

Abstract
Collagen-related peptide (CRP) stimulates powerful activation of platelets through the GPVI-FcR γ-chain complex. We have combined proteomics and traditional biochemistry approaches to study the proteome of CRP-activated platelets, focusing in detail on tyrosine phosphorylation. In two separate approaches, phosphotyrosine immunoprecipitations followed by 1D-PAGE, and 2-DE, were used for protein separation. Proteins were identified by MS. By following these approaches, 96 proteins were found to undergo post-translational modification in response to CRP in human platelets, including 11 novel platelet proteins such as Dok-1, SPIN90, osteoclast stimulating factor 1, and β-Pix. Interestingly, the type I transmembrane protein G6f was found to be specifically phosphorylated on Tyr-281 in response to platelet activation by CRP, providing a docking site for the adapter Grb2. G6f tyrosine phoshporylation was also found to take place in response to collagen, although not in response to the G protein-coupled receptor agonists, thrombin and ADP. Further, we also demonstrate for the first time that Grb2 and its homologue Gads are tyrosine-phosphorylated in CRP-stimulated platelets. This study provides new insights into the mechanism of platelet activation through the GPVI collagen receptor, helping to build the basis for the development of new drug targets for thrombotic disease.
Keywords: G6f, Gads, GPVI-signaling, Grb2, Platelet-proteomics
1 Introduction
Activation of platelets by collagens of the subendothelium is a critical event in prevention of excessive blood loss following tissue injury. Platelets express two major receptors for collagen, the integrin α2β1 and the immunoglobulin GPVI [1]. GPVI is the major receptor mediating platelet activation by collagen [1]. Collagen-related peptide (CRP) is a synthetic peptide based on the repeat sequence, Gly-Pro-HydroxyPro (GPO), which enables it to adopt the stable triple-helical structure found in collagen [2]. When cross-linked via N-terminal and C-terminal lysine or cysteine residues, CRP adopts the typical collagen quaternary structure, becoming a powerful platelet activator. CRP activates GPVI in platelets, stimulating a similar pattern of increase in tyrosine phosphorylation to native collagen [3, 4].
In recent years, proteomics has become a key tool in the analysis of signaling cascades in human platelets [5]. The separation of proteins using zoom 2-DE or sample pre-fractionation followed by 1D-PAGE, and their identification by MS has proven to be an efficient way to analyze the proteome of basal and activated platelets, including identification of post-translational modifications such as phosphorylation. 2-DE enables the separation of thousands of proteins at a time according to their isoelectric point (pI) and mass [6]. After protein staining, a detailed image analysis allows detection of proteins which can then be excised from the gel, trypsinized and analyzed by LC-MS/MS. We have recently applied this technology to the investigation of the proteomes from un-activated and thrombin receptor-activating peptide (TRAP)-activated human platelets [7, 8].
The present study was designed to identify novel phosphorylated proteins in CRP-activated platelets in order to improve our knowledge on platelet regulation by GPVI. The proteome of CRP-activated platelets was analyzed in detail by using two complementary separation procedures, namely phosphotyrosine immunoprecipitation followed by 1D-gel electrophoresis and MS, and by 2-DE and MS. By using these two approaches, 96 proteins were found to undergo post-translational modification in response to CRP. Strikingly, 11 of these proteins had not previously been identified in platelets, including β-Pix, and SPIN90, which undergo tyrosine phosphorylation upon platelet stimulation with CRP. In addition, the recently identified transmembrane immunoglobulin G6f was found to undergo tyrosine phosphorylation in response to platelet activation by CRP and collagen, leading to the recruitment of the adapter Grb2 to the plasma membrane. We speculate that many of these new signaling events play important roles in platelet activation by GPVI.
2 MATERIALS AND METHODS
2.1 Reagents, antibodies and suppliers
Agarose-conjugated and non-conjugated anti-phosphotyrosine monoclonal antibody (mAb) (clone: 4G10) and anti-Gads polyclonal antibody were purchased from Upstate Biotechnology Inc. (NY, USA). Anti-Grb2 polyclonal antibody, normal mouse IgG conjugated to agarose and normal rabbit IgG were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-β-Pix polyclonal antibody was from Chemicon International (Temecula, CA, USA). G6f rabbit polyclonal antiserum was raised from CovalAB UK (Cambridge, UK) against the following peptides: 153RMDSVTWQEGKGPV166, and 266GRDASIPQFKPEIQ279. A second G6f rabbit polyclonal antiserum was raised from Eurogentec (Liège, Belgium) against the following peptides: 259QRVRGAPGRDASIPQF274 and 284IHLARLGPPAHKPR297. Pro-Q diamond phosphoprotein gel stain was purchased from Molecular Probes (Invitrogen Ltd, Paisley, UK).
Unless specifically stated, the suppliers of other chemicals and instruments were the same as described previously [9] or were obtained from Sigma (St. Louis, MO, USA). In order to generate a positive control for G6f expression in platelets, the ORF of G6f with a C-terminal Myc tag was cloned into the pEF6 vector in frame with the Myc tag (Invitrogen), and transiently transfected into HEK 293T cells at 40-50% confluence using calcium phosphate precipitation reagents and standard protocols.
2.2 Platelet preparation and activation with CRP
Platelets were isolated by an established method that limits contamination from other blood cells as previously described [5, 9]. This includes taking only the upper third of the platelet rich plasma, the use of leukocyte removal filters and manual inspection for the presence of contaminating leukocytes. Washed platelets were prepared as previously described using acid citrate dextrose (ACD) solution (117 mM sodium citrate, 111 mM glucose, 78 mM citric acid) as anticoagulant [9]. Platelets were resuspended in Tyrodes-HEPES (134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, 5 mM glucose, 1 mM MgCl2, 1 mM EGTA pH 7.3) at 1 × 109 platelets/ml, and incubated at room temperature for 30 min. For the tyrosine phosphorylation studies, 10 μM indomethacin and 2 U/ml apyrase were included. One milliliter aliquots of platelets were warmed at 37°C for 10 min prior to stimulation with CRP (final concentration: 10 μg/ml). Unless specifically stated, stimulations were carried out at 37°C for 90 seconds with constant stirring at 1200 rpm. Unstimulated (basal) samples were treated with CRP diluent (0.01 M acetic acid containing 0.1% fatty acid free BSA) alone. For the 2-DE analysis, platelets were frozen in liquid nitrogen following all stimulations.
2.3 Two-dimensional gel electrophoresis
Proteins were extracted from frozen platelet suspensions by TCA/acetone protein precipitation and delipidation, as described previously [8]. For pI 4-7 2-DE gels, pellets were resuspended in 375 μl sample buffer (5 M urea, 2 M thiourea, 2 mM tributyl-phosphine, 65 mM DTT, 65 mM CHAPS, 0.15 M NDSB-256, 1 mM sodium vanadate, 0.1 mM sodium fluoride, 1 mM benzamidine). For pI 6-11 2-DE gels, protein pellets were resuspended in 500 μl of the same sample buffer containing 10% isopropanol. Ampholytes (Servalyte 4-7 and 6-11, respectively) were added to the sample to a final concentration of 1.6% (v/v). 2-DE gel electrophoresis was carried out as described previously.9 Following electrophoresis, the gels were fixed in 40 % (v/v) ethanol:10 % (v/v) acetic acid and stained with the fluorescent dye OGT MP17 as described previously [8].
2.4 Differential image analysis
Scanned images were processed with a custom version of MELANIE II (Bio-Rad Laboratories Ltd., Hemel Hempstead, UK). Four pI 4-7 gels and four pI 6-11 gels were prepared in independent experiments for both basal and CRP-activated platelets. Internal calibration of the 2-DE gel images with regard to pI and molecular weight was carried out as described previously [9]. For differential image analysis, two synthetic gel images, one for each of the two applied analytical pI ranges of 4-7 and 6-11, were generated by means of accurate spot matching, as previously described [8]. These synthetic images contained all protein features detected in basal and stimulated platelets. Only features present in at least three out of four individual gels belonging to either the basal or CRP group were considered for differential analysis. Differential expression of a protein present in both the basal and CRP gels was considered significant when the fold change was at least 2 and the p value (probability) was no more than 0.05 after rank sum test was applied on %V values.
2.5 Immunoprecipitation and western blotting
Basal and stimulated platelets (8 × 108/ml, 500 μl) were lysed as described previously [8]. The amount of each antibody added to the lysates for immunoprecipitation was as follows: 10 μg of monoclonal anti-phosphotyrosine antibody conjugated to agarose, 5 μg of a rabbit polyclonal anti-Gads, and 5 μg of a rabbit polyclonal anti-Grb2. In the case of G6f, 10 μl of antibody serum was added to the lysates. Normal mouse IgG conjugated to agarose and normal rabbit IgG were used as negative controls for the immunoprecipitations. Before the addition of the antibodies, samples were pre-cleared with 30 μl of Protein A-Sepharose (50% w/v in TBS-T [20 mM Tris, 137 mM NaCl and 0.1% (v/v) Tween 20, pH 7.6)]) at 4°C for 30 min with end-over-end mixing. The antibodies were then added and samples rotated overnight at 4°C. In all cases, with the exception of phoshpotyrosine immunoprecipitations, twenty microlitres of Protein A-Sepharose (50% w/v in TBS-T) was added to the samples and they were mixed end-over-end for 1 hour at 4°C. Pellets were washed once in 1 × lysis buffer and two times in TBS-T before addition of 2 × Laemmli sample buffer (4% [w/v] SDS, 10% [v/v] 2-mercaptoethanol, 20% [v/v] glycerol, 50 mM Tris, pH 6.8). Proteins were resolved on 4-12% NuPAGE Bis-Tris gels (Invitrogen Ltd, Paisley, UK), using MOPS (Invitrogen) as running buffer, and transferred on to PVDF membranes (Amersham Biosciences; Buckinghamshire, UK). The membranes were blocked in 10% BSA (w/v) in TBS-T overnight at 4°C, then incubated for 2 hours at room temperature with the primary antibodies rabbit anti-Gads (1:1000), rabbit anti-Grb2 (1:1000), rabbit anti-G6f (1:1000), rabbit anti-β-Pix (1:1000), or mouse anti-phosphotyrosine mAb (1:1000). Following washes in TBS-T, the blots were incubated for 1 hour with horseradish peroxidase-labelled anti-rabbit or anti-mouse polyclonal antibodies (1:10,000), respectively. Membranes were washed again and developed using an enhanced-chemiluminescence system (ECL, Amersham). Whenever necessary, blots were stripped by incubation for 30 min at 80°C in stripping buffer (TBS-T, 2% [w/v] SDS and 1% [v/v] 2-mercaptoethanol) and re-blotted.
2.6 Phosphoprotein gel staining
For MS experiments, 6 identical immunoprecipitations were pooled together and loaded on the same lane on 4-12% NuPAGE Bis-Tris gels, using MOPS as running buffer. Following electrophoresis, the gels were fixed overnight in 40 % (v/v) ethanol:10% (v/v) acetic acid and stained with Pro-Q diamond phosphoprotein gel stain following Invitrogen recommendations. When necessary, gels were re-stained for total protein with the fluorescent dye OGT MP17. Bands of interest were excised for protein digestion and MS analysis.
2.7 Mass spectrometric analysis
Protein features chosen for MS analysis were in-gel digested with trypsin, and peptides extracted as described previously [8, 10]. MS analysis was carried out using a Q-TOF 1 (Micromass, Manchester, UK) coupled to a CapLC (Waters, Milford, MA, USA) configured with a 300 μm id/5 mm C18 precolumn and a 75 μm id/25 cm C18 PepMap analytical column (LC packings, San Francisco, CA, USA). Tryptic peptides were eluted to the mass spectrometer using a 45 min 5-95% acetonitrile gradient containing 0.1% formic acid at a flow rate of 200 nl/min. Spectra were acquired in an automatic data dependent fashion with a 1 sec survey scan followed by three 1 sec MS/MS scans of the most intense ions. The selected precursor ions were excluded from further analysis for 2 min. The MS/MS spectra were converted into pkl files using Mass Lynx 3.4. The database search was performed with the MASCOT 1.8 search tool (Matrix science, London, UK) screening SWISS-PROT (release 45.2 of 23-Nov-2004) and TrEMBL (release 28.2 of 23-Nov-2004). Searches were restricted to the human taxonomy allowing carbamidomethyl cysteine as a fixed modification and both oxidized methionine and tyrosine phosphorylation as potential variable modifications. Both the precursor mass tolerance and the MS/MS tolerance were set at 0.5 Da. Positive identification was only accepted when the data satisfied the following criteria: (i) MS data were obtained for at least 80% y-ions series of a peptide comprising at least eight amino acids and no missed tryptic cleavage sites; (ii) MS data with more than 50% y-ions were obtained for two or more different peptides comprising at least eight amino acids long and no more than one missed tryptic cleavage site.
3 RESULTS
3.1 Tyrosine phosphoproteome analysis of CRP-stimulated platelets
We investigated in detail the GPVI signaling cascade using a combination of phosphotyrosine immunoprecipitation and 1D-gel electrophoresis followed by MS analysis. In these studies, we avoided the effect of the secondary mediators ADP and thromboxanes through inclusion of indomethacin and apyrase. In addition, EGTA was included to prevent aggregation and thereby facilitate recovery of tyrosine phosphorylated proteins. Time-course and dose-response experiments were carried out to verify the optimal conditions to achieve maximal tyrosine-phosphorylation induced by CRP (not shown). A concentration of 10 μg/ml of CRP for 90 sec was selected for subsequent investigations.
Phosphotyrosine immunoprecipitated proteins from basal and CRP-stimulated platelets were separated on 4-12% NuPage Bis-Tris gels and stained with the specific phosphoprotein gel stain Pro-Q Diamond, which detects all sites of protein phosphorylation (Fig. 1A). Equivalent gels were run in parallel and stained for total protein with a fluorescent dye as described in the Materials and Methods (Fig. 1A). An anti-phosphotyrosine western blot was run alongside for comparison (Fig. 1B). Immunoprecipitations using a control mAb (mouse IgG) was also run alongside (not shown).
Figure 1.
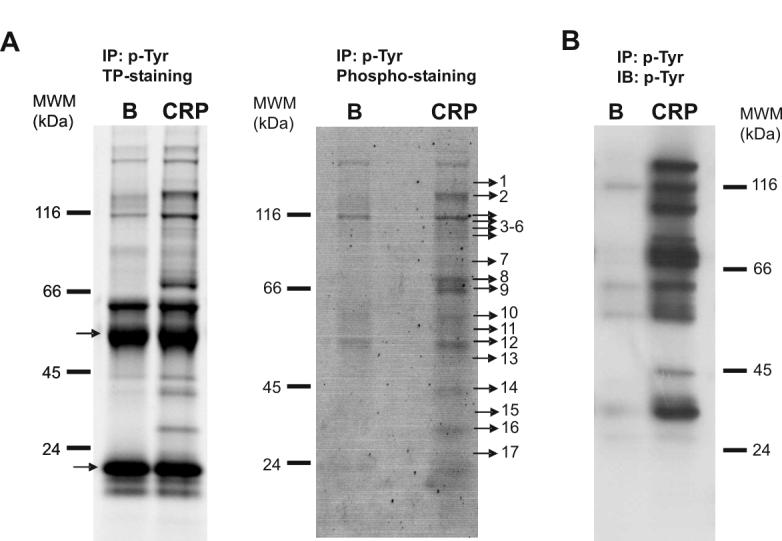
Tyrosine phosphoproteome analysis of CRP-stimulated platelets. (A) left panel, total protein staining of a 4-12% NuPAGE Bis-tris gel showing all the proteins immunoprecipitated with the 4G10 antibody in Basal and CRP-stimulated platelets (bands corresponding to IgG chains are indicated by arrows); right panel, phosphoprotein staining of an equivalent 4-12% NuPAGE Bis-tris gel showing tyrosine phosphorylated proteins obtained after 4G10 immunoprecipitation of Basal and CRP-stimulated platelets. Indicated bands were excised from the gel and analyzed. Proteins identified are reported in Table 1. (B) Similar to (A) but the gel was blotted instead of stained, followed by an anti-4G10 western blot to visualize the tyrosine-phosphorylated proteins. B, basal platelets; CRP, platelets stimulated with CRP (10 μg/mL, 90 sec); IP, immunoprecipitation; IB, immuno blot; TP, total protein. Images represent at least three independent experiments.
Specific bands corresponding to tyrosine-phosphorylated proteins or equivalent regions in the control and basal gels were excised and analyzed by LC-MS/MS. This approach identified 30 different proteins specifically recruited to a phospho-tyrosine signaling complex in response to CRP (see Table 1 and Supplementary Table 1). Further, the majority of the proteins are known to play a significant role in the GPVI signaling cascade, namely ADAP, Btk, c-Cbl, Dok-2, Fes, Fyn, Gads, Hck, Lyn, p38α mapkinase, PLCγ2, SHIP, SKAP-130, SLP76, Src, Syk and Vav1. Significantly, all of these proteins, with the exception of Gads, are known to be regulated by tyrosine phosphorylation. This is the first time that a significant number of proteins in the GPVI signaling cascade have been identified in platelets using a proteomics approach.
Table 1.
List of proteins identified following tyrosine-phosphoproteome analysis of CRP-stimulated human platelets. Proteins from basal and CRP-stimulated platelets (10 μg/ml, 90 sec) were immunoprecipitated using an antibody to phosphotyrosine. Immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and analyzed by MS. Band numbers correspond to Figure 1.
| Protein Name | ANa) | Band No. | Functionb) |
|---|---|---|---|
| Proteins known to be tyrosine phosphorylated by GPVI | |||
| α-actinin 1 | P12814 | 4 | Actin-binding protein. Tyrosine-phosphorylated in activated platelets. |
| ADAP [Adhesion- and degranulation-promoting adapter protein] (SLAP-130, FYB) | O15117 | 3 | No recognized function in GPVI signaling. Binds to SKAP-Hom and SLP-76. |
| Btk [Bruton’s tyrosine kinase] | Q06187 | 7 | Critical role in activation of PLCγ2 and possibly Ca2+ entry by GPVI. |
| c-Cbl [E3 ubiquitin protein ligase] | P22681 | 4 | Feedback inhibition of GPVI signaling. |
| Cortactin | Q14247 | 7 | Role in GPVI signaling is unknown; shown to regulate actin polymerization in other cells. |
| Dok-2 [Downstream of tyrosine kinases] | O60496 | 13 | Function in the GPVI cascade is not known. |
| Fes | P07332 | 5 | GPVI signaling is maintained in Fes-knockout mice. |
| Fyn | P06241 | 11 | Src family kinase: stimulates FcR γ-chain ITAM phosphorylation. |
| Hck [Hemopoietic cell kinase] | P08631 | 11 | Src family kinases regulate multiple steps in GPVI cascade. |
| Lyn | P07948 | 12 | Src family kinase which mediates phosphorylation of FcR -chain ITAM and feedback regulation of GPVI. |
| p38α MAP kinase | Q16539 | 14 | Serine-threonine kinase. Implicated in regulation of cPLA2. |
| PLCγ2 [Phospholipase C γ2] | P16885 | 2 | Generates second messengers 1,2-diacylglycerol and IP3. |
| SHIP [SH2 containing inositol-5-phosphatase] | O00145 | 2 | Hydrolyses PI 3-kinase product, PI 3,4,5P3 to PI 3,4P2. Feedback inhibition of GPVI signaling. |
| SKAP-HOM [Src kinase-associated phosphoprotein 55-related protein] | O75563 | 13 | GPVI signaling is maintained in SKAP-HOM knockout mice. |
| SLP-76 [SH2 domain-containing leucocyte protein of 76 kDa] | Q13094 | 9 | Critical role in activation of PLCγ2 by GPVI. Constitutively bound to Gads. |
| Src | P12931 | 10 | Src family kinases regulate multiple steps in GPVI cascade |
| Syk [Spleen tyrosine kinase] | P43405 | 8 | Critical role in GPVI signaling: binds to phosphorylated ITAM in GPVI-FcRγ-chain complex. |
| Vav 1 | P15498 | 4 | Play a critical role in GPVI mediated activation of PLCγ2. |
| Proteins not previously known to be tyrosine phosphorylated by GPVI | |||
| 14-3-3 protein ε | P42655 | 17 | Member of 14-3-3 family. |
| 14-3-3 protein ζ/δ | P29312 | 17 | Member of 14-3-3 family. |
| Annexin A2 | P07355 | 15 | Calcium/phospholipid-binding protein. |
| Annexin A5 | P08758 | 15 | Calcium/phospholipid-binding protein. |
| DIP-1 [Dia interacting protein-1] (SPIN90) | Q9NZQ3 | 6 | Adapter protein implicated in regulation of actin polymerization. |
| Gads [Grb2-related adaptor protein 2] | O75791 | 14 | Supports activation of PLCγ2 by GPVI. Constitutively bound to SLP-76. Tyrosine phosphorylation shown in this study. |
| G6f | Q7Z5H2 | 16 | Immunoglobulin membrane protein of unknown function. Binds Grb2. Phosphorylated on Y281 by GPVI (present study). |
| Myosin-9 | P35579 | 1 | Regulation of cell contractility. |
| PSTPIP2 [Proline-serine-threonine phosphatase-interacting protein 2] | Q9H939 | 14 | Involved in the regulation of actin polymerisation |
| RSP-1 [Ras suppressor protein 1] | Q15404 | 17 | Inhibits Ras signaling |
| Tight junction protein ZO-2 | Q9UDY2 | 2 | Plays a role in tight junctions. Contains one SH3 domain. |
| β-Pix [Rho guanine nucleotide exchange factor 7] | Q14155 | 7 | Interacts with PAK kinases through the SH3 domain. Interacts with c-Cbl. |
AN indicates SWISS-PROT accession number.
function either known or predicted by homology via SwissProt and NCBI.
This study also identified eleven additional proteins which have not been previously shown to be phosphorylated on tyrosine in response to stimulation by GPVI. It is important to bear in mind, however, that some of these proteins may have been pulled out as part of a complex rather than having undergone tyrosine phosphorylation downstream of GPVI. This list of proteins includes several proteins that were identified in GPVI-stimulated platelets for the first time, including β-Pix, the SH3 adapter protein SPIN90 (also known as DIP-1) and the type I membrane protein G6f (see Table 1 and Supplementary Table 1). The presence of tyrosine phosphorylated β-Pix was confirmed by western blot analysis (see Figure 2). The presence of tyrosine-phosphorylated G6f in CRP-stimulated platelets is analyzed in detail below.
Figure 2.
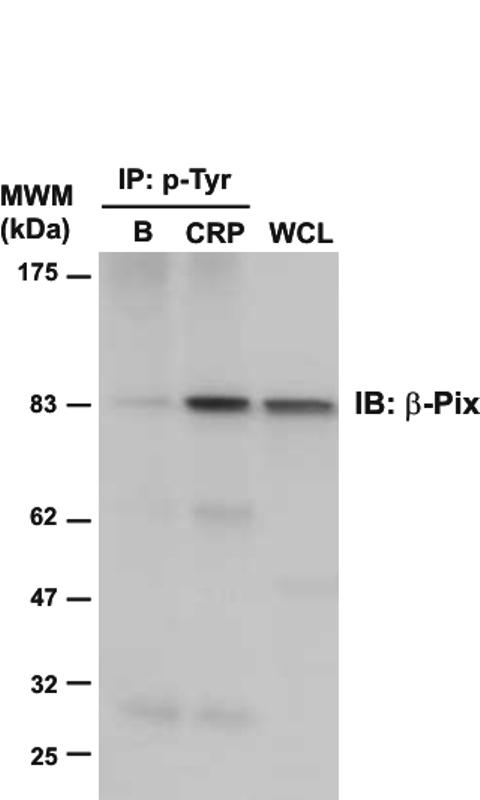
Immunodetection of β-Pix in phosphotyrosine immunoprecipitates of CRP-stimulated platelets. Whole cell lysates and phosphotyrosine immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using an anti-β-Pix antibody. B, basal platelets; CRP, platelets stimulated with CRP (10 μg/mL, 90 sec); IP, immunoprecipitation; IB, immuno blot; WCL, whole cell lysate. Images represent at least three independent experiments.
3.2 Differential proteome analysis of basal versus CRP-activated platelets by 2-DE
In order to gain a more complete picture of the biochemical events that follow platelet activation with CRP, we undertook a 2-DE-based proteomics differential analysis comparing basal and CRP-stimulated platelets. Platelets were again stimulated with 10 μg/ml of CRP for 90 sec in the presence of EGTA to prevent aggregation. For these studies, inhibitors of the actions of ADP and thromboxanes were not included in order to maximize changes in post-translational processing, and increase the chance of identifying new proteins involved in platelet activation by CRP.
Using 1 ml of 1×109 platelets, we were able to obtain approximately 1 mg of protein for subsequent 2-DE analysis. Protein extraction by cell lysis in liquid nitrogen followed by TCA/acetone precipitation and delipidation resulted in highly reproducible 2-DE proteome maps for both the 4-7 and 6-11 narrow range pH gradients (see Figure 3). To identify CRP-induced changes in the platelet proteome, image analysis of four independent experiments was performed following conservative criteria to avoid misidentifications due to gel-to-gel variations, as described in the methods section. A mean of 1386 ± SE6 and 502 ± SE5 (n = 4 in both cases) protein features were found in basal platelets on pI 4-7 gels and pI 6-11 gels, respectively. Following stimulation by CRP, the total numbers of resolved protein features were 1348 ± SE9 and 515 ± SE9 (n = 4 in both cases) for the pI 4-7 and 6-11 analytical ranges, respectively. In total, 117 differentially regulated protein features were detected of which 75 and 42 were found in the pI 4-7 and 6-11 analytical ranges, respectively. 111 (95%) of the differentially regulated protein features were successfully identified by LC-MS/MS. They correspond to 72 different open reading frames (ORFs). The full list of identified proteins can be found in the Supplementary Table 2, which includes information on their mass, pI, fold change and function. The corresponding compiled data for the Mascot MS searches is shown in Supplementary Table 3. The specific location on the gels of all the signaling proteins identified is shown in Figure 3. Overall, there was a shift of many proteins present towards a more acidic region after CRP-stimulation, which is consistent with protein phosphorylation. This is exemplified by pleckstrin, which has six potential phosphorylation sites for protein kinase C [11]. Pleckstrin is present as an array of spots under basal conditions which move to the left upon stimulation by CRP (see Supplementary Table 2 and Fig. 4A). The presence of multiple features suggests the presence of several distinct forms of phosphorylated pleckstrin.
Figure 3.


Synthetic 2-DE gel images representing all protein features present in the basal versus CRP analysis. (A) pI 4-7 range. (B) pI 6-11 range. The figure shows the location on the 2D gels of the features corresponding to the signaling proteins successfully identified in the analysis. The synthetic images represent groups of eight gels (four basal and four CRP) for the two pH ranges. The original gels were used for the differential analysis. P1, CLP-36; P2, ADAP; P3, RGS10; P4, Dok-2; P5, SKAP-HOM; P6, Gads; P7, MHC class I HLA-A protein; P8, Heat Shock 27 kDa protein; P9, Pleckstrin; P10, Src; P11, p21-Rac1; P12, Annexin VII; P13, Grb2; P14, RKIP; P15, Crk-like protein; P16, Rho GDP-dissociation inhibitor 2; P17, adenylyl cyclase-associated protein 1; P18, MEK1; P19, ILK-1; P20, Ras suppressor protein 1; P21, p38α MAP kinase; P22, OSF1; P23, MacGAP; P24, Dok-1; P25, RGS18; P26, Drebrin F. More information on all these proteins can be obtained from the Supplementary Table 2.
Figure 4.
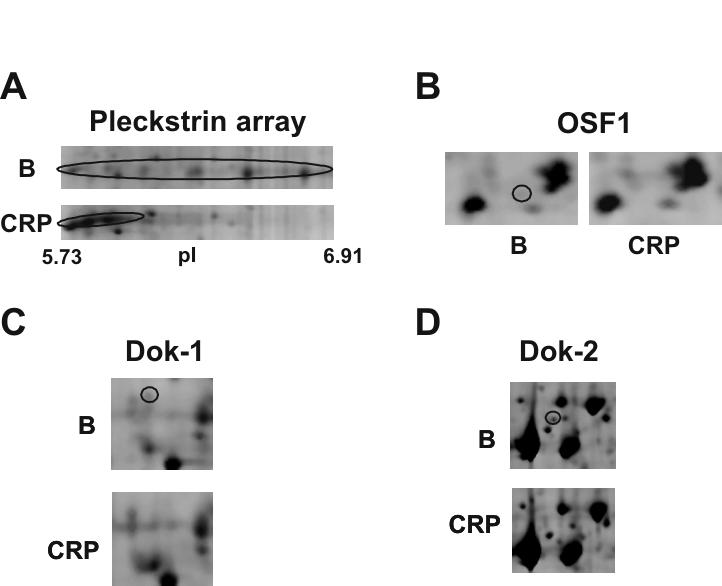
Close-up images of representative 2-DE gel images showing changes in a selection of signaling proteins that have been identified by differential analysis. (A) Pleckstrin. (B) OSF1. (C) Dok-1. (D) Dok-2. B indicates 2D gel corresponding to basal platelets; and CRP, 2D gel corresponding to CRP activated platelets (10 μg/mL, 90 sec). Circled areas indicate the differentially regulated protein feature.
A high proportion of the proteins identified through 2-DE differential analysis are signaling (36%) and cytoskeletal (14%) proteins, consistent with the critical role played by these two groups in mediating platelet function and their regulation by phoshorylation. To our knowledge, 9 of the identified proteins have not previously been described in platelets, including the adapters Dok-1, and OSF1 (see Supplementary Table 2 and Fig. 4B, C). Many of the other identified signaling proteins were already known to participate in GPVI signaling, including ADAP (previously known as SLAP-130), Crk-l, Dok-2 (see Fig. 4D), Gads, Grb2, MEK, p38α mapkinase, pleckstrin, Rac1, SKAP-HOM and Src. Interestingly, only six of the proteins that had been identified in the tyrosine-phosphoproteome analysis of CRP-stimulated platelets were also identified using the 2-DE approach, namely ADAP, Dok-2, Gads, p38α mapkinase, SKAP-HOM, and Src.
3.3 Identification of G6f as a tyrosine phosphorylated transmembrane protein able to bind Grb2 in GPVI-stimulated platelets
This study has demonstrated tyrosine phosphorylation of several, novel signaling proteins in GPVI-activated platelets. This includes the type I transmembrane protein G6f, a member of the immunoglobulin superfamily whose ligand is not yet known [12]. Significantly, G6f was observed using mass spectrometry to undergo tyrosine phosphorylation on its cytosolic tail at position 281 following CRP-stimulation (see Supplementary Table 1). Interestingly, tyrosine phosphorylation of G6f at this site has been recently shown to mediate binding to Grb2 in a pervanadate-treated cell line model [12].
Two antipeptide antibodies were raised against distinct regions of the G6f cytosolic tail to enable further characterization of phosphorylation of the immunoglobulin protein and its possible association with Grb2 and Gads. Verification of the efficacy of the two antibodies was achieved using control and G6f-transfected 293T cells (not shown). Western blotting using either antibody confirmed that G6f is precipitated from CRP stimulated-platelets by the antiphosphotyrosine mAb 4G10, peaking at 90 sec (Fig. 5A and not shown). Furthermore, direct immunoprecipitation of G6f and western blotting for phosphotyrosine confirmed that the novel Ig transmembrane protein undergoes tyrosine phosphorylation downstream of GPVI (Fig. 5B and not shown). Consistent with this, G6f also undergoes marked tyrosine phosphorylation in response to collagen, although not in response to the G protein-coupled receptor agonists, thrombin and ADP (Fig. 5C). Tyrosine phosphorylation of G6f by GPVI is also independent of secondary mediators (Fig. 5C). G6f was also detected following immunoprecipitation of the cytosolic adapter Grb2, in parallel with its extent of tyrosine phosphorylation (Fig. 5D). On the other hand, the converse experiment did not work i.e. Grb2 was not present in G6f immunoprecipitates, most likely because the recognition site of the two anti-G6f antibodies overlaps with the putative Grb2-binding site. These results confirm that G6f undergoes tyrosine phosphorylation upon activation by CRP or collagen and that this leads to its association with Grb2.
Figure 5.
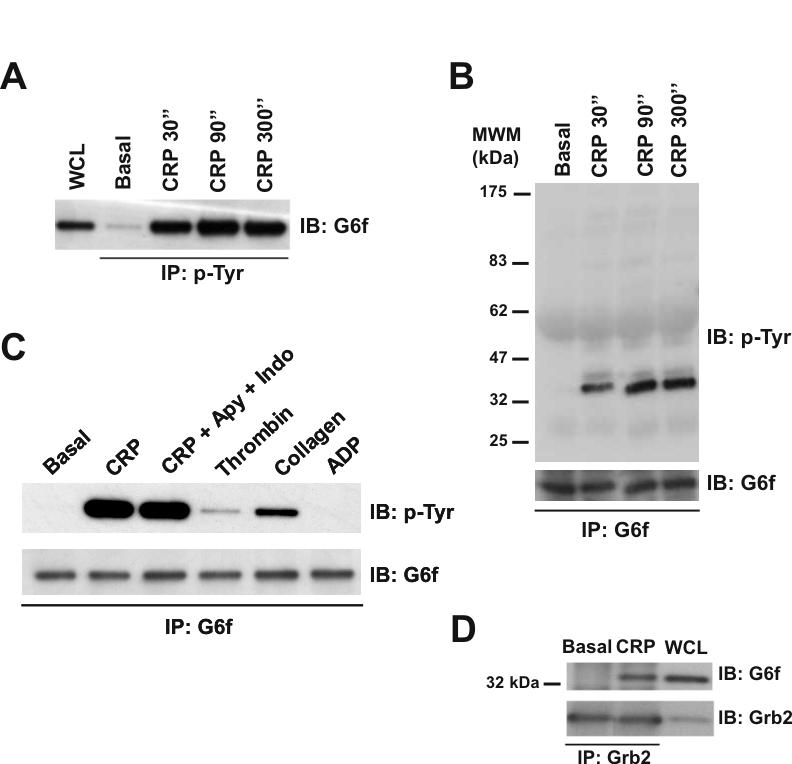
G6f is phosphorylated in Tyr-281 and is able to bind Grb2 in GPVI-stimulated platelets. (A) Tyrosine-phosphorylated G6f is recruited into the GPVI signaling cascade upon platelet stimulation with CRP (10 μg/mL), with maximum levels of phosphorylated G6f being observed after 90 sec stimulation. Whole cell lysates and phosphotyrosine immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using an anti-G6f antibody. (B) Time-course experiments for CRP-platelet stimulation confirm that G6f is tyrosine-phosphorylated upon platelet-activation with CRP (10 μg/mL). G6f immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using anti-4G10 and then anti-G6f antibodies. (C) G6f undergoes tyrosine-phosphorylation downstream of GPVI but not by the G protein-coupled receptor agonists thrombin and ADP. G6f immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using anti-4G10 and anti-G6f antibodies. All platelets had EGTA added to prevent aggregation; only where specified have apyrase (2 U/mL) and indomethacin (10 μM) been added. Platelets were stimulated for 90 sec with CRP (10 μg/mL), collagen (30 μg/mL), thrombin (1 U/mL), and ADP (100 μM). (D) G6f co-immunoprecipitates with Grb2 in CRP-stimulated platelets. Whole cell lysate and Grb2 immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using anti G6f and then anti-Grb2 antibodies. Platelets were stimulated with CRP (10 μg/mL, 90 sec). IP, immunoprecipitation; IB, immuno blot; WCL, whole cell lysate. Images represent at least three independent experiments.
The possibility that G6f is a novel receptor for collagen and CRP was investigated by measurement of adhesion to immobilized collagen/CRP and tyrosine phosphorylation of G6f in transfected 293T, Jurkat and DT40 cells. There was no significant difference in adhesion between mock- and G6f-transfected cells on collagen or CRP, or detection of tyrosine phosphorylation of G6f in response to stimulation by collagen or CRP (data not shown). Confirmation of expression of G6f at the cell surface was achieved by confocal microscopy using a specific antibody (not shown). These results suggest that G6f is a novel membrane adapter protein that is regulated downstream of GPVI rather than a novel receptor for collagen or CRP.
3.4 Tyrosine phosphorylation of Gads and Grb2 in CRP-stimulated platelets
Additional studies were carried out to investigate whether the two related adapters Gads and Grb2 undergo tyrosine phosporylation downstream of GPVI. As discussed above, Gads was identified in the 4G10 precipitation studies as well as in the 2-DE analysis. Indeed, in the 2-DE differential analysis Gads was represented by two protein features separated by 0.37 pI units (Fig. 6A). The disappearance of the protein feature at pI 6.29 together with the appearance of the feature at pI 5.92 strongly suggests that Gads is phosphorylated to a high stoichiometry in response to GPVI receptor stimulation by CRP. To directly address this possibility, Gads was immunoprecipitated using a specific antibody and probed for tyrosine phosphorylation. As shown in Figure 6B, a band that co-migrated with Gads was clearly identified following immunoprecipitation of Gads, along with major tyrosine phosphorylated bands at 38, 60 and 75 kDa. The identity of the 76 kDa band was confirmed as the adapter SLP-76 by MS, while the 38 kDa band is most likely LAT, bearing in mind that Gads forms a complex with the two proteins in haematopoietic cells [13]. We were not however able to detect the presence of G6f in the immunoprecipitates, indicating that the membrane adapter asscociates specifically with Grb2.
Figure 6.
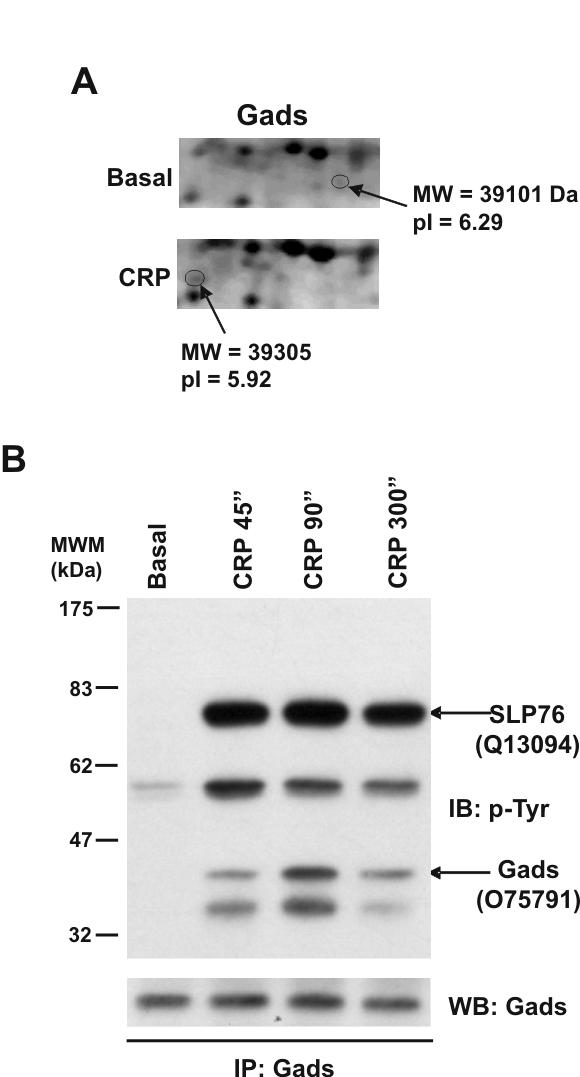
Gads is tyrosine phosphorylated in response to CRP in platelets. (A) Close-up image of representative 2D gel images showing the Gads features. (B) Gads is tyrosine phosphorylated in response to CRP in human platelets, with maximum phosphorylation levels observed after 90 sec stimulation. Gads immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using anti-4G10 or anti-Gads antibodies. MS/MS analysis of the bands indicated by arrows, from gels run alongside the blots, allowed the identification of SLP-76 and Gads in the same signaling complex. Swiss-Prot accession numbers are shown in brackets before the protein names. B, basal platelets; CRP, platelets stimulated with CRP (10 μg/mL, 90 sec); IP, immunoprecipitation; IB, immuno blot; WCL, whole cell lysate. Images represent at least three independent experiments.
Grb2 was identified in the 2-DE study as a feature that is up-regulated upon CRP-stimulation (Fig. 7A). However, a feature corresponding to Grb2 that was down-regulated was not identified, most likely because it underwent less than a 2-fold decrease, the cutoff for selection. Grb2 was also identified in 4G10 immunoprecipitates from CRP-stimulated platelets (Fig. 7B) and direct confirmation of tyrosine phosphorylation of Grb2 was achieved by immunoprecipitation and reprobing for phosphotyrosine (Fig. 7C). Grb2 has not previously been shown to undergo phosphorylation in platelets.
Figure 7.
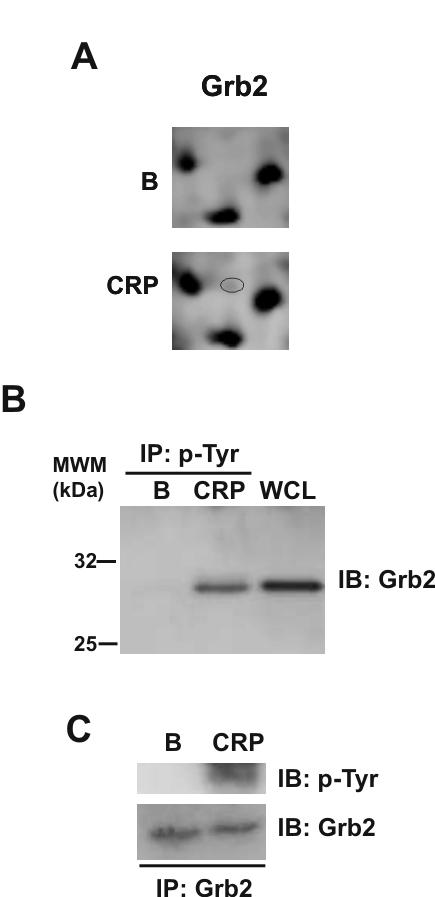
Grb2 is tyrosine phosphorylated in response to CRP in platelets. (A) Close-up image of representative 2D gels showing up-regulation of a Grb2 protein feature upon CRP stimulation (B) Tyrosine-phosphorylated Grb2 is recruited into the GPVI signaling cascade upon platelets stimulation with CRP. Whole cell lysates and phosphotyrosine immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using anti-Grb2 antibodies. (C) Grb2 is tyrosine phosphorylated in response to CRP in human platelets. Grb2 immunoprecipitated proteins were separated by 4-12% NuPAGE Bis-Tris gels and immunoblotted using anti-4G10 or anti-Grb2 antibodies. B, basal platelets; CRP, platelets stimulated with CRP (10 μg/mL, 90 sec); IP, immunoprecipitation; IB, immuno blot; WCL, whole cell lysate. Images represent at least three independent experiments.
4 Discussion
The proteome of CRP-stimulated platelets was analyzed to further investigate the intracellular signaling events that take place upon stimulation of the collagen receptor, GPVI. Analysis of the tyrosine-phosphoproteome of CRP-stimulated platelets was achieved by enrichment of samples using a specific antiphosphotyrosine antibody. The list of identified proteins included four members of the Src family of protein tyrosine kinases and members of three further families of tyrosine kinases, namely Btk, Fes, and Syk. In addition, this approach identified a number of adapter proteins, namely ADAP, Cbl, Dok-2, Gads, SKAP-HOM and SLP-76, and the effector enzymes PLCγ2, SHIP and Vav1. Significantly, all of these proteins were previously known to play a role in signaling by GPVI and, with the exception of Gads, were known to be regulated by tyrosine phosphorylation [13-15]. Interestingly, several other tyrosine phosphorylated proteins in the GPVI signaling cascade were not identified using this approach, including LAT, the FcR γ-chain and Grb2, which was identified using 2-DE by comparison of basal and CRP-stimulated platelets. This may in part reflect a low level of expression of some of these proteins in platelets e.g. LAT, or the stochastic nature of the approach. We also identified eleven additional proteins that were previously not known to undergo tyrosine phosphorylation downstream of GPVI in platelets. Of these, eight were previously known to be present in high levels in platelets and additional studies are required to confirm whether their presence is due to tyrosine phosphorylation or their association with other tyrosine-phosphorylated proteins. The other three proteins, β-Pix, G6f, and SPIN90, are discussed further below.
We have previously used the 2-D gel-based differential approach to analyze the proteome of TRAP-activated platelets. This had successfully identified a number of novel signaling proteins and phosphorylation events, including the adapter protein Dok-2 which undergoes tyrosine phosphorylation [8]. We have now used this strategy to analyze the proteome of CRP-stimulated platelets, in order to obtain information complementary to the tyrosine phosphoproteome analysis-based approach. The much greater number of proteins identified with the 2-DE approach compared to the phosphotyrosine immunoprecipitation-based approach (72 vs. 30) can be attributed to the fact that the 2-DE approach detects changes mediated by additional methods to tyrosine phosphorylation, such as phosphorylation on serine and threonine residues, as well as to the absence of inhibitors of secondary mediators. Significantly, the 2-DE differential analysis approach led to the identification of several novel platelet signaling proteins, including Dok-1 and OSF1. Interestingly, OSF1 contains one SH3-domain, three potential tyrosine phosphorylation sites and is able to bind Src and Cbl [16, 17]. Further research is needed to investigate the precise role of this and other newly identified proteins in platelet activation by GPVI.
Although the 2-DE differential analysis allowed the identification of 72 proteins that undergo post-translational modifications in response to CRP, there were a number of notable omissions including the majority of the proteins that were identified using the antiphosphotyrosine antibody approach. This could be a consequence of a number of factors, including a low level of expression of the tyrosine phosphorylated proteins, as is the case for many signaling proteins, their co-localization with other, more highly expressed proteins on the gel, changes in spot volume below the selected threshold of 2-fold, or inability of certain proteins to be properly resolved by 2-DE because of problems with solubility during the IEF. Indeed, this result validates the use of an affinity step in the mapping of the GPVI-stimulated proteome. It is widely recognised that one or more concentration steps are required to identify proteins that are expressed at low level using mass spectrometry.
One of the proteins identified by both the 2-DE-based and the phosphotyrosine immunoprecipitation-based approach was the Grb2-related adapter protein Gads. To our knowledge, this is the first report of tyrosine phosphorylation of Gads in platelets or in other haematopoietic cells. The NetPhos 2.0 server [18] predicts two tyrosine-phoshorylation sites for Gads at positions 45 and 207 in the protein sequence, corresponding to one of the SH3 domains and a glutamine / proline-rich region, respectively. Similarly, we were able to demonstrate that the related adapter Grb2 was also tyrosine-phosphorylated in response to CRP-stimulation in human platelets. There are two potential tyrosine phosphorylation sites in Grb2 at positions 134 and 160 in the protein sequence, corresponding to the SH2 domain and one of the SH3 domains, respectively. This opens a new line of research on the regulation and possible roles of Gads and Grb2 in the GPVI signaling cascade [19, 20].
As mentioned above, one of the striking findings of this study was the detection of the majority of the known tyrosine phosphorylated proteins that make up the GPVI signaling cascade in platelets using an antiphosphotyrosine antibody and 1-DE. This not only verifies the approach, but it also brings into speculation the potential importance of the other proteins in the GPVI signaling cascade that were detected using this approach. In particular, this approach identified two proteins that had not been previously reported in platelets, namely β-Pix [21, 22] and SPIN90 [23-27] and G6f, which was recently identified by MS in a proteome analysis of glycosylated proteins in human platelets [28]. In the present study, we used specific antibodies to demonstrate tyrosine phosphorylation of G6f, β-Pix and SPIN90 (data not shown) in platelets downstream of GPVI.
The SH3 adapter protein SPIN90 has been shown to bind to the adapter Nck, which is implicated in receptor tyrosine kinase signaling, Ras/MAP kinase pathways and cytoskeletal reorganization. The amino acid sequence of the SH3 domain of SPIN90 has the highest homology with those of Fyn, Yes, and c-Src [23]. Interestingly, in vitro experiments have shown that SPIN90 forms a complex with Wiscott-Aldrich syndrome protein (WASP), β-Pix and Nck, which is crucial for stable cell adhesion to the extracellular matrix [24]. In addition, SPIN90 binds to Grb2 and Src [25]. β-Pix is a key regulator in the activation of p38 MAP kinase and p21-activated kinase (PAK) [21], and is also able to bind c-Cbl [22]. Thus, β-Pix and SPIN90 are not only able to bind to each other to stabilize cell adhesion to the extracellular matrix, but also are able to bind other key proteins on the GPVI signaling cascade, such as c-Cbl, Grb2, and Src suggesting that they may play a critical role in signaling by the collagen receptor.
G6f protein is a type I transmembrane protein belonging to the Ig superfamily and which was initially identified from the human erythroleukaemia cell line K562 [12]. The G6f gene is located in the class III region of the MHC and lies in a cluster of genes encoding cell-surface molecules. The intracellular tail of G6f is 40 amino acids long and contains one tyrosine residue (Y281), which has previously been shown to be phosphorylated after treatment of cells with pervanadate. Indeed, as for the NetPhos 2.0 server [18], that is the only potential tyrosine phosphorylation site in G6f. In the present study, we have shown using MS that G6f also undergoes tyrosine phosphorylation at that site in response to platelet activation by CRP. Tyrosine phosphorylation is most likely mediated downstream of GPVI rather than through direct binding to CRP, as expression of G6f in three different cell lines failed to confer adhesion to CRP or its tyrosine phosphorylation in response to stimulation by the synthetic collagen.
The G6f phosphotyrosine residue is found in a consensus-binding motif (YXN) for the SH2 domain of Grb2. Consistent with this, a previous study has shown an interaction of full-length Y281-phosphorylated G6f with full length Grb2 in a pervanadate-treated cell line model [12]. In our study, we show that G6f is immunoprecipitated with an antibody to Grb2 following platelet activation by CRP. Potentially, however, the role of G6f in recruiting Grb2 to the membrane may be redundant with that of the transmembrane adapter LAT, bearing in mind that the phosphorylated sequence in G6f (Y281ENI) is similar to a Grb2-binding motif found in human LAT (Y226ENL) and that a phosphorylated 38 kDa platelet protein, later identified as LAT [13], binds to Grb2 in platelets [29]. Our data now indicate that G6f also forms part of this 38 kDa band, suggesting that both G6f and LAT are major Grb2 docking proteins in CRP-activated platelets. Grb2 lies upstream of regulation of Erk1/2 mapkinases which have been shown to play a role in the regulation of cPLA2 in platelets [30] and are also vital for megakaryocyte growth and development [31]. It is of interest to establish whether the roles of G6f and LAT are redundant in these two pathways.
In conclusion, we have used two distinct proteomic approaches to identify novel signaling proteins and phosphorylation events that could play a relevant role in the GPVI-based signaling cascade in platelets. Significantly, these have led to identification of two largely distinct sets of proteins thereby emphasizing the importance of both methodologies. The list of identified proteins includes several that are anticipated to play key roles in signaling by the collagen receptor, including β-Pix, SPIN90 and G6f. We demonstrated that the transmembrane protein G6f is tyrosine phosphorylated in its intracellular tail and is able to bind Grb2 in CRP-stimulated platelets. In addition, we also demonstrated tyrosine phosphorylation of Grb2 and the related adapter Gads. We believe that the data presented in this study and the further understanding of the role of all the newly identified platelet proteins and phosphorylation events will contribute to a better understanding of the GPVI signaling pathway, and help to build the basis for the identification of new drug targets that may help to treat thrombotic diseases.
Supplementary Material
Acknowledgements
This work was funded by The Oxford Glycobiology Institute Endowment. NZ is a Senior Research Fellow at Linacre College, Oxford (UK). AG is a Parga Pondal Fellow (Xunta de Galicia, Spain). SPW holds a British Heart Foundation Chair and acknowledges The Wellcome Trust and BHF for support. YAS holds a Birmingham University Research Fellowship that is funded through the BHF. Ethical approval for this study was granted by the Central Oxford Research Ethics Committee (No:00.231).
Glossary
Abbreviations:
- GPVI
Glycoprotein VI
- CRP
collagen-related peptide
- ADAP
Adhesion-and degranulation promoting adapter protein
- Btk
Bruton’s tyrosine
- Dok
Downstream of tyrosine kinases
- Hck
Hematopoietic cell kinase
- PLCγ2
Phospholipase C γ2
- SHIP
SH2 containing inositol-5-phosphatase
- SKAP-HOM
Src kinase-associated phosphoprotein 55-related protein
- SLP-76
SH2 domain-containing leucocyte protein of 76 kDa
- Syk
Spleen tyrosine kinase
- Gads
Grb2-related adapter protein 2
- DIP-1
Dia interacting protein-1
- RGS
Regulator of G-protein signaling
- OSF1
Osteoclast stimulating factor 1
- RKIP
Raf kinase inhibitor protein
- ILK
Integrin-linked protein kinase.
References
- [1].Nieswandt B, Watson SP. Blood. 2003;102:449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- [2].Morton LF, Hargreaves PG, Farndale RW, Young RD, Barnes MJ. Biochem. J. 1995;306:337–344. doi: 10.1042/bj3060337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kehrel B, Wierwille S, Clemetson KJ, Anders O, et al. Blood. 1998;91:491–499. [PubMed] [Google Scholar]
- [4].Asselin J, Gibbins JM, Achison M, Lee YH, et al. Blood. 1997;89:1235–1242. [PubMed] [Google Scholar]
- [5].García A, Watson SP, Dwek RA, Zitzmann N. Mass Spectrom. Rev. 2005;24:918–930. doi: 10.1002/mas.20047. [DOI] [PubMed] [Google Scholar]
- [6].Görg A, Weiss W, Dunn MJ. Proteomics. 2004;4:3665–3685. doi: 10.1002/pmic.200401031. [DOI] [PubMed] [Google Scholar]
- [7].García A, Prabhakar S, Brock CJ, Pearce AC, et al. Proteomics. 2004;4:656–668. doi: 10.1002/pmic.200300665. [DOI] [PubMed] [Google Scholar]
- [8].García A, Prabhakar S, Hughan S, Anderson TW, et al. Blood. 2004;103:2088–2095. doi: 10.1182/blood-2003-07-2392. [DOI] [PubMed] [Google Scholar]
- [9].O'Neill EE, Brock CJ, von Kriegsheim AF, Pearce AC, et al. Proteomics. 2002;2:288–305. doi: 10.1002/1615-9861(200203)2:3<288::aid-prot288>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- [10].Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, et al. Proc. Natl. Acad. Sci. U. S. A. 1996;93:14440–14445. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tyers M, Haslam RJ, Rachubinski RA, Harley CB. J. Cell. Biochem. 1989;40:133–145. doi: 10.1002/jcb.240400202. [DOI] [PubMed] [Google Scholar]
- [12].De Vet ECJM, Aguado B, Campbell RD. Biochem. J. 2003;375:207–213. doi: 10.1042/BJ20030293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Asazuma N, Wilde JI, Berlanga O, Leduc M, et al. J. Biol. Chem. 2000;(43):33427–33434. doi: 10.1074/jbc.M001439200. [DOI] [PubMed] [Google Scholar]
- [14].Watson SP, Asazuma N, Atkinson B, Berlanga O, et al. Thromb. Haemost. 2001;86:276–288. [PubMed] [Google Scholar]
- [15].Best D, Pasquet S, Littlewood TJ, Brunskill S, et al. Br. J. Haematol. 2001;112:609–615. doi: 10.1046/j.1365-2141.2001.02624.x. [DOI] [PubMed] [Google Scholar]
- [16].Reddy S, Devlin R, Menaa C, Nishimura R, et al. J. Cell. Physiol. 1998;177:636–645. doi: 10.1002/(SICI)1097-4652(199812)177:4<636::AID-JCP14>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- [17].Szymkiewicz I, Destaing O, Jurdic P, Dikic I. FEBS Letters. 2004;565:33–38. doi: 10.1016/j.febslet.2004.03.100. [DOI] [PubMed] [Google Scholar]
- [18].Blom N, Gammeltoft S, Brunak S. J. Mol. Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- [19].Pasquet JM, Gross B, Quek L, Asazuma N, et al. Mol. Cell. Biol. 1999;19:8326–8334. doi: 10.1128/mcb.19.12.8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Judd BA, Myung PS, Obergfell A, Myers EE, et al. J. Exp. Med. 2002;195:705–717. doi: 10.1084/jem.20011583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee J, Jung ID, Chang WK, Park CG, et al. Exp. Cell. Res. 2005;307:315–328. doi: 10.1016/j.yexcr.2005.03.028. [DOI] [PubMed] [Google Scholar]
- [22].Wu WJ, Tu S, Cerione RA. Cell. 2003;114:715–725. doi: 10.1016/s0092-8674(03)00688-3. [DOI] [PubMed] [Google Scholar]
- [23].Lim CS, Park ES, Kim DJ, Song YH, et al. J. Biol. Chem. 2001;276:12871–12878. doi: 10.1074/jbc.M009411200. [DOI] [PubMed] [Google Scholar]
- [24].Lim CS, Kim SH, Jung JG, Kim JK, Song WK. J. Biol. Chem. 2003;278:52116–52123. doi: 10.1074/jbc.M310974200. [DOI] [PubMed] [Google Scholar]
- [25].Satoh S, Tominaga T. J. Biol. Chem. 2001;276:39290–39294. doi: 10.1074/jbc.M107026200. [DOI] [PubMed] [Google Scholar]
- [26].Oda A, Ochs HD, Druker BJ, Ozaki K, et al. Blood. 1998;92:1852–1858. [PubMed] [Google Scholar]
- [27].Kim DJ, Kim SH, Lim CS, Choi KY, et al. J. Biol. Chem. 2006;281:617–625. doi: 10.1074/jbc.M504450200. [DOI] [PubMed] [Google Scholar]
- [28].Lewandrowski U, Moebius J, Walter U, Sickmann A. Mol. Cell. Proteomics. 2006;5:226–233. doi: 10.1074/mcp.M500324-MCP200. [DOI] [PubMed] [Google Scholar]
- [29].Robinson A, Gibbins J, Rodríguez-Liñares B, Finan PM, et al. Blood. 1996;88:522–530. [PubMed] [Google Scholar]
- [30].Borsch-Haubold AG, Kramer RM, Watson SP. Eur. J. Biochem. 1997;245:751–759. doi: 10.1111/j.1432-1033.1997.t01-1-00751.x. [DOI] [PubMed] [Google Scholar]
- [31].Kamata T, Kang J, Lee TH, Wojnowski L, et al. Blood. 2005;106:833–840. doi: 10.1182/blood-2004-11-4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.