

Abstract
The binding kinetics between cell surface receptors and extracellular biomolecules is critical to all intracellular and intercellular activity. Modeling and prediction of receptor-mediated cell functions are facilitated by measurement of the binding properties on whole cells, ideally indicating the subcellular locations or cytoskeletal associations that may affect the function of bound receptors. This dual need is particularly acute vis à vis ligand engineering and clinical applications of antibodies to neutralize pathological processes. Here, we map individual receptors and determine whole-cell binding kinetics by means of functionalized force imaging, enabled by scanning probe microscopy and molecular force spectroscopy of intact cells with biomolecule-conjugated mechanical probes. We quantify the number, distribution, and association/dissociation rate constants of vascular endothelial growth factor receptor-2 with respect to a monoclonal antibody on both living and fixed human microvascular endothelial cells. This general approach to direct receptor imaging simultaneously quantifies both the binding kinetics and the nonuniform distribution of these receptors with respect to the underlying cytoskeleton, providing spatiotemporal visualization of cell surface dynamics that regulate receptor-mediated behavior.
Keywords: cell surface, mechanical imaging, vascular endothelial growth factor receptor
Molecular receptors at the living cell surface drive critical cell behaviors ranging from adhesion to differentiation, primarily by means of structural/functional changes induced by binding to extracellular molecules or ligands. Both the receptor location and the kinetics of ligand binding are important to the understanding of receptor-driven functions within cells, but few experimental approaches provide simultaneous access to spatial, temporal, and intermolecular force dynamics in individual, whole cells (1). Such quantification is crucial to understanding how cells within or among subpopulations may respond differentially to the same ligand [e.g., drug responsivity (2) and differentiation (3)] and how ligand binding can depend on clustering of multiple molecules [e.g., synapse formation (4)] or cytoskeletal association [e.g., focal adhesion formation (5)]. Several impressive experimental approaches including flow cytometry, immunocytochemical staining, Förster resonance energy transfer (FRET) and fluorescence recovery after photobleaching (FRAP) are based on optical signals that require either fluorophore-labeling or genetic modification of cell surface proteins (1). Binding affinity and kinetics among ligands and cell surface receptors are typically extracted from time course monitoring of total radio- or fluorophore-labeled ligand levels in the presence of unlabeled ligand counterparts, and thus the spatial distribution of active receptors during such competitive ligand binding is not accessed. Measurement of intermolecular interaction forces and associated binding kinetics of several antibody–antigen and ligand–receptor pairs has been demonstrated through atomic force microscopy (AFM) on purified proteins adhered to flat, rigid surfaces (6–11) and through discretized “blind” mapping of adhesion forces between the ligand-coated AFM probe and the cell surface (12–18). However, leveraging such piconewton-scale molecular interaction forces to image individual receptors and infer ligand-binding kinetics on intact, topographically rough cells has remained challenging.
In addition to the fundamental understanding of cell signaling enabled by direct imaging and kinetic analysis, ligand-binding affinity as quantified by the equilibrium dissociation constant KD is pertinent to clinical therapies that regulate signal transduction through direct receptor binding. Vascular endothelial growth factor receptors (VEGFRs), transmembrane receptor tyrosine kinases primarily expressed by vascular endothelial cells (VECs) (19), are key targets because of the apparent role of these receptors in mechanosensory functions such as focal adhesion turnover, actin cytoskeletal remodeling, and angiogenesis (20–23). Intermittent blocking of VEGFR2 in VE cells promotes rapid blood vessel regression in animal models (3), but new strategies to inhibit/promote this signaling require enhanced understanding of VEGFR2 distribution and binding kinetics with biological or synthetic molecules. Here we develop and apply an approach through which receptor location and binding kinetics to extracellular biomolecules are achieved at the single-molecule and single-cell levels, respectively. Through this chemomechanical imaging on living and fixed VECs, we find that available VEGFR2s are nonuniformly distributed, in close spatial association with the underlying cortical cytoskeleton, and that equilibrium rate constants can be accessed on intact cells to correlate binding affinity with subcellular location.
Results
Determination of Receptor Location and Binding Specificity.
Direct mechanical imaging of cell surface receptor location can elucidate nonuniform distributions of receptors with respect to other structural features and provides access to whole-cell binding kinetics. By maintaining constant or intermittent contact with a cantilevered probe while scanning the cell surface, AFM feedback voltages create image contrast through differential height or stiffness (Fig. 1A) or through reversible adhesion between molecules tethered to the scanning probe and molecules bound to rigid, flat surfaces (11, 24). By displacing the probe normal to the surface at discrete points and analyzing the force-displacement responses, interaction force spectra also can be measured on rigid surfaces and on chemically fixed or living cells to construct two-dimensional (2D) grids of pixels indicating either stiff/compliant or strong/weak binding regions (e.g., refs. 25–27). Although such molecular interactions are far from equilibrium, the spectrum of piconewton-scale unbinding or rupture forces between probe-bound ligands and adsorbed monolayers of purified receptors has been used to estimate the equilibrium dissociation rate koff between antigen/antibody and ligand/receptor pairs (28, 29) and has been reported to agree reasonably well with surface plasmon resonance measurements of population-averaged rates for rigidly bound, purified proteins (30). However, pointwise acquisition of such profiles on cell surfaces thus far has proven to be too spatially coarse [pixels of ≈500 nm size (23, 27)] or slow [e.g., 45 min to acquire 32 × 32 pixels of 20 nm size (12)] to resolve both the nanoscale location and binding kinetics of individual cell surface receptors (31).
Fig. 1.

Chemomechanical imaging of individual VEGFR2 receptors on fixed HUVEC surfaces. (A) Phase image of cell body and periphery. (B) Recognition image over indicated area in A shows strong binding events between the anti-VEGFR2 functionalized probe and the cell surface as discrete, dark spots (e.g., circled) that are ostensibly VEGFR2. (C and D) The capacity to block these binding events by addition of 5 μg/ml soluble anti-VEGFR2 supports this binding specificity between the probe and VEGFR2, as the number of recognition events decreases with time postblocking of 12 (C) and 60 (D) min. (F–I) Corresponding height images (F–H) indicate the position of four cytoskeletal bundles (shown only in H for clarity, as blue bands reconstructed from height traces such as I). These stiff, subsurface bundles can be correlated with receptor position and show that cell topography is stable over this time scale. Bundles are identified as F-actin through structural correlations between fluorescence optical images (FITC–phalloidin stains F-actin) and conventional AFM height images (E). (Scale bars: 10 μm, A and E; 500 nm, B–D and F–I.) Imaging in Hepes buffer at 27°C at scan rates of 10 μm/sec (A) or 1 μm/sec (B–D and F–I).
To both visualize and measure the binding kinetics of VEGFR2 receptors on VECs, we used chemomechanical imaging of chemically fixed and living human umbilical vein endothelial cells (HUVECs) that endogenously express VEGFR2. By scanning the cell surface with a magnetically driven oscillating, cantilevered probe to which monoclonal anti-VEGFR2 antibodies were tethered at a concentration of approximately one antibody per probe [see Materials and Methods and supporting information (SI) Fig. 6], retardation of full-amplitude oscillations indicative of piconewton-scale unbinding force between the probe and the cell surface creates image contrast (32) in the form of punctate, dark regions of diameters ranging 45.9 ± 8.9 nm (Figs. 1 A–D and 2B and C); see http://www.pnas.org/cgi/content/full/0702668104/DC1SI Text regarding image resolution. This molecular recognition imaging has been demonstrated for rigid surface-bound molecular pairs (33), so we refer to these regions of strong binding as recognition sites, which are assumed as putative receptor locations that can be confirmed through demonstration of binding specificity. We demonstrate specificity of this interaction through competitive binding, introduction of the soluble anti-VEGFR2 to the imaging solution; binding of these soluble antibodies to VEGFR2 on the cell surface should block specific interaction forces between the anti-VEGFR2 probe and the cell over time scales comparable with those used for immunocytochemical staining. Fig. 1 B–D shows this competitive inhibition over 60 min postblocking, as the number of observable binding sites in these images is diminished without concurrent degradation of the cell surface topography (Fig. 1 F–H). In contrast, the number of recognition sites did not decrease over the same imaging duration upon the addition of 5 μg/ml monoclonal anti-CD31, an antibody specific to these cells as confirmed by flow cytometry and immunocytochemistry (Fig. 2); and no recognition sites were observed in repeated, identical experiments on human NIH 3T3 fibroblasts that do not express VEGFR2, as confirmed by flow cytometry (SI Figs. 7 and 8). The specificity of this antibody for VEGFR2 in these cells is further supported by flow cytometry for HUVECs cultured under identical conditions (SI Fig. 8). Note that, in these oscillatory interaction imaging modes, the output voltage signals of Fig. 1 B–D are related to, but not convertible to, force or displacement in a straightforward manner.
Fig. 2.

Confirmation of anti-VEGFR2 binding specificity to VEGFR2 receptors on cell surfaces. (A and B) After imaging fixed HUVECs with anti-VEGFR2-functionalized probe [phase image (A) and recognition image in region of interest (B)], a soluble antibody against a different HUVEC receptor was added. (C) Thirty minutes after addition of anti-CD31 (5 μg/ml), no competitive blocking of recognition events was observed, indicating that recognition events are specific between the cell VEGFR2 and probe-bound anti-VEGFR2. (Inset) Corresponding height image indicates the position of cytoskeletal bundles beneath the cell membrane; as in Fig. 1, bundle edges (white lines) were constructed from height traces such as E. (D) Output voltage scale for B and C shows recognition signal compared with background in a line scan over a region including three binding events (receptors). B–E show that receptors are nonuniformly distributed near cytoskeletal bundles beneath the plasma membrane. (Scale bars: 10 μm, white; 500 nm, black.) Scan rates: 10 μm/sec, A; 1 μm/sec, B and C.
Analysis of fixed-cell images such as Figs. 1B and 2B indicate 1.47 ± 0.38 × 105 VEGFR2 per cell (n = 60; see Materials and Methods). This determination among individual cells agrees well with HUVEC population-averaged measurements through radiolabeled ligands [1.1 × 105 (34) and 1.5 × 105 VEGFR2 per cell (35)]. Additionally, this nanoscale imaging indicates the nonuniform receptor distribution over ≈2 μm2 regions and the close cytoskeletal association of these receptors (Figs. 1 B–I and 2 B–E).
Binding Kinetics Analysis.
Imaging by means of intermolecular forces also gives access to ligand-binding affinities on individual cells. To determine the dissociation rate koff between the probe-bound antibody and cell surface receptors, we acquired force–displacement spectra on imaged cell regions such as Fig. 1B. This enabled us to efficiently sample unbinding or rupture forces FR at recognition sites (ostensible receptor locations) before and after blocking with soluble antibody. As shown in Fig. 3A, each force–displacement retraction profile represents (on average) a single ligand–receptor unbinding event from which rupture force FR and unbinding width l (proportional to the characteristic unbinding time τ) are determined (8, 36). Fig. 3B shows the distribution of these FR, with maxima at 32.5 ± 2.5 and 64.1 ± 5.4 pN; these significantly exceed nonspecific unbinding forces measured at cell surface regions of low-recognition image contrast or after blocking (Fig. 3D; 12.5 ± 2.1 pN). Multiple force maxima indicate a nonzero probability of binding two receptors (homodimers) with a single antibody or, more likely, binding of two antibodies on the probe to a pair of closely spaced receptors (28). From these FR and τ values acquired on cells, the equilibrium dissociation rate koff can be determined directly through Bell's model (36, 37) (see Materials and Methods). For anti-VEGFR2/VEGFR2 on HUVECs, we found that koff = 1.05 ± 0.6 × 10−4 s−1.
Fig. 3.

Force spectroscopy analysis on fixed HUVECs to extract koff. (A) Representative force–displacement profile for specific ligand–receptor unbinding between the anti-VEGFR2 probe and imaged receptor recognition sites. (B) Distribution of >600 rupture forces measured at recognition sites, indicating maxima of 33 and 64 pN. (C and D) Representative profiles at >400 nonrecognition sites on the cell surface indicate a nonspecific rupture force level of 13 pN. Effective loading rate: 11.7 nN/sec. Bond lifetime τ in A is proportional to the binding displacement and is used to calculate binding kinetics (see Materials and Methods).
To determine the equilibrium association rate kon, we imaged cell surfaces during competitive inhibition with soluble anti-VEGFR2. The number of observable binding sites in images such as Fig. 1 B–D decreased with time as soluble antibodies bound to VEGFR2 on the cell surface and blocked probe–receptor binding. We analyzed this temporal increase in bound receptors according to a monovalent binding kinetic model (1) to obtain kon = 5.83 ± 1.48 × 104 s−1·M−1, corresponding to an equilibrium dissociation constant KD = koff/kon of 1.80 ± 0.87 × 10−9 M (n = 6).† Here, koff was assumed from force spectroscopy, rather than a best fit to the response in Fig. 4(see SI Text for comparison). Deviations from the model at early times postblocking are attributed in part to the model assumption of uniformly distributed ligand; in practice, diffusion of the ligand upon injection is required. These binding kinetics measured directly on intact cells represent the rate at which an ensemble of receptors on an individual cell surface is occupied and can plausibly differ from kinetics measured as cell population-averages or on purified proteins. However, we note that KD is well within the range of antibody–antigen interactions (KD = 10−4 to 10−12 M) measured by various approaches for purified antigens (38, 39), and the rates koff and kon compare well with those inferred from surface plasmon resonance for anti-VEGFR2/purified humanVEGFR2 (40) (see SI Text).
Fig. 4.

Receptor imaging during competitive inhibition. Receptor recognition sites in images such as Fig. 1B decrease with time after addition of soluble anti-VEGFR2 (5 μg/ml, 27°C). As the number of observable sites decreases during blocking, the number of receptors bound by soluble antibodies correspondingly increases (red filled circles). Kinetic constants kon and KD can be determined by application of a binding kinetic model for which koff is assumed from independent force spectroscopy experiments (blue line), or by a least-squares best fit to the experimental data (pink line). See SI Text for detailed calculation of binding kinetics.
Visualization of Receptors on Living Cell Surfaces.
Although biological receptor diffusivity and internalization are typically retarded in kinetic and structural analysis through processes such as chemical fixation (41–43), we note that this imaging through reversible intermolecular binding also provides direct access to receptor dynamics on living cell surfaces. Fig. 5 shows specific, punctate unbinding events between an anti-VEGFR2-functionalized probe and the living HUVEC surface: un/binding events are detectable as marked phase lag of cantilevered probe oscillation over the compliant, mechanically heterogeneous surface of the living cell.
Fig. 5.

Receptor imaging on living HUVEC surface. (A) Portion of living cell imaged with anti-VEGFR2-functionalized probe in magnetic AC mode at 27°C, phase image. (B and C) Ligand–receptor binding results in punctate image contrast (circled regions are subset of observed receptors) in phase lag images that is competitively inhibited with soluble anti-VEGFR2 antibody (data not shown). Time lapse between B and C is 30 min. Note mechanical contrast and displacement of underlying cytoskeletal actin (normal to arrow) over this time scale. These images indicate 1.32 ± 0.44 × 105 receptors per cell (n = 6). Scan rates: 10 μm/sec, A; 1 μm/sec, B and C. (Scale bars: 10 μm, A; 500 nm, B and C.)
In contrast to fixed-cell surface imaging, here the position and number of putative receptors vary over time in the absence of competitive binding. This variation ostensibly is due to lateral diffusion along and recycling through the membrane. The diffusion coefficients D measured by fluorescence recovery after photobleaching for other receptors over cell membrane areas of comparable size [0.001–0.1 μm2/sec (1)] are comparable with imaging scan rates (here ≈0.02 μm2/sec at 2 min per image). This means that a receptor could diffuse across the region of Fig. 5B over a period t ≈ 〈x2〉/4D ranging from 6 sec to 10 min; thus, receptors may not be observed in sequential images of the same region acquired minutes apart. However, in sequential images such as Fig. 5 B and C, we observed that receptors adjacent to cortical cytoskeletal filaments displaced only 178 ± 49 nm (n = 12) with respect to the moving cytoskeleton. This compartmentalized motion near filaments is consistent with the root mean squared displacement of other membrane proteins (30–700 nm) that has been attributed to cortical cytoskeletal confinement and steric hindrance (41, 43, 44).
Discussion
Through this functionalized force imaging approach, we found that individual VEGFR2 can be imaged on intact, fixed, and living cell surfaces with molecular resolution through reversible, intermolecular binding events (Fig. 1). We also showed that the specificity of these binding events can be demonstrated through competitive binding of soluble molecules (Figs. 1–4) and associated control experiments (SI Figs. 6–8). Because the number of receptors per cell compared well with that estimated from cell population-averaged approaches, we infer that the efficiency of this force-based imaging approach is sufficiently high to provide an accurate depiction of receptor location. Further, we found that the equilibrium binding kinetics could be measured on an individual cell basis through a combination of molecular force spectroscopy of putative receptors (koff) and real-time image acquisition during competitive binding (kon). Moreover, our approach provides the opportunity to correlate binding kinetics with structural features such as cytoskeletal association of the receptor.
From direct analysis of corresponding recognition and height images (e.g., Figs. 1 and 2), we can consider the distribution of individual VEGFR2 with respect to the center and apparent edge of subsurface cytoskeletal bundles; the staining and dimensions of these bundles are consistent with linear bundles of actin filaments termed stress fibers. The majority of recognition sites were located directly adjacent to these underlying cytoskeletal bundles: 61% were above the cytoskeletal bundles (i.e., “on” 2D projections of the bundle height traces); 34% were located within 72 ± 49 nm from apparent bundle edges and thus within the subsurface bundle width; and <5% were observed at distances of >500 nm from bundle edges. The observed VEGFR2 were uniformly distributed along the bundle length and width (bundle diameter-normalized distance of 0.53 ± 0.31 from the bundle center). This finding supports the current hypothesis that VEGFR2 function is intimately related to that of transmembrane integrin complexes that transmit force from the extracellular matrix to the actin cytoskeleton (19, 45, 46). These results also lay the groundwork for important and open questions, including whether this imaging approach and/or the binding kinetics are altered in mechanically stiff regions of cell surfaces; such work is ongoing. However, reasonable agreement of VEGFR2/cell with cell population-averaged levels (34, 35) suggests that total receptor number is not grossly underestimated.
We note that, although functionalized force imaging can identify the existence and distribution of receptors, full analysis of binding kinetics requires that the diffusion and recycling of receptors must be suppressed to maintain a constant receptor population, e.g., through light fixation of the membrane proteins. Despite the potential to alter binding kinetics through modification of membrane protein structure, such chemomechanical imaging provides the capacity to compare ligand-binding properties for a given receptor in the presence of drug ant/agonists or among cells within an inherently mixed population (e.g., tumors or differentiating progenitor cells). Further, chemical fixation is a well accepted approach to enable lower resolution visualization (optics-based imaging) of cell structure and gross spatial distributions of proteins. In fact, our claim of antibody–receptor binding specificity is supported by standard immunocytochemical staining practices: incubation of primary antibodies with fixed cells over the same duration as our competitive binding experiments (≈60 min) is considered sufficient to saturate receptors.
The dual access to chemically informed, subcellular structure and to ligand–receptor binding kinetics enabled by this imaging approach allows us to ask new questions about how colocalization of subcellular structures affects receptor function and physiological/pathological cell processes. In the present case, we observed that VEGFR2 is accessible to functionalized force imaging and that these imaged receptors are spatially associated with the underlying cytoskeleton. However, this is a general and versatile approach for interrogation of other receptors or molecules presented at the cell surface; it is limited chiefly by the capacity to functionalize probe surfaces with active biomolecules including proteins, polysaccharides, and synthetic drugs. We anticipate that the fundamental measurements of individual cell surface molecules and their ligand-binding properties enabled by this approach will enable predictions of key dynamic interactions between extracellular molecules and the intact cell surface, especially as these relate to ligand-induced clustering and the association of transmembrane receptors with mechanically dynamic structures such as the cytoskeleton.
Materials and Methods
Cell Culture.
HUVECs were cultured in endothelial basal medium-2 (EBM-2; Cambrex Bioscience, East Rutherford, NJ). 3T3 fibroblast cells (ATCC, Manassas, VA) were cultured in DMEM, 10% calf serum, 1% penicillin-streptomycin, and 1% glutamine (Invitrogen, Carlsbad, CA) on tissue culture-treated coverslips (Nalge Nunc, Rochester, NY).
Functionalized Force Imaging.
Living and fixed [3% formaldehyde and 0.1% glutaraldehyde (Sigma-Aldrich, St. Louis, MO) followed by rinsing with 0.05 M Tris buffer, pH 7.4] HUVECs were imaged in TopMAC mode within a fluid cell (PicoPlus scanning probe microscope; Agilent/Molecular Imaging, Palo Alto, CA) (11), by using backside magnetically coated Si3N4 cantilevers functionalized with monoclonal anti-VEGFR2 antibody (see SI Text). Living HUVECs were rinsed well and imaged in Hepes containing Ca2+ at 27°C. All images were acquired by using a closed loop piezoscanner for which positional stability was better than 0.3 nm/min, as confirmed with elapsed imaging of avidin adsorbed to mica. All measurements are expressed as average ± standard deviation.
The number of binding events per cell was determined by direct image analysis (summing of the number of closed regions at least one order of magnitude darker than background threshold) of a given cell area, obtaining 22 ± 6 sites per 1.5 × 1.5 μm2 recognition image, or 9.8 sites per μm2 in fixed HUVECs (n = 11); these regions were considered representative of the cell surface, as supported by immunohistochemistry shown in SI Fig. 8B. Surface area per cell was determined by 3D analysis through magnetic AC mode images of 60 images including at least one HUVEC, where for each cell the area was calculated as the sum of an ellipsoid surface area (based on known maximum cell height and long/short axis of the organelle-containing region) and an annulus surface area of the comparably flat cell body perimeter: 10,400 ± 2,700 μm2 (n = 60). The probability density function of molecular force spectroscopy indicated a finite probability of encountering either one receptor (33 pN, 58.4%) or two receptors (64 pN, 42.6%). The total number of receptors per cell then was determined as the product of the probability-weighted number of sites per cell surface area and the average cell surface area (1.47 ± 0.38 × 105 VEGFR2 per cell).
Molecular Force Spectroscopy.
Directly after scanning probe microscopy imaging in MAC mode, molecular force spectroscopy was conducted on 600 locations of strong binding (dark in recognition image) and 400 locations of weak binding (light in recognition image) for each cell area imaged (n = 11). Cantilever force constant (nanonewtons per nanometer) and photodiode optical lever sensitivity (nanometers per volt) were determined experimentally in air for each cantilever before functionalization (47) and confirmed as unchanged at the conclusion of each experiment. Unbinding or rupture force FR was determined for each event from the calibrated force–displacement response (36), and nonspecific unbinding events were excluded from the calculated probability density functions. Topographic images were recorded subsequently to verify nondestructive interrogation of the surface.
Supplementary Material
Acknowledgments
K.J.V.V. thanks T. Johnson and G. Kada (Molecular Imaging, Tempe, AZ) for technical assistance, and P. Hinterdorfer, L. Chtcheglova, D. A. Lauffenburger, and D. J. Irvine for insightful discussions. This work was supported by the National Science Foundation Nanoscale Exploratory Research, the Center for the Integration of Medicine and Innovative Technology, and the Hugh Hampton Young Memorial Foundation (S.L.). K.J.V.V. was supported by the Beckman Foundation Young Investigators Program.
Abbreviations
- AFM
atomic force microscopy
- VEGFR
vascular endothelial growth factor receptor
- VEC
vascular endothelial cell
- HUVEC
human umbilical vein endothelial cell.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0702668104/DC1.
The off-rate, on-rate, and dissociation constant determined from a best fit of this competitive binding response result in a difference of only −24%, −55%, and +41%, respectively, from those calculated above: koff = 7.98 ×10−5 s−1, kon = 2.60×104 M−1·s−1, and KD = 3.07 ×10−9 M. See SI Text for detailed comparison of these kinetic analyses.
References
- 1.Lauffenburger DA, Linderman JJ. Receptors: Models for Binding, Trafficking, and Signaling. Oxford: Oxford Univ Press; 1993. [Google Scholar]
- 2.Rotsch C, Radmacher M. Biophys J. 2000;78:520–535. doi: 10.1016/S0006-3495(00)76614-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamba T, Tam BYY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O'Brien SM, Davis RB, Gowen LC, et al. Am J Physiol. 2006;290:H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- 4.Vyas YM, Maniar H, Lyddane CE, Sadelain M, Dupont B. J Immunol. 2004;173:1571–1578. doi: 10.4049/jimmunol.173.3.1571. [DOI] [PubMed] [Google Scholar]
- 5.Avalos AM, Arthur WT, Schneider P, Quest AFG, Burridge K, Leyton L. J Biol Chem. 2004;279:39139–39145. doi: 10.1074/jbc.M403439200. [DOI] [PubMed] [Google Scholar]
- 6.Browning-Kelley ME, Wadu-Mesthrige K, Hari V, Liu GY. Langmuir. 1997;13:343–350. [Google Scholar]
- 7.Li L, Chen S, Oh S, Jiang S. Anal Chem. 2002;74:6017–6022. doi: 10.1021/ac0258148. [DOI] [PubMed] [Google Scholar]
- 8.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Nature. 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 9.Moy VT, Florin EL, Gaub HE. Science. 1994;266:257–259. doi: 10.1126/science.7939660. [DOI] [PubMed] [Google Scholar]
- 10.Schwesinger F, Ros R, Strunz T, Anselmetti D, Guntherodt H-J, Honegger A, Jermutus L, Tiefenauer L, Pluckthun A. Proc Natl Acad Sci USA. 2000;97:9972–9977. doi: 10.1073/pnas.97.18.9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stroh C, Wang H, Bash R, Ashcroft B, Nelson J, Gruber H, Lohr D, Lindsay SM, Hinterdorfer P. Proc Natl Acad Sci USA. 2004;101:12503–12507. doi: 10.1073/pnas.0403538101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupres V, Monozzi FD, Locht C, Clare BH, Abbott NL, Cuenot S, Bampard C, Raze D, Duprene YF. Nat Methods. 2005;2:515–520. doi: 10.1038/nmeth769. [DOI] [PubMed] [Google Scholar]
- 13.Gad M, Itoh A, Ikai A. Cell Biol Int. 1997;21:697–706. doi: 10.1006/cbir.1997.0214. [DOI] [PubMed] [Google Scholar]
- 14.Horton M, Charras G, Lehenkari P. J Recept Signal Transduction. 2002;22:169–190. doi: 10.1081/rrs-120014594. [DOI] [PubMed] [Google Scholar]
- 15.Kim H, Arakawa H, Hatae N, Sugimoto Y, Matsumoto O, Osada T, Ichikawa A, Ikai A. Ultramicroscopy. 2006;106:652–662. doi: 10.1016/j.ultramic.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Lehenkari PP, Horton MA. Biochem Biophys Res Commun. 1999;259:645–650. doi: 10.1006/bbrc.1999.0827. [DOI] [PubMed] [Google Scholar]
- 17.Madl J, Rhode S, Stangl H, Stockinger H, Hinterdorfer P, Schutz GJ, Kada G. Ultramicroscopy. 2006;106:645–651. doi: 10.1016/j.ultramic.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 18.Puntheeranurak T, Wildling L, Gruber HJ, Kinne RKH, Hinterdorfer P. J Cell Sci. 2006;119:2960–2967. doi: 10.1242/jcs.03035. [DOI] [PubMed] [Google Scholar]
- 19.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 20.Izumi Y, di Tomaso E, Hooper A, Huang P, Huber J, Hicklin DJ, Fukumura D, Jain RK, Suit HD. Cancer Res. 2003;63:747–751. [PubMed] [Google Scholar]
- 21.Posadas E, Davidson B, Kohn E. Curr Opin Oncol. 2004;6:478–484. doi: 10.1097/00001622-200409000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Rakhmilevich A, Hooper A, Hicklin D, Sondel P. Mol Cancer Ther. 2004;3:969–976. [PubMed] [Google Scholar]
- 23.Almqvist N, Bhatia R, Primbs G, Desai N, Banerjee S, Lal R. Biophys J. 2004;86:1753–1762. doi: 10.1016/S0006-3495(04)74243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson NH, Fritz M, Radmacher M, Cleveland JP, Schmidt CF, Hansma PK. Biophys J. 1996;70:2421–2431. doi: 10.1016/S0006-3495(96)79812-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charras G, Horton M. Biophys J. 2002;82:2970–2981. doi: 10.1016/S0006-3495(02)75638-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikai A, Afrin R, Itoh A, Thogersen HC, Hayashi Y, Osada T. Colloids Surf B. 2002;23:165–171. [Google Scholar]
- 27.Pfister G, Stroh CM, Perschinka H, Kind M, Knoflach M, Hinterdorfer P, Wick G. J Cell Sci. 2005;118:1587–1594. doi: 10.1242/jcs.02292. [DOI] [PubMed] [Google Scholar]
- 28.Baumgartner W, Hinterdorfer P, Ness W, Raab A, Vestweber D, Schindler H, Drenckhahn D. Proc Natl Acad Sci USA. 2000;97:4005–4010. doi: 10.1073/pnas.070052697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kienberger F, Ebner A, Gruber HJ, Hinterdorfer P. Acc Chem Res. 2006;39:29–36. doi: 10.1021/ar050084m. [DOI] [PubMed] [Google Scholar]
- 30.Green RJ, Davies J, Davies MC, Roberts CJ, Tendler SJ. Biomaterials. 1997;18:405–413. doi: 10.1016/s0142-9612(96)00141-x. [DOI] [PubMed] [Google Scholar]
- 31.Van Vliet K, Hinterdorfer P. Nano Today. 2006;1:18–25. [Google Scholar]
- 32.Saxton MJ. Biophys J. 2001;81:2226–2240. doi: 10.1016/S0006-3495(01)75870-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinterdorfer P, Dufrene YF. Nat Methods. 2006;3:347–355. doi: 10.1038/nmeth871. [DOI] [PubMed] [Google Scholar]
- 34.Waltenberger J, Claesson-Welsh L, Siegbahnll A, Shibuya M, Heldin C-H. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 35.Scheidegger P, Weiglhofer W, Suarez S, Console S, Waltenberger J, Pepper MS, Jaussi R, Ballmer-Hofer K. Biochem J. 2001;353:569–578. doi: 10.1042/0264-6021:3530569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baumgartner W, Hinterdorfer P, Schindler H. Ultramicroscopy. 2000;82:85–95. doi: 10.1016/s0304-3991(99)00154-0. [DOI] [PubMed] [Google Scholar]
- 37.Bell GI. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 38.Dill K, Lin M, Poteras C, Fraser C, Hafeman DG, Owicki JC, Olson JD. Anal Biochem. 1994;217:128–138. doi: 10.1006/abio.1994.1093. [DOI] [PubMed] [Google Scholar]
- 39.Hinterdorfer P, Baumgartner W, Gruber HJ, Schilcher K, Schindler H. Proc Natl Acad Sci USA. 1996;93:3477–3481. doi: 10.1073/pnas.93.8.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu D, Jimenez Z, Zhang H, Bohlen P, Witte L, Zhu Z. Int J Cancer. 2002;97:393–399. doi: 10.1002/ijc.1634. [DOI] [PubMed] [Google Scholar]
- 41.Murase K, Fujiwara T, Umemura Y, Suzuki K, Iino R, Yamashita H, Saito M, Murakoshi H, Ritchie K, Kusumi A. Biophys J. 2004;86:4075–4093. doi: 10.1529/biophysj.103.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritchie K, Shan X-Y, Kondo J, Iwasawa K, Fujiwara T, Kusumi A. Biophys J. 2005;88:2266–2277. doi: 10.1529/biophysj.104.054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sako Y, Kusumi A. J Cell Biol. 1995;129:1559–1574. doi: 10.1083/jcb.129.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheetz MP. Annu Rev Biophys Biomol Struct. 1993;22:417–431. doi: 10.1146/annurev.bb.22.060193.002221. [DOI] [PubMed] [Google Scholar]
- 45.Choquet D, Felsenfeld DP, Sheetz MP. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 46.Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K. Cancer Res. 2000;60:203–212. [PubMed] [Google Scholar]
- 47.Cleveland JP, Manne S, Bocek D, Hansma PK. Rev Sci Instrum. 1993;64:403–405. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
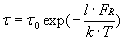{kind=link}
{kind=link}
{kind=link}
{kind=link}
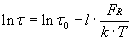{kind=link}
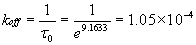{kind=link}
{kind=link}
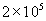{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}