

Summary
The highly directional and tightly regulated recombination reaction used to site-specifically excise the bacteriophage λ chromosome out of its E. coli host chromosome requires the binding of six sequence-specific proteins to a 99 bp segment of the phage att site. To gain structural insights into this recombination pathway, we measured 27 FRET distances between eight points on the 99 bp regulatory DNA bound with all six proteins. Triangulation of these distances using a metric matrix distance-geometry algorithm provided coordinates for these eight points. The resulting path for the protein-bound regulatory DNA, which fits well with the genetics, biochemistry, and X-ray crystal structures describing the individual proteins and their interactions with DNA, provides a new structural perspective into the molecular mechanism and regulation of the recombination reaction and illustrates a design by which different families of higher-order complexes can be assembled from different numbers and combinations of the same few proteins.
Introduction
The phage λ site-specific recombinase, Int, is responsible for integrating the viral genome into the chromosome of its E. coli host when growth conditions and prospects for phage propagation are poor and then excising the viral chromosome when growth conditions are more favorable or when survival of the host is threatened (Campbell, 1962). It belongs to the Tyrosine recombinase family, whose members participate in a wide variety of biological functions such as chromosome segregation, chromosome copy number control, gene expression, conjugative transposition, gene dissemination, and viral integration and excision (Azaro and Landy, 2002; Grainge and Jayaram, 1999; Hallet and Sherratt, 1997; Van Duyne, 2002).
The λ Int pathway differs fromthat ofmany other family members, such as Cre and Flp, in that it is highly directional and tightly regulated by virtue of its response to three accessory DNA-bending proteins: the phage-encoded Xis protein, which stimulates excisive and inhibits integrative recombination; the host-encoded Fis protein, which stimulates excisive recombination when Xis is limiting; and the host-encoded IHF protein, which is required for both reactions but inhibits excision at high concentrations. The intracellular concentration of Xis is tightly regulated by the elegant circuitry governing the switch between the lytic and lysogenic pathways of thephage. Intracellular levels of IHFarehighestwhencells enter stationary phase while levels of Fis peak as cells emerge from stationary phase into logarithmic growth. The host-encoded IHF and Fis proteins are both important global regulators of DNA expression and replication, and it has been proposed that their intracellular levels signal conditions that are more appropriate for recombination in one direction or the other (Ball et al., 1992; Bushman et al., 1985; Nilsson et al., 1992; Thompson et al., 1987).
Integrative recombination between specific sites on the phage (attP) and bacterial (attB) chromosomes generates an integrated prophage with hybrid sites at the left (attL) and right (attR) phage-host DNA junctions. Excisive recombination between attL and attR regenerates attP and attB on the respective disjoined chromosomes (Campbell, 1962) (see Figure 1). The chemistry and mechanics of the DNA exchanges are similar for all Tyrosine recombinase family members: two ordered pairs of transesterification reactions are executed by a tetrameric complex of recombinases that first generate a four-way DNA junction (Holliday recombination intermediate) and then resolve it to recombinant DNA products. This chemistry is executed at four ‘‘core’’ Int-binding sites by the carboxy-terminal domain of Int, which corresponds to the Cre and Flp proteins.
Figure 1. Pathways of Integrative and Excisive Recombination Catalyzed by λ Int.
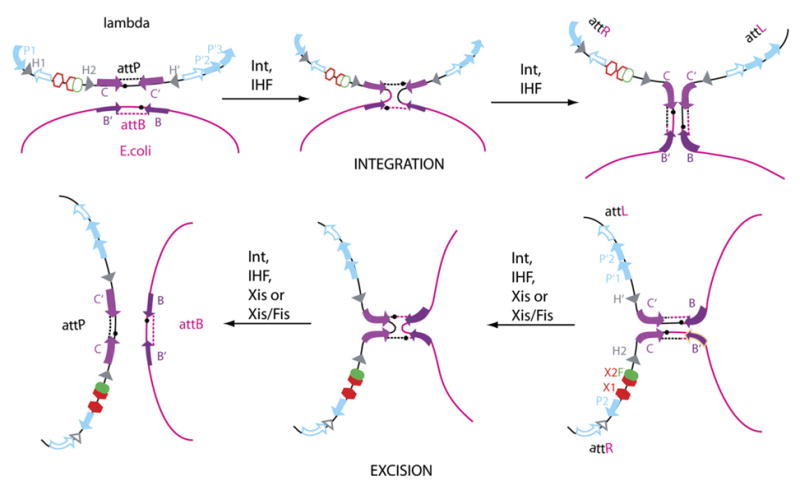
Recombination between attP on the phage chromosome and attB on the bacterial chromosome generates an integrated prophage bounded by the hybrid sites attL and attR (top row). Recombination between attL and attR regenerates the two chromosomes (bottom row, right to left). Both reactions proceed via a single-strand-exchange Holliday junction intermediate (center panels) and use different but overlapping sets of protein binding sites (filled symbols). DNA cleavage and ligation reactions are carried out on each DNA strand (at the site of the small black circles) by the carboxy-terminal domain of Int bound at inverted repeat core sites (lavender arrows) flanking 7 bp regions of homology. The amino-terminal domain of Int binds at arm sites (blue arrows), P1, P′2, and P′3 for integration and P2, P′1, and P′2 for excision. Binding sites for the host-encoded accessory protein IHF (gray triangles) are H1, H2, and H′ for integration and at H2 and H′ for excision. Additional accessory proteins for excision are the phage-encoded Xis, which binds at X1-X2 (red hexagons), and the host-encoded Fis protein, which binds at F (green oval).
Int differs from family members Cre and Flp in having an additional (amino-terminal) domain that binds with high affinity to five ‘‘arm’’ binding sites distributed throughout the att sites. Int bridging between the two classes of sites is assisted by DNA bends induced by the accessory proteins bound to specific sites located between arm and core sites (Figure 1). Two overlapping subsets of the 16 protein binding sites are used for integrative and excisive recombination, and this differential usage of protein binding sites confers directionality and regulation through the formation of integration- or excision-specific higher-order ‘‘intasome’’ complexes (reviewed in Azaro and Landy, [2002]).
Of the four att site arms, the P-arm in attR is the most complex. During excisive recombination, this 99 bp segment is populated with Int and three types of accessory DNA-bending proteins: one IHF, one Fis dimer, and, as will be shown here, three Xis protomers (Figure 1). Crystal structures have been determined for IHF, Xis, and the amino-terminal domain of Int bound to their cognate DNA sites and for unliganded Fis (Biswas et al., 2005; Kostrewa et al., 1991; Rice et al., 1996; Sam et al., 2004; Yuan et al., 1991). However, despite the X-ray crystal structures, genetics, and biochemistry describing the individual proteins and their interactions with DNA, we still lack a global view of how they work in concert to shape the overall DNA path. This is not surprising, since there are very few large multi-protein-DNA complexes for which the DNA trajectory has been determined experimentally.
To determine the DNA path, we used a previously described mapping method based on the triangulation of in-gel FRET measurements (TIF) (Radman-Livaja et al., 2005). Twenty-seven FRET distances between eight points on the 99 bp P-arm DNA bound with the Int N domain and all the accessory proteins were measured in gello. The triangulation of the measured distances provided Cartesian coordinates for these eight points and consequently yielded a unique path for the protein-bound regulatory DNA. The 3D contour of this complex curve, which fits well with all of the relevant structures and biochemistry, is discussed in relation to the mechanics and regulation of the recombination reaction.
Results
P-Arm Six-Protein Complex Assembly
As the first step toward determining the trajectory of the protein-bound P-arm DNA, it was necessary to ascertain whether a unique and electrophoretically stable protein-DNA complex could be assembled on the 99 bp DNA fragment containing the binding sites for all of the excision-relevant proteins: IHF (H2), Fis (F), Xis (X1-X2), and the amino-terminal domain of λ Int (P2) (Figure 2A). An amino-terminal domain of Int, N75 (residues 1–75), has been used for binding the P2 site in order to avoid artifacts arising from an intact Int with an unbound carboxy-terminal DNA binding domain (Sarkar et al., 2001, 2002).
Figure 2. P-Arm DNA-Six-Protein Complex Assembly.
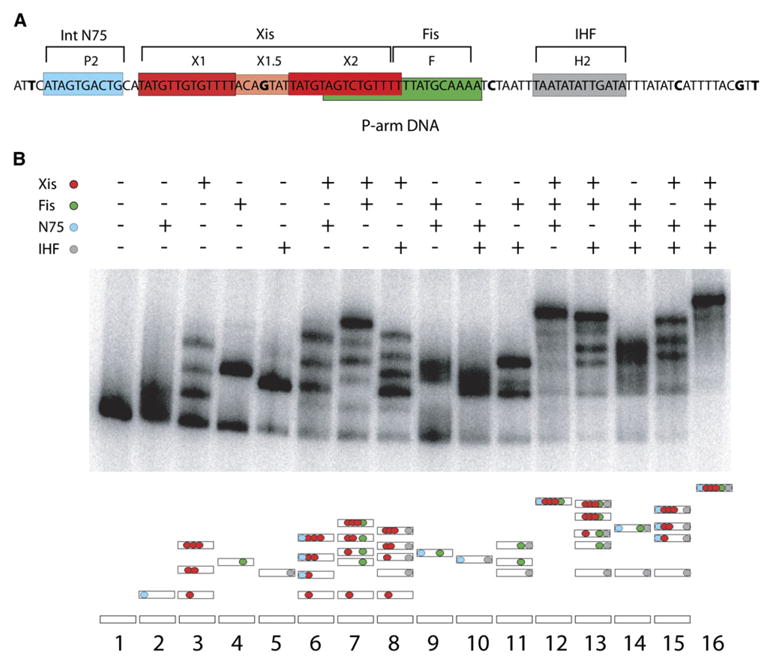
(A) Diagram of the 99 bp P-arm DNA and six-protein binding sites. N75 is the amino-terminal domain of Int (residues 1–75), and the X2 and F sites overlap. The bases that have been changed to As are shown in bold type.
(B) Gel retardation assay in 7% polyacrylamide of 32P-labeled 99 bp regulatory P-arm DNA bound by N75 (0.11μM), Xis (0.82 μM), Fis (0.10 μM), and/or IHF (0.30 μM), as indicated (see Experimental Procedures). The schematic representation of the proteins present in each gel band (bottom panel) is color coded in accord with the top panel (see also Figure 5 and text).
The same concentrations used in each of the single-protein binding reactions (lanes 1–5 of Figure 2B) were used in all of the subsequent multiprotein binding reactions (lanes 6–16 of Figure 2B). The DNA binding cooperativity between Xis and Int N domain and between Xis and Fis is clearly evident in the two-protein binding reactions (lanes 6 and 7, respectively) and even more dramatically for Xis, Fis, and Int N domain in the three-protein binding reactions (lane 12) (Cho et al., 2002; Sarkar et al., 2002; Swalla et al., 2003; Warren et al., 2003). However, the most important point of the gel-shift experiments is that the 99 bp P-arm DNA forms an electrophoretically unique and stable complex that is dependent upon the presence of all four accessory proteins (lane 16, Figure 2B), as confirmed by western blotting with antibodies against each of the proteins (data not shown).
FRET Measurements
To determine the DNA path in the 99 bp P-arm DNA-protein complex (lane 16 of Figure 2B), we used a previously developed in-gel FRET mapping method (TIF) based on the triangulation of FRET-measured pairwise distances (Radman-Livaja et al., 2005). In the present work, the pairwise distances were between eight points on the DNA substrate; the positions of donor (Bodipy-Fl) and acceptor (TAMRA) dyes are shown in Figure 3A. To homogenize the influence of the neighboring sequence on the spectral properties of the dyes (Nazarenko et al., 2002), single base changes were introduced where necessary to provide the same ATA context for each dye (except for the dye at fF, where the wild-type TTT sequence was retained) (see Figure 3A and Experimental Procedures). In all cases, excisive recombination with substrates containing dyes and the altered flanking bases was comparable to reactions using unmodified wild-type DNA substrates (data not shown).
Figure 3. FRET Measurements in the P-Arm DNA-Six-Protein Complex.
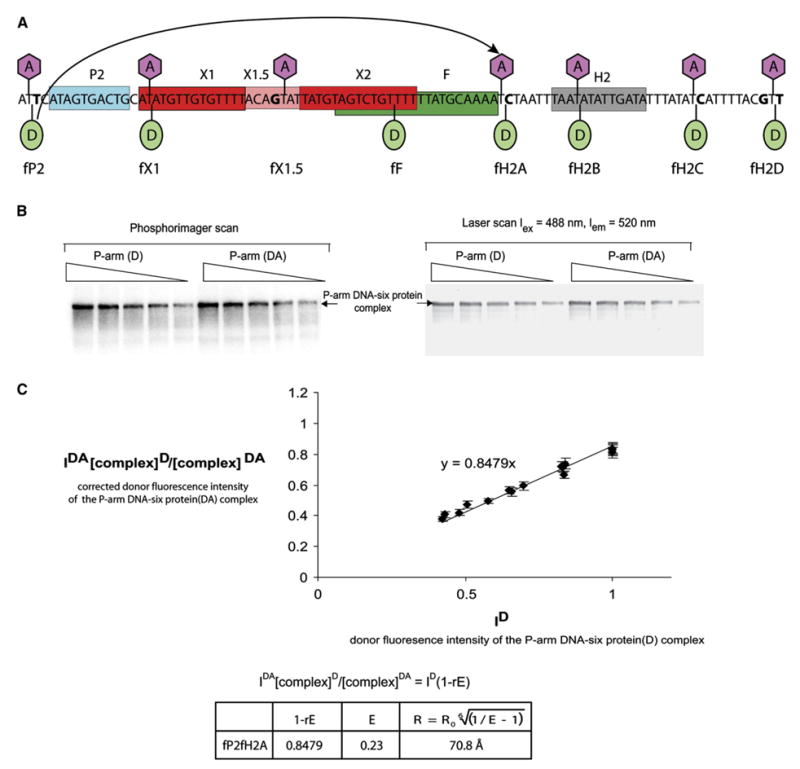
(A) Map and labels of the fluorescent dye positions. An acceptor dye, TAMRA, was attached to C5 of the indicated top-strand thymines (lavender hexagons). A donor dye, Bodipy-Fl, was attached to the DNA phosphate backbone of the 32P-labeled bottom strand at the indicated positions (green ovals) (see Experimental Procedures). Short-hand labels for each of the dye positions are shown in the bottom row. The donor (fP2)-acceptor (fH2A) pair used for the example in (B) is connected by an arrow.
(B) Example of FRET data collection using measurement of the distance between positions fP2 and fH2A (see [A]). Protein-DNA complexes were formed with DNA containing either donor-only at position fP2 (D) or donor + acceptor at positions fP2 and fH2A, respectively (DA). Following separation by gel electrophoresis, the six-protein complex (lane 16, Figure 2B) was analyzed in a Typhoon laser scanner to quantify the fluorescence (right panel) and then dried to quantify the 32P-labeled DNA in a phosphorimager (left panel) (see Experimental Procedures).
(C) A plot showing the correlation between the donor fluorescence intensity (IDA) of the donor-plus acceptor-labeled P-arm(y axis) and the donor fluorescence intensity (ID) of the donor-only-labeled P-arm(x axis). The IDA values shown on the y axis have been corrected for differences in the amounts of donor-only and donor + acceptor complexes by multiplying by [complex]D/[complex]DA, as determined from the radioactivity scans. All of the values in the plot have been normalized to 1 by dividing by the highest donor intensity of donor-only complex bands. Each experimental point on the graph represents the average of five corrected IDA values, and the bars show the standard deviation. The line was constructed from data collected in at least three independent experiments. The correlation between IDA and ID is described by the equation shown above the table where E is the efficiency of energy transfer and r is the fraction of acceptor-labeled P-arm DNA (r for fP2H2A = 0.65). The distance between the donor and the acceptor dyes, R, for fP2fH2A was calculated using R0 = 57.9 Å (see Experimental Procedures, Table 1, and Table S2).
The efficiencies of energy transfer between donor and acceptor dyes at various positions were determined from the extent of donor fluorescence quenching in complexes containing the acceptor compared with donor fluorescence in complexes without acceptor. A radioactively labeled P-arm with the donor dye attached to the bottom strand at one point and the acceptor dye attached to the top strand at another point (Parm[ DA] in Figure 3B) was mixed with all four proteins in the binding buffer. In a parallel reaction, a radioactively labeled P-arm carrying only the donor dye (P-arm[D] in Figure 3B) was also mixed with the four bending proteins as described in Experimental Procedures. After native PAGE, the fluorescence intensities of the donor in the complexes with and without acceptor were obtained from a laser scan of the gel using a 488 nm blue laser for excitation and a 520 nm emission filter. The total amount of complex formed was determined by quantifying the radioactivity in a Phosphoimager scan of the same gel (Figure 3B). The efficiencies of energy transfer and distances between each donor-acceptor position in the P-arm DNA-six-protein complex were calculated as described in the Experimental Procedures and Figure 3C and are summarized in Table 1.
Table 1.
FRET Measurements
| Donor | Acceptor | DNA Substrate | Protein-Bound | dd (Å) | Ea (%) | Rb (Å) | Dc(Å) | Curvaturee (°) |
|---|---|---|---|---|---|---|---|---|
| fP2 | fX1 | P-arm | All | 53.2 | 47.8 < 51.9 < 61.1 | 51.0 < 54.3 < 55.8 | 51.7 ± 1.1 | NA |
| fP2 | fX1.5 | P-arm | All | 109.0 | 12.5 < 20.0 < 28.8 | 65.0 < 70.4 < 77.3 | 74.8 ± 3.1 | NA |
| fP2 | fH2A | P-arm | All | 210.0 | 12.3 < 23.0 < 29.4 | 67.0 < 70.8 < 80.3 | 78.1 ± 2.1 | NA |
| fP2 | fH2B | P-arm | All | 243.9 | 15.3 < 20.8 < 33.0 | 64.5 < 71.6 < 76.2 | 70.3 ± 4.0 | NA |
| fP2 | fH2C | P-arm | All | 291.4 | 13.9 < 22.6 < 38.6 | 63.0 < 76.5 < 79.0 | 66.6 ± 3.9 | NA |
| fP2 | fH2D | P-arm | All | 325.3 | 7.7 < 31.4 < 37.0 | 62.0 < 64.7 < 86 | 68.5 ± 5.3 | NA |
| fX1 | fX1.5 | P-arm | All | 59.0 | 43.7 < 51.6 < 58.8 | 52.4 < 55.0 < 58.0 | 53.6 ± 1.8 | NA |
| fX1 | fH2A | P-arm | All | 159.3 | 9.4 < 12.5 < 27.9 | 69.0 < 81.5 < 86.0 | 78.2 ± 4.7 | NA |
| fX1 | fH2B | P-arm | All | 193.1 | 9.6 < 15.0 < 25.2 | 67.0 < 74.6 < 81.2 | 73.6 ± 5.0 | NA |
| fX1 | fH2C | P-arm | All | 240.5 | 3.1 < 6.7 < 10.6 | 83.0 < 88.0 < 98.6 | 96.5 ± 2.4 | NA |
| fX1 | fH2D | P-arm | All | 274.4 | 7.1 < 9.6 < 11.3 | 80.0 < 82.4 < 87.0 | 85.4 ± 2.1 | NA |
| fF | fX1 | P-arm | All | 112.2 | 9.5 < 13.5 < 20.4 | 69.3 < 75.2 < 80.4 | 72.7 ± 3.7 | NA |
| fF | fX1.5 | P-arm | All | 54.3 | 31.7 < 50.0 < 57.5 | 50.2 < 52.8 < 60.0 | 54.5 ± 3.9 | NA |
| fF | fH2A | P-arm | All | 51.2 | 58.8 < 64.7 < 78.3 | 43.7 < 48.9 < 51.0 | 44.5 ± 1.4 | NA |
| fF | fH2B | P-arm | All | 85.0 | 12.3 < 17.2 < 29.5 | 60.7 < 68.2 < 72.8 | 71.3 ± 1.9 | NA |
| fF | fH2C | P-arm | All | 132.4 | 4.5 < 11.6 < 15.7 | 74.10 < 78.6 < 93.2 | 83.2 ± 5.4 | NA |
| fF | fH2D | P-arm | All | 166.0 | 30.2 < 54.0 < 58.0 | 54.0 < 55.5 < 65.5 | 59.5 ± 4.5 | NA |
| fH2A | fX1.5 | P-arm | All | 104.0 | 5.7 < 8.9 < 29.0 | 64.0 < 81.2 < 88.0 | 79.6 ± 4.3 | NA |
| fH2A | fH2B | P-arm | All | 37.5 | 77.7 < 86.3 < 93.7 | 35.7 < 38.7 < 42.7 | 36.0 ± 0.7 | NA |
| fH2A | fH2C | P-arm | All | 82.3 | 44.4 < 50.0 < 62.7 | 48.8 < 53.2 < 55.2 | 50.6 ± 2.2 | NA |
| fH2A | fH2D | P-arm | All | 115.5 | 62.5 < 65.6 < 76.7 | 44.2 < 48.4 < 49.5 | 46.1 ± 2.1 | NA |
| fH2B | fX1.5 | P-arm | All | 139.1 | 0 < 4.9 < 6.1 | 83.0 < 86.0 < 105 | 96.9 ± 4.1 | NA |
| fH2B | fH2C | P-arm | All | 59.0 | 35.0 < 60.7 < 76.2 | 45.00 < 51.5 < 62.0 | 54.4 ± 3.4 | NA |
| fH2B | fH2D | P-arm | All | 82.3 | 13.8 < 21.1 < 34.0 | 61.0 < 68.0 < 74.0 | 70.4 ± 2.1 | NA |
| fH2C | fX1.5 | P-arm | All | 186.3 | 0 < 4.5 < 10.5 | 74.0 < 86.3 < 103.0 | 95.5 ± 4.4 | NA |
| fH2C | fH2D | P-arm | All | 37.9 | 87.1 < 90.0 < 94.5 | 32.8 < 37.0 < 38.9 | 35.1 ± 2.5 | NA |
| fH2D | fX1.5 | P-arm | All | 216.8 | 12.3 < 21.1 < 32.0 | 63.7 < 70.0 < 78.0 | 67.7 ± 3.9 | NA |
| fX1 | fX1.5 | P2X1 | Xis | 59.0 | 38.8 < 44.9 < 50.3 | 60.0 > 57.5 > 55.5 | NA | 0 < 25.9 < 39.7 |
| fP2 | fX1.5 | P2X1 | Xis + N75 | 109.0 | 10.3 < 14.4 < 19.57 | 80.2 > 75.2 > 70.8 | NA | 85.3 < 92.8 < 99 |
| fH2A | fX1.5 | X2F | Fis | 104.0 | 4.0 < 7.6 < 11.3 | 93.6 > 83.4 > 77.6 | NA | 51.7 < 73.4 < 83.5 |
| fH2A | fX1.5 | X2F | Fis + Xis | 104.0 | 6.2 < 11.0 < 23.4 | 86.5 > 78.2 > 67.0 | NA | 67.4 < 82.5 < 99.8 |
| fH2A | fX1 | P-arm | 3*Xis | 159.3 | 3.9 < 8.0 < 13.3 | 100 > 88.5 > 80.0 | NA | 102 < 113 < 120 |
| fH2A | fX1 | P-arm | 2*Xis | 159.3 | 0 < 2.7 < 11.9 | 159.3.0 > 107.3 > 84.0 | NA | 0 < 95.3 < 116 |
| fH2A | fX1 | P-arm | Xis | 159.3 | 0 < 6.0 | 159.3.0 > 95.2 | NA | 0 < 107 |
E is the efficiency of energy transfer between donor and acceptor obtained from plots as shown in Figure 3. The upper and lower limits were obtained from error bars.
R is the distance between the donor and acceptor. For donor-acceptor pairs with E = 0, the distance was estimated to be 2*R0, with a range between 1.5*R0 and ∝.
D is the average distance between the donor and acceptor positions obtained from sets of 3D maps with coordinates calculated by using the 27 R distance constraints.
d is the distance between donor and acceptor on straight B-DNA ( , where nbp is the number of base pairs between donor and acceptor and 3.4 and 20 are the distance between adjacent base pairs and the double helix diameter in Å, respectively).
Curvature was calculated from the formula curvature = (180 − 2* sin −1[R/d]), where d is the length in Å between donor and acceptor in a straight B-DNA substrate. The bend center was assumed to be at d/2 for curvature calculations.
Coordinate Determination
A metric matrix distance-geometry computer program, routinely employed for NMR structure determination (Havel, 1990), was used with minor modifications to calculate the Cartesian coordinates for the eight dye positions from the 27 distance ranges measured by FRET and summarized in Table 1 (see Experimental Procedures). This procedure has been shown to provide robust searching of all possible topologies with no bias or compaction of structures (Havel, 1993). Therefore, we are confident that all structures consistent with the FRET data have been generated. The program was run 1200 times, producing 86 conformations consistent with the FRET data: 30 of one chirality and 56 of the opposite chirality. The chirality shown in Figure 4 was chosen based on its compatibility with the previously published X-ray structures of the tetrameric Int-Holliday junction-arm DNA complexes (Biswas et al., 2005) (see Figure 6B and Discussion).
Figure 4. The P-Arm DNA Trajectory Calculated from 27 FRET-Determined Distances by Using a Metric Matrix Distance Geometry Algorithm.
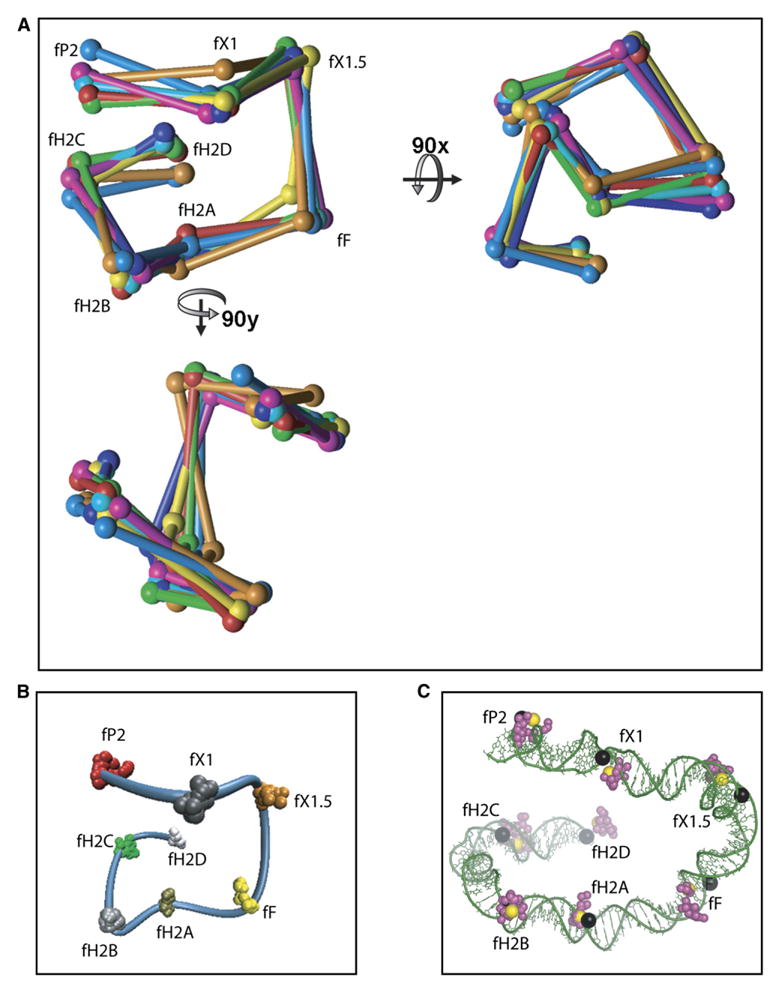
(A) Superposition of seven DNA paths randomly chosen from the 30 paths generated by the distance geometry program using the data from Table 1. The coordinates (spheres) of seven different computer-generated paths (each a different color) have been connected by straight lines. Labels refer to the dye positions shown in Figure 3.
(B) The 30 sets of coordinates generated from 1200 iterations of the distance geometry program are superimposed on the NAB-generated path of one set. The tight clustering of 30 points for each dye coordinate illustrates the power of triangularization.
(C) The DNA path from the model in Figure 6A is shown without the proteins but with the dye coordinates (magenta spheres) for comparison to the NAB-generated path in Figure 6B. The black spheres represent the positions of the dyes in the DNA sequence. The coordinates of the structure used to generate the NAB DNA tube are shown as yellow spheres.
Figure 6. Models of the P-Arm DNA-Six-Protein Regulatory Complex and the Tetrameric Int-Holliday Junction Excisive Recombination Intermediate.
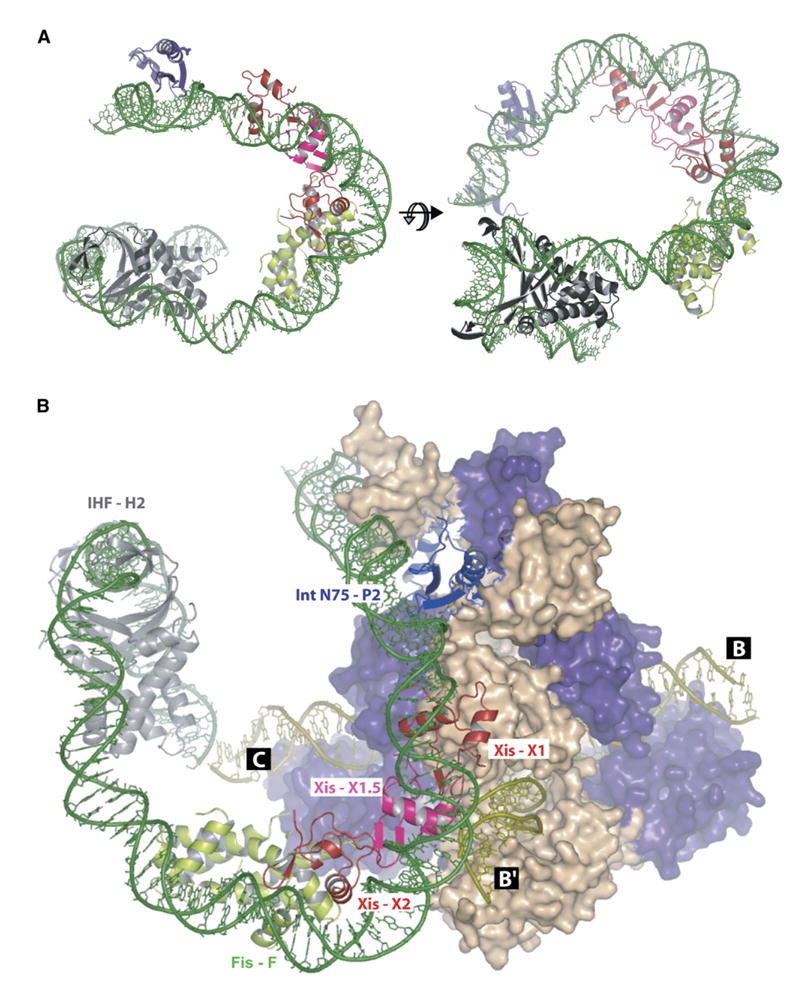
(A) A model of the P-arm DNA bound to the N domain of integrase (N75, purple) and accessory factors Xis (Xis1 or Xis2 [red] and Xis1.5 [pink]), Fis (green), and IHF (gray). The model was built by fitting the protein-DNA cocrystal structures of IHF (Rice et al., 1996), Xis (Sam et al., 2004), and Int (Biswas et al., 2005) and a model of the Fis crystal structure bound to DNA (Kostrewa et al., 1991; Yuan et al., 1991) to the 99 bp P-arm DNA path from Figure 4. The sequences between the protein-binding sites were created using the Insight II program. The final P-arm DNA was energy minimized using the program CNS to fix the backbone breaks and base planarity.
(B) The model in (A) was docked on the crystal structure of the tetrameric lambda integrase bound to a Holliday junction recombination intermediate and oligonucleotides encoding the P′1 and P′2 arm-type Int-binding sites (Biswas et al., 2005) to create a model of the excisive recombination complex. The Int bound to the P2 site (purple) bridges and binds to the C core site of the Holliday junction DNA (olive). Its partner (purple) bridges the P′1 arm and B core sites. The other pair of partner Ints (wheat) also bridge arm- and core-type sites. The P′ arm is not included in this figure, and the C′ core site is not visible in this view (see Figure 1 and Biswas et al., [2005]).
Seven randomly chosen structures are shown super-imposed in Figure 4A. To illustrate the DNA path, a DNA ‘‘tube’’ connecting eight dye positions from one of the structures was generated using NAB (Macke and Case, 1998), which interpolates a point every 3.34 Å between the dye positions (Figure 4B). The dye positions from the other 29 structures are shown superimposed on the DNA tube in Figure 4B.
Xis and Fis Binding to the P-Arm
To resolve a long-standing dichotomy between the delineation of two Xis consensus recognition sequences (see Figure 1) (Numrych et al., 1991; Yin et al., 1985) and the observation of three Xis-dependent DNA complexes in gel shift experiments (Figure 2B) (Sam et al., 2002; Thompson and Landy, 1988), it was necessary to investigate the stoichiometry and patterns of Xis binding to the P-arm DNA. His-tagged Xis was incubated with a fluorescently labeled 99 bp P-arm DNA fragment, and the three Xis complexes were separated by gel electrophoresis. After quantifying the DNA in each band by laser scanning with an emission filter at 520 nm, the gel was stained with a His-tag-specific dye and rescanned with a 580 nm emission filter specific for the dye.
If the ratio of protein-stained and fluorescent DNA band intensities of the fastest migrating Xis complex is assumed to correspond to an Xis/DNA ratio of 1:1, the Xis-to-DNA ratios for the second and third higher-order Xis-DNA complexes are consistent with the binding of two and three Xis molecules, respectively (Figure 5A).
Figure 5. Xis and Fis Binding to the P-Arm.
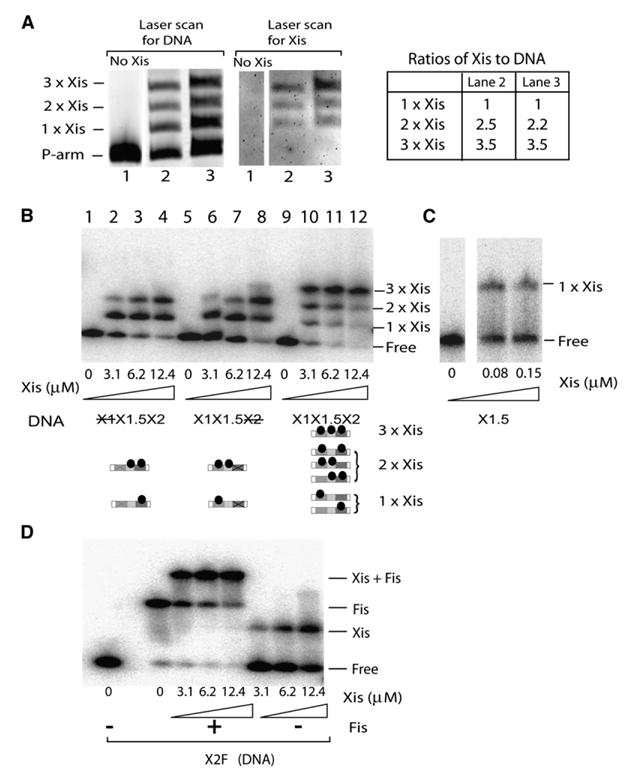
(A) Stoichiometry of Xis binding to the P-arm DNA. Xis was incubated with Bodipy-Fl labeled-P-arm DNA (99 bp) and electrophoresed on a native polyacrylamide gel. The gel was scanned in a Typhoon imager with an emission filter for 520 nm to quantify the DNA in each band (left panel) and then processed with a His-tag-specific stain followed by a second Typhoon scan with an emission filter for 580 nm (middle panel) to quantify the amount of Xis. The DNA/Xis ratios in the two slower-migrating bands were normalized to the ratio for the fastest-migrating band (right panel).
(B) Gel-retardation assays of Xis binding to three minimized 47 bp Xis-binding DNA fragments containing either wild-type sequences (X1X1.5X2), a scrambled X1 (X1X1.5X2), or a scrambled X2 (X1X1.5X2) site (the DNA sequences can be found in Table S1). Bottom panel is a schematic representation of the proteins in each gel band.
(C) Xis binding, in the absence of carrier herring sperm DNA, to a 19 bp DNA fragment containing only X1.5 DNA.
(D) Simultaneous binding of Xis and Fis (1.1 μM) to a minimized 39 bp DNA fragment containing the X2 and F sites.
The pattern of Xis site occupancy was further investigated using three different 47 bp fragments of the P-arm containing only the Xis binding sites (we refer to the DNA sequence between X1 and X2 as X1.5): one oligonucleotide consisted of the wild-type sequence, and each of the other two had either a mutated X1 site or a mutated X2 site (Figure 5B). Mutation of either X1 or X2 obliterates or greatly diminishes the formation of the slowest-migrating complex (lanes 2–4 and 6–8, Figure 5B), suggesting that in the wild-type the P-arm contains Xis protomers bound to X1, X1.5, and X2.
When both X1 and X2 are mutated (data not shown) or deleted (Figure 5C), Xis binds to X1.5 only in the absence of carrier DNA (Figure 5C), suggesting that the X1.5 sequence is a weak binding site to which Xis is ‘‘recruited’’ through cooperative interactions with the Xis molecules bound specifically at sites X1 and/or X2. The virtual disappearance of the single Xis complex in lane 11 of Figure 5B compared to lanes 4 and 8 suggests that optimal cooperative binding of Xis to the P-arm involves binding to all three Xis sites. We propose that the Xis bound to X1.5 is a protein-interaction ‘‘bridge’’ that stabilizes and promotes binding of Xis to X1 and X2 and is responsible for the previously observed cooperativity of binding at these two rather distantly spaced sites (Bushman et al., 1984; Sam et al., 2002).
The gel mobility shift patterns in Figure 2B suggest that, contrary to previous assumptions, Xis and Fis bind simultaneously to the overlapping X2/F sites (compare lanes 3, 4, 7, 12, 13, and 16). This was confirmed in a gel mobility shift experiment with a small, 39 bp DNA fragment containing only the overlapping X2 and F sites (Figure 5D). The presence of the unique band that is dependent on both Xis and Fis indicate that the two proteins bind to X2/F simultaneously and cooperatively. Thus, the mechanism by which Fis promotes excision at limiting Xis concentrations (Thompson et al., 1987) is not by stimulating Xis binding to X1 but probably through cooperative interactions with Xis bound to X2, which then promotes Xis binding to X1.5 and X1.
Taken together, the data in Figures 2B and 5 suggest that the complete P-arm DNA-protein complex (lane 16, Figure 2B) contains three Xis molecules in addition to one Int N domain, one IHF dimer, and one Fis dimer.
Xis and Fis DNA Bending in the P-Arm
To investigate the extent of Xis-induced P-arm bending, we used FRET to measure the distance between points fX1 and fH2A (see Figure 3A) in P-arm DNA bound with one, two, or three Xis molecules (Table 1). While there is no significant curvature detected for the singly bound Xis, the doubly and triply bound complexes show increasing curvature of approximately 95° and 115°, respectively. Likewise, when a single Xis is bound to a small DNA fragment, the fX1-to-fX1.5 distance measured by FRET does not deviate significantly from linear DNA (the calculated curvature of approximately 40° is indistinguishable from linear DNA). Thus, the FRET data, in agreement with the cocrystal structure of Xis bound to an X2 site, indicate that a single Xis does not induce a significant DNA bend (Sam et al., 2004). These data are consistent with a model in which the DNA bending induced by multiple adjacently bound Xis protomers is the result of protein-protein interactions that enhance the small DNA bends introduced by individual Xis molecules bound to X1, X1.5, and X2. However, because of caveats associated with both crystallography and FRET data on the singly bound Xis, we cannot rule out the possibility that the overall curvature results only from an additive effect of small individual bends induced by each of the bound Xis molecules.
Protein-protein interactions involving Xis are responsible for the bend induced between P2 and X1 when bound by N75 and Xis, respectively. Since N75 bound to P2 does not bend the DNA (Table 1 and Thompson and Landy, [1988]), the 55° increase in DNA curvature upon binding N75 to an Xis-DNA complex (Table 1) is most likely due to the previously described interactions between these two proteins (Cheng et al., 2000; Numrych et al., 1992; Sam et al., 2002; Sarkar et al., 2002; Swalla et al., 2003; Warren et al., 2003).
A potential Xis interaction that does not significantly increase DNA bending is that involving Fis. A small DNA fragment containing the X2/F sites and bound by Fis shows only a marginal (less than 10°) increase of curvature upon binding of Xis (Table 1), although their combined action may modify the direction of the DNA path, as discussed further below.
Modeling the P-Arm Regulatory Complex
The NAB-generated DNA trajectory (Figure 4B) was used as the starting point for modeling individual proteins into the P-arm DNA-protein regulatory complex in accord with data and conclusions presented above. The PDB coordinates for the protein-DNA cocrystal structures of IHF (Rice et al., 1996), Xis (Sam et al., 2004), and the amino-terminal domain of Int (bound to a cognate arm site) (Biswas et al., 2005) were used along with the coordinates of a model of the Fis-DNA complex (Hengen et al., 2003) based on the crystal structure of unliganded Fis (Yuan et al., 1991). The DNAs in these structures were laid out along the NAB P-arm trajectory in register with the locations of the dyes, and, where necessary, the DNA sequences in the crystal structures were manually changed to the corresponding DNA sequence in P-arm DNA: H′ to H2 for IHF, X2 to X1 and X1.5 for Xis, P′1 to P2 for the Int N domain, and a Fis consensus sequence to F for Fis. The resulting P-arm DNA was then energy minimized in the presence of the bound proteins using CNS (Brunger et al., 1998) (Figures 4C and 6A).
In the model, Arg63, which is present within the unstructured C-terminal region of Xis bound to X1, is within reach of residue E47 of Int bound at P2, consistent with genetic analyses of interactions between them (Cheng et al., 2000; Swalla et al., 2003; Warren et al., 2003). It is interesting that, in the docking model (Figure 6B), Arg63 of Xis is also close to E47 of the Int bound at P′2, although the importance of this is yet to be determined.
The three Xis protomers lie along the inside face of a smooth curve that must be due in large part to protein-protein interactions, because the binding of a single Xis induces only minimal curvature (Sam et al., 2004) (Table 1). A candidate for one element in these interactions is an arginine-rich patch at the interface of the protomers. This would be consistent with the finding by Sam et al. (2004) that the single Xis mutants R11A, R13A, and R14A all have anomalous electrophoretic mobilities, which we propose are due to deficiencies in DNA bending as a result of defects in protein-protein interactions.
The modeling also readily accommodates our finding that Xis binds X2 at the same time that Fis binds the overlapping F site (Figure 5D). The DNA segment between the F site and the H2 IHF site is incorporated as a relatively undistorted B form DNA. The only crystal structure that could not be fit closely to the FRET-based DNA trajectory was IHF complexed with its H′ binding site (see Discussion).
From the X-ray crystal structure of tetrameric Int bridging four core sites and four arm sites, the Int with an amino-terminal domain bound to P2 is predicted to have its carboxy-terminal domain bound to the C core site during excision (Biswas et al., 2005). Docking of the P-arm DNA-protein model to the X-ray crystal structure of the tetrameric Int complex accommodates such a bridge, although the fit is not perfect (Figure 6B) (see Discussion).
Discussion
Many eukaryotic and prokaryotic DNA-bending proteins bind specifically to adjacent DNA sequences and form defined protein-DNA complexes that effect the reactions performed by DNA-processing enzymes such as polymerases, nucleases, and recombinases. Often, the same DNA-bending protein is involved in several different cellular processes. For example, two of the proteins studied here, IHF and Fis, have roles in DNA condensation, transcription, and recombination (Johnson et al., 2005). The results reported here suggest how different multiples of the same bending protein and/or different combinations of the same group of bending proteins could be used to construct different nucleic acid trajectories with presumably different properties.
Determining how protein interactions influence DNA shape is hampered by the fact that traditional NMR and X-ray crystallographic approaches to studying DNA trajectories in very large complexes are extremely difficult and often not feasible. Examples of a few recent studies using alternative methods to model DNA trajectories are the following: cryo-EM combined with electron spectroscopic imaging (ESI) of the Mu transpososome I (Yuan et al., 2005), site-specific DNA-protein photocrosslinking of RNA polymerase II-transcription factors-promoter complex (Forget et al., 2004), and site-directed mutagenesis of DNA-binding surfaces of PepA (Reijns et al., 2005). In this study, we have used TIF distance measurements to determine the 3D path of the 99 bp λ phage P-arm DNA bound with six regulatory proteins. This approach, which has a small initial investment, can be readily applied to a wide range of systems involving large protein-nucleic acid complexes.
Role of Protein-Protein Interactions in Shaping the P-Arm DNA Trajectory
The DNA path of the P-arm segment bound with the Int-N domain, Xis, Fis, and IHF consists of three major bends: the bend between the P2 and X1 sites, the bend between X1 and X2/F sites, and the bend at the H2 site (Figures 4 and 6). The data presented here and discussed below indicate that the first of these bends results primarily from the interactions between DNAbound proteins, the second could be due to proteinprotein interactions, and the third results from the properties of an individual protein binding to its cognate site.
The results in Figure 5 and Table 1 show that the P-arm DNA complex contains three Xis molecules (binding at X1, X1.5, and X2) and not two, as previously thought (Sam et al., 2002; Thompson and Landy, 1988). These results are consistent with genetic studies showing that point mutants in what is now identified as the X1.5 site impaired Xis and Fis binding. Additionally, the small amount of DNA curvature (20°–50°) measured by FRET for the singly bound Xis is consistent with that observed in the cocrystal structure of Xis bound to an X2 oligonucleotide (approximately 25°).
In addition to the intermolecular interactions between Xis molecules, the Xis bound at X1 also interacts and binds cooperatively with the amino-terminal domain of Int bound at the adjacent P2 site (Cheng et al., 2000; Numrych et al., 1992; Sam et al., 2002; Sarkar et al., 2002; Swalla et al., 2003; Warren et al., 2003). We propose that these Xis-Int interactions are responsible for the bend between X1 and P2. This is based upon the fact that Xis (discussed above) or Int (Table 1 and Thompson and Landy, [1988]) binding alone each generate only a minimal bend, but when bound simultaneously (at X1 and P2) they generate approximately 95° of curvature (Table 1). It is interesting that this is approximately the same amount of curvature generated by two bound Xis protomers.
The unstructured C-terminal tail (residues 52–72) of Xis has been predicted to interact with E47 and the α-helical residues 51–55 of Int (Bushman et al., 1985; Numrych et al., 1992; Sarkar et al., 2002; Warren et al., 2003; Wu et al., 1998). E47 of Int and L52 of Xis (the last residue in the crystal structure used for modeling [Sam et al., 2004]) are ~27 Å apart in our model. This distance is compatible with the length of the unstructured C-terminal tail of Xis, assuming that residues 59–65 adopt an α helix upon contacting Int, as has been proposed (Sam et al., 2002; Wu et al., 1998).
Unlike the DNA bend-generating interactions between Xis protomers and between Xis and Int protomers, Xis and Fis do not synergistically increase DNA curvature, although their combined action may modify the direction of the DNA path. In contrast to earlier notions that Xis and Fis compete for binding to the P-arm DNA (Thompson and Landy, 1988), we have shown here that Xis and Fis bind simultaneously to the overlapping X2 and F sites (Figure 5D and Table 1). Since the directions of the Fis bend and the bend introduced by the three Xis on the full P-arm DNA are out of phase, there is no significant difference in overall curvature between three bound Xis molecules (approximately 113°) and the curvature of the X1-F region in the P-arm model (approximately 103°) (Figure 6), consistent with gel retardation analyses of circularly permuted DNAs (Thompson and Landy, 1988). A mutagenesis analysis has excluded involvement of the Xis C-terminal tail in interactions with Fis, which is in agreement with the Xis and Fis orientations in the model in Figure 6 (Numrych et al., 1992).
The third large bend in the P-arm DNA-protein complex is not due to protein-protein interactions but rather to the intrinsic properties of IHF binding to DNA on its own. The contour of the IHF-induced bend at H2 (approximately 130°), measured here in the context of the entire regulatory P-arm DNA complex (Figure 6), is similar but not indentical to the IHF-induced bend of H′ as seen in the cocrystal structure (160°) (Rice et al., 1996) and measured in solution by FRET (145°–150°) (Lorenz et al., 1999). It will be interesting to determine whether the observed difference in bending between the H2 and H′ complexes is due to the few differences (Hales et al., 1996) between the two DNA sequences or to the difference between isolated binding sites on short DNAs and binding sites in the context of a higher-order complex.
In summary, the model shown in Figure 6 suggests a mechanism for P-arm DNA folding: IHF bends the DNA at the H2 site; at limiting Xis concentration, Fis bound to the F site recruits Xis, which binds to the X2 site; the X2-bound Xis facilitates Xis binding to X1.5 and X1; finally, the X1-bound Xis helps Int binding to the weak P2 site. This ensemble of six proteins—Fis, three Xis, Int, and IHF—shape the P-arm DNA through a combination of protein-protein interactions and the intrinsic bending properties of individual proteins.
The Role of the P-Arm Complex in Excisive Recombination
Recent crystal structures of the Int tetramer bound to its cognate core and arm sites, one of a HJ intermediate and the other of a poststrand exchange intermediate, have provided critical views of how Int recognizes and bridges two different binding sites via its amino- and carboxy-terminal DNA-binding domains (Biswas et al., 2005). However, these extremely informative structures came with two caveats based on the experimental limitations. The first was that two copies of the P-arm Int-binding sites had to be used, instead of one copy each of P- and P′-arm sequences. The second was that the arm-type Int-binding sites had to be added in trans, as short oligonucleotides, disconnected from the core-type Int sites and lacking the intervening DNA and accessory DNA-bending proteins. The experiments presented here provide the missing critical view of the regulatory P-arm DNA bound by the accessory DNAbending proteins.
To dock the P-arm model to the Int tetramer crystal structure, the DNA at the junction between the H2 site and the C core site on the HJ has to be distorted (Figure 6). This may be related to, but is not entirely explained by, the earlier finding that the region between the H2 and C sites is not in a B-DNA form and has intrinsic curvature (Snyder et al., 1989a, 1989b; Thompson and Landy, 1988; Thompson et al., 1988). One particularly interesting explanation for the imperfect docking of the P-arm complex to the crystal structure is that the trajectory of P2-arm DNA determined here and the approximately square planar tetrameric Int-DNA complexes determined from the crystal structures reflect different intermediates along the reaction pathway.
Topological tangle analysis of lambda recombination reactions predicts that the synaptic complex and the product complex adopt a crossed X shape, rather than a parallel planar one (Crisona et al., 1999). The same study also proposes that the HJ intermediate switches from an X-shape nonplanar conformation to a square planar one (seen in all of the crystal structures of Int family members thus far) (Guo et al., 1997; Chen et al., 2000). Nonplanar HJ/Int complexes have also been observed in a FRET study of HJ recombination intermediates (Radman-Livaja et al., 2005). Very similar observations and arguments have been made for the topology and geometry of Flp and Cre recombination intermediates (Vetcher et al., 2006).
Incorporation of the P-arm DNA regulatory complex into a complete intasome, along with its multiple arm-core Int bridges, could either drive the synaptic and HJ intermediates into a nonplanar conformation or could drive some other conformational change associated with progress of the reaction. In either case, the implied conformational changes required for perfect docking suggest that the P-arm DNA regulatory complex affords a mechanical device for propelling recombination in one direction in an Xis/Fis-dependent (regulated) pathway. Beyond its role in the λ recombination reaction, the architecture of the P-arm DNA regulatory complex highlights how a small number of bending proteins, which induce DNA bends of varying magnitude depending on the number of bound protomers, could be combinatorally assembled to make a large number of different complexes whose features and functions would be expected to vary widely.
Experimental Procedures
Protein and Oligonucleotide Preparation
Proteins Int-N75 (the amino-terminal domain of λ Int, residues spanning 1–75), Xis, Fis, and IHF were purified as described previously (Lynch et al., 2003; Pan et al., 1996; Sarkar et al., 2002; Warren et al., 2005). Oligonucleotides were synthesized and PAGE or HPLC purified by Operon Technologies (Alameda, California) (for sequences, see Table S1 in the Supplemental Data available with this article online). The donor dye Bodipy-Fl was attached to the DNA phosphate backbone at the indicated positions of the bottom strand via a 5C linker. The acceptor dye TAMRA was attached to C5 of thymine at indicated positions of the top strand via a 6C linker. The same bottom strand, containing the donor dye and radioactively labeled at the 5′ terminus with 32P (using T4 polynucleotide Kinase [New England Biosciences, Massachusetts]), was used to make both donor only or donor plus acceptor DNAs, thus ensuring the same specific activity of 32P-labeling in the two substrates.
In-Gel FRET
Reaction mix for the full P-arm complex contained Int-N75 (0.11 μM), Xis (0.82 μM), Fis (0.10 μM), and IHF (0.30 μM) in buffer A (10mMTris-HCl [pH 7.5], 50mMNaCl, 1 mg/ml BSA, 2.5mMDTT, 0.1 mg/ml herring sperm DNA, 1 mM EDTA). When present alone, Xis was at 0.82 μM. For complexes with the 39 bp X2F DNA, Xis and/or Fis was present at 3.1 μM and 1.1 μM, respectively. For complexes with the 40 bp P2X1 DNA, N75 and/or Xis was present at 0.5 μM and 6.2 μM, respectively. All reactions contained donor only or donor plus acceptor DNA at 12 nM and were incubated at 25° for 20 min prior to electrophoresis on a 7% polyacrylamide gel at 10 V/cm. Fluorescence intensities of the donor in the complexes with and without acceptor were obtained from a laser scan of the gel using a 488 nm blue laser for excitation and a 520 nm emission filter (Typhoon, Amersham Biosciences). The amount of radiolabeled complex in each band was determined from a Phosphorimager (Fuji BAS-2500) analysis of the same gel after drying.
FRET Calculations
The efficiency of energy transfer (E) was determined from the extent of donor fluorescence quenching in doubly labeled (donor and acceptor) P-arm DNA bound with six proteins compared with donor-only-labeled P-arm bound with six proteins. The determinations of the efficiency of energy transfer E and FRET calculations were performed as described previously (Radman-Livaja et al., 2005) and in Supplemental Data. R0, the distance between the donor and acceptor that gives 50% FRET (the Förster distance), was measured (Wu and Brand, 1994) as described in Supplemental Data, and the values for each donor-acceptor pair can be found in Table S2. Steady-state anisotropies (r) were measured for all donor-labeled and acceptor-labeled P-arms unbound and in the context of the P-arm-protein complex (see Supplemental Data).
Coordinate Determination and Structure Modeling
As the last step in TIF, structures are generated by a metric matrix distance-geometry program (written by D.F.M.) incorporating the method of random metrization of Havel (Havel, 1993). The upper and lower distances from the FRET measurements were placed into an 8 × 8 matrix. An atom pair (i, j) was selected randomly and a distance between the upper and lower values randomly chosen. The remaining distances were then adjusted to this distance employing triangular inequality, a procedure called bound smoothing. The next atom pair was randomly chosen and the procedure repeated until the matrix was complete. One thousand two hundred trials produced 86 structures: 30 of one and 56 of the other chirality. The small number of successful structures derives from the lack of constraints on the initial distances chosen (not required to be consistent with all other distances). The Cartesian coordinates were calculated from the three Eigenvectors with the largest Eigenvalues and then optimized by using a steepest-descent algorithm against the distance matrix. A unique structure is determined with 27 out of 28 possible distances.
Excision and Gel Retardation Assays
Excision assays were performed as described previously and typically contained 36 nM unlabeled attL site (128 bp) and 12 nM labeled attR site (145 bp) (DNA sequences of the excision substrates can be found in Table S1). Gel retardation assays were carried out as described previously (Sarkar et al., 2002).
To determine the stoichiometry of Xis binding, (His-tagged) Xis (0.82 μM) was incubated with Bodipy-Fl-labeled P-arm DNA (99 bp). Following electrophoresis, the gel was scanned twice, first using a 488 nm laser for excitation and a 520 nm emission filter to quantify the DNA, and then, after staining with Invision His-tag In-gel Stain (Invitrogen), by using a 532 nm green laser for excitation and a 580 nM emission filter to quantify the Xis in each band.
Supplementary Material
Acknowledgments
We thank Christine Lank for technical assistance, Joan Boyles for manuscript preparation, David Filman for assistance with the DNA modeling and energy minimization, and members of the A.L. laboratory for helpful comments and suggestions. This work was supported by research grants from the National Institutes of Health (to A.L., D.F.M, and Tom Ellenberger [T.B.]) and a fellowship from the Jeane B. Kempner Fund (T.B.).
Footnotes
Supplemental Data
Supplemental Data include two tables, supplemental text, and Supplemental References and can be found with this article online at http://www.molecule.org/cgi/content/full/24/4/569/DC1/.
References
- Azaro MA, Landy A. λ Integrase and the λ Int family. In: Craig NL, Craigie R, Gellert M, Lambowitz A, editors. In Mobile DNA II. 2. Washington, DC: ASM Press; 2002. pp. 118–148. [Google Scholar]
- Ball CA, Osuna R, Ferguson KC, Johnson RC. Dramatic changes in Fis levels upon nutrient upshift in Escherichia coli. J Bacteriol. 1992;174:8043–8056. doi: 10.1128/jb.174.24.8043-8056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by λ integrase. Nature. 2005;435:1059–1066. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Bushman W, Yin S, Thio LL, Landy A. Determinants of directionality in lambda site-specific recombination. Cell. 1984;39:699–706. doi: 10.1016/0092-8674(84)90477-x. [DOI] [PubMed] [Google Scholar]
- Bushman W, Thompson JF, Vargas L, Landy A. Control of directionality in lambda site-specific recombination. Science. 1985;230:906–911. doi: 10.1126/science.2932798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AM. Episomes. In: Caspari EW, Thoday JM, editors. In Advances in Genetics. New York: Academic Press; 1962. pp. 101–145. [Google Scholar]
- Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell. 2000;6:885–897. [PubMed] [Google Scholar]
- Cheng Q, Swalla BM, Beck M, Alcaraz R, Jr, Gumport RI, Gardner JF. Specificity determinants for bacteriophage Hong Kong 022 integrase: analysis of mutants with relaxed core-binding specificities. Mol Microbiol. 2000;36:424–436. doi: 10.1046/j.1365-2958.2000.01860.x. [DOI] [PubMed] [Google Scholar]
- Cho EH, Gumport RI, Gardner JF. Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex. J Bacteriol. 2002;184:5200–5203. doi: 10.1128/JB.184.18.5200-5203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisona NJ, Weinberg RL, Peter BJ, Sumners DW, Cozzarelli NR. The topological mechanism of phage λ integrase. J Mol Biol. 1999;289:747–775. doi: 10.1006/jmbi.1999.2771. [DOI] [PubMed] [Google Scholar]
- Forget D, Langelier MF, Therien C, Trinh V, Coulombe B. Photo-cross-linking of a purified preinitiation complex reveals central roles for the RNA polymerase II mobile clamp and TFIIE in initiation mechanisms. Mol Cell Biol. 2004;24:1122–1131. doi: 10.1128/MCB.24.3.1122-1131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainge I, Jayaram M. The integrase family of recombinase: organization and function of the active site. Mol Microbiol. 1999;33:449–456. doi: 10.1046/j.1365-2958.1999.01493.x. [DOI] [PubMed] [Google Scholar]
- Guo F, Gopaul DN, Van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- Hales LM, Gumport RI, Gardner JF. Examining the contribution of a dA+dT element to the conformation of Escherichia coli integration host factor-DNA complexes. Nucleic Acids Res. 1996;24:1780–1786. doi: 10.1093/nar/24.9.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallet B, Sherratt DJ. Transposition and site-specific recombination: adapting DNA cut-and-paste mechanisms to a variety of genetic rearrangements. FEMS Microbiol Rev. 1997;21:157–178. doi: 10.1111/j.1574-6976.1997.tb00349.x. [DOI] [PubMed] [Google Scholar]
- Havel TF. The sampling properties of some distance geometry algorithmsapplied to unconstrained polypeptide chains: a study of 1830 independently computed conformations. Biopolymers. 1990;29:1565–1585. doi: 10.1002/bip.360291207. [DOI] [PubMed] [Google Scholar]
- Havel TF. An evaluation of computational strategies for use in the determination of protein structure from distance constraints obtained by nuclear magnetic resonance. Prog Biophys Mol Biol. 1993;56:43–78. doi: 10.1016/0079-6107(91)90007-f. [DOI] [PubMed] [Google Scholar]
- Hengen PN, Lyakhov IG, Stewart LE, Schneider TD. Molecular flip-flops formed by overlapping Fis sites. Nucleic Acids Res. 2003;31:6663–6673. doi: 10.1093/nar/gkg877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC, Johnson LM, Schmidt JW, Gardner JF. Major nucleoid proteins in the structure and function of the Escherichia coli chromosome. In: Higgins NP, editor. In The Bacterial Chromosome. Washington, DC: ASM Press; 2005. pp. 65–132. [Google Scholar]
- Kostrewa D, Granzin J, Koch C, Choe HW, Labahn J, Kahmann R, Saenger W. Three-dimensional structure of the E. coli DNA-binding protein FIS. Nature. 1991;349:178–180. doi: 10.1038/349178a0. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Hillisch A, Goodman SD, Diekmann S. Global structure similarities of intact and nicked DNA complexed with IHF measured in solution by flourescence resonance energy transfer. Nucleic Acids Res. 1999;27:4619–4625. doi: 10.1093/nar/27.23.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TW, Mattis AN, Gardner JF, Rice PA. Integration host factor: putting a twist on protein-DNA recognition. J Mol Biol. 2003;330:493–502. doi: 10.1016/s0022-2836(03)00529-1. [DOI] [PubMed] [Google Scholar]
- Macke TJ, Case DA. Modeling unusual nucleic acid structures. In: Leontes NB, SantaLucia J Jr, editors. In Molecular Modeling of Nucleic Acids. Washington, DC: American Chemical Society; 1998. pp. 379–393. [Google Scholar]
- Nazarenko I, Pires R, Lowe B, Obaidy M, Rashtchian A. Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res. 2002;30:2089–2095. doi: 10.1093/nar/30.9.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson L, Verbeek H, Vijgenboom E, van Drunen C, Vanet A, Bosch L. FIS-dependent trans activation of stable RNA operons of Escherichia coli under various growth conditions. J Bacteriol. 1992;174:921–929. doi: 10.1128/jb.174.3.921-929.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numrych TE, Gumport RI, Gardner JF. A genetic analysis of Xis and FIS interactions with their binding sites in bacteriophage lambda. J Bacteriol. 1991;173:5954–5963. doi: 10.1128/jb.173.19.5954-5963.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numrych TE, Gumport RI, Gardner JF. Characterization of the bacteriophage lambda excisionase (Xis) protein: the C-terminus is required for Xis-integrase cooperativity but not for DNA binding. EMBO J. 1992;11:3797–3806. doi: 10.1002/j.1460-2075.1992.tb05465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan CQ, Finkel SE, Cramton SE, Feng JA, Sigman DS, Johnson RC. Variable structures of Fis-DNA complexes determinedby flankingDNA-protein contacts. J Mol Biol. 1996;264:675–695. doi: 10.1006/jmbi.1996.0669. [DOI] [PubMed] [Google Scholar]
- Radman-Livaja M, Biswas T, Mierke D, Landy A. Architecture of recombination intermediates visualized by In-gel FRET of λ integrase-Holliday junction-arm-DNA complexes. Proc Natl Acad Sci USA. 2005;102:3913–3920. doi: 10.1073/pnas.0500844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns M, Lu Y, Leach S, Colloms SD. Mutagenesis of PepA suggests a new model for the Xer/cer synaptic complex. Mol Microbiol. 2005;57:927–941. doi: 10.1111/j.1365-2958.2005.04716.x. [DOI] [PubMed] [Google Scholar]
- Rice PA, Yang SW, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA u-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- Sam M, Papagiannis C, Connolly KM, Corselli L, Iwahara J, Lee J, Phillips M, Wojciak JM, Johnson RC, Clubb RT. Regulation of directionality in bacteriophage lambda site-specific recombination: structure of the Xis protein. J Mol Biol. 2002;324:791–805. doi: 10.1016/s0022-2836(02)01150-6. [DOI] [PubMed] [Google Scholar]
- Sam MD, Cascio D, Johnson RC, Clubb RT. Crystal structure of the excisionase-DNA complex from bacteriophage lambda. J Mol Biol. 2004;338:229–240. doi: 10.1016/j.jmb.2004.02.053. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Radman-Livaja M, Landy A. The small DNA binding domain of λ Int is a context-sensitive modulator of recombinase functions. EMBO J. 2001;20:1203–1212. doi: 10.1093/emboj/20.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, Azaro MA, Aihara H, Papagiannis C, Tirumalai RS, Nunes-Düby SE, Johnson RC, Ellenberger T, Landy A. Differential affinity and cooperativity functions of the amino-terminal 70 residues of λ integrase. J Mol Biol. 2002;324:775–789. doi: 10.1016/s0022-2836(02)01199-3. [DOI] [PubMed] [Google Scholar]
- Snyder UK, Thompson JF, Landy A. Phasing of protein-induced DNA bends in a recombination complex. Nature. 1989a;341:255–257. doi: 10.1038/341255a0. [DOI] [PubMed] [Google Scholar]
- Snyder UK, Thompson JF, Landy A. Phasing of protein-induced DNA bends in a recombination complex. Nature 342, 255–257, Nature. 1989b;342(6426):1989. doi: 10.1038/341255a0. [DOI] [PubMed] [Google Scholar]
- Swalla BM, Cho EH, Gumport RI, Gardner JF. The molecular basis of cooperative DNA binding between lambda integrase and excisionase. Mol Microbiol. 2003;50:89–99. doi: 10.1046/j.1365-2958.2003.03687.x. [DOI] [PubMed] [Google Scholar]
- Thompson JF, Landy A. Empirical estimation of protein-induced DNA bending angles: applications to λ site-specific recombination complexes. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JF, Moitoso de Vargas L, Koch C, Kahmann R, Landy A. Cellular factors couple recombination with growth phase: characterization of a new component in the λ site-specific recombination pathway. Cell. 1987;50:901–908. doi: 10.1016/0092-8674(87)90516-2. [DOI] [PubMed] [Google Scholar]
- Thompson JF, Mark HF, Franz B, Landy A. Functional and structural characterization of stable DNA curvature in lambda attP. In: Olson WK, Sarma MH, Sarma RH, Sundaralingam M, editors. In DNA Bending and Curvature. Guilderland, NY: Adenine Press; 1988. pp. 119–128. [Google Scholar]
- Van Duyne GD. A structural view of tyrosine recombinase site-specific recombination. In: Craig NL, Graigie R, Gellert M, Lambowitz AM, editors. In Mobile DNA II. Washington, DC: ASM Press; 2002. pp. 93–117. [Google Scholar]
- Vetcher AA, Lushnikov AY, Navarra-Madsen J, Scharein RG, Lyubchenko YL, Darcy IK, Levene SD. DNA topology and geometry in Flp and Cre recombination. J Mol Biol. 2006;357:1089–1104. doi: 10.1016/j.jmb.2006.01.037. [DOI] [PubMed] [Google Scholar]
- Warren D, Sam M, Manley K, Sarkar D, Lee SY, Abbani M, Clubb RT, Landy A. Identification of the λ integrase surface that interacts with the Xis accessory protein reveals a residue that is also critical for homomeric dimer formation. Proc Natl Acad Sci USA. 2003;100:8176–8181. doi: 10.1073/pnas.1033041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren D, Lee SY, Landy A. Mutations in the aminoterminal domain of lambda-integrase have differential effects on integrative and excisive recombination. Mol Microbiol. 2005;55:1104–1112. doi: 10.1111/j.1365-2958.2004.04447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- Wu Z, Gumport RI, Gardner JF. Defining the structural and functional roles of the carboxyl region of the bacteriophage lambda excisionase (Xis) protein. J Mol Biol. 1998;281:651–661. doi: 10.1006/jmbi.1998.1963. [DOI] [PubMed] [Google Scholar]
- Yin S, Bushman W, Landy A. Interaction of λ site-specific recombination protein Xis with attachment site DNA. Proc Natl Acad Sci USA. 1985;82:1040–1044. doi: 10.1073/pnas.82.4.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan HS, Finkel SE, Feng JA, Kaczor-Grzeskowiak M, Johnson RC, Dickerson RE. The molecular structure of wild-type and a mutant Fis protein: relationship between mutational changes and recombinational enhancer function or DNA binding. Proc Natl Acad Sci USA. 1991;88:9558–9562. doi: 10.1073/pnas.88.21.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JF, Beniac DR, Chaconas G, Ottensmeyer FP. 3D reconstruction of the Mu transposase and the type 1 transpososome: a structural framework for Mu DNA transposition. Genes Dev. 2005;19:840–852. doi: 10.1101/gad.1291405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.