

Abstract
Misfolding and self-assembly of proteins in nanoaggregates of different sizes and morphologies (nanoensembles, primary nanofilaments, nanorings, filaments, protofibrils, fibrils, etc.) is a common theme unifying a number of human pathologies termed protein misfolding diseases. Recent studies highlight increasing recognition of the public health importance of protein misfolding diseases, including various neurodegenerative disorders and amyloidoses. It is understood now that the first essential elements in the vast majority of neurodegenerative processes are misfolded and aggregated proteins. Altogether, the accumulation of abnormal protein nanoensembles exerts toxicity by disrupting intracellular transport, overwhelming protein degradation pathways, and/or disturbing vital cell functions. In addition, the formation of inclusion bodies is known to represent a major problem in the production of recombinant therapeutic proteins. Formulation of these therapeutic proteins into delivery systems and their in vivo delivery are often complicated by protein association. Thus, protein folding abnormalities and subsequent events underlie a multitude of human pathologies and difficulties with protein therapeutic applications. The field of medicine therefore can be greatly advanced by establishing a fundamental understanding of key factors leading to misfolding and self-assembly responsible for various protein folding pathologies. This article overviews protein misfolding diseases and outlines some novel and advanced nanotechnologies, including nanoimaging techniques, nanotoolboxes and nanocontainers, complemented by appropriate ensemble techniques, all focused on the ultimate goal to establish etiology and to diagnose, prevent, and cure these devastating disorders.
Keywords: misfolding, protein aggregation, conformational disease, partially folded intermediate, nanomedicine
Protein Misfolding Diseases
Amyloid Deposits
The first documented description of macroscopic abnormalities in liver (lardaceous and waxy liver) and spleen (spongy and “white stone” containing spleens) associated with what is currently known as proteinaceous misfoldings was probably reported as early as in 1639.1 However, the official history of observed amyloidosis is generally thought to have started in 1854, when the German physician Rudolph Virchow introduced and popularized the term “amyloid” to denote a macroscopic tissue abnormality in cerebral corpora amylacea and the waxlike deposits in spleen, liver and kidney.2 Soon thereafter, Friedreich and Kekulé extracted amyloid-rich segments from the spleen of a patient with amyloidosis, performed direct chemical analyses of the extracted material, and came to the definitive conclusion that the main substance was protein in nature.3 This was later confirmed by Hanssen, who showed that amyloids are digestible with pepsin.4 Understanding the role of abnormal deposits in the development of neurodegenerative disorders began with a report by Alois Alzheimer in 1907,5 which described senile plaques and neurofibrillary tangles in the brain of a middle-aged woman with memory deficits and a progressive loss of cognitive function. This brief report is considered the first published description of Alzheimer’s disease (AD).6 The neuropathological hallmarks of Parkinson’s disease (PD), Lewy bodies (LBs), and Lewy neuritis (LNs) were described shortly thereafter in 1912 by Friederich Lewy.7
Amyloidogenic Proteins
The nature of amyloid protein was a long-standing puzzle that was unresolved until relatively recently. Through the application of immunohistochemical methods, it was found that amyloids are not a singular substance of some unspecific degenerative origin,8 but in fact approximately 20 distinct proteins are involved in amyloidoses (extracellular deposits). A number of diseases are also associated with the accumulation of fibrillar intracellular deposits or are associated with the buildup of nonfibrillar deposits.9–14 It should also be remembered that the amyloid, deposited in tissues, does not only contain the fibrils made from a single, pure protein species, but also includes additional proteins such as serum amyloid P component (SAP), glycosaminoglycans, and proteoglycans.
Protein Misfolding Diseases
The current list of protein misfolding disorders includes, but is not limited to, numerous neurodegenerative diseases such as PD, Down’s syndrome, AD, and Huntington’s disease; transmissible encephalopathies; Gerstmann–Straussler–Scheinker disease; fatal familial insomnia; multiple system atrophy; numerous synucleinopathies and taupathies; amyotrophic lateral sclerosis; and familial encephalopathy with neuroserpin inclusion bodies, as well as monoclonal protein systemic amyloidosis, reactive systemic amyloidosis, familial amyloidotic polyneuropathy, hereditary apoAI amyloidosis, hereditary lysozyme amyloidosis, senile systemic amyloidosis, isolated atrial amyloidosis, hereditary cerebral amyloid, Finnish hereditary amyloidosis, hereditary fibrinogen α-chain amyloidosis, insulin-related amyloid, cataracts, medullary carcinoma of the thyroid, late-onset diabetes mellitus, symptomatic (haemodialysis-related) β2 microglobulin amyloidosis, arthritis, and many other systemic, localized, and familial amyloidoses.11,15–17
It is accepted now that many neurodegenerative disorders are associated with the accumulation of protein deposits in the central nervous system (CNS). Neurodegenerative diseases are generally disabling and often fatal. They are also irreversible, since they involve the death and atrophy of neurons in the brain and neurons cannot be regenerated, and therefore, health care options are limited to only symptomatic treatments. Age-related neurodegenerative disease is considered to be the largest health care challenge of the 1990s and has been referred to as “the silent epidemic”, since these diseases affect the majority of the geriatric population but are still poorly recognized due to the social isolation of those who suffer from these diseases. In the United States, 5 million people suffer from AD, and this figure is predicted to jump to 16 million by 2050. Health care costs for the AD patients exceed $100 billion a year, according to the Alzheimer’s Association. In addition to AD, about 1.5 million people in the United States are afflicted with PD. Other neurodegenerative diseases, such as multiple sclerosis and amyotrophic lateral sclerosis, disable additional thousands of individuals.
A vast majority of amyloidoses are multisystemic diseases, which affect several organs or systems, and are therefore often called generalized or systemic. Although amyloid proteins can deposit in any organ that has an unrestricted blood supply, accumulations of amyloid deposits in certain organs will cause illness much faster than others. The primary organs in which accumulations of amyloid deposits are found are the kidney, heart, digestive tract, liver, skin, joints, eye, and peripheral nerves. As can be seen from the lists presented above, amyloidosis covers a large group of diseases defined by the presence of insoluble protein deposits in different tissues and organs. Thus, the diagnosis of amyloidosis is mostly histological, and the classification of amyloidoses relies on a range of clinical diagnostic tests and the identities of the amyloid protein.
Molecular Mechanisms of Protein Misfolding Diseases
It is generally recognized that in disease terms the amyloidoses are defined as disorders of protein metabolism, acquired or inherited, in which normally soluble proteins and polypeptides are deposited in a particular fibrillar form in the extracellular spaces. Because they are highly stable, amyloid deposits usually persist and accumulate, ultimately resulting in organ failure and death. Amyloid pathogenesis centers on the off-pathway folding of the various amyloid fibril precursor proteins. These proteins exist in two radically different conformational states, the normal soluble form and a number of highly abnormal fibrillation/aggregation-prone conformations. It has been noted that, although such amyloidogenic conformations may be transient, they may have a rather long lifetime.18,19 A great advance was achieved when it was shown that fibrils can be formed in vitro with properties similar to those of amyloid fibrils extracted from the affected organs.20,21 This finding offers a unique opportunity to study the aggregation process in general, and the protein fibrillogenesis in particular, using a wide arsenal of modern biophysical approaches developed for analysis of protein structure, conformational stability, and folding. Currently, amyloid fibrils have been formed in vitro from several disease-associated and disease-unrelated proteins and peptides [reviewed in ref 14]. There is an increasing belief that the ability to form fibrils is a generic property of the polypeptide chain; that is, many proteins, perhaps all, are potentially able to form amyloid fibrils under appropriate conditions. This means that amyloidogenic polypeptides are unrelated in terms of sequence or native structure. Prior to fibrillation, they may be globular proteins rich in β-sheet, α-helix, β-helix, or both α-helices and β-sheets or they may be natively unfolded (i.e., intrinsically unstructured) proteins. Despite this diversity, the fibrils from different pathologies display many common properties and have similar morphologies. This suggests that the common characteristic of these amyloidogenic proteins is conformational plasticity, which would allow them to refold in response to the structural context.9,14,22 On the basis of the results of extensive structural studies on several amyloidogenic proteins, a general hypothesis of fibrillogenesis has been formulated: structural transformation of a polypeptide chain into a partially folded conformation, which might in fact represent a highly dynamic ensemble of nonfolded forms, represents an important prerequisite for protein fibrillation.14 Therefore, deciphering the interplay between protein folding and misfolding/aggregation is crucial for better understanding of the molecular mechanisms of protein conformational diseases.10,11,14,23,24 In fact, it has been emphasized that the use of the current understanding of the protein folding energy landscapes, which are determined using an array of traditional biophysical methods, theory, and simulation, already provided important answers to some of the key questions in protein misfolding diseases providing potential basis for the development of future therapeutic strategies based on a full biophysical description of the combined folding and aggregation free-energy surface.23
The formation of amyloid-like fibrils does not represent the only pathological hallmark of “conformational” or protein misfolding diseases. In several neurodegenerative disorders and numerous in vitro experiments, the misfolded proteins are composed of amorphous aggregates, which are cloud-like inclusions without defined structure. Also, soluble oligomers represent another alternative final product of the aggregation process for some proteins under particular conditions. The choice between these three forms of aggregation–fibrils, amorphous aggregates, or oligomers–is determined by the amino acid sequence (possibly modified by mutations) and by peculiarities of the protein environment. Irrespective of the form of aggregation, a first stage of the aggregation process is assumed to be protein misfolding. Misfolding is taken to be the structural transformation of soluble proteins into a “sticky” aggregation-prone precursor or intermediate(s), which are structurally different for different proteins. Furthermore, an intermediate might contain a different amount of ordered structure, even for the same protein undergoing different aggregation processes. The variations in the amount of the ordered structure in the amyloidogenic precursor might be responsible for the formation of fibrils with distinct morphologies.25 There are several studies favoring the idea that deposited proteinacous inclusions, such as senile plaques in AD brains or LBs and LNs neurites in PD brains, are not toxic, but that some protofibrillar structures are responsible for the toxicity.26–30 Currently, the amount of evidence supporting this concept is rapidly growing, and this view is accepted by the majority of the scientific community.
It has been emphasized that protein conformational diseases are generally multifactorial disorders, where pathogenic misfolded and aggregated forms of a protein can arise in a multitude of different ways.31–33 This includes, but is not limited to, such factors as an intrinsic propensity of specific proteins to gain pathologic (misfolded) conformations,14 which can be enhanced by single mutations (e.g., hereditary amyloidoses,34–38 familial cases of PD39,40 and AD41) or by proteolytic digestion.42–45 In addition to the intrinsic amyloidogenic potential of the pathogenic protein, other factors may act synergistically in amyloid-forming protein misfolding. For example, quite often the interactions (or impaired interactions) of a pathogenic protein with some endogenous factors (e.g., chaperones, intracellular or extracellular matrixes, other proteins) can change its conformation and increase its propensity to misfold and aggregate.31 Similarly, conformational changes in a given protein can be induced by internal or external toxins, which might facilitate misfolding and aggregation.46 Finally, malfunction and impaired functioning of different systems (e.g., proteasomes and other proteolytic systems, the antioxidant defense system, and post-translational modifications) might change a protein’s conformation, facilitating its misfolding and promoting aggregation.47,48 It is important to keep in mind that all these mechanisms can act independently or in association with one another.
Nanomedicine for Protein Misfolding Disorders
Despite obvious progress, there still remain many unanswered questions concerning protein misfolding diseases. An important set of questions is related to better understanding the structural and mechanical properties of nanoaggregates. What is the precise organization of different nanostructures? How stable are they? How are they formed? Furthermore, little is known about the genetic or environmental factors that determine individual susceptibility to protein misfolding or the factors that govern anatomical distribution of protein deposits and clinical effects. Two crucial questions unify all protein misfolding disorders. First, why do natively soluble proteins, both intrinsically disordered and intrinsically ordered, undergo misfolding into conformations that dramatically change their molecular interactions? Second, how does the misfolded protein look like? There are additional important unanswered questions. If almost any protein can fibrillate in vitro, then why only a small number of unrelated proteins form amyloids in vivo? How do misfolded or aggregated proteins lead to the cell death, tissue degradation, and development of pathology? What are the toxic protein species, and are the aggregates themselves toxic? What underlies the selective vulnerability of proteins that are often ubiquitously expressed throughout different tissues? Finally, a number of problems are associated with the requirements for targeted delivery of drugs to the affected organs and even to the individual cells in these organs.
A majority of current experimental tools and techniques are ensemble approaches. They provide a population-based picture of a system, where structural information is averaged over the ensemble of all conformations present in the system during the time of measurements, and thus do not allow for the unambiguous measurement of protein interaction forces or the kinetics of interconversion among different protein conformations. The ability to measure these parameters is critical for the achievement of a quantitative understanding of protein misfolding and aggregation at the nanoscale level. Thus, new nanomedicine tools and approaches are crucial for understanding the protein misfolding phenomenon. Some concepts and ideas related to the application of nanomedicine for probing protein misfolding diseases were outlined in our recent review.49 The most critical points from that review are summarized below and used to build a more comprehensive story focused on the detailed description of different nanomedicine tools and approaches.
The causative agent of protein misfolding diseases is an improperly folded protein molecule, which either lost its function, gained toxic function, or became involved in aggregation. This structural transformation directly or indirectly alters the physiology of its host cell. In principle, the severity and progression of conformational diseases can be altered by cellular factors that recognize and attempt to ameliorate the harmful effects of the disease-causing misfolded protein. Specific proteins that cause each of the conformational diseases have to be studied in order to (1) better understand the mechanistic foundations of cellular processes that precede, accompany, and follow protein misfolding; (2) elucidate factors that mediate quality control; and (3) understand why a single misfolded protein can impact cell viability. All of this, together with the majority of the questions outlined in the preceding paragraph, is related to the understanding and potential treatment of a given pathology on a molecular level. Thus, we believe that the best approach to answer these questions and to solve these problems is by using nanoimaging tools that are already available or currently in development. Nanomedicine, which developed out of nanotechnology, is an emerging field of medicine with novel applications. It usually refers to highly specific medical intervention at the molecular scale for curing disease or repairing damaged tissues, such as bone, muscle, or nerve. Thus, some of the major goals of nanomedicine in application to protein misfolding diseases are to better visualize nanoaggregate structure, to understand the protein misfolding phenomenon, to develop novel approaches for diagnostics, and ultimately to prevent or treat diseases mediated by protein misfolding. It is generally appreciated that novel approaches are necessary to determine the molecular basis of protein misfolding and aggregate formation in vitro and in vivo. This fundamental knowledge then can be used to formulate a predictive molecular model that incorporates the cellular nanomachinery and physiological processes involved in protein misfolding disorders. In this way, we can establish crucial insights enabling the development of new therapeutic advances and novel nanotechnologies for diagnosis.
Solutions to the unsolved problems related to protein misfolding diseases can be combined into three major groups: (i) a better description of these nanostructures and their components, (ii) a deeper understanding of the molecular mechanisms underlying protein misfolding and aggregate formation, and (iii) the ability to deliver treatments to these nanotargets. These solutions will likely be obtained by (i) elaboration of nanoimaging tools for enhanced structural analysis, (ii) a nanotoolbox with different nanotools for conformational and mechanistic analysis, (iii) and nanocontainers for targeted drug delivery.
Looking at the Protein Misfolding and Aggregation with the Use of Nanoimaging Tools
There are two obvious applications of nanoimaging: first, to gain more detail on misfolded and aggregated protein structures; and second, to follow the protein misfolding and aggregation process at the level of a single molecule or a single supramolecular ensemble. Several tools that have been already developed for these purposes are considered below.
Focusing on the Structure of Aggregated Proteins
It is now recognized that amyloid fibrils, since they are the end point of an ordered misfolding and assembly/aggregation process or possible precursors of cytotoxic, smaller oligomeric species, are an important target for three-dimensional (3D) structural study. For a long time, the major foci of studies on protein misfolding diseases were the structural characterization of deposits and the identification of proteins causing these disorders. Thus, the development of this field was driven by the advances in visualization techniques, the improvements of approaches to purify and identify amyloids, and the evolution of methodologies for the identification and characterization of involved proteins. Investigation of the nature of amyloids began with observation of the abnormal macroscopic appearance of some disease tissues, followed by the utilization of light microscopy and histopathologic dyes, such as thioflavin and Congo red.50 The application of polarization light microscopy revealed that amyloids are not structurally amorphous but possess ordered submicroscopic structure.51 Using electron microscopy (EM), Cohen and Calkins showed that amyloids that are hyaline and structureless under the light microscope actually have a characteristic fine fibrillar ultrastructure, which suggests a specific supramolecular organization of the constituent molecules.52 Subsequent EM studies showed that, irrespective of origin, the amyloid fibrils may be composed of even thinner subelements, designated protofibrils.53–55
Currently, there are two major methods for preparing macromolecular assemblies for EM: negative staining and cryo-EM. In negative staining EM, the sample is dried onto a support film and treated with a solution of heavy metal stain. Although the negative staining approach is proven to provide good contrast of the outer surface of structures, the resulting image may be distorted by the staining and dehydration processes. In contrast, specimens for cryo-EM are preserved in the native, hydrated state by flash-freezing a thin layer of solution, which is then imaged at low temperature. The 3D structure of assemblies with helical symmetry can be determined from EM images,56,57 where, in theory, a single view of a long helical fibril is sufficient. However, in practice, the extreme sensitivity of biological macromolecules to electron beam damage requires that any given area receive only very low exposure. This provides individual images with a low signal-to-noise ratio so that images of different fibrils must be averaged together to obtain a sufficient amount of signal.58
A survey of protofilament arrangement in a wide range of fibrils suggested that fibrils contained five or six protofilaments of ~30 Å in diameter.59 The first successful reconstruction of an amyloid fibril 3D structure at ~25 Å resolution using cryo-EM was reported by the Saibil group in 1999.60 According to this study, the model fibril grown from an SH3 (Src homology 3) domain is formed by four protofilaments, each with a 20 × 40 Å cross section, wound around a hollow core 50 Å in diameter at its widest part.60 This hierarchical view of amyloid fibril assembly was further developed using atomic force microscopy (AFM), which showed that for all proteins studied there are at least three fibrillar species that vary in diameter. The smallest, protofilaments, have a uniform height, whereas the larger species, protofibrils and fibrils, have morphologies that are indicative of the intertwining of multiple protofilaments. In all cases, protofilaments intertwine to form protofibrils, and protofibrils intertwine to form fibrils.61 The next advance in the understanding of the nature of amyloid fibrils was achieved when isolated amyloid protein fibrils were analyzed by X-ray diffraction.15,21,62 This analysis revealed that proteinaceous amyloid fibrils are characterized by broadly similar, though not very detailed, diffraction patterns dominated by meridional reflections at ~4 ± 7 Å and equatorial reflections at 10 ± 12 Å. These scattering profiles are consistent with an ordered core structure where the polypeptide backbone assumes the cross-β-pleated sheet conformation runs parallel to the fibril axis and constituent β-strands are oriented perpendicular to the fibril.15 The β-pleated sheet structured fibril seems to be the basis of the unusual resistance of all kinds of amyloid to degradation and, therefore, the in vivo accumulation of amyloid material.8 On the basis of the results of X-ray diffraction analysis, M. F. Perutz proposed the water-filled nanotubes model of amyloid fibrils in which polypeptide chains fold into β-strands, which form a cylindrical sheet of 3 nm diameter, which in turn folds into a hollow cylinder filled with water.63 In particular, structural models based on the results of the solid state NMR spectroscopy were recently reported for amyloid fibrils formed by the Aβ40 peptide64–66 and a protein fragment comprised of residues 10–39 of the yeast prion protein Ure2p (Ure2p10–39).67 The modern hierarchical view of amyloid deposits is depicted in Figure 1. In-depth analysis of the modern state-of-art approaches in the field of amyloid fibril structure analysis is present in several recent reviews.68–72
Figure 1.

Modern, hierarchical view of protein misfolding and amyloid fibrils: intra- and extracellular inclusions in different tissues associated with misfolding diseases (A) are proteinaceous deposits (B). These deposits are comprised of amyloid fibrils, crude structures which can be resolved by EM or AFM (C). Cryo-EM analysis provides a detailed view of fibrils as an intertwined bundle of several filaments (D). Modeling based on X-ray diffraction and EM/AFM analyses gives the ultrastructure of amyloid fibrils and filaments as cross-β spines (E). The first structure of a model amyloid fibril comprised of a short peptide derived from the yeast prion protein was determined at atomic resolution (F) (for more details see Figure 3). There are at least 20 different proteins associated with protein misfolding diseases. An example of one of the amyloidogenic polypeptides, bovine prion protein, is shown in panel G. Panels D and E are modified from ref 220 with the permission of authors. Panel F is adapted with permission from ref 130. Copyright 2005 Macmillan Publishers Ltd: Nature. Panel G is a generous gift of Prof. Wutrich.
Understanding the Structural Peculiarities of Misfolded Proteins
Misfolded proteins differ from native proteins by their increased propensity to interact with each other leading to the formation of nanoaggregates. The crude structure of individual protein molecules within well-ordered nanoaggregates (i.e., nanoassemblies) was elucidated by traditional structural techniques, including X-ray crystallography, NMR, circular dichroism, fluorescence, Raman spectroscopy, and IR spectroscopy [reviewed in refs 73, 74]. Low-resolution methods such as electron microscopy and atomic force microscopy (AFM) have also provided useful data regarding the morphologies of self-assembled aggregates. It is important to note, however, that all studies discussed thus far have primarily focused on the end point of the aggregation process: amyloid fibrils and other nanoaggregates. Missing in this picture is the structure of a misfolded protein prior to and during the aggregation process. Numerous attempts to gain this information using conventional structural biology methods fail primarily because none of the approaches typically used in protein fibrillation studies are capable of detecting these misfolded conformations. All these approaches are ensemble-oriented techniques that do not distinguish between the conformational changes of individual protein molecules and those induced in protein molecule ensembles induced by protein–protein interactions. There are two major directions to address these questions: (i) techniques based on the single-molecule detection and (ii) traditional approaches, applied to the physically isolated protein molecules. These nanotools are considered below.
(i) Single-Molecule Biophysics Nanotools
The major advantage of probing single molecules is that this approach enables the intimate detection and quantitative characterization of conformational states of a single molecule, including transient conformations. Single-molecule methods provide the ability to identify misfolded states, establish the sequence of misfolding/aggregation pathways, and discriminate the formation of oligomeric nanostructures from fibrillation under a variety of conditions, in different cellular components, and in the presence of different metabolites. These methods may help answer several outstanding questions. What is the time scale for the protein misfolding? How homogeneous are misfolded states for individual proteins? What are the dynamics of misfolded protein molecules at each misfolded state? These fundamental questions can be explored through the use of single-molecule fluorescence microscopy utilizing the Förster (i.e., fluorescence) resonance energy transfer (FRET) phenomenon. It has become increasingly clear that state-of-the-art single-molecule FRET offers a powerful new approach to understand the dynamics of a protein structure, enabling evaluation of molecules as they fluctuate over time and space.75–84 This technique allows observation of the dynamic behavior of individual protein molecules at various folding states, exploration of heterogeneity between molecules, and determination of their mechanisms of interaction. These studies can be extended to the intracellular environment where individual molecules can be viewed as they move within the cell, carry out specific functions, or behave as components of larger systems.
It has been emphasized that FRET is one of a few physical methods that are able to provide direct information on intra-and intermolecular distances in solution.85 The first attempts in this direction were based on the analysis of FRET between extrinsic fluorophores covalently bound to protein functional groups. These covalent modifications were shown to produce significant labeling heterogeneity and induce structural alterations.86,87 Fortunately, toward the end of the 1960s, it was found that the reaction of tyrosine (Tyr) with tetranitromethane results in the formation of 3-nitrotyrosine [Tyr(NO2)].88 Tyr-(NO2) absorbs radiation in the wavelength range where Tyr and Trp emit fluorescence (300–450 nm), and it is essentially nonfluorescent. Therefore, Tyr(NO2) may act as an effective energy acceptor in resonance energy transfer studies on proteins containing Trp and Tyr residues,89 where this donor–acceptor pair has a critical Förster distance (ro) of 26 Å.90
Structural and dynamic properties of several proteins in folded, partially folded, and natively unfolded states were assessed implementing FRET between Trp and Tyr(NO2).85,89,91–94 For example, the structure and dynamics of the natively unfolded protein α-synuclein, which is known to play a key role in the pathogenesis of PD,31 has been recently analyzed by time-resolved Trp-Tyr(NO2) FRET under a variety of conditions, including physiological pH (7.4) solutions, acidic pH (4.4) solutions, and in the presence of sodium dodecyl sulfate (SDS) micelles.94 To this end, six different Trp-Tyr(NO2) donor–acceptor pairs were introduced in the protein via site-directed mutagenesis, which included the constructs Phe4Trp/Ala19Tyr-(NO2), Ala19Tyr(NO2)/Tyr39Trp, Phe4Trp/Tyr39(NO2), Val74Tyr-(NO2)/Phe94Trp, Phe94Trp/Tyr136(NO2), and Phe4Trp/Tyr136-(NO2). Analysis of the donor–acceptor distance distributions revealed that SDS micelles induced compaction of the N-terminal region and expansion of the acidic C-terminus of α-synuclein. In acidic solutions, there was an increase of collapse in the C-terminal region.94
An analogous approach was used to analyze the aggregation and fibrillation of α-synuclein.95 Here, a Tyr125Trp/Tyr133Phe/Tyr136Phe mutant of α-synuclein was used, where the mutant contained one Tyr, Tyr39, chemically modified to Tyr39(NO2) and one Trp, Trp125. It has been confirmed with FRET that the aggregation and fibrillation of α-synuclein involves multiple oligomeric intermediates and competing pathways. It has also been shown that the average distance between Tyr39 and Trp125 decreases from 24.9 Å in the monomer to 21.9 Å in the early oligomer, and further decreased to 18.8 Å in the late oligomer. 95
FRET between Trp and Tyr(NO2) has also been recently applied to analyze native protein–protein interaction in the hirudin–thrombin system.96 In this system, the fluorescence of thrombin was reduced as a result of the binding of nitrated hirudin due to energy transfer between the Trp residues of thrombin and the single Tyr(NO2) of hirudin.96 Thus, the incorporation of Tyr(NO2) can be effectively used not only to analyze the structure and dynamics of a protein undergoing folding and misfolding, but also to detect protein–protein interactions with a sensitivity in the low-nanomolar range, to uncover subtle structural features at the protein–protein interface, and to obtain reliable Kd values for the study of structure–activity relationships.96
Extensive application of FRET methods utilizing intrinsically fluorescent proteins, such as yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP), has revealed that FRET between YFP and CFP can be used to demonstrate interaction between proteins to which they are linked.97 When two proteins stably interact, fluorescent excitation of CFP (donor) at 440 nm excites YFP (acceptor) at 480 nm to fluoresce at 535 nm, with a Förster critical distance (50% efficiency) of 5 nm.98 A recent study of the Ca2+/calmodulin-dependent protein kinase II (CaMK-II) oligomerization has successfully utilized this technique.99 CaMK-II is a highly conserved serine/threonine protein kinase encoded by four genes (α, β, γ, and δ) which are known to produce at least 38 alternatively spliced variants.100,101 These alternatively spliced variants assemble into an oligomer of approximately 12 subunits that assumes a unique morphology.102–105 CaMK-II oligomers are formed by a core of oligomerization domains linked to outwardly facing N-terminal catalytic domains via the variable domains.102–105 The 29–218 amino acid V domain is encoded by the alternative use of up to eight exons per CaMK-II gene. To analyze CaMK-II oligomerization in vivo, the catalytic domains of α, β, and δ CaMK-IIs were replaced with CFP or YFP, and the efficiency of FRET was analyzed.99 FRET occurred only when individual CFP- and YFP-linked CaMK-II subunits were coexpressed, but not when they were expressed separately and then mixed. It has been shown that the α, β, and δ CaMK-II subunits are able to freely hetero-oligomerize when coexpressed and that longer lengths of the variable domain place amino termini of CFP- and YFP-linked CaMK-II subunits further apart. It has also been established that FRET can be used to identify the cellular localization of CaMK-II hetero-oligomers.99 This study clearly shows the power and advantages of FRET for the analysis of protein–protein interactions and oligomerization processes. Most single-molecule FRET studies are performed with one donor–acceptor pair, a so-called two-color FRET that is limited to two points in a particular system. The structural information obtained with FRET can be extended by a three-color FRET approach. The feasibility of the three-color method at the single-molecule level has been demonstrated recently.106,107 Although this approach represents a recent development in FRET technology that may or may not be easily applied to the misfolding problem, the utilization of three-color FRET might increase the information content of this technique.
(ii) Assessing Caged Proteins by Conventional Biophysical Techniques
Under particular circumstances, misfolded conformations and associated conformational transitions can be studied by conventional spectroscopic techniques, including UV–vis absorption, fluorescence, and CD. These traditional biophysical methods can potentially provide crucial structural information for individual protein molecules, if these molecules are physically separated from each other. Such physical separation can be achieved either by protein encapsulation in silica glass using the sol–gel technique or by covalent attachment of protein molecules to chemically modified glass surfaces (Figure 2). For numerous proteins, sol–gel glass encapsulation provides relatively mild conditions, which do not generally perturb structure or function, at least to a significant degree.108–115 The only know exception is apomyoglobin, whose encapsulated form was shown to be highly unfolded.112 It is believed that the size of protein-occupied pores in these gels is of the same order of magnitude as the diameter of the protein. Furthermore, it has been suggested that the protein might dictate the pore size during the gelation process.116 The exact size of a protein-occupied pore is unknown, but the average pore diameter in a wet, aged glass is expected to be over 100 Å. The pore size can be dramatically reduced by solvent evaporation from the newly gelated sol–gel samples, leading to the formation of denser xerogels, which may occupy only 10%–15% of the original volume.111,112 Silica glasses are highly porous materials, and, as a result, solvent and small solute molecules can diffuse into the interconnected pores of the silica matrix, but the larger protein molecules are prevented from escaping into the surrounding buffer. Thus, separated, trapped proteins may be analyzed by many of the same spectroscopic techniques (e.g., near- and far-UV circular dichroism, fluorescence) used to monitor the structure of macromolecules in solution.111,112 Protein caging in the silica glasses is therefore an attractive approach, which may help solve the problem of protein misfolding by allowing detailed structural analysis of the isolated protein molecules. This tool has the potential to provide crucial information on the conformational behavior of a protein molecule unperturbed by the influence of other protein molecules, since caged polypeptides are physically isolated. The method of the sol–gel glass encapsulation has been successfully applied to the analysis of protein unfolding under a variety of environmental conditions,111,112 and analogous studies on aggregating proteins are currently underway.
Figure 2.
Individual protein molecules can be physically separated via encapsulation in silica glass using the sol–gel technique.
Nanotools for Conformational and Mechanistic Analyses of Misfolded Proteins
Advances over the past few years have demonstrated that single-molecule biophysics techniques can visualize intermediates, follow molecular scale events in real time, and probe a wide range of intermolecular interactions. We believe that exploiting new and established single-molecule methods, in conjunction with complementary ensemble techniques and a set of theoretical approaches enabling quantitative analysis of experimental data, represents a novel nanotool box for analysis of transient conformations of proteins. This nanotool box will provide the scientific resources needed to develop innovative nanomedicine approaches for understanding, diagnosing, preventing, and curing protein misfolding diseases. Obviously, a majority of the techniques described in the previous sections as specific tools for the structural analysis of misfolded proteins can also be used to follow structural changes accompanying transitions to and from misfolded conformations.
Fatal Attraction: Studying Intermolecular Interactions between Misfolded Proteins
It is well-established that AFM can be used to probe mechanical stability of proteins.117 Recently, it has been proposed that AFM might represent a useful tool for probing misfolded proteins, particularly to probe single-molecule interactions. AFM was used to study misfolding-induced interactions in three unrelated and structurally distinctive proteins, α-synuclein, amyloid β-peptide (Aβ), and lysozyme.118 The interprotein interaction, being minimal at neutral pH, increases sharply with a decrease in pH, that is, under the conditions known to induce misfolding of these proteins.118 This study showed that misfolded proteins undergo a dramatic increase in the strength of interprotein interactions relative to native protein, which eventually promotes the formation of protein aggregates.118,119 These studies illustrate the power of nanomechanical tools for unraveling molecular mechanisms of protein misfolding, aggregation, and fibrillation. Force spectroscopy has also been able to detect increases in interprotein interactions immediately following a change in experimental conditions, that is, well before any macroscopic manifestations of protein aggregation. This opens several new avenues into the application of force spectroscopy for protein misfolding disease studies. We believe that this technique can be potentially used as a novel diagnostic tool to detect the development of pathological conditions at very early stages via advance detection of misfolded conformations with an increased predisposition for participation in intermolecular interactions.
Direct Observations of Fibril Growth. 1. Total Internal Reflection Fluorescence Microscopy
Although convincing structural models of amyloid fibrils are emerging, the precise kinetic mechanisms of fibrillation are still unknown. Thus, novel tools enabling the direct observation of individual fibril growth are important to obtain further insight into the kinetic mechanism of fibril formation. Recently, direct observation of individual fibril growth was reported.120,121 In these studies, seed-dependent amyloid fibril growth of Aβ1–40 was visualized in real-time at the single fibril level using total internal reflection fluorescence microscopy (TIRFM), which used an amyloid-specific fluorescence dye thioflavin T (ThT) as an extrinsic fluorophore. Seed fibrils were mixed with monomeric Aβ1–40 peptide, ThT was added to the sample, mixtures were deposited on the surface of a quartz glass slide, and fibrils were observed every 2 min under TIRFM. This approach allowed observation of novel fibrillation events. At first, only the ThT fluorescent spots of the seeds were observed, and later, the growth of fibrils occurred concomitantly at many seeds. Once started, unidirectional growth continued, producing remarkably long fibrils. Thus, real-time kinetics of fibril formation on a glass surface can be clearly visualized without requiring covalent fluorophore labeling. One of the advantages of TIRFM is that the exact length of fibrils can be obtained via the selective monitoring of fibrils lying along the slide glass, which makes kinetic analysis straightforward. TIRFM revealed that fibril growth is a highly cooperative process extending blunt ends at a constant rate.121
2. In Situ AFM Analysis of Amyloidogenic Proteins
Recently, in situ tapping mode AFM was successfully applied to continuously monitor the self-assembly of several amyloidogenic proteins.122,123 In the case of α-synuclein, different aggregation modes were observed depending on experimental conditions, that is, pH, temperature, protein concentration, polyamine concentration, and the supporting substrate. For example, the prevalent aggregated form of this protein at neutral pH in the presence of spermidine or spermine was spheroid with a height of 5.9 ± 1.0 nm. These spheroidal structures share some characteristics with spheroids reported for several amyloidogenic proteins, which are linked to neurotoxicity. In rare cases, fibril growth from spheroids or preformed aggregates was observed.123 On the other hand, fibrils were easily formed at pH 5.0. Under these conditions, the initial, relatively rapid fibrillation was followed by the incorporation of the formed fibrils into amorphous aggregates, suggesting that amyloid fibril surfaces act as nucleation sites in amorphous aggregation.123 In situ tapping mode AFM applied to the visualization of protein aggregation has several important advantages such as the abilities to continuously monitor individual aggregate species, to provide high-resolution images of prefibrillar aggregates, and to enable temperature control. In situ tapping mode AFM also offers the opportunity to apply various solution conditions, which is a crucial advantage considering the strong solution dependence of protein aggregation.123
An Integrated Approach: Studying the Protein Misfolding Problem at Multiple Levels
Misfolding is a complex phenomenon that can be facilitated, impeded, or prevented by interactions with other proteins, various intracellular metabolites, and intracellular nanomachines that control protein folding. Elucidating the role of these interactions requires the study of the protein misfolding phenomenon at all levels: atomic, molecular, ensemble, and cellular.
1. Atomic Level
The innovative thrust of studies on protein misfolding diseases should be focused at the characterization of misfolded conformations and protein–protein interactions by single-molecule force measurement approaches that span multiple orders of magnitude in applied force and time scale. The force microscopy data, translated into prominent barriers in the energy landscape that governs the stability of protein conformation and the strength of pairwise interactions, should be used to provide the essential experimental foundation for molecular modeling and theoretical analysis of misfolded conformations and for elucidating the underlying mechanisms in protein misfolding. At the atomic level, computer modeling approaches may help to reveal interactions and refolding patterns that can be used to construct dynamic maps of folding and misfolding pathways. Both experimental and theoretical studies of non-native protein conformations have yielded dramatic developments during the past 10 years. Numerous computational approaches have recently been elaborated for generating models of denatured and partially folded proteins and performing dynamic analysis of conformational transitions.124 The majority of work in this area uses molecular dynamics (MD) techniques, which can be divided into three main categories: (i) techniques that use elevated temperatures in the simulations, (ii) approaches that add extra term(s) to the MD force field, and (iii) methods where changes are made to the initial protein–solvent system.124
Recently, significant progress has been achieved in understanding the structure of amyloid fibril at the atomic level both from experimental studies and from computer modeling. It has been shown that a seven-residue peptide segment, GNNQQNY, derived from the yeast protein Sup35 (a protein whose fibril formation represents the basis of protein-based inheritance and prion-like infectivity in yeast125–129) is not only able to assemble into the amyloid-like fibrils but can also form closely related microcrystals, from which the atomic structure of the cross-β spine was determined.130 The atomic structure of this cross-β spine is shown in Figure 3. It can be seen that GNNQQNY molecules are extended and are hydrogen-bonded to each other forming standard parallel β-sheets, where each GN-NQQNY molecule represent a single β-strand. The overall structure of a fibril is a double β-sheet, which is stabilized by side chains protruding from the two sheets via a dry, tightly self-complementing steric zipper.130 The shape complementarity of this dry interface is unusually tight in comparison with other protein interfaces that are thought to result from the extensive use of van der Waals interactions for double β-sheet stabilization rather than hydrogen bonds, which are common in protein interfaces.130 This remarkable complementarity between sheets might provide a physical background for outstanding conformational stability of amyloid fibrils. On the basis of these observations, it has been concluded that one can find three levels of organization within the fibrils. The first level is the alignment of GNNQQNY molecules to form a β-sheet. The second level is the self-complementation of two sheets to form the pair-of-sheets structure with a dry interface. Finally, in the third level, pair-of-sheets structures interact to form a fibril. Because the self-complementation of two sheets in the second level involves van der Waals forces rather than hydrogen bonding, the patterns of bonding are less specific than those of the first level. This means that there is a possibility to form some alternative interfaces, which could give rise to fibril polymorphism and prion strains.130
Figure 3.
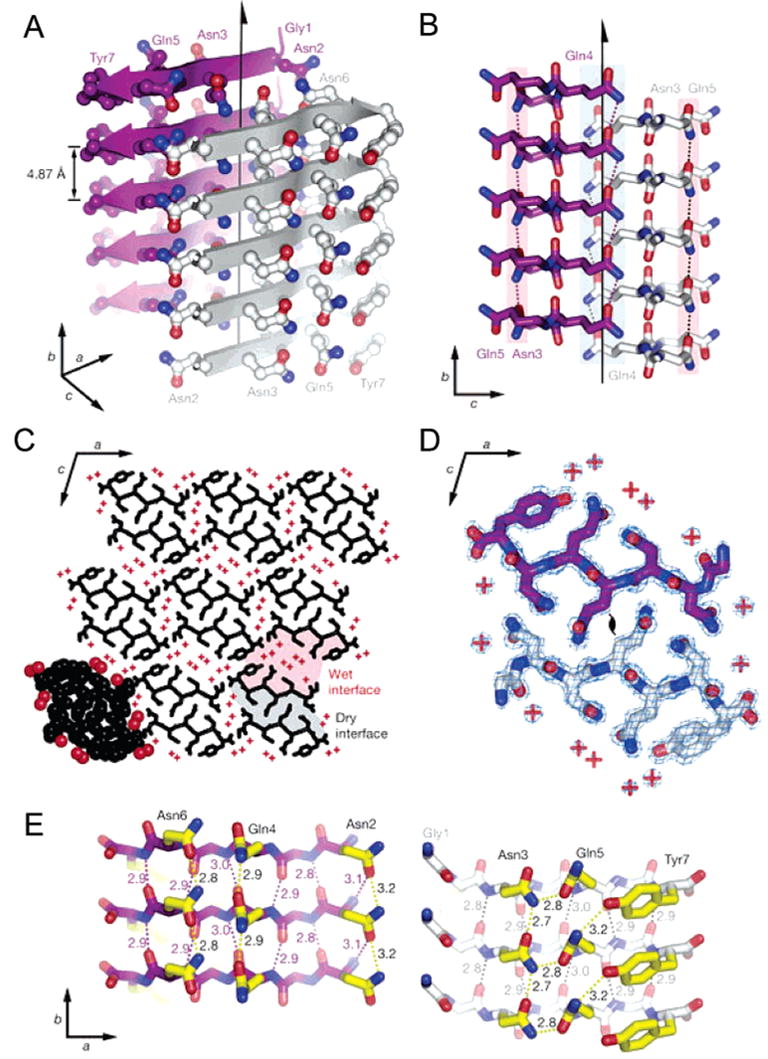
Structure of a GNNQQNY-based amyloid fibril. (A) The pair-of-sheets structure, showing the backbone of each β-strand as an arrow, with side chains protruding. The dry interface is between the two sheets, with the wet interfaces on the outside surfaces. The side chains of Asn2, Gln4, and Asn6 point inward, forming the dry interface. The 21 screw axis of the crystal is shown as the vertical line. It rotates one of the strands of the near sheet 180°about the axis and moves it up 4.87 Å so that it is superimposed on one of the strands of the far sheet. (B) The steric zipper viewed edge on (down the a axis). Note the vertical shift of one sheet relative to the other, allowing interdigitation of the side chains emanating from each sheet. The amide stacks of the dry interface are shaded in gray at the center, and those of the wet interface are shaded in pale red on either side. (C) The GNNQQNY crystal viewed down the sheets (from the top of panel A, along the b axis). Six rows of β-sheets run horizontally. Peptide molecules are shown in black, and water molecules are red plus signs. The atoms in the lower left unit cell are shown as spheres representing van der Waals radii. (D) The steric zipper. This is a close-up view of a pair of GNNQQNY molecules from the same view as panel C, showing the remarkable shape complementarity of the Asn and Gln side chains protruding into the dry interface. 2Fo – Fc electron density is shown, and the position of the central screw axis is indicated. (E) Views of the β-sheets from the side (down the c axis), showing three β-strands with the inter-strand hydrogen bonds. Side-chain carbon atoms are yellow. Backbone hydrogen bonds are shown by purple or gray dots and side-chain hydrogen bonds by yellow dots. Hydrogen bond lengths are noted in angstroms. The views of the interfaces are close to the views of panel A. The left-hand set is viewed from the center of the dry interface; the right-hand set is viewed from the wet interface. Note the amide stacks in both interfaces. Reprinted with permission from ref 130. Copyright 2005 Macmillan Publishers Ltd: Nature.
In addition to this breakthrough in the experimental determination of the atomic-level structures of the cross-β spine, significant progress has recently been made in the development of various atomic models of amyloid fibrils. Several types of models have been developed to explain the conversion of proteins from soluble to fibrous forms, which includes refolding, natively disordered, and gain-of-interaction models. The gain-of-interaction models may additionally be subdivided into direct stacking, cross-β spine, three-dimensional domain swapping, and three-dimensional domain swapping with a cross-β spine. Although no single model accounts adequately for all properties of all fibrils, some models explain many properties of a range of different fibrils [reviewed in ref 68].
2. Understanding Protein Misfolding at the Levels of Molecules and Ensembles of Molecules
At the molecular level, single-molecule imaging and interaction approaches can identify intracellular metabolites and extracellular molecules critically involved in protein misfolding. Those effectors that robustly stimulate the misfolding could be candidates for the development of diagnostic tools. Compounds that work in the opposite way may represent potential therapeutics. It has been recently shown that some fragments of the Aβ peptide stimulate the aggregation of the full-length Aβ peptide, suggesting that these fragments can be considered as the intracellular enhancers of Aβ misfolding and aggregation.131 On the other hand, fibrillation of α-synuclein was shown to be inhibited by interaction with β- or γ-synucleins, suggesting that that β- and γ-synucleins may act as regulators of α-synuclein fibrillation in vivo, in effect acting as chaperones to minimize the aggregation of α-synuclein.19
Various studies have shown that protein misfolding is promoted by oxidative damage.132,133 A couple of questions about this phenomenon have yet to be answered. How do different antioxidant proteins affect the conformation and structural dynamics of pathogenic proteins? Do antioxidant proteins prevent the misfolding of previously oxidized and nonoxidized amyloidogenic proteins incubated in the presence of reactive oxygen species? We believe that these questions can be answered by the application of the force measuring nanotools described above.
Finally, it is known that protein folding in vivo is controlled by intracellular machines, and therefore, elucidating the principles of protein misfolding in vivo should include detailed consideration of the participation of chaperones and proteasomes. The interaction of monomeric or oligomeric forms of misfolded proteins with chaperones and cochaperones has the potential to perturb the balance between refolding, degradation, and cellular signaling controlled by these complexes. Misfolded protein species are notoriously resistant to soluble proteases and may inhibit or sequester chaperone systems. Alzheimer’s β-peptide interacts with human hsp70 and the cochaperone BAG-1, and such interactions may be exacerbated by oligomeric and/or fibrillar forms of Aβ. Alteration of the relative populations of fibrillar and spherical forms of oligo-meric huntingtin exon I by hsp70-hsp40 chaperones has also been demonstrated.134 The use of single-molecule imaging and probing techniques, such as AFM, single-molecule FRET, and cross correlation fluorescence spectroscopy, together with such tools as conformation-specific antibodies as well as techniques for introducing protein complexes into cells will help to better understand the role of chaperone interaction with the misfolded proteins. By continuously following fibrils or oligomers and their interactions with chaperones at the single-molecule level, one can learn if the pathway to potentially toxic oligomers goes through monomers or whether soluble oligomers can form directly from fibrils.
3. Evaluating the Dynamics of the Amyloidogenic Proteins
The next crucial question is related to understanding the changes in protein dynamics associated with amyloidogenesis. There are two major approaches to this problem, limited proteolysis and the hydrogen–deuterium exchange, which in combination with mass spectrometry constitute complementary strategies to investigate the protein regions that are solvent-exposed and/or highly flexible. These two approaches are briefly outlined below. It has been emphasized that folded proteins with rigid 3D structure are highly resistant to proteolysis, as native conformations of such proteins provide some stereochemical barriers to enzymatic attack.135 Furthermore, it is known that proteolysis occurs predominantly in unfolded or disordered regions of proteins. For example, trypsin should cleave a polypeptide at nearly every lysine–X and arginine–X bond (with the partial exception of proline at X), assuming that about 5–10% of the peptide bonds in a typical protein are susceptible to proteolytic attack. However, in native (or near-native) conditions, trypsin will cut only a limited number of such bonds (or on occasions none at all) in a native protein fold. This means that the structure and dynamics of the substrate protein are playing crucial roles in limiting the proteolysis.136,137 Interestingly, the analysis of X-ray crystallographic structures of proteases with small protein inhibitors, such as BPTI, revealed that the inhibitor reactive site loop bound into the enzyme active site in the manner of a ‘perfect’ substrate.138,139 Using this canonical conformation of the inhibitor reactive site loops as a template, Thornton and co-workers showed that limited proteolytic sites are quite different in structure from the idealized inhibitor loops, and they must therefore undergo a conformational change in order to enter the proteinase active site.140 Furthermore, for several proteins, it has been shown that local unfolding of at least 13 residues is needed for a set of observed cut sites to properly fit into trypsin’s active site.136,137 It has been also established that limited proteolytic sites are typically found at flexible loop regions (as indicated by crystallographic temperature factors or B-values) that are also exposed to the solvent,140–142 and are notably absent in regions of regular secondary structure, especially β-sheets.136,143,144 Therefore, protease digestion is much faster in flexible regions and thus can be used to map flexible and nonflexible regions. Furthermore, when limited proteolysis experiments are performed using a series of proteases with different specificities, the pattern of preferential cleavage sites can identify the exposed regions of the protein.135 For example, Fontana and co-workers showed that apomyoglobin is digested many orders of magnitude faster than myoglobin, and the sites of digestion from several different proteases all mapped to a flexible region.143,144 The application of this technique to monitor conformational changes occurring in protein structure in passing from the native state to the misfolded state and to the amyloid fibril-helped identification of the regions possessing variations in structural dynamics associated with the amyloidogenesis.145–148
Similarly, variations in solvent accessibility and conformational dynamics of different protein regions associated with amyloid fibril formation can be assessed by analyzing the changes in accessibility of their backbone amide hydrogen atoms to hydrogen–deuterium exchange.149–153
4. Controlling Protein Misfolding with Conformation-Specific Antibodies
It has been shown recently that protein misfolding can be modulated by conformation-specific antibodies. There is growing evidence that misfolded and aggregated conformations of different proteins might share certain structural features, including β-sheets, β-strands, and β-turns.154 Antibodies specific for such aberrant conformations can recognize inclusions composed of various and unrelated disease-associated proteins, suggesting common structural characteristics among disorders.155 Another antibody, A11, was shown to recognize a structure that appears to be unique to soluble oligomeric forms of APP, α-synuclein, prion, poly-glutamine, and some non-neuronal prefibrils, where recognition is independent of amino acid sequence.156 Such structural similarities may permit the development of a generic, conformation-specific antibody that generally recognizes misfolded protein conformations as well as a conformation-specific antibody for specific misfolded proteins. In addition, antibodies not only inhibit but also reverse, both in vitro and in vivo, fibril formation by amyloidogenic peptides or proteins,157–160 suggesting that antibodies (and single-domain binding fragments) may lead to a potentially powerful method to treat protein misfolding diseases.161 Antibodies expressed intracellularly, termed intrabodies, have already been investigated as treatments for a variety of conditions, including HIV infection,162,163 tumor growth,164,165 and tissue transplantation.166 Finally, such an antibody can be used as a powerful nanoprobe for the detection of misfolded protein conformations in both in vivo and in vitro studies.
Morphological Diversity and Toxicity of Aggregated Proteins
The morphologies of aggregates formed under different conditions vary significantly and may be the consequence of divergent misfolded conformations.167,168 One illustrative example is prion protein, for which different “strains” are known to produce different diseases each with unique clinical and pathologic characteristics.169–174 Another example is poly-glutamine and polyalanine expansions in ataxin7 that were shown to result in different types of aggregation and levels of toxicity. Ataxin-100Q produces fibrillar inclusions, whereas ataxin-90A aggregates to small and amorphous inclusions, which are more toxic to mesencephalic neurons, suggesting that toxicity was determined by the type of aggregate.175
There is rapidly growing evidence that the pathogenesis of protein misfolding diseases is not a result of inclusions or other visible protein aggregates, which represent an end stage of a multistep molecular association cascade. Rather, earlier steps in the series of conformational changes and protein–protein interactions are directly tied to pathogenesis. There is now increased understanding of the pathways involved in protein association, and some recent clues have emerged as to the molecular mechanisms of cellular toxicity.154 A growing body of experimental data indicates that precursors of amyloid fibrils are cytotoxic because they form channels in the lipid bilayer.176 The interaction of amyloid peptides with membranes was assessed by measuring the current through planar lipid membranes induced by homogeneous preparations of soluble monomers, spherical oligomers, and fibrils from Aβ40 and Aβ42, α-synuclein, IAPP, polyglutamine, and prion protein (PrP 106–126). In all cases, only the spherical amyloid oligomers induced currents that increased proportional to the amyloid peptide concentration; neither monomeric nor fibrillar forms increased the current.161 In addition, membrane currents are shut down by addition of anti-oligomer antibodies. Heterogeneous channels are also observed with prion PrP (82–146),177 while another study suggests that sterols are required for the formation of Aβ40 channels.178 Taken together, these results suggest that the cylindrical structures assembled from either the Arctic APP, different PD-linked α-synuclein mutants, or serum amyloid A might be the channels inserted in the lipid bilayer. Attempts were made to directly image porous structures. α-Synuclein protofibrils have been shown to form pore-like assemblies on the surface of brain-derived vesicles.179 Donut-shaped protrusions were observed by AFM on lipid bilayers after reconstitution of Aβ42 in dioleoylphosphocholine (DOPC).180 At higher magnification, these “donuts” were seen to contain 4–6 distinct protrusions. Aβ42 also produced membrane currents when integrated in a planar lipid membrane.180 Although experiments with human amylin (hA) showed induction of dye release by the hA peptide, no pores were visible by negative stain EM.181 Thus, these data led to the conclusion that small protein aggregates are toxic species of misfolded proteins.182 One of the hypotheses is that oligomers with pore-like morphologies change the permeability of membranes.179,180,183 Although pore formation was detected by AFM,180,183,184 the detailed structure of these critical nanostructures has not been elucidated. Thus, a combination of experimental and computer modeling approaches is needed to fill this gap and to establish a nanoscale model for pores in membranes resulting from misfolded proteins. The aims in this endeavor are the following:
(i) development of methods for assembling the pore-like structures of misfolded proteins suitable for high-resolution AFM imaging and probing,
(ii) study of the structure of pores using atomic force microscopy,
(iii) study of the conduction of single channels using conductive cantilevers,
(iv) probing the membrane topology of pores using AFM,
(v) assessing the high resolution 3D structure of pores by electron crystallography,
(vi) refining structural models of membrane pore formation using molecular dynamics simulations in micelles and bilayers, and
(vii) building a comprehensive model for the formation of pore-like structures applicable to various misfolded proteins.
Nanocages for the Delivery of Macromolecular Systems into the Cell and to the Brain
Delivery of water-soluble chaperones, antioxidant proteins, antibody constructs, or other biomacromolecules into cells is essential for many of the experiments outlined above. Many pharmaceutical agents, including various large molecules (proteins, enzymes, antibodies) and even drug-loaded pharmaceutical nanocarriers, need to be delivered intracellularly to exert their therapeutic action inside the cytoplasm or into the nucleus or other specific organelles, such as lysosomes, mitochondria, or endoplasmic reticulum.185 Furthermore, once a better understanding of the role of misfolded aggregates in disease progression is acquired, the ability to deliver macro-molecular constructs to the site of the disease is likely to become essential for development of efficacious modalities for early detection and therapy of protein misfolding diseases. The barriers to successful delivery of diagnostic or therapeutic biomacromolecules into a cell are well-known and discussed in the literature.186 For example, the receptor-mediated en-docytosis of pharmaceutical agents, drug carriers, and DNA187 results in their lysosomal delivery and significant degradation.185 Several approaches have been developed for the direct intra-cytoplasmic drug delivery that bypasses the endocytic pathway. This includes but is not limited to liposomes,188–190 immunoliposomes,191 micelles,192 lipoplexes/polyplexes,193 and cell-penetrating proteins or peptides (CPPs) that can translocate through the cellular membranes enhancing the delivery of CPP-modified molecules inside the cell.194
Even more challenging is the task of delivery of biomacromolecules to the brain, which requires crossing the blood brain barrier (BBB). There is a tremendous need to enhance delivery of macromolecular systems to the brain to treat diseases of the CNS. Some examples include PD and AD,195–198 lysosomal storage diseases,198,199 and human obesity.200,201 The BBB restricts and controls the exchange of peptides and regulatory proteins between the CNS and the blood. Some peptides and regulatory proteins cross the BBB by saturable or nonsaturable mechanisms, whereas most others cannot cross, which hinders their use as a therapeutic agent. Several innovative drug delivery approaches have emerged recently that may lead to solving this fundamental problem. These approaches include (1) chemical modification of proteins (e.g., chaperones and antibodies) with amphiphilic block copolymers, (2) incorporation of these proteins into immunomicelles and immunoliposomes targeted to the brain, and (3) incorporation of these proteins into nanogels or polyelectrolyte complex micelles.
The first approach, chemical modification, is based on a finding that covalently coupling proteins to amphiphilic block copolymers enhances the penetration of these proteins into the cell cytoplasm and across the BBB.202,203 The block copolymers of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) can assume different conformations depending on the polarity of the solution. In aqueous solution, the more hydrophobic PPO chains are compact, while the hydrophilic PEO chains are exposed to water. The same molecule, if placed in a nonpolar solvent, changes the conformation such that the hydrophilic chains become hidden from the external solution while the hydrophobic chains become exposed to the solvent. Furthermore, in an aqueous solution above the critical micelle concentration (cmc), these molecules self-assemble into “polymer micelles” of nanoscale size (20–40 nm). The micelles have a core–shell structure with the core formed of collapsed PPO chains and a shell of hydrophilic PEO chains. The micelles are in equilibrium with the single chains of the block copolymer (“unimers”). The block copolymers can be administered to the body in a micellar form, which then serves as a depot producing more unimers upon dissolution in the body fluids.204 The micelles can be also assembled via cooperative electrostatic interactions of polyion chains. For example, Figure 4 schematically represents a protein delivery nanocontainer in which the core is formed by the polyion complex of proteins and synthetic polyelectrolytes of opposite charge. Here, the chains of PEO–PPO–PEO triblock copolymers, which are grafted to the poly-electrolyte within the core, form a micelle-like structure surrounding the core.
Figure 4.
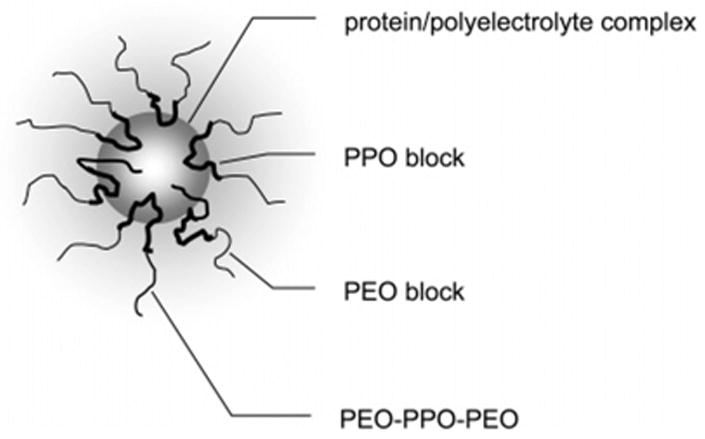
Nanocontainers for protein delivery. The core represents the polyion complex of proteins and synthetic polyelectrolytes of opposite charge. The PEO–PPO–PEO chains grafted to the polyelectrolyte within the core form a micelle-like structure around the core, with the hydrophobic PPO blocks near the core and the hydrophilic PEO blocks forming the exterior shell. Additional nonmodified PEO–PPO–PEO chains fill in to optimize the stability of the micelle-like structure.
Because of the amphiphilic character, these block copolymers cross lipid membranes. For example, triblock copolymers PEO–PPO–PEO, called Pluronic, were shown to accumulate within cells.203 On the basis of these observations, the block copolymers were further evaluated for delivery of biomacromolecules into cells. By the use of a model impermeable protein, horseradish peroxidase (HRP), it has been demonstrated that point modification of this protein with Pluronic enables translocation of this protein into the cytoplasm of the brain microvessel endothelial cells (BMVEC) that form the BBB (see Figure 5).203 Furthermore, this study demonstrated that such modification increased the rate of transport across the BBB and facilitated accumulation within the brain.203 In a mouse animal model, the modified HRP exhibited an increased permeability in the BBB and same clearance as unmodified HRP. As a result, the uptake of the modified protein by the brain was almost doubled. Therefore, we believe that application of such modifications will provide a tool for the delivery of potential therapeutic molecules to the brain and, perhaps, can be used for early diagnostics and bioimaging of the disease sites by targeting antigens hidden behind the BBB.
Figure 5.
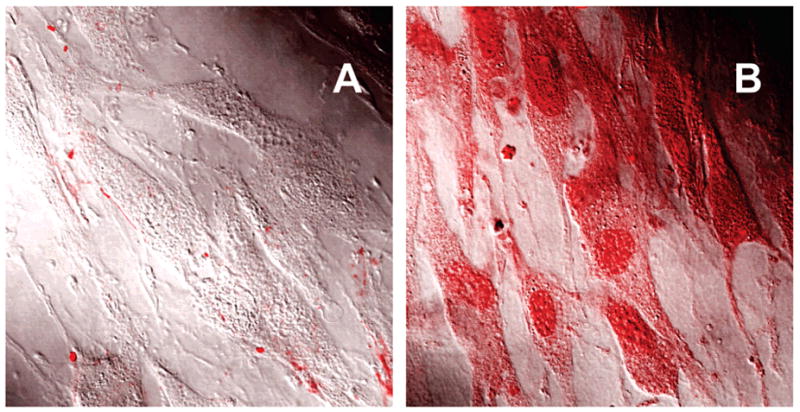
Intracellular localization of native HRP (A) and HRP-P85 (B) in living bovine brain microvessel endothelial cells (BBMEC) visualized by confocal laser scanning microscopy. HRP was labeled with Alexa Fluor 594.203
The second approach employs the use of immunomicelles and immunoliposomes for drug delivery to the brain. One of the early studies of targeted drug delivery to the brain used Pluronic micelles as carriers for CNS drugs.205 These micelles (having an average diameter of ca. 15–30 nm) were conjugated with either polyclonal antibodies against brain α2-glycoprotein or insulin to target the receptors at the lumenal side of BMVEC. Both the antibody- and insulin-conjugated micelles were shown to effectively deliver a drug or a fluorescent probe incorporated into the micelles to the brain tissue in vivo.205 Therefore, by the incorporation of therapeutic molecules (e.g., conjugated to Pluronic) into immunomicelles, it may be possible to further enhance delivery of such molecules to the brain.
An alternative approach is to entrap such molecules into immunoliposomes. The surface of the liposomes is often covered by the poly(ethylene glycol) (PEG, same as PEO) to increase their circulation time in the body. Attachment of targeting moieties to PEGylated liposomes can direct them to the BBB. Thus, efficient delivery of PEG–liposomes conjugated with transferrin (Tf) to the post-ischemic cerebral endothelium was achieved in rats.206 PEGylated immunoliposomes were also successfully employed to target and transfect brain tissues.207 These liposomes were directed to Tf receptor-rich tissues, such as brain, liver, and spleen by a monoclonal antibody, OX26, that was linked to the free termini of the PEG chains. Similar immunoliposome constructs using an OX26 Tf receptor monoclonal antibody were also used to deliver doxorubicin,208 digoxin,209 biotinylated oligonucleotides,210 and neurotrophin peptides211 to the brain. OX26-conjugated liposomes were selectively distributed to BMVEC but avoided choroids plexus epithelium, neurons, and glia.212 On the basis of these results, the PEGylated immunoliposomes are a promising approach for the delivery of macromolecular systems to the brain.
The third approach is based on incorporation of these proteins into nanogels or polyion complex micelles. Nanogel is a new family of hydrophilic carrier systems that was recently developed for targeted delivery of biomacromolecules and other drug molecules to the brain.213 Nanogel consists of a nanoscale size polymer network with cross-linked ionic chains, for example, polyethyleneimine (PEI), and nonionic PEO chains. It swells in aqueous solution and collapses upon binding of a drug through ionic interactions. Because of the effect of PEO chains, the collapsed nanogel forms stable dispersions with particles size of ca. 80 nm. Nanogel can spontaneously absorb biomacromolecules including oligonucleotides (ODNs), plasmid DNA, proteins, and small charged molecules. A key advantage is that nanogel displays very high loading capacities, 40–60% of “payload” by weight, which cannot be achieved by conventional nanocarrier systems. The transport of ODNs incorporated in nanogel particles across an in vitro model of the BBB was recently reported.213 To enhance delivery across the BBB, the surface of nanogels was modified by either transferrin or insulin.213 Both peptides were shown to increase transcellular permeability of the nanogel and enhance delivery of ODNs across an in vitro model of the BBB. Overall, nanogel was shown to be a promising carrier for CNS drug delivery, but is, however, in the early development state.
Polyion complex micelles (also termed “block ionomer complexes”) are formed as a result of the reaction of a double hydrophilic ionic block copolymer (“block ionomers”) with biomacromolecules of opposite charge, including oligonucleotides, plasmid DNA, and proteins.214–216 For example, polyion complex micelles were prepared by reacting trypsin or lysozymes, which are positively charged at physiological conditions, with an anionic block copolymer, PEG–poly(aspartic acid).215,216 Such complexes spontaneously assemble into nanosized particles (<100 nm) with core-shell architecture. The core contains polyion complexes of the biopolymer molecules and the ionic block of the copolymer. The shell is formed by the nonionic block. Similarly to the nanogel system, an advantage of this approach is that it does not chemically modify the protein groups and the proteins can be released in the native form within the target cells. In addition, the polyion complex micelles display a higher loading capacity compared to immunoliposomes, and there is no loss of the incorporated macromolecules due to the polyion coupling and self-assembly processes. These nanomaterials were shown to efficiently deliver DNA molecules in vitro and in vivo into a cell and release them at the site of action.214,217–219 To improve stability of the complexes in the body, the biopolymer and block ionomer can be additionally cross-linked with each other,215,216 or the polyion chains of the block ionomer within the core can be cross-linked using degradable links. The advantages of such systems in drug delivery applications include simplicity and versatility of the design allowing the incorporation of a considerable amount of different biomacromolecules. Furthermore, block ionomer complexes are environmentally responsive nanomaterials that enable biopolymer release in response to an external stimulus, such as change of pH (acidification), concentration, and chemical structure of elementary salt, which allows optimizing the release characteristics at the target site. Although this enabling technology has not been applied to protein delivery to the brain, it provides considerable opportunity to address this problem, since the polyion complex micelles can be easily combined with targeting antibodies similar to those used in immunoliposomes. Notably, the nanogel system, which is fundamentally related to the polyion complex micelles, has already been shown to successfully deliver incorporated biomacromolecules (e.g., ODN) to the brain in vivo.213
Concluding Remarks
The last couple of decades witnessed a remarkable improvement in our understanding of the pathology of numerous human diseases at the molecular level. In many cases, the etiology of a disease is traced to a key protein, and it is recognized now that many devastating disorders, including neurodegenerative diseases, amyloidoses, cataracts, arthritis, and type 2 diabetes, belong to the family of so-called protein misfolding or conformational diseases. These disorders, while representing a group of heterologous disorders, are united by a molecular mechanism where an underlying host protein undergoes a change in its native conformation. The mentioned conformational changes are accompanied by loss of normal function, gain of new and often toxic function, aggregation, and misfolding. These proteins could be misfolded or predisposed to misfolding as a result of point mutations, alternative splicing, or other genetic alterations. For example, such proteins might contain mutations that primarily decrease protein conformational stability but do not affect their expression or function. This mutation-induced conformational instability can intermittently cause the affected protein to partially unfold, then misfold, and then undergo aggregation causing cumulative cell damage. Alternatively, even normal proteins can undergo some post-translational conformational alterations induced by changes in their environments or as a result of toxic insult. The gradual accumulation of protein aggregates and the acceleration of their formation by stress might explain the characteristic late or episodic onset of the clinical disease. The causative agents of these diseases are smaller than bacteria and viruses. As subcellular disorders, protein conformation diseases have to be studied at the molecular level. Thus, the etiology and pathology of misfolding diseases appeal for the development of novel tools for their diagnosis and analysis, and require elaboration of new techniques for their cure. Traditionally, many diseases are diagnosed symptomatically, based on the result of physical examinations. These approaches fail to recognize the early stages of the vast majority of protein misfolding diseases, as obvious symptoms usually develop late. For example, the symptoms of one of the neurodegenerative disorders, PD, become obvious only when about 80% of the cells of the substantia nigra have already died. Diagnosis is further complicated by the fact that PD is just one of several neurologic movement disorders that produce similar symptoms. In some of these diseases, patients quickly become totally disabled, whereas in others, the disease progresses extremely slowly, and finally, in some cases, illness is chronic but may develop more severe symptoms as time goes on. Unfortunately, there are no available laboratory tests to definitively diagnose PD. Conclusions are made based on systematic neurological examinations, which include testing reflexes and analyzing muscle strength throughout the body, as well as coordination, balance, and other details of movement. The possibility of other movement disorders is excluded by specific tests, such as blood tests, urine tests, computed tomography (CT), or magnetic resonance imaging (MRI) of the brain. Although none of these tests actually diagnose PD, they may help reveal the development of some other conditions that could be responsible for the symptoms. This example illustrates a vital need for the development of novel diagnosis tools for protein misfolding diseases. We believe that diagnoses, detailed analyses, as well as potential cures of these disorders, considering the nanoscale of their causative agents, rely heavily on nanomedical approaches. In this respect, the existing and to-be-developed nanotools should play a role similar to that of the light microscope in the development of micro- and cellular biology.
Acknowledgments
The authors express their deepest gratitude to Christopher J. Oldfield for careful reading and editing the manuscript. This work was supported in part by the Programs of the Russian Academy of Sciences for the “Molecular and cellular biology” and “Fundamental science for medicine” (to V.N.U.); the grants from NIH (GM62235 and 1-PN1 EY016593-01), NATO (LST.CLG.980194), the M. J. Fox Foundation for Parkinson’s Research (CFT03) (all to Y.L.L.); and the grants from NIH (NS051335 and NS36229) and NSF (DMR 0513699) (all to A.V.K.).
References
- 1.Cohen AS. General introduction and a brief history of the amyloid fibril. In: Marrink J, Van Rijswijk MH, editors. Amyloidosis. Martinus Nijhoff; Dordrecht, The Netherlands: 1986. pp. 3–19. [Google Scholar]
- 2.Virchow R. Cellulose-Frage. Virchows Arch Pathol Anat. 1854;6:416–426. [Google Scholar]
- 3.Friedreich NK, Zur Amyloidfrage A. Virchows Arch Pathol Anat. 1859;15:50–65. [Google Scholar]
- 4.Hanssen O. Ein Beitrag zur Chemie der amyloiden Entartung. Biochem Z. 1908;13:185–198. [Google Scholar]
- 5.Alzheimer A. Über eine eigenartige Eskrankung der Nirnrinde. Allg Z Psychiatr Psych-Gerichtl. 1907;64:146–148. [Google Scholar]
- 6.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 7.Lewy F. Paralysis agitans. In: Lewandowski M, Abelsdorff G, editors. Pathologische Anatomie Handbuch der Neurologie. Springer Vertag; Berlin, Germany: 1912. pp. 920–933. [Google Scholar]
- 8.Westermark P. Amyloidosis and amyloid proteins: brief history and definitions. In: Sipe JD, editor. Amyloid Proteins The Beta Sheet Conformation and Disease. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2005. pp. 3–28. [Google Scholar]
- 9.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 10.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 11.Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 12.Bellotti V, Mangione P, Stoppini M. Biological activity and pathological implications of misfolded proteins. Cell Mol Life Sci. 1999;55:977–991. doi: 10.1007/s000180050348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rochet JC, Lansbury PT., Jr Amyloid fibrillogenesis: themes variations. Curr Opin Struct Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 14.Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Sunde M, Blake CC. From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. Q Rev Biophys. 1998;31:1–39. doi: 10.1017/s0033583598003400. [DOI] [PubMed] [Google Scholar]
- 16.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein deposits as the molecular basis of amyloidosis: I. Systemic Amyloidoses. Med Sci Monit. 1999;5:1001–1012. [Google Scholar]
- 17.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein deposits as the molecular basis of amyloidosis: II. Localized amyloidosis and neurodegenerative disordres. Med Sci Monit. 1999;5:1238–1254. [Google Scholar]
- 18.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 19.Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 20.Glenner GG, Ein D, Eanes ED, Bladen HA, Terry W, Page DL. Creation of “amyloid” fibrils from Bence Jones proteins in vitro. Science. 1971;174:712–714. doi: 10.1126/science.174.4010.712. [DOI] [PubMed] [Google Scholar]
- 21.Glenner GG, Eanes ED, Bladen HA, Linke RP, Termine JD. Beta-pleated sheet fibrils. A comparison of native amyloid with synthetic protein fibrils. J Histochem Cytochem. 1974;22:1141–1158. doi: 10.1177/22.12.1141. [DOI] [PubMed] [Google Scholar]
- 22.Perutz MF. Amyloid fibrils. Mutations make enzyme polymerize. Nature. 1997;385:773–775. doi: 10.1038/385773a0. [DOI] [PubMed] [Google Scholar]
- 23.Jahn TR, Radford SE. The Yin and Yang of protein folding. FEBS J. 2005;272:5962–5970. doi: 10.1111/j.1742-4658.2005.05021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 25.Smith DP, Jones S, Serpell LC, Sunde M, Radford SE. A systematic investigation into the effect of protein destabilisation on beta 2-microglobulin amyloid formation. J Mol Biol. 2003;330:943–954. doi: 10.1016/s0022-2836(03)00687-9. [DOI] [PubMed] [Google Scholar]
- 26.Selkoe DJ. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 27.Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci US A. 2002;99:13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 30.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-synuclein especially the Parkinson’s disease-associated mutants forms pore-like annular tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 31.Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurode-generative disorders. J Biomol Struct Dyn. 2003;21:211–234. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- 32.Stefani M. Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim Biophys Acta. 2004;1739:5–25. doi: 10.1016/j.bbadis.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Gregersen N, Bross P, Vang S, Christensen JH. Protein misfolding and human disease. Annu Rev Genomics Hum Genet. 2006 doi: 10.1146/annurev.genom.7.080505.115737. in press. [DOI] [PubMed] [Google Scholar]
- 34.Ikeda S. Cardiac amyloidosis: heterogenous pathogenic backgrounds. Intern Med (Tokyo) 2004;43:1107–1114. doi: 10.2169/internalmedicine.43.1107. [DOI] [PubMed] [Google Scholar]
- 35.Munar-Ques M, Saraiva MJ, Viader-Farre C, Zabay-Becerril JM, Mulet-Ferrer J. Genetic epidemiology of familial amyloid polyneuropathy in the Balearic Islands (Spain) Amyloid. 2005;12:54–61. doi: 10.1080/13506120500032741. [DOI] [PubMed] [Google Scholar]
- 36.van der Hilst JC, Simon A, Drenth JP. Hereditary periodic fever and reactive amyloidosis. Clin Exp Med. 2005;5:87–98. doi: 10.1007/s10238-005-0071-6. [DOI] [PubMed] [Google Scholar]
- 37.Pepys MB, Hawkins PN, Booth DR, Vigushin DM, Tennent GA, Soutar AK, Totty N, Nguyen O, Blake CC, Terry CJ, et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362:553–557. doi: 10.1038/362553a0. [DOI] [PubMed] [Google Scholar]
- 38.Gambetti P, Parchi P, Chen SG. Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia. Clin Lab Med. 2003;23:43–64. doi: 10.1016/s0272-2712(02)00065-3. [DOI] [PubMed] [Google Scholar]
- 39.Nussbaum RL, Polymeropoulos MH. Genetics of Parkinson’s disease. Hum Mol Genet. 1997;6:1687–1691. doi: 10.1093/hmg/6.10.1687. [DOI] [PubMed] [Google Scholar]
- 40.Kruger R. Genes in familial parkinsonism and their role in sporadic Parkinson’s disease. J Neurol. 2004;251(Suppl 6):VI/2–6. doi: 10.1007/s00415-004-1602-x. [DOI] [PubMed] [Google Scholar]
- 41.Imbimbo BP, Lombard J, Pomara N. Pathophysiology of Alzheimer’s disease. Neuroimaging Clin N Am. 2005;15:727–753. doi: 10.1016/j.nic.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 42.Ratnaswamy G, Koepf E, Bekele H, Yin H, Kelly JW. The amyloidogenicity of gelsolin is controlled by proteolysis and pH. Chem Biol. 1999;6:293–304. doi: 10.1016/S1074-5521(99)80075-1. [DOI] [PubMed] [Google Scholar]
- 43.Bellotti V, Gallieni M, Giorgetti S, Brancaccio D. Dynamic of beta(2)-microglobulin fibril formation and reabsorption: the role of proteolysis. Semin Dial. 2001;14:117–122. doi: 10.1046/j.1525-139x.2001.00030.x. [DOI] [PubMed] [Google Scholar]
- 44.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 45.de Laureto PP, Frare E, Battaglia F, Mossuto MF, Uversky VN, Fontana A. Protein dissection enhances the amyloidogenic properties of alpha-lactalbumin. FEBS J. 2005;272:2176–2188. doi: 10.1111/j.1742-4658.2005.04638.x. [DOI] [PubMed] [Google Scholar]
- 46.Uversky VN. Neurotoxicant-induced animal models of Parkin-son’s disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res. 2004;318:225–241. doi: 10.1007/s00441-004-0937-z. [DOI] [PubMed] [Google Scholar]
- 47.Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinefelter G, Cookson MR, Greenamyre JT. Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis. 2006;22:404–420. doi: 10.1016/j.nbd.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, Wang X. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. 2006;20:362–364. doi: 10.1096/fj.05-4869fje. [DOI] [PubMed] [Google Scholar]
- 49.Lyubchenko YL, Sherman S, Shlyakhtenko LS, Uversky VN. Nanoimaging for protein misfolding and related diseases. J Cell Biochem. 2006 doi: 10.1002/jcb.20989. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 51.Divry P, Florkin M. Sur les proprietes optiques de l′amyloide. C R Soc Biol. 1927;97:1808–1810. [Google Scholar]
- 52.Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. 1959;183:1202–1203. doi: 10.1038/1831202a0. [DOI] [PubMed] [Google Scholar]
- 53.Shirahama T, Cohen AS. High-resolution electron microscopic analysis of the amyloid fibril. J Cell Biol. 1967;33:679–708. doi: 10.1083/jcb.33.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shirahama T, Cohen AS. Reconstitution of amyloid fibrils from alkaline extracts. J Cell Biol. 1967;35:459–464. doi: 10.1083/jcb.35.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shirahama T, Cohen AS. A brief review of the ultrastructure of amyloid. In: Marrink J, Van Rijswijk MH, editors. Amyloidosis. Martinus Nijhoff: Dordrecht; The Netherlands: 1986. pp. 51–57. [Google Scholar]
- 56.DeRosier DJ, Klug A. Reconstruction of three-dimensional structures from electron micrographs. Nature. 1968;217:130–134. doi: 10.1038/217130a0. [DOI] [PubMed] [Google Scholar]
- 57.Stewart M. Computer image processing of electron micrographs of biological structures with helical symmetry. J Electron Microsc Tech. 1988;9:325–358. doi: 10.1002/jemt.1060090404. [DOI] [PubMed] [Google Scholar]
- 58.Saibil HR. Macromolecular structure determination by cryo-electron microscopy. Acta Crystallogr, D: Biol Crystallogr. 2000;56:1215–1222. doi: 10.1107/s0907444900010787. [DOI] [PubMed] [Google Scholar]
- 59.Serpell LC, Sunde M, Benson MD, Tennent GA, Pepys MB, Fraser PE. The protofilament substructure of amyloid fibrils. J Mol Biol. 2000;300:1033–1039. doi: 10.1006/jmbi.2000.3908. [DOI] [PubMed] [Google Scholar]
- 60.Jimenez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khurana R, Ionescu-Zanetti C, Pope M, Li J, Nielson L, Ramirez-Alvarado M, Regan L, Fink AL, Carter SA. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys J. 2003;85:1135–1144. doi: 10.1016/S0006-3495(03)74550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonar L, Cohen AS, Skinner MM. Characterization of the amyloid fibril as a cross-beta protein. Proc Soc Exp Biol Med. 1969;131:1373–1375. doi: 10.3181/00379727-131-34110. [DOI] [PubMed] [Google Scholar]
- 63.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci USA. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid-state NMR reveals a pH-dependent antiparallel beta-sheet registry in fibrils formed by a beta-amyloid peptide. J Mol Biol. 2004;335:247–260. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 65.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid-state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Amyloid fibril formation by A beta 16–22, a seven-residue fragment of the Alzheimer’s beta-amyloid peptide, and structural characterization by solid-state NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 67.Chan JC, Oyler NA, Yau WM, Tycko R. Parallel beta-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 2005;44:10669–10680. doi: 10.1021/bi050724t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nelson R, Eisenberg D. Recent atomic models of amyloid fibril structure. Curr Opin Struct Biol. 2006;16:260–265. doi: 10.1016/j.sbi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 69.Makin OS, Serpell LC. Examining the structure of the mature amyloid fibril. Biochem Soc Trans. 2002;30:521–525. doi: 10.1042/bst0300521. [DOI] [PubMed] [Google Scholar]
- 70.Makin OS, Serpell LC. Structures for amyloid fibrils. FEBS J. 2005;272:5950–5961. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- 71.Makin OS, Serpell LC. X-ray diffraction studies of amyloid structure. Methods Mol Biol. 2005;299:67–80. doi: 10.1385/1-59259-874-9:067. [DOI] [PubMed] [Google Scholar]
- 72.Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC. Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci USA. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dobson CM. Experimental investigation of protein folding and misfolding. Methods. 2004;34:4–14. doi: 10.1016/j.ymeth.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 74.Dobson CM. Structural biology: prying into prions. Nature. 2005;435:747–749. doi: 10.1038/435747a. [DOI] [PubMed] [Google Scholar]
- 75.Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weiss S. Fluorescence spectroscopy of single biomolecules. Science. 1999;283:1676–1683. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- 77.Weiss S. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat Struct Biol. 2000;7:724–729. doi: 10.1038/78941. [DOI] [PubMed] [Google Scholar]
- 78.Deniz AA, Dahan M, Grunwell JR, Ha T, Faulhaber AE, Chemla DS, Weiss S, Schultz PG. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of Forster distance dependence and subpopulations. Proc Natl Acad Sci USA. 1999;96:3670–3675. doi: 10.1073/pnas.96.7.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S. Single-molecule protein folding: diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc Natl Acad Sci USA. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ha T, Ting AY, Liang J, Caldwell WB, Deniz AA, Chemla DS, Schultz PG, Weiss S. Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc Natl Acad Sci USA. 1999;96:893–898. doi: 10.1073/pnas.96.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tokunaga M, Kitamura K, Saito K, Iwane AH, Yanagida T. Single molecule imaging of fluorophores and enzymatic reactions achieved by objective-type total internal reflection fluorescence microscopy. Biochem Biophys Res Commun. 1997;235:47–53. doi: 10.1006/bbrc.1997.6732. [DOI] [PubMed] [Google Scholar]
- 82.Smith DE, Babcock HP, Chu S. Single-polymer dynamics in steady shear flow. Science. 1999;283:1724–1727. doi: 10.1126/science.283.5408.1724. [DOI] [PubMed] [Google Scholar]
- 83.Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, Herschlag D, Chu S. A single-molecule study of RNA catalysis and folding. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 84.Rhoades E, Cohen M, Schuler B, Haran G. Two-state folding observed in individual protein molecules. J Am Chem Soc. 2004;126:14686–14687. doi: 10.1021/ja046209k. [DOI] [PubMed] [Google Scholar]
- 85.Tcherkasskaya O, Ptitsyn OB. Direct energy transfer to study the 3D structure of non-native proteins: AGH complex in molten globule state of apomyoglobin. Protein Eng. 1999;12:485–490. doi: 10.1093/protein/12.6.485. [DOI] [PubMed] [Google Scholar]
- 86.Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum Press; New York: 1983. [Google Scholar]
- 87.Cheung HC. Resonance energy transfer. In: Lakowicz JR, editor. Topics in Fluorescence Spectroscopy. Plenum Press; New York: 1991. pp. 127–176. [Google Scholar]
- 88.Lundblad RL. Chemical Reagents for Protein Modification. CRC Press; Boca Raton, Florida: 1991. [Google Scholar]
- 89.Borkman RF, Phillips SR. Tyrosine-to-tryptophan energy transfer and the structure of calf gamma-II Crystallin. Exp Eye Res. 1985;40:819–826. doi: 10.1016/0014-4835(85)90127-7. [DOI] [PubMed] [Google Scholar]
- 90.Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 91.Rischel C, Thyberg P, Rigler F, Poulsen FM. Time-resolved fluorescence studies of the molten globule state of apomyoglobin. J Mol Biol. 1996;257:877–885. doi: 10.1006/jmbi.1996.0208. [DOI] [PubMed] [Google Scholar]
- 92.Uversky VN, Fink AL. Do protein molecules have a native-like topology in the pre-molten globule state. Biochemistry (Moscow, Russ Ed) 1999;64:552–555. [PubMed] [Google Scholar]
- 93.Tcherkasskaya O, Uversky VN. Denatured collapsed states in protein folding: example of apomyoglobin. Proteins. 2001;44:244–254. doi: 10.1002/prot.1089. [DOI] [PubMed] [Google Scholar]
- 94.Lee JC, Langen R, Hummel PA, Gray HB, Winkler JR. Alpha-synuclein structures from fluorescence energy-transfer kinetics: implications for the role of the protein in Parkinson’s disease. Proc Natl Acad Sci USA. 2004;101:16466–16471. doi: 10.1073/pnas.0407307101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kaylor J, Bodner N, Edridge S, Yamin G, Hong DP, Fink AL. Characterization of oligomeric intermediates in alpha-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha-synuclein. J Mol Biol. 2005;353:357–372. doi: 10.1016/j.jmb.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 96.De Filippis V, Frasson R, Fontana A. 3-Nitrotyrosine as a spectroscopic probe for investigating protein protein interactions. Protein Sci. 2006;15:976–986. doi: 10.1110/ps.051957006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berney C, Danuser G. FRET or no FRET: a quantitative comparison. Biophys J. 2003;84:3992–4010. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lantsman K, Tombes RM. CaMK-II oligomerization potential determined using CFP/YFP FRET. Biochim Biophys Acta. 2005;1746:45–54. doi: 10.1016/j.bbamcr.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 100.Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 101.Tombes RM, Faison MO, Turbeville JM. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene. 2003;322:17–31. doi: 10.1016/j.gene.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 102.Kanaseki T, Ikeuchi Y, Sugiura H, Yamauchi T. Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy. J Cell Biol. 1991;115:1049–1060. doi: 10.1083/jcb.115.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kolodziej SJ, Hudmon A, Waxham MN, Stoops JK. Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. J Biol Chem. 2000;275:14354–14359. doi: 10.1074/jbc.275.19.14354. [DOI] [PubMed] [Google Scholar]
- 104.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tet-radecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–1251. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 105.Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN. Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:12484–12494. doi: 10.1074/jbc.M313597200. [DOI] [PubMed] [Google Scholar]
- 106.Hohng S, Joo C, Ha T. Single-molecule three-color FRET. Biophys J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Clamme JP, Deniz AA. Three-color single-molecule fluorescence resonance energy transfer. ChemPhysChem. 2005;6:74–77. doi: 10.1002/cphc.200400261. [DOI] [PubMed] [Google Scholar]
- 108.Brennan JD. Using intrinsic fluorescence to investigate proteins entrapped in sol–gel derived materials. Appl Spectrosc. 1999;53:106A–121A. [Google Scholar]
- 109.Gill I, Ballesteros A. Bioencapsulation within synthetic polymers (Part 1): sol–gel encapsulated biologicals. Trends Biotechnol. 2000;18:282–296. doi: 10.1016/s0167-7799(00)01457-8. [DOI] [PubMed] [Google Scholar]
- 110.Gill I, Ballesteros A. Bioencapsulation within synthetic polymers (Part 2): non-sol–gel protein-polymer biocomposites. Trends Biotechnol. 2000;18:469–479. doi: 10.1016/s0167-7799(00)01493-1. [DOI] [PubMed] [Google Scholar]
- 111.Eggers DK, Valentine JS. Crowding and hydration effects on protein conformation: a study with sol–gel encapsulated proteins. J Mol Biol. 2001;314:911–922. doi: 10.1006/jmbi.2001.5166. [DOI] [PubMed] [Google Scholar]
- 112.Eggers DK, Valentine JS. Molecular confinement influences protein structure and enhances thermal protein stability. Protein Sci. 2001;10:250–261. doi: 10.1110/ps.36201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dave BC, Dunn B, Valentine JS, Zink JI. Sol–gel encapsulation methods for biosensors. Anal Chem. 1994;66:1120A–1127A. [Google Scholar]
- 114.Shen C, Kostic NM. Kinetics of photoinduced electron-transfer reactions within sol–gel silica glass doped with zinc cytochrome c. Study of electrostatic effects in confined liquids. J Am Chem Soc. 1997;119(9):1304–1312. [Google Scholar]
- 115.Badjic J, Kostic NM. Effects of encapsulation in sol–gel silica glass on esterase activity, conformational stability, and unfolding of bovine carbonic anhydrase II. Chem Mater. 1999;11:3671–3679. [Google Scholar]
- 116.Lan EH, Dave BC, Fukuto JM, Dunn B, Zink JI, Valentine JS. Synthesis of sol–gel encapsulated heme proteins with chemical sensing properties. J Mater Chem. 1999;9:45–53. [Google Scholar]
- 117.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 118.McAllister C, Karymov MA, Kawano Y, Lushnikov AY, Mikheikin A, Uversky VN, Lyubchenko YL. Protein interactions and misfolding analyzed by AFM force spectroscopy. J Mol Biol. 2005;354:1028–1042. doi: 10.1016/j.jmb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 119.Kransnoslobodtsev AV, Shlyakhtenko LS, Ukraintsev E, Zaikova TO, Keana JF, Lyubchenko YL. Nanomedicine and protein misfolding diseases. Nanomedicine. 2005;1:300–305. doi: 10.1016/j.nano.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ban T, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J Biol Chem. 2003;278:16462–16465. doi: 10.1074/jbc.C300049200. [DOI] [PubMed] [Google Scholar]
- 121.Ban T, Hoshino M, Takahashi S, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of A beta amyloid fibril growth and inhibition. J Mol Biol. 2004;344:757–767. doi: 10.1016/j.jmb.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 122.Kowalewski T, Holtzman DM. In situ atomic force microscopy study of Alzheimer’s beta-amyloid peptide on different substrates: new insights into mechanism of beta-sheet formation. Proc Natl Acad Sci USA. 1999;96:3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hoyer W, Cherny D, Subramaniam V, Jovin TM. Rapid self-assembly of alpha-synuclein observed by in situ atomic force microscopy. J Mol Biol. 2004;340:127–139. doi: 10.1016/j.jmb.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 124.Smith LJ. Computational methods for generating models of denatured and partially folded proteins. Methods. 2004;34:144–150. doi: 10.1016/j.ymeth.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 125.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog iSaccharomyces cerevisiae . Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 126.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 127.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 128.Serio TR, Lindquist SL. Protein-only inheritance in yeast: something to get [PSI+]-ched about. Trends Cell Biol. 2000;10:98–105. doi: 10.1016/s0962-8924(99)01711-0. [DOI] [PubMed] [Google Scholar]
- 129.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 130.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liu R, McAllister C, Lyubchenko Y, Sierks MR. Residues 17–20 and 30–35 of beta-amyloid play critical roles in aggregation. J Neurosci Res. 2004;75:162–171. doi: 10.1002/jnr.10859. [DOI] [PubMed] [Google Scholar]
- 132.Dawson TM, Dawson VL. Molecular pathways of neurode-generation in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 133.Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P. Alpha-synuclein and Parkinson’s disease. FASEB J. 2004;18:617–626. doi: 10.1096/fj.03-0338rev. [DOI] [PubMed] [Google Scholar]
- 134.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 135.Zappacosta F, Pessi A, Bianchi E, Venturini S, Sollazzo M, Tramontano A, Marino G, Pucci P. Probing the tertiary structure of proteins by limited proteolysis and mass spectrometry: the case of Minibody. Protein Sci. 1996;5:802–813. doi: 10.1002/pro.5560050502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hubbard SJ, Eisenmenger F, Thornton JM. Modeling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci. 1994;3:757–768. doi: 10.1002/pro.5560030505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hubbard SJ, Beynon RJ, Thornton JM. Assessment of conformational parameters as predictors of limited proteolytic sites in native protein structures. Protein Eng. 1998;11:349–359. doi: 10.1093/protein/11.5.349. [DOI] [PubMed] [Google Scholar]
- 138.Laskowski M, Jr, Kato I. Protein inhibitors of proteinases. Annu Rev Biochem. 1980;49:593–626. doi: 10.1146/annurev.bi.49.070180.003113. [DOI] [PubMed] [Google Scholar]
- 139.Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur J Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- 140.Hubbard SJ, Campbell SF, Thornton JM. Molecular recognition. Conformational analysis of limited proteolytic sites and serine proteinase protein inhibitors. J Mol Biol. 1991;220:507–530. doi: 10.1016/0022-2836(91)90027-4. [DOI] [PubMed] [Google Scholar]
- 141.Fontana A, Fassina G, Vita C, Dalzoppo D, Zamai M, Zambonin M. Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry. 1986;25:1847–1851. doi: 10.1021/bi00356a001. [DOI] [PubMed] [Google Scholar]
- 142.Novotny J, Bruccoleri RE. Correlation among sites of limited proteolysis, enzyme accessibility and segmental mobility. FEBS Lett. 1987;211:185–189. doi: 10.1016/0014-5793(87)81433-3. [DOI] [PubMed] [Google Scholar]
- 143.Fontana A, Zambonin M, Polverino de Laureto P, De Filippis V, Clementi A, Scaramella E. Probing the conformational state of apomyoglobin by limited proteolysis. J Mol Biol. 1997;266:223–230. doi: 10.1006/jmbi.1996.0787. [DOI] [PubMed] [Google Scholar]
- 144.Fontana A, Polverino de Laureto P, De Filippis V, Scaramella E, Zambonin M. Probing the partly folded states of proteins by limited proteolysis. Folding Des. 1997;2:R17–26. doi: 10.1016/S1359-0278(97)00010-2. [DOI] [PubMed] [Google Scholar]
- 145.Myers SL, Thomson NH, Radford SE, Ashcroft AE. Investigating the structural properties of amyloid-like fibrils formed in vitro from beta2-microglobulin using limited proteolysis and electrospray ionisation mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:1628–1636. doi: 10.1002/rcm.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Monti M, Amoresano A, Giorgetti S, Bellotti V, Pucci P. Limited proteolysis in the investigation of beta2-microglobulin amyloidogenic and fibrillar states. Biochim Biophys Acta. 2005;1753:44–50. doi: 10.1016/j.bbapap.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 147.Polverino de Laureto P, Taddei N, Frare E, Capanni C, Costantini S, Zurdo J, Chiti F, Dobson CM, Fontana A. Protein aggregation and amyloid fibril formation by an SH3 domain probed by limited proteolysis. J Mol Biol. 2003;334:129–141. doi: 10.1016/j.jmb.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 148.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structural features of the Abeta amyloid fibril elucidated by limited proteolysis. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 149.Kheterpal I, Zhou S, Cook KD, Wetzel R. Abeta amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc Natl Acad Sci USA. 2000;97:13597–13601. doi: 10.1073/pnas.250288897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kheterpal I, Lashuel HA, Hartley DM, Walz T, Lansbury PT, Jr, Wetzel R. Abeta protofibrils possess a stable core structure resistant to hydrogen exchange. Biochemistry. 2003;42:14092–14098. doi: 10.1021/bi0357816. [DOI] [PubMed] [Google Scholar]
- 151.Kheterpal I, Wetzel R, Cook KD. Enhanced correction methods for hydrogen exchange-mass spectrometric studies of amyloid fibrils. Protein Sci. 2003;12:635–643. doi: 10.1110/ps.0225703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 153.Kheterpal I, Chen M, Cook KD, Wetzel R. Structural differences in Abeta amyloid protofibrils and fibrils mapped by hydrogen exchange–mass spectrometry with on-line proteolytic fragmentation. J Mol Biol. 2006;361:785–795. doi: 10.1016/j.jmb.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 154.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 155.O’Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci USA. 2002;99:1485–1490. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Glabe CG. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci. 2004;29:542–547. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 157.Emadi S, Liu R, Yuan B, Schulz P, McAllister C, Lyubchenko Y, Messer A, Sierks MR. Inhibiting aggregation of alpha-synuclein with human single chain antibody fragments. Biochemistry. 2004;43:2871–2878. doi: 10.1021/bi036281f. [DOI] [PubMed] [Google Scholar]
- 158.Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 159.Dumoulin M, Last AM, Desmyter A, Decanniere K, Canet D, Larsson G, Spencer A, Archer DB, Sasse J, Muyldermans S, Wyns L, Redfield C, Matagne A, Robinson CV, Dobson CM. A camelid antibody fragment inhibits the formation of amyloid fibrils by human lysozyme. Nature. 2003;424:783–788. doi: 10.1038/nature01870. [DOI] [PubMed] [Google Scholar]
- 160.Zhou C, Emadi S, Sierks MR, Messer A. A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed alpha-synuclein. Mol Ther. 2004;10:1023–1031. doi: 10.1016/j.ymthe.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 161.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 162.Tewari D, Notkins AL, Zhou P. Inhibition of HIV-1 replication in primary human T cells transduced with an intracellular anti-HIV-1 p17 antibody gene. J Gene Med. 2003;5:182–189. doi: 10.1002/jgm.336. [DOI] [PubMed] [Google Scholar]
- 163.Marasco WA, LaVecchio J, Winkler A. Human anti-HIV-1 tat sFv intrabodies for gene therapy of advanced HIV-1-infection and AIDS. J Immunol Methods. 1999;231:223–238. doi: 10.1016/s0022-1759(99)00159-3. [DOI] [PubMed] [Google Scholar]
- 164.Wheeler YY, Kute TE, Willingham MC, Chen SY, Sane DC. Intrabody-based strategies for inhibition of vascular en-dothelial growth factor receptor-2: effects on apoptosis, cell growth, and angiogenesis. FASEB J. 2003;17:1733–1735. doi: 10.1096/fj.02-0942fje. [DOI] [PubMed] [Google Scholar]
- 165.Wheeler YY, Chen SY, Sane DC. Intrabody and intrakine strategies for molecular therapy. Mol Ther. 2003;8:355–366. doi: 10.1016/s1525-0016(03)00183-7. [DOI] [PubMed] [Google Scholar]
- 166.Beyer WE, Palache AM, Luchters G, Nauta J, Osterhaus AD. Seroprotection rate, mean fold increase, seroconversion rate: which parameter adequately expresses seroresponse to influenza vaccination? Virus Res. 2004;103:125–132. doi: 10.1016/j.virusres.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 167.Wood SJ, Maleeff B, Hart T, Wetzel R. Physical, morphological and functional differences between ph 5.8 and 7.4 aggregates of the Alzheimer’s amyloid peptide Abeta. J Mol Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- 168.Zurdo J, Guijarro JI, Jimenez JL, Saibil HR, Dobson CM. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J Mol Biol. 2001;311:325–340. doi: 10.1006/jmbi.2001.4858. [DOI] [PubMed] [Google Scholar]
- 169.Clarke AR, Jackson GS, Collinge J. The molecular biology of prion propagation. Philos Trans R Soc London, Ser B: Biol Sci. 2001;356:185–195. doi: 10.1098/rstb.2000.0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hill AF, Collinge J. Strain variations and species barriers. Contrib Microbiol. 2001;7:48–57. doi: 10.1159/000060375. [DOI] [PubMed] [Google Scholar]
- 171.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 172.Jackson GS, Collinge J. The molecular pathology of CJD: old and new variants. Mol Pathol. 2001;54:393–399. [PMC free article] [PubMed] [Google Scholar]
- 173.Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem. 2004;73:617–656. doi: 10.1146/annurev.biochem.72.121801.161837. [DOI] [PubMed] [Google Scholar]
- 174.Johnson RT. Prion diseases. Lancet Neurol. 2005;4:635–642. doi: 10.1016/S1474-4422(05)70192-7. [DOI] [PubMed] [Google Scholar]
- 175.Latouche M, Fragner P, Martin E, El Hachimi KH, Zander C, Sittler A, Ruberg M, Brice A, Stevanin G. Polyglutamine and polyalanine expansions in ataxin7 result in different types of aggregation and levels of toxicity. Mol Cell Neurosci. 2006;31:438–445. doi: 10.1016/j.mcn.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 176.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiologi-cal changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bahadi R, Farrelly PV, Kenna BL, Kourie JI, Tagliavini F, Forloni G, Salmona M. Channels formed with a mutant prion protein PrP(82–146) homologous to a 7-kDa fragment in diseased brain of GSS patients. Am J Physiol: Cell Physiol. 2003;285:C862–872. doi: 10.1152/ajpcell.00077.2003. [DOI] [PubMed] [Google Scholar]
- 178.Micelli S, Meleleo D, Picciarelli V, Gallucci E. Effect of sterols on beta-amyloid peptide (AbetaP 1–40) channel formation and their properties in planar lipid membranes. Biophys J. 2004;86:2231–2237. doi: 10.1016/S0006-3495(04)74281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. Interactions among alpha-synuclein, dopamine, and biomem-branes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23:23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- 180.Lin H, Bhatia R, Lal R. Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 181.Green JD, Kreplak L, Goldsbury C, Li Blatter X, Stolz M, Cooper GS, Seelig A, Kistler J, Aebi U. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. J Mol Biol. 2004;342:877–887. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 182.Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627–638. doi: 10.7326/0003-4819-140-8-200404200-00047. [DOI] [PubMed] [Google Scholar]
- 183.Bhatia R, Lin H, Lal R. Fresh and globular amyloid beta protein (1–42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. FASEB J. 2000;14:1233–1243. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- 184.Ding TT, Lee SJ, Rochet JC, Lansbury PT., Jr Annular alpha-synuclein protofibrils are produced when spherical protofi-brils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 185.Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 186.Noguchi H, Matsumoto S. Protein transduction technology: a novel therapeutic perspective. Acta Med Okayama. 2006;60:1–11. doi: 10.18926/AMO/30757. [DOI] [PubMed] [Google Scholar]
- 187.Varga CM, Wickham TJ, Lauffenburger DA. Receptor-mediated targeting of gene delivery vectors: insights from molecular mechanisms for improved vehicle design. Biotechnol Bioeng. 2000;70:593–605. doi: 10.1002/1097-0290(20001220)70:6<593::aid-bit1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 188.Winterhalter M, Lasic DD. Liposome stability and formation: experimental parameters and theories on the size distribution. Chem Phys Lipids. 1993;64:35–43. doi: 10.1016/0009-3084(93)90056-9. [DOI] [PubMed] [Google Scholar]
- 189.Lasic DD. Liposomes: From Physics to Applications. Elsevier Sci. Publ; Amsterdam: 1993. [Google Scholar]
- 190.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discovery. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 191.Khaw BA, Vural I, daSilva J, Torchilin VP. Use of cytosk-eleton-specific immunoliposomes for preservation of cell viability and gene delivery. STP Pharma Sci. 2000;10:279–283. [Google Scholar]
- 192.Torchilin VP, Weissig V. Polymeric micelles for the delivery of poorly soluble drugs. In: Park K, Mrsny RJ, editors. Controlled Drug Delivery: Designing Technologies for the Future. American Chemical Society; Washington, DC: 2000. pp. 297–313. [Google Scholar]
- 193.Elouahabi A, Ruysschaert JM. Formation and intracellular trafficking of lipoplexes and polyplexes. Mol Ther. 2005;11:336–347. doi: 10.1016/j.ymthe.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 194.Kerkis A, Hayashi MA, Yamane T, Kerkis I. Properties of cell penetrating peptides (CPPs) IUBMB Life. 2006;58:7–13. doi: 10.1080/15216540500494508. [DOI] [PubMed] [Google Scholar]
- 195.Brinton RD. A women’s health issue: Alzheimer’s disease and strategies for maintaining cognitive health. Int J Fertil Women’s Med. 1999;44:174–185. [PubMed] [Google Scholar]
- 196.Gozes I. Neuroprotective peptide drug delivery and development: potential new therapeutics. Trends Neurosci. 2001;24:700–705. doi: 10.1016/s0166-2236(00)01931-7. [DOI] [PubMed] [Google Scholar]
- 197.Gozes I, Brenneman DE, Geppetti P, Kastin AJ, Mains RE, Moody TW, Seroogy K, Spier AD, Zimmermann M. Neuropeptides: brain messengers of many faces. Trends Neurosci. 2001;24:687–690. doi: 10.1016/s0166-2236(00)02001-4. [DOI] [PubMed] [Google Scholar]
- 198.Kroll RA, Pagel MA, Muldoon LL, Roman-Goldstein S, Fiamengo SA, Neuwelt EA. Improving drug delivery to intracerebral tumor and surrounding brain in a rodent model: a comparison of osmotic versus bradykinin modification of the blood-brain and/or blood-tumor barriers. Neurosurgery. 1998;43:879–886. doi: 10.1097/00006123-199810000-00090. discussion 886–879. [DOI] [PubMed] [Google Scholar]
- 199.Downs-Kelly E, Jones MZ, Alroy J, Cavanagh KT, King B, Lucas RE, Baker JC, Kraemer SA, Hopwood JJ. Caprine mucopolysaccharidosis IIID: a preliminary trial of enzyme replacement therapy. J Mol Neurosci. 2000;15:251–262. doi: 10.1385/JMN:15:3:251. [DOI] [PubMed] [Google Scholar]
- 200.Banks WA, Lebel CR. Strategies for the delivery of leptin to the CNS. J Drug Targeting. 2002;10:297–308. doi: 10.1080/10611860290031895. [DOI] [PubMed] [Google Scholar]
- 201.Banks WA. Is obesity a disease of the blood-brain barrier? Physiological, pathological, and evolutionary considerations. Curr Pharm Des. 2003;9:801–809. doi: 10.2174/1381612033455350. [DOI] [PubMed] [Google Scholar]
- 202.Kabanov AV, Batrakova EV. New technologies for drug delivery across the blood brain barrier. Curr Pharm Des. 2004;10:1355–1363. doi: 10.2174/1381612043384826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Batrakova EV, Vinogradov SV, Robinson SM, Niehoff ML, Banks WA, Kabanov AV. Polypeptide point modifications with fatty acid and amphiphilic block copolymers for enhanced brain delivery. Bioconjugate Chem. 2005;16:793–802. doi: 10.1021/bc049730c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kabanov AV, Alakhov VY. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev Ther Drug Carrier Syst. 2002;19:1–72. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.10. [DOI] [PubMed] [Google Scholar]
- 205.Kabanov AV, Batrakova EV, Melik-Nubarov NS, Fedoseev NA, Dorodnich TY, Alakhov VY, Chekhonin VP, Nazarova IR, Kabanov VA. A new class of drug carriers: micelles of poly-(oxyethylene)-poly(oxypropylene) block copolymers as micro-containers for drug targeting from blood in brain. J Controlled Release. 1992;22:141–158. [Google Scholar]
- 206.Omori N, Maruyama K, Jin G, Li F, Wang SJ, Hamakawa Y, Sato K, Nagano I, Shoji M, Abe K. Targeting of post-ischemic cerebral endothelium in rat by liposomes bearing polyethylene glycol-coupled transferrin. Neurol Res. 2003;25:275–279. doi: 10.1179/016164103101201508. [DOI] [PubMed] [Google Scholar]
- 207.Shi N, Zhang Y, Zhu C, Boado RJ, Pardridge WM. Brain-specific expression of an exogenous gene after i.v. administration. Proc Natl Acad Sci USA. 2001;98:12754–12759. doi: 10.1073/pnas.221450098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci USA. 1996;93:14164–14169. doi: 10.1073/pnas.93.24.14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Huwyler J, Cerletti A, Fricker G, Eberle AN, Drewe J. Bypassing of P-glycoprotein using immunoliposomes. J Drug Targeting. 2002;10:73–79. doi: 10.1080/10611860290007559. [DOI] [PubMed] [Google Scholar]
- 210.Wu D, Song BW, Vinters HV, Pardridge WM. Pharmaco-kinetics and brain uptake of biotinylated basic fibroblast growth factor conjugated to a blood-brain barrier drug delivery system. J Drug Targeting. 2002;10:239–245. doi: 10.1080/10611860290022679. [DOI] [PubMed] [Google Scholar]
- 211.Wu D, Boado RJ, Pardridge WM. Pharmacokinetics and blood-brain barrier transport of [3H]-biotinylated phospho-rothioate oligodeoxynucleotide conjugated to a vector-mediated drug delivery system. J Pharmacol Exp Ther. 1996;276:206–211. [PubMed] [Google Scholar]
- 212.Gosk S, Vermehren C, Storm G, Moos T. Targeting anti-transferrin receptor antibody (OX26) and OX26-conjugated lip-osomes to brain capillary endothelial cells using in situ perfusion. J Cereb Blood Flow Metab. 2004;24:1193–1204. doi: 10.1097/01.WCB.0000135592.28823.47. [DOI] [PubMed] [Google Scholar]
- 213.Vinogradov SV, Batrakova EV, Kabanov AV. Nanogels for oligonucleotide delivery to the brain. Bioconjugate Chem. 2004;15:50–60. doi: 10.1021/bc034164r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nguyen HK, Lemieux P, Vinogradov SV, Gebhart CL, Guerin N, Paradis G, Bronich TK, Alakhov VY, Kabanov AV. Evaluation of polyether-polyethyleneimine graft copolymers as gene transfer agents. Gene Ther. 2000;7:126–138. doi: 10.1038/sj.gt.3301052. [DOI] [PubMed] [Google Scholar]
- 215.Jaturanpinyo M, Harada A, Yuan X, Kataoka K. Preparation of bionanoreactor based on core–shell structured polyion complex micelles entrapping trypsin in the core cross-linked with glutaraldehyde. Bioconjugate Chem. 2004;15:344–348. doi: 10.1021/bc034149m. [DOI] [PubMed] [Google Scholar]
- 216.Harada A, Kataoka K. Novel polyion complex micelles entrapping enzyme molecules in the core. 2. Characterization of the micelles prepared at nonstoichiometric mixing ratios. Langmuir. 1999;15:4208–4212. [Google Scholar]
- 217.Roy S, Zhang K, Roth T, Vinogradov S, Kao RS, Kabanov A. Reduction of fibronectin expression by intravitreal administration of antisense oligonucleotides. Nat Biotechnol. 1999;17:476–479. doi: 10.1038/8654. [DOI] [PubMed] [Google Scholar]
- 218.Harada-Shiba M, Yamauchi K, Harada A, Takamisawa I, Shimokado K, Kataoka K. Polyion complex micelles as vectors in gene therapy–pharmacokinetics and in vivo gene transfer. Gene Ther. 2002;9:407–414. doi: 10.1038/sj.gt.3301665. [DOI] [PubMed] [Google Scholar]
- 219.Junghans M, Loitsch SM, Steiniger SC, Kreuter J, Zimmer A. Cationic lipid-protamine-DNA (LPD) complexes for delivery of antisense c-myc oligonucleotides. Eur J Pharm Biopharm. 2005;60:287–294. doi: 10.1016/j.ejpb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 220.Cohen-Krausz S, Saibil HR. Three-dimensional structural analysis of amyloid fibrils by electron microscopy. In: Uversky VN, Fink AL, editors. Protein Misfolding, Aggregation and Conformational Diseases. Part A: Protein Aggregation and Conformational Diseases. Springer; New York: 2006. In press. [Google Scholar]