid flow on cAMP were performed on FSK-stimulated IBDUs.
To address whether cholangiocyte cilia are involved in the flow-induced [Ca2+]i and cAMP signaling responses, experiments were performed on IBDUs deciliated by chloral hydrate. Chlorate hydrate deciliates different cell types presumably by destabilizing the junction between the ciliary axoneme and the basal body but does not cause other significant structural and/or functional alterations in deciliated cells, and at present is the only effective pharmacological tool for removal of cilia.17, 25 Images in Figure 4 show that compared to control (Figure 4A), cholangiocyte cilia are no longer present in IBDUs treated with chloral hydrate (4 mM) for 24 hours (Figure 4B). Importantly, the morphology of IBDUs assessed by SEM and TEM, and their viability assessed by trypan blue exclusion, were not affected by chloral hydrate. In addition, we found that IBDUs treated with chloral hydrate respond to ATP by a 38.4±5.2% increase in [Ca2+]i i.e., similarly to untreated IBDUs, in which ATP induced a 42.3±4.3% increase in [Ca2+]i, suggesting that chloral hydrate did not affect [Ca2+]i signaling stimulated by ATP via non-ciliary apically located P2Y receptors.23 Moreover, cholangiocytes treated with chloral hydrate responded to forskolin by a 3.8 fold increase in cAMP (i.e., 10.34±3.08 pmol/μg DNA compared to 2.73±0.58 pmol/μg DNA in control IBDUs) (Figure 4D). Thus, these data taken together suggest that cholangiocytes treated with chloral hydrate lose cilia but still respond to exogenous stimuli (i.e., ATP and FSK) by changes in [Ca2+]i and cAMP levels.
Figure 4.

Chloral hydrate removes cholangiocyte cilia and abolishes the flow-induced changes in [Ca2+]i and cAMP.(A) An SEM image of the lumen of IBDUs incubated for 24 hours under control conditions shows normal primary cilia. (B) An SEM image of the lumen of IBDUs treated for 24 hours with 4 mM chloral hydrate shows the absence of normal primary cilia. (Bars 1 μm). (C) The flow-induced increase in Fluo-4 fluorescence [flow(-chloral hydrate)] was abolished by removal of cholangiocyte cilia by chloral hydrate [flow(+chloral hydrate)]. (D)Forskolin stimulated a 3.8 increase in cAMP level in cholangiocytes of non-perfused IBDU treated with chloral hydrate [No flow(FSK+chloral hydrate)] compared to the basal level of cAMP. No fluid flow-induced decrease in cAMP level was observed in microperfused IBDU stimulated with FSK and treated with chloral hydrate [Flow(FSK+chloral hydrate)]. (n=4-6 IBDUs in each group, *, P<0.05; NS indicates that the difference is not significant).
Functional studies revealed that in IBDUs treated with chloral hydrate, no flow-induced increase in [Ca2+]i (i.e., 2.7±1.0% compared to 41.1±3.3% in control, Figures 4C) and no flow-induced decrease in cAMP (i.e., 9.90±0.95 pmol/μg DNA compared to 10.34±3.08 pmol/μg DNA in control, Figures 4D) occurred suggesting that cholangiocyte cilia are likely involved in transmission of luminal mechanical flow stimuli into intracellular signaling responses.
The flow-induced changes in [Ca2+]i and cAMP are dependent on the ciliary-associated proteins, PC-1 and PC-2
Given the mechanosensory properties of PC-1 and a Ca2+ channel properties of PC-2 observed in primary cilia of renal tubular epithelial cells, we addressed whether these proteins are localized to cholangiocyte cilia and, if yes, whether they are involved in the flow-induced changes in [Ca2+]i and cAMP. Using highly specific, affinity-purified antibodies to PC-1, PC-2, and the ciliary axoneme marker, acetylated α-tubulin, we found that both PC-1 and PC-2 are localized to cholangiocyte cilia (Figure 5).
Figure 5.
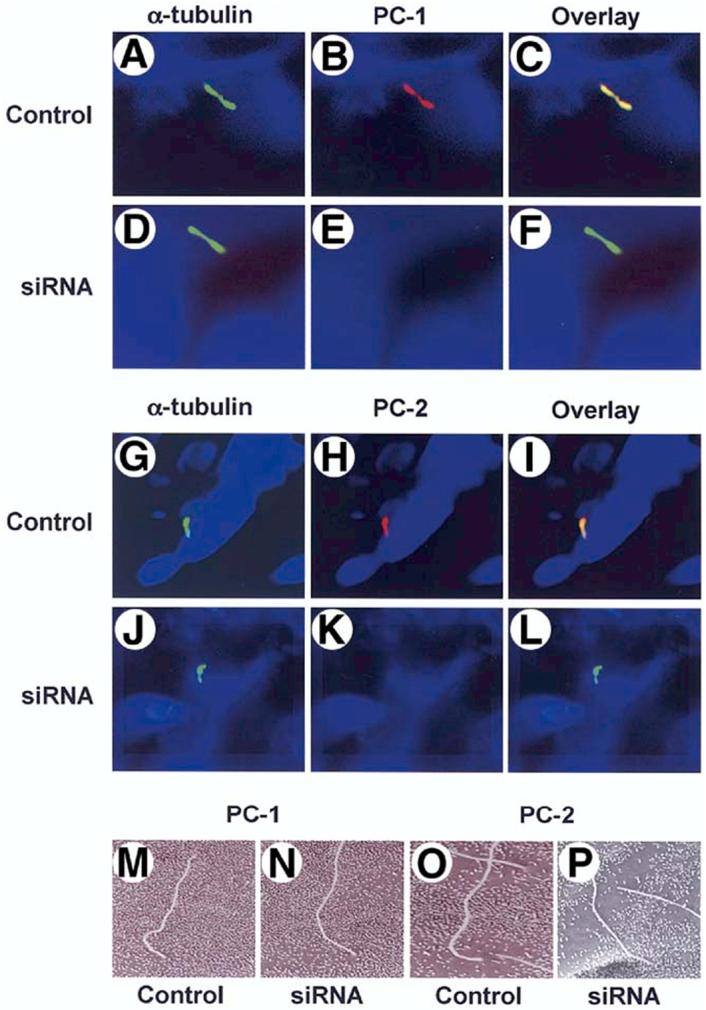
PC-1 and PC-2 in cholangiocyte cilia. Cholangiocytes of isolated IBDUs were stained with an antibody to a ciliary marker acetylated α-tubulin (green, A, D, G, J) and antibodies to PC-1 (red, B, E) and PC-2 (red, H, K). When the two images overlay co-localization of acetylated α-tubulin and PC-1 and PC-2 was seen (yellow, C, I) indicating that both polycystins are expressed in cholangiocyte cilia. In IBDUs incubated 24 hours with siRNAs against PC-1 (D-F) and PC-2 (J-L) no red staining (E, K) and overlay images in green (F, L) show that PC-1 and PC-2 do not present in cholangiocyte cilia after exposure of IBDUs to siRNAs. Nuclei were visualized by staining with DAPI (blue). The SEM images of control IBDUs (M, O) and IBDUs incubated with siRNAs against PC-1 (N) and PC-2 (P) show no difference in the morphology of cholangiocyte cilia.
To explore whether PC-1 and PC-2 contribute to the flow-induced changes in [Ca2+] i and cAMP in cholangiocytes, the expression of Pkd1 and Pkd2, the genes encoding PC-1 and PC-2, respectively, was inhibited by siRNAs. On the mRNA level, the expression of PC-1 and PC-2 was inhibited by siRNAs against Pkd1 and Pkd2 by 65±4% and 86±5%, respectively. In a different model, these siRNAs also dramatically suppressed the expression of PC-1 and PC-2 on the protein level shown by western blotting (V. E. Torres, unpublished observations). By confocal microscopy, IBUDs transfected with siRNAs against Pkd1 and Pkd2 showed no PC-1 and PC-2 in cholangiocyte cilia (Figure 5). siRNAs did not affect the morphology of cilia (Figure 5) but affected their functions (Figure 6). The flow-induced Fluo-4 fluorescence increase in cholangiocytes exposed to scrambled control siRNAs (i.e., by 34.3±5.5%) was not different from the Fluo-4 fluorescence increase in perfused normal IBDUs (i.e., by 37.5±3.1%). However, in IBDUs transfected with siRNAs against PC-1 and PC-2, the flow-induced Fluo-4 fluorescence was reduced by 2.5-fold and 5.2-fold, respectively (i.e., to 14.1±5.4% and 6.6±3.0% compared to 34.3±5.5% in control, Figure 6A), suggesting that both PC-1 and PC-2 are involved in the fluid flow-induced increase in [Ca2+]i.
Figure 6.

PC-1 and PC-2 are involved in cholangiocyte responses to luminal flow. (A) An increase in Fluo-4 fluorescence in response to luminal fluid flow [No siRNAs and scrambled (Scr) siRNAs] was abolished by siRNAs to both, PC-1 and PC-2 suggesting the importance of a mechanoreceptor, PC-1, and a Ca2+ channel, PC-2, in the flow-induced increase in [Ca2+]i. (B, C) Forskolin stimulated an increase in cAMP levels in cholangiocytes of non-perfused IBDUs treated with scrambled siRNAs [No flow (control, FSK)] and siRNAs to PC-1 [No flow (PC-1 siRNA, FSK)] (B) and PC-2 [No flow (PC-2 siRNA, FSK)] (C) compared to the basal levels of cAMP. Luminal fluid flow induced a decrease in cAMP levels in cholangiocytes of microperfused IBDUs transfected with scrambled siRNAs [Flow (control, FSK)] but not with siRNAs to PC-1 [Flow (PC-1 siRNA, FSK)] and PC-2 [Flow (PC-2 siRNA, FSK)] suggesting the involvement of both PC-1 and PC-2 in the fluid flow-induced decrease in cAMP (n=3-5 IBDUs in each group, *, P<0.05; NS indicates that the difference is not significant).
The effects of inhibition of PC-1 and PC-2 on the fluid flow-induced decrease in cAMP levels in cholangiocytes are shown in Figures 6B and 6C. In non-perfused IBDUs transfected with scrambled siRNAs and siRNAs to PC-1, FSK induced a 4.5-fold increase in cAMP levels. Luminal fluid flow induced a 3.4-fold decrease in cAMP in cholangiocytes transfected with scrambled siRNAs, whereas in IBDUs transfected with siRNAs to PC-1 the fluid-flow-induced decrease in cAMP was significantly abolished. Similar changes in a cAMP response to luminal fluid-flow were observed in cholangiocytes transfected with scrambled siRNAs and siRNAs to PC-2. Compared to the basal cAMP level, FSK induced a 6-fold cAMP increase in cholangiocytes transfected with scrambled siRNAs, and a 4-fold increase in cAMP in cholangiocytes transfected with siRNAs to PC-2. Luminal fluid flow induced a 3.9-fold decrease in cAMP level in cholangiocytes transfected with scrambled siRNAs, whereas in cholangiocytes transfected with siRNAs to PC-2 the fluid-flow-induced decrease in cAMP was abolished. These findings suggest that both PC-1 and PC-2 are crucial for the flow-induced suppression of cAMP signaling.
The fluid flow-induced decrease in cAMP requires extracellular Ca2+ (Figure 7). In non-perfused IBDUs incubated in Ca2+-free KRB, FSK stimulated a 3.9-fold increase in cAMP. Perfusion of IBDUs with Ca2+-free KRB did not induce a cAMP decrease, suggesting the importance of extracellular Ca2+ in the flow-induced suppression of cAMP signaling in cholangiocytes.
Figure 7.
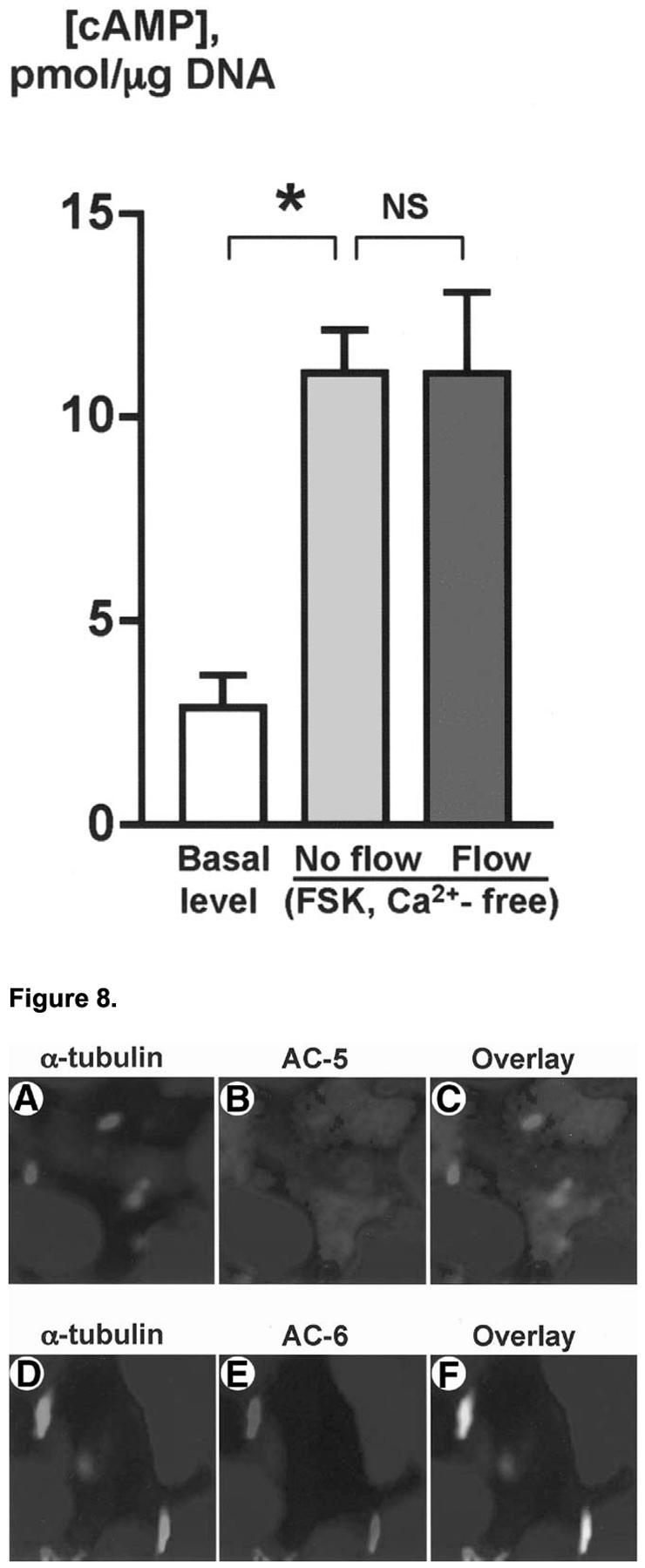
Effects of luminal fluid flow on cAMP in cholangiocytes depend on extracellular Ca2+. Forskolin stimulated an increase in cAMP level in cholangiocytes of non-perfused IBDUs incubated in Ca2+-free KRB [No flow (Ca2+-free, FSK)] compared to the basal level of cAMP. No fluid flow-induced decrease in cAMP level was observed in IBDUs stimulated with FSK and perfused with Ca2+-free KRB [Flow (Ca2+-free, FSK)] suggesting the importance of extracellular Ca2+ in the fluid flow-induced suppression of FSK-stimulated cAMP signaling. (n=4-8 IBDUs in each group, *, P<0.001; NS indicates that the difference is not significant).
Adenylyl cycalse isoform 6 (AC6) is present in cholangiocyte cilia and is involved in the effects of luminal fluid flow on cAMP signaling
The results showing that the flow-induced decrease in cAMP depends on extracellular Ca2+ and the presence of Ca2+ entry channel, PC-2, in cilia, suggested to us that Ca2+-inhibitable isoforms of AC may account for the ciliary-mediated suppression of cAMP signaling in cholangiocytes. To address this issue, we assessed for the expression of two known Ca2+-inibitable ACs (i.e., AC5 and AC6) in cholangiocytes and cholangiocyte cilia. It has been reported in preliminary studies26 that rat cholangiocytes express mRNA for seven of known ten isoforms of AC, including AC5 and AC6. We confirmed these data by RT-PCR (data not shown), and by using highly specific, affinity-purified antibodies to AC5, AC6, and acetylated α-tubulin, demonstrated that both AC5 and AC6 are expressed in cholangiocytes. Moreover, one of them, i.e., AC6, was found for the first time to be localized to cholangiocyte cilia (Figure 8).
Figure 8.
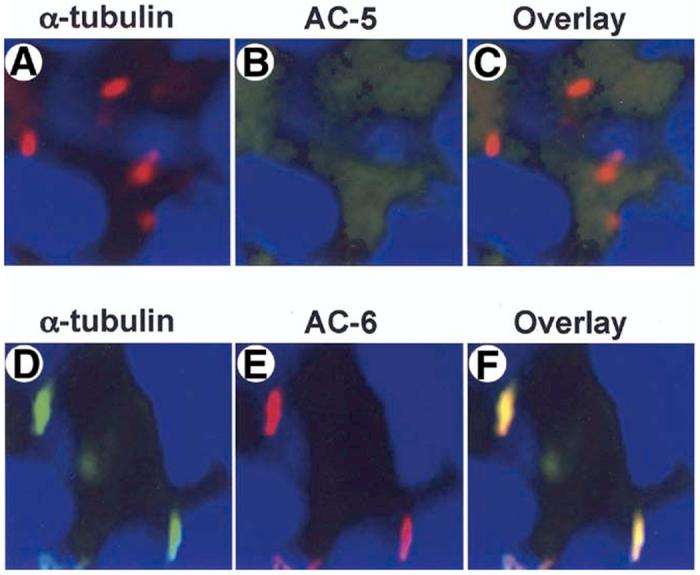
AC6 is expressed in cholangiocyte cilia. Cholangiocytes of isolated IBDUs were stained with an antibody to ciliary marker, acetylated a-tubulin (red) (A, D) and antibodies to AC5 and AC6 (green) (B, E). When the two images were overlay (C, F) co-localization of acetylated a-tubulin and AC6 (yellow, F) but not AC5 (red, C) was seen, indicating that Ca2+-inhibitable AC6 is expressed in cholangiocyte cilia. Nuclei were visualized by staining with DAPI (blue).
To address the involvement of AC5 and/or AC6 in the flow-induced decrease in cAMP, studies were performed on IBDUs transfected with siRNAs to Adcy5 and Adcy6, the genes encoding AC5 and AC6, respectively (Figure 9A). Incubation of IBDUs for 24 hours with siRNAs to AC5 and AC6 resulted in decreased levels of AC5 and AC6 mRNA by 83±3% and 92±4%, respectively.
Figure 9.

AC6 is involved in the fluid flow-induced decrease in cAMP. (A) Incubation of IBDUs for 24 hours with siRNAs to AC5 and AC6 resulted in significantly decreased levels of AC5 and AC6 mRNA compared to control IBDUs incubated with scrambled siRNAs. (B, C) Forskolin stimulated an increase in cAMP levels in cholangiocytes of non-perfused IBDUs treated with scrambled siRNAs [No flow (control, FSK)] and siRNAs to AC5 [No flow (AC5 siRNA, FSK)] (B) and AC6 [No flow (AC6 siRNA, FSK]) (C) compared to the basal levels of cAMP. Luminal fluid flow induced a decrease in cAMP levels in cholangiocytes of microperfused IBDUs transfected with scrambled siRNAs [Flow(control, FSK)] and with siRNAs to AC5 [Flow (AC5 siRNA, FSK)] but not with siRNA to AC6 [Flow (AC6 siRNA, FSK)] suggesting the involvement of AC6 in the fluid flow-induced decrease in cAMP (n=4-8 IBDUs in each group, *, P< 0.05; NS indicates that the difference is not significant).
The effects of inhibition of AC5 and AC6 on the fluid flow-induced decrease in cAMP in cholangiocytes are shown in Figures 9B and 9C. In non-perfused IBDUs transfected with scrambled siRNAs, FSK induced a 4.9-fold increase in cAMP, whereas in non-perfused IBDUs transfected with siRNAs to AC5, FSK stimulated only a 3.6-fold increase in cAMP. This finding suggests that about 25% of the total increase in FSK-stimulated cAMP level depends on AC5. Scrambled siRNAs and siRNAs to AC6 caused similar effects on the FSK-stimulated cAMP increase in cholangiocytes of non-perfused IBDUs; FSK induced a 5.3-fold increase in cAMP in cholangiocytes transfected with scrambled siRNAs, whereas in cholangiocytes transfected with siRNAs to AC6, FSK induced a 4.2 increase in cAMP suggesting that about 22% of the total increase in FSK-stimulated cAMP level depends on AC6. Thus, these data suggest that AC5 and AC6 contribute equally to the FSK-stimulated cAMP increase in cholangiocytes.
In contrast, the contribution of AC5 and AC6 to the fluid flow-induced decrease in cAMP differs. Luminal fluid flow induced a 2.5-fold decrease in cAMP in cholangiocytes transfected with siRNAs to AC5, whereas in IBDUs transfected with siRNAs to AC6, the fluid-flow-induced decrease in cAMP was abolished. In positive controls (i.e., IBDUs transfected with scrambled siRNAs) luminal fluid flow induced a 3.3-fold decrease in cAMP levels for AC5 and a 3.6-fold decrease for AC6.
Thus, these data suggest that in cholangiocytes, Ca2+-inhibitable AC6 is primarily responsible for the flow-induced ciliary-mediated decrease in cAMP. Potentially, other ACs may be also involved in this phenomenon; however, it is likely that AC6 plays a central role in the cAMP signaling network through which cholangiocyte cilia transduce mechanical stimuli.
Discussion
The key findings reported here relate to the functions of primary cilia in intrahepatic bile ducts. We demonstrated for the first time that: (i) luminal fluid flow induces in cholangiocytes of microperfused IBDUs [Ca2+]i and cAMP signaling responses through the bending of primary cilia; (ii) the flow-induced increase in [Ca2+]i reflects both extracellular and intracellular Ca2+ stores; (iii) PC-1 and PC-2 are localized to cholangiocyte cilia and account for the flow-induced increase in [Ca2+]i; (iv) the flow-induced decrease in cAMP is secondary to an increase in [Ca2+]i and is dependent on PC-2 and extracellular Ca2+; and (v) Ca2+-inhibitable AC6 is localized to cholangiocyte cilia accounting for the flow-induced decrease in cAMP. Collectively, these data support the concept that cholangiocyte cilia may function as sensory organelles that transmit luminal fluid flow stimuli into integrated [Ca2+]i and cAMP signaling.
In experiments described in this study, cholangiocytes were shown to respond to luminal flow by an increase in [Ca2+]i and a decrease in cAMP when a flow rate of 3 μl/sec was applied. Since bile flow measured in the rat common bile duct is about of 0.3 μl/sec, a question regarding the physiologic relevance of our observations may be justifiably raised. Regarding this issue, at least three points are worth making. First, we believe that our results are proof of concept that flow-induced mechanical stimuli can affect cholangiocyte function via ciliary-associated mechanisms. Second, the in vivo bile flow rate measured at the tip of the common bile duct almost certainly does not accurately reflect bile flow rates in the intrahepatic biliary system; indeed, current technology does not permit direct measurements of bile flow rates along the intrahepatic biliary system. Third, it has been previously shown in experimental models that mechanical forces greater than biomechanical forces of a physiological range disassemble cilia.27 Our data showing that luminal fluid flow bends cholangiocyte cilia but do not cause any damage in these organelles suggest that we developed a suitable model for addressing the mechanosensory function of cholangiocyte cilia.
Hypothetically, an increase in [Ca2+]i and a decrease in cAMP observed in microperfused IBDUs may be induced by several mechanical forces. Cholangiocytes, like endothelial cells and renal tubular epithelial cells, may experience at least three types of mechanical forces that originate from variations in fluid (bile) flow (i.e., hydrostatic pressure, circumferential stretch, and fluid flow-induced drag forces), and potentially may exhibit [Ca2+]i transient if exposed to any of them.20, 21, 28, 29 However, in intrahepatic bile ducts in vivo and in in vitro microperfused IBDU, in which the distal end is open, hydrostatic pressure and circumferential stretch are minimal. Thus, the primary mechanical forces to which the luminal surface of intrahepatic bile ducts and cholangiocyte cilia are exposed are bile (fluid)-flow induced drag forces that physically can affect cilia.20, 21 In our model, such forces bent cilia through an angle of approximately 70° in the middle portion of the axoneme; the findings are consistent with observations of others demonstrating that chondrocyte primary cilia are bent by mechanical forces in situ in the middle part of an axoneme through an angle of approximately 90°.30
The mechanosensory function of cholangiocyte cilia depends on PC-1, PC-2 and both extracellular and intracellular Ca2+ sources. The initial increase in [Ca2+]i originates from the entry of extracellular Ca2+ via PC-2 which in turn stimulates Ca2+-dependent release of Ca2+ from intracellular stores. In epithelial cells, the mechanisms of Ca2+ release from intracellular stores in response to fluid flow remains uncertain. In MDCK cells, this mechanism involves activation of IP3 receptors,16 whereas in cultured mouse renal epithelial cells, it involves activation of ryanodine receptors.18 Cholangiocytes do not express ryanodine receptors, but express three isoforms of IP3 receptors that are responsible for [Ca2+]i signaling.31 Since in thapsigargin-treated cholangiocytes, the only partial suppression of the flow-induced increase in [Ca2+]i was observed, mechanisms other than activation of IP3 receptors may contribute to the flow-induced ciliary-mediated release of Ca2+ from intracellular stores. However, currently, such mechanisms remain unknown.
The mechanosensory functions of cholangiocyte cilia are not limited to transmitting luminal mechanical flow stimuli into [Ca2+]i signaling response but also occur through cAMP signaling. To our knowledge, this study is the first demonstration of the expression of AC in primary cilia and of the involvement of the cAMP-signaling pathway in ciliary flow sensing in any epithelial cell type. Since the flow-induced decrease in cAMP was dependent on extracellular Ca2+ and the presence of PC-2 and AC6 in cholangiocytes and cilia, it suggested to us that at a molecular level, this may occur through the Ca2+-dependent inhibition of AC6.
AC6 is one of six membrane-bound ACs (i.e., AC4-AC9) expressed in rat cholangiocytes.26 In different cell types, AC6 is activated by hormones through G-protein-coupled receptors and is inhibited by extracellular Ca2+ in micromollar concentrations.32 Recent developments show that AC6 is co-localized with Ca2+ entry channels in discrete domains of the plasma membrane (i.e., “lipid rafts”) where its activity is inhibited by extracellular Ca2+.33 Our data suggest that an analogous mechanism may exist in cholangiocyte cilia where extracellular Ca2+ entering into the cell via PC-2 may inhibit AC6 activity.
The physiological and pathophysiological implications of the flow-induced ciliary-mediated increase in [Ca2+]i and decrease in cAMP remain unclear. Our findings that PC-1, PC-2 and AC6 are localized to cholangiocyte cilia suggest the existence on the apical plasma membrane of a previously unknown regulatory mechanism that may provide a molecular explanation for the initiation and cessation of ductal bile secretion. In a working model depicted on Figure 10, we present the concept that cholangiocyte cilia function as sensory organelles located on the apical plasma membrane that monitor changes in bile flow within intrahepatic bile ducts and adjust cholangiocyte functional response (i.e., ductal bile secretion) to such changes.
Figure 10.

Working model of coordinated regulation of cholangiocyte secretion. Cholangiocytes possess numerous transporters, exchangers and channels necessary for ductal bile formation. The functions of these proteins on the apical plasma membrane are regulated by different regulatory molecules via specific receptors and the cAMP and [Ca2+]i signaling pathways. Secretin induces cholangiocyte bicarbonate rich fluid secretion via the cAMP-PKA signaling pathway by activation of apical CFTR Cl- channel resulting in extrusion of Cl ions, which in turn stimulate Cl-/HCO-3 exchanger and subsequent secretion of HCO-3. Secreted bicarbonate ions drive passive AQP1-mediated water transport in response to established osmotic gradients. Secretin-stimulated cholangiocyte secretion may be potentiated or inhibited by different regulatory molecules (e.g., somatostatin, acetylcholine, nucleotides, bile acids, etc). Primary cilia located on the apical plasma membrane may represent a novel regulatory mechanism that is involved in coordinated regulation of cholangiocyte secretion (see text for details). +, activation; -, inhibition.
Physiologically, both canalicular and ductal bile secretion increase in response to a meal resulting in an increase in bile flow in the biliary tree. In the digestive phase, an increase in ductal bile secretion is stimulated by secretin, which is released into the portal circulation from endocrine S cells in the duodenum and jejunum. We and others have previously demonstrated that secretin stimulates ductal bile secretion via the cAMP-signaling pathway and its action may be potentiated or terminated by other regulatory molecules (i.e., hormones, regulatory peptides, neurotransmitters, etc.) via both cAMP and [Ca2+]i signaling.35-40 Hormonal regulation of ductal bile secretion occurs primarily on the cholangiocyte basolateral plasma membrane by activation of specific receptors. However, it is now evident that ductal bile secretion is also controlled via regulatory mechanisms on the apical cholangiocyte plasma membrane. The regulatory molecules that may affect cholangiocyte function on their apical side include but are not limited to bile acids,41 nucleotides,23, 42 glucose, 43-45 etc. In the model that we propose, ductal bile secretion may also be regulated by luminal bile flow via apically-located ciliary-associated mechanisms. Physiologically, bile flow in intrahepatic bile ducts is pulsatile and its changes may alter the mechanical forces to which cholangiocyte cilia are exposed. Given that cholangiocyte cilia contain a mechanoreceptor, PC-1, and Ca2+ channel, PC-2, proteins thought to form a functional complex when a primary cilium is bent,18, 19 luminal bile flow may bend cholangiocyte cilia resulting in an increase in [Ca2+]i which in turn suppresses cAMP signaling via ciliary-associated AC6. Functionally, luminal bile flow via activation of [Ca2+]i signaling may terminate both global and local cAMP signaling (i.e., turn off regulation) initially activated by secretin and other regulatory molecules (i.e., turn on regulation), thus, providing a coordinated regulation of ductal bile secretion and fast functional adjustments of intrahepatic ductal system to physiological needs.
Thus, our study demonstrates that cholangiocyte cilia may function as fluid (bile) flow sensors in intrahepatic bile ducts. The precise mechanisms of a mechanosensory function of cholangiocyte cilia and the physiological and pathophysiological implications of this phenomenon remain to be fully characterized; however, our initial experiments show that cholangiocyte cilia may have a significant role in coordinated regulation of ductal bile formation.
Acknowledgements
We thank Dr. M. Dan Dragomir-Daescu for helpful discussions and Ms. D. Hintz for secretarial assistance.
Footnotes
This work was supported by grant DK24031 to Dr. N. F. LaRusso from the National Institutes of Health and by the Mayo Foundation
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wheatley DN. Primary cilia in normal and pathological tissues. Pathobiology. 1995;63:222–238. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- 2.Praetorius HA, Spring KR. A physiological view of the primary cilium. Ann Rev Physiol. 2005;67:515–529. doi: 10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- 3.Davenport JR, Yoder BK. An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol. 2005;289:F1159–F1169. doi: 10.1152/ajprenal.00118.2005. [DOI] [PubMed] [Google Scholar]
- 4.McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 5.Yost HJ. Left-right asymmetry: nodal cilia make and catch a wave. Curr Biol. 2003;13:R808–R809. doi: 10.1016/j.cub.2003.09.051. [DOI] [PubMed] [Google Scholar]
- 6.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to the kidney cilia and the ciliary level is elevated in ORPK mice with polycystic kidney disease. Curr Biol. 2002;12:R378–R380. doi: 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 7.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 8.Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso NF, Torres VE, Harris PC. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet. 2003;12:2703–2710. doi: 10.1093/hmg/ddg274. [DOI] [PubMed] [Google Scholar]
- 9.Menezes LF, Cai Y, Nagasawa Y, Silva AM, Watkins ML, Da Silva AM, Somlo S, Guay-Woodford LM, Germino GG, Onuchic LF. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 2004;66:1345–55. doi: 10.1111/j.1523-1755.2004.00844.x. [DOI] [PubMed] [Google Scholar]
- 10.Masyuk TV, Huang BQ, Ward CJ, Masyuk AI, Yuan D, Splinter PL, Punyashthiti R, Ritman EL, Torres VE, Harris PC, LaRusso NF. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, Harris PC, Larusso NF. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165:1719–1730. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774–782. doi: 10.1002/hep.20431. [DOI] [PubMed] [Google Scholar]
- 13.Tahvanainen E, Tahvanainen P, Kaariainen H, Hockerstedt K. Polycystic liver and kidney diseases. Ann Med. 2005;37:546–555. doi: 10.1080/07853890500389181. [DOI] [PubMed] [Google Scholar]
- 14.Pan J, Wang Q, Snell WJ. Cilium-generated signaling and cilia-related disorders. Lab Invest. 2005;85:452–463. doi: 10.1038/labinvest.3700253. [DOI] [PubMed] [Google Scholar]
- 15.Vogel G. News focus: Betting on cilia. Science. 2005;310:216–218. doi: 10.1126/science.310.5746.216. [DOI] [PubMed] [Google Scholar]
- 16.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 17.Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol. 2003;191:69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- 18.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 19.Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26:844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM. Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol. 2003;285:F998–F1012. doi: 10.1152/ajprenal.00067.2003. [DOI] [PubMed] [Google Scholar]
- 21.Liu W, Murcia NS, Duan Y, Weinbaum S, Yoder BK, Schwiebert E, Satlin LM. Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am J Physiol Renal Physiol. 2005;289:F978–F88. doi: 10.1152/ajprenal.00260.2004. [DOI] [PubMed] [Google Scholar]
- 22.Masyuk AI, Gong AY, Kip S, Burke MJ, LaRusso NF. Perfused rat intrahepatic bile ducts secrete and absorb water, solute, and ions. Gastroenterology. 2000;119:1672–1680. doi: 10.1053/gast.2000.20248. [DOI] [PubMed] [Google Scholar]
- 23.Dranoff JA, Masyuk AI, Kruglov EA, LaRusso NF, Nathanson MH. Polarized expression and function of P2Y ATP receptors in rat bile duct epithelia. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1059–G1067. doi: 10.1152/ajpgi.2001.281.4.G1059. [DOI] [PubMed] [Google Scholar]
- 24.Splinter PL, Masyuk AI, LaRusso NF. Specific inhibition of AQP1 water channels in isolated rat intrahepatic bile duct units by small interfering RNAs. J Biol Chem. 2003;278:6268–6274. doi: 10.1074/jbc.M212079200. [DOI] [PubMed] [Google Scholar]
- 25.Dunlap K. Localization of calcium channels in Paramecium caudatum. J Physiol. 1977;271:119–133. doi: 10.1113/jphysiol.1977.sp011993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melero S, Strazzabosco M, Spirli C, Fiorotto R, Glaser S, Francis H, Alpini G. Expression and regulation of adenylyl cyclase isoforms (ACs) in rat cholangiocytes; effects of lipopolysaccaride (LPS) and α-naphthylisothiocyanate (ANIT) treatment. Hepatology. 2003;38:687A. [Google Scholar]
- 27.Iomini C, Tejada K, Mo W, Vaananen H, Piperno G. Primary cilia of human endothelial cells disassemble under laminar shear stress. J Cell Biol. 2004;164:811–817. doi: 10.1083/jcb.200312133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traub O, Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler Thromb Vasc Biol. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- 30.Jensen CG, Poole CA, McGlashan SR, Marko M, Issa ZI, Vujcich KV, Bowser SS. Ultrastructural, tomographic and confocal imaging of the chondrocyte primary cilium in situ. Cell Biol Int. 2004;28:101–110. doi: 10.1016/j.cellbi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 31.Hirata K, Dufour JF, Shibao K, Knickelbein R, O′Neill AF, Bode HP, Cassio D, St-Pierre MV, Larusso NF, Leite MF, Nathanson MH. Regulation of Ca2+ signaling in rat bile duct epithelia by inositol 1,4,5-trisphosphate receptor isoforms. Hepatology. 2002;36:284–296. doi: 10.1053/jhep.2002.34432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chabardes D, Imbert-Teboul M, Elalouf JM. Functional properties of Ca2+-inhibitable type 5 and type 6 adenylyl cyclases and role of Ca2+ increase in the inhibition of intracellular cAMP content. Cell Signal. 1999;11:651–663. doi: 10.1016/s0898-6568(99)00031-5. [DOI] [PubMed] [Google Scholar]
- 33.Cooper DM. Molecular and cellular requirements for the regulation of adenylate cyclases by calcium. Biochem Soc Trans. 2003;31:912–915. doi: 10.1042/bst0310912. [DOI] [PubMed] [Google Scholar]
- 34.Alpini G, Roberts S, Kuntz SM, Ueno Y, Gubba S, Podila PV, LeSage G, LaRusso NF. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology. 1996;110:1636–1643. doi: 10.1053/gast.1996.v110.pm8613073. [DOI] [PubMed] [Google Scholar]
- 35.Fitz JG. Regulation of cholangiocyte secretion. Semin Liver Disease. 2002;22:241–249. doi: 10.1055/s-2002-34502. [DOI] [PubMed] [Google Scholar]
- 36.Boyer JL. Bile duct epithelium: frontiers in transport physiology. Am J Physiol. 1996;270:G1–G5. doi: 10.1152/ajpgi.1996.270.1.G1. [DOI] [PubMed] [Google Scholar]
- 37.Baiocchi L, LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte bile secretion. J Hepatol. 1999;31:179–191. doi: 10.1016/s0168-8278(99)80180-9. [DOI] [PubMed] [Google Scholar]
- 38.Kanno N, LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte bicarbonate secretion. Am J Physiol Gastrointest Liver Physiol. 2001;281:G612–625. doi: 10.1152/ajpgi.2001.281.3.G612. [DOI] [PubMed] [Google Scholar]
- 39.Gigliozzi A, Fraioli F, Boyer JL. Hormonal regulation of cholangiocyte secretion. In: Alpini G, Alvaro D, Marzioni M, LeSage G, LaRusso N, editors. The pathophysiology of the biliary epithelia. Lamdes Bioscience, Eurekah; Georgetown: 2004. pp. 89–95. [Google Scholar]
- 40.Masyuk AI, Masyuk TV, LaRusso NF. In: Physiology of the gastrointestinal tract. Johnson LR, editor. Academic Press; 2006. pp. 1505–1533. [Google Scholar]
- 41.Alpini G, Glaser S, Francis H, Marzioni M, Venter J, Phinizy J, LeSage G. Bile acids interactions with cholangiocytes. In: Alpini G, Alvaro D, Marzioni M, LeSage G, LaRusso N, editors. The pathophysiology of the biliary epithelia. Lamdes Bioscience, Eurekah; Georgetown: 2004. pp. 112–126. [Google Scholar]
- 42.Schlenker T, Romac JM, Sharara AI, Roman RM, Kim SJ, LaRusso N, Liddle RA, Fitz JG. Regulation of biliary secretion through apical purinergic receptors in cultured rat cholangiocytes. Am J Physiol. 1997;273:G1108–G1117. doi: 10.1152/ajpgi.1997.273.5.G1108. [DOI] [PubMed] [Google Scholar]
- 43.Guzelian P, Boyer JL. Glucose reabsorption from bile. Evidence for a biliohepatic circulation. J Clin Invest. 1974;53:526–535. doi: 10.1172/JCI107586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lira M, Schteingart CD, Steinbach JH, Lambert K, McRoberts JA, Hofmann AF. Sugar absorption by the biliary ductular epithelium of the rat: evidence for two transport systems. Gastroenterology. 1992;102:563–571. doi: 10.1016/0016-5085(92)90104-7. [DOI] [PubMed] [Google Scholar]
- 45.Masyuk AI, Masyuk TV, Tietz PS, Splinter PL, LaRusso NF. Intrahepatic bile ducts transport water in response to absorbed glucose. Am J Physiol Cell Physiol. 2002;283:C785–C791. doi: 10.1152/ajpcell.00118.2002. [DOI] [PubMed] [Google Scholar]