

Abstract
The HIV (human immunodeficiency virus) integrase has a crucial role in viral replication. Moreover, it has no cellular homologue in humans. Hence, it is considered as an attractive drug target. Many inhibitors against the integrase protein has been designed and discussed. The Y-3 inhibitor (4-acetyl amino-5-hydroxy naphthalene - 2, 7- disulfonic acid) is already known to inhibit HIV-1 integrase. However, it is not suitable as a drug like candidate due to its high cyto-toxicity. In this report, a pharmacophore model for HIV integrase is described using the already known Y-3 inhibitor binding site. Fourteen compounds chemically related to the Y-3 inhibitor were generated using the described pharmacophore model and reported. Subsequent computational analysis showed that these compounds have interactions with the Y3 binding site and their possible utility as an integrase inhibitor is discussed.
Keywords: integrase, Y3 inhibitor, docking, chemical compounds
Background
AIDS (acquired immuno-deficiency syndrome) is a major epidemic caused by HIV (human immuno-deficiency virus) and significant efforts are being made for years to develop drugs against the dreadful disease. The HIV genome encodes for the protease, reverse transcriptase and integrase enzymes. The HIV integrase contains three functional domains. [1] Interestingly, HIV integrase has no sequence homologue in the human host and hence, it is considered as a potential drug target. [2] The integrase protein has three domains, namely, the N-terminal domain stabilized by Zn2+ [3,4 ,5], the DNA binding C-terminal domain [6] and the catalytic domain consisting of residues Asp64, Asp116, and Glu152 forming the active site. The catalytic triad is essential for enzymatic activity with metal cofactors. [4,5] The structure of the C-terminal and N-terminal domains of the HIV-1 integrase were solved by NMR (nuclear magnetic resonance) [7,8,9], whereas, the catalytic domain of HIV integrase was determined by x-ray crystallography. [10,11] A number of natural products inhibiting integrase have been reported, recently. [12] The macromolecular crystallography laboratory identified three structurally related inhibitors called Y-1, Y-2 and Y-3. Among them, Y-3 (Figure 1) showed maximum integrase inhibition. [13] Y-3 interacts with the flexible loop of the enzyme's active site and induces loop conformation change. Pre-incubation of integrase with metal cations did not prevent Y3 inhibition. [14] Pharmacophores describing interactions between integrase and ligands are important in drug screening. Inhibitors with high potency, solubility and reduced toxicity are generally considered for clinical trials. [13] Here, we describe the design of potential inhibitors for HIV integrase using a pharmacophore model.
Figure 1.
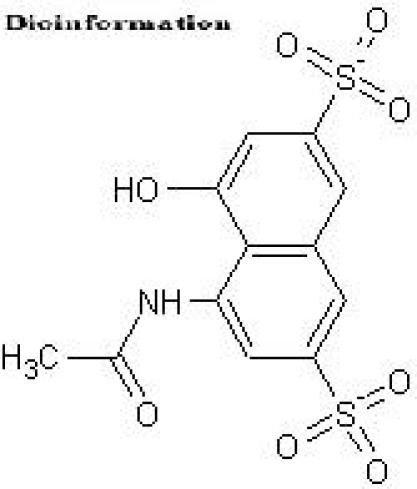
Structure of the Y-3 inhibitor
Methodology
Integrase catalytic domain structure
The three dimensional structure of the catalytic domain of the HIV-1 integrase protein (PDB ID: 1BI4) was obtained from PDB (Protein Data Bank). [15] This domain consists of a five-stranded beta-sheet with six surrounding helices.
Pharmacophore model
We analyzed the binding pockets of the integrase catalytic domain using PASS (a tool that uses geometry to characterize regions of buried volume in proteins) to identify positions likely to represent binding sites based on size, shape and volume buried [16 ] (Figure 2). LigBuilder (a program for structure-based drug design) was used to build ligand molecules within the binding pocket of the HIV-1 integrase catalytic domain. [17] A pharmacophore model for receptor active site was derived using the POCKET module in LigBuilder. It should be noted that the previously proposed pharmacophore models were based on already known HIV-1 integrase inhibitors. [18,19 ]
Figure 2.
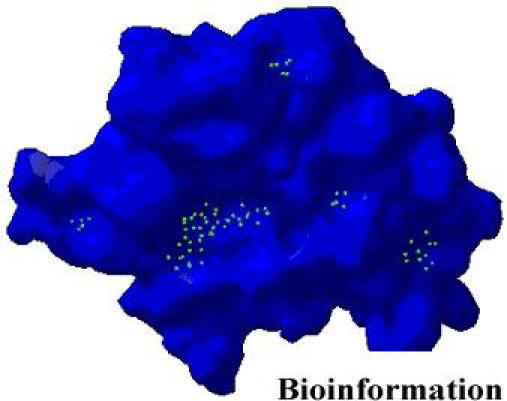
Cavities predicted by PASS for HIV-1 integrase
Inhibitor design
The 3D database searching methods that search for inhibitors in known compounds have been described elsewhere. [20,21,22] A dynamic pharmacophore model (screen a database of chemical compounds) for HIV-1 integrase was also available. [23, 24,25] Here, we describe a pharmacophore model using receptor active site and key interaction sites for Y-3 inhibitor as a seed compound. The model was used for constructing novel ligand molecules within the constraints of the target intergrase using the LigBuilder program. A total of 3000 ligand molecules were constructed using the GROW module in LigBuilder.
Compounds generation
An initial population was generated based on the seed structure and the parent molecules are copied into the mating pool. Thus, a new population was generated by performing structural manipulations to the molecules in the mating pool. The generated molecules were analyzed using the PROCESS module in LigBuilder. A set of 500 molecules meeting the chemical criteria setup in the parameter file in PROCESS module were filtered and then converted into viewable Mol2 formatted files. LigBuilder estimates binding affinities using the SCORE v2.08 algorithm [26] and LogP values using the XLOGP v2.0 algorithm [27] for each compound. These 500 molecules were then clustered into 21 groups based on structural similarity. The compounds were then tested for Lipinski's Rule of Five using the Molinspiration server. [28]
Ligand binding
The AutoDock3.0/ADT [29] program was used to investigate ligand binding to HIV-1 integrase using a grid encompassing the active site surface. The AutoDock program combines a rapid energy evaluation through pre-calculated grids of affinity potentials with a variety of search algorithms to find suitable binding positions. The affinity and electrostatic maps were computed with a grid spacing of 0.375 Å and the grid points in x, y and z axis were set to 58x58x56. The search was based on the Lamarckian genetic algorithm and the results were analyzed using binding energy. The cluster analysis was performed based on RMSD (root mean square deviation). For each ligand, a docking experiment consisting of 10 simulations was performed. The ligand molecules were then ranked in order of increasing docking energies and grouped into clusters of similar conformation. The Swiss-PdbViewer was then used to generate images of protein structures docked with potential compounds.
Results and Discussion
A static pharmacophore model was developed using LigBuilder based on the crystal structure of HIV-1 integrase bound with the Y-3 inhibitor (4-acetylamino-5-hydroxynaphthalene-2, 7-disulfonic acid). Figure 3 and Figure 4, shows the interaction between Y-3 inhibitor and active site residues Asp64, Asp116, Glu152 in the catalytic domain of integrase. In Figure 5, nitrogen atoms (blue) represent hydrogen-bond donor sites; oxygen atoms (red) represent hydrogen-bond acceptor sites; and carbon atoms (green) represent hydrophobic sites. Using the pharmacophore model, 500 out of the initial 3000 molecules satisfying the minimum chemical criteria were identified for further analysis (Table 1). The 500 compounds were then grouped in 21 different clusters using structural similarity. A molecule from each cluster was chosen based on binding affinity to integrase. Fourteen (structures given in Figure 7) of the twenty-one molecules satisfied the Lipinski's rule of five with zero violations (Table 2). A useful parameter for predicting drug transport properties is TPSA (Topological Molecular polar surface area) and it is determined for these molecules. A topological parameter is number of rotatable bonds and it describes the molecular flexibility of these compounds. This parameter was calculated for the fourteen molecules that satisfy the ‘rule-of-5’ and it is found that all the molecules have rotatable bonds in the range of 2-6 (Table 3).
Figure 3.
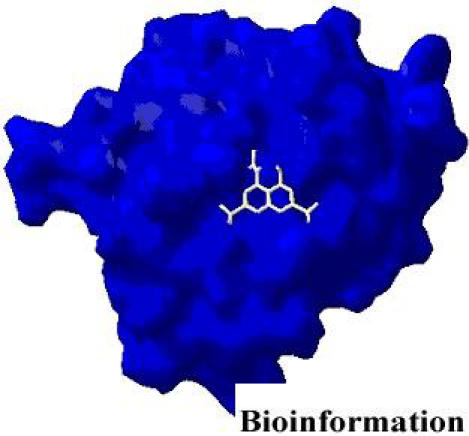
Y3 inhibitor interacting with HIV-1 integrase
Figure 4.
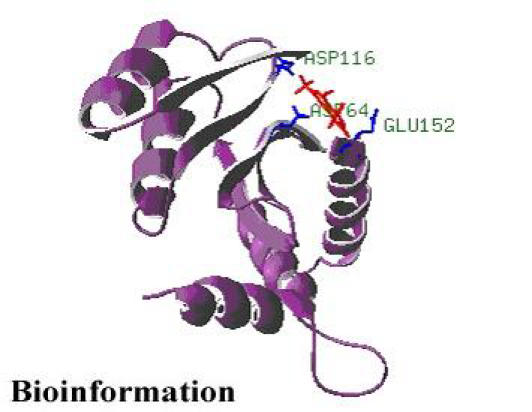
Y3 inhibitor interacting with HIV-1 integrase
Figure 5.
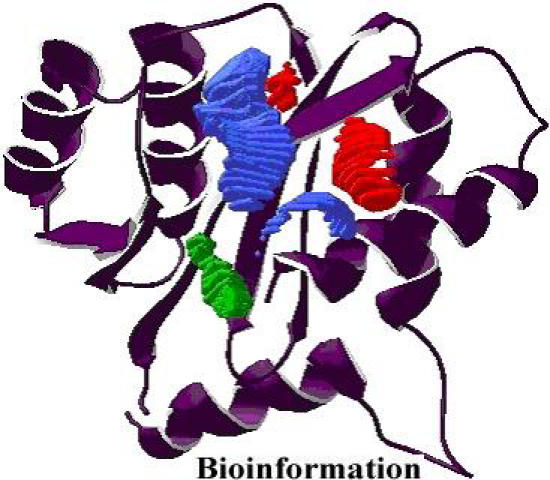
A pharmacophore model for integrase
Table 1. Chemical criteria used for PROCESS module in LigBuilder.
| Maximal Molecular weight | 500 |
| Minimal Molecular weight | 50 |
| Maximal LogP | 5.0 |
| Minimal LogP | -5.0 |
| Maximal PKD | 5.0 |
| Minimal PKD | 1.0 |
| Similarity cutoff | 1.0 |
| Number of molecules satisfied the criteria | 500 |
Figure 7.

Two dimensional structures of fourteen ligand molecules
Table 2. Lipinski’s rule of five calculated by Molinspiration.
| Molecule | MiLogp | Mw | H acceptor | H donor | Violations |
|---|---|---|---|---|---|
| Y-3 | -0.719 | 359.337 | 9 | 2 | 0 |
| 1 | 0.906 | 481.504 | 10 | 1 | 0 |
| 2 | 1.131 | 493.515 | 10 | 0 | 0 |
| 3 | 1.317 | 489.527 | 9 | 0 | 0 |
| 4 | -0.103 | 453.45 | 10 | 0 | 0 |
| 5 | -0.799 | 498.491 | 12 | 2 | 1 |
| 6 | 1.196 | 489.527 | 9 | 0 | 0 |
| 7 | -0.188 | 465.461 | 10 | 0 | 0 |
| 8 | -0.374 | 496.475 | 12 | 1 | 1 |
| 9 | 0.858 | 481.504 | 10 | 0 | 0 |
| 10 | 0.979 | 455.466 | 10 | 0 | 0 |
| 11 | 1.137 | 479.488 | 10 | 0 | 0 |
| 12 | -1.764 | 483.436 | 13 | 1 | 1 |
| 13 | 0.293 | 467.477 | 10 | 0 | 0 |
| 14 | 0.948 | 495.531 | 10 | 0 | 0 |
| 15 | -0.574 | 467.433 | 11 | 1 | 1 |
| 16 | -0.558 | 468.465 | 11 | 1 | 1 |
| 17 | 1.324 | 481.504 | 10 | 0 | 0 |
| 18 | 0.672 | 479.488 | 10 | 0 | 0 |
| 19 | -0.762 | 498.447 | 13 | 2 | 1 |
| 20 | -0.857 | 482.492 | 11 | 1 | 1 |
| 21 | 1.136 | 489.483 | 10 | 0 | 0 |
Table 3. Molecular properties for ligand molecules.
| Molecule | TPSA | Atoms | Rotatable bonds | Volume |
|---|---|---|---|---|
| Y-3 | 151.752 | 23 | 3 | 257.413 |
| 1 | 152.197 | 32 | 2 | 368.637 |
| 2 | 141.203 | 33 | 4 | 385.03 |
| 3 | 131.969 | 33 | 3 | 382.513 |
| 4 | 149.04 | 30 | 3 | 340.148 |
| 5 | 131.969 | 33 | 5 | 387.431 |
| 6 | 149.04 | 31 | 2 | 350.387 |
| 7 | 149.04 | 33 | 6 | 394.675 |
| 8 | 149.04 | 33 | 4 | 371.617 |
| 9 | 149.04 | 30 | 5 | 350.373 |
| 10 | 149.04 | 32 | 5 | 372.242 |
| 11 | 149.04 | 31 | 3 | 356.683 |
| 12 | 149.04 | 32 | 4 | 373.402 |
| 13 | 149.04 | 32 | 4 | 367.189 |
| 14 | 149.04 | 32 | 6 | 378.428 |
All docking calculations were carried out using AutoDock3.0/ADT. Each of the fourteen molecules was docked into the active site of integrase that is fixed with flexible ligands. A smaller grid, concentrating on the catalytic domain of integrase was used for 10 simulations. The docked conformation with the lowest free binding of energy was then probed for further analysis. The docking results revealed a consistent set of binding characteristics for the 14 molecules with HIV-1 integrase (Figure 6). AutoDock calculated RMSD value, free energy and the docked energy values for the resulting fourteen structures (Table 4). Thus, we demonstrate the building of small-molecule inhibitors for HIV-1 integrase using a pharmacophore model.
Figure 6.
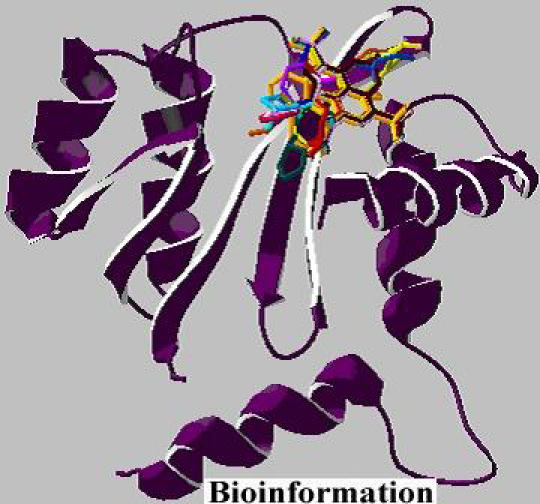
Docked complex between HIV-1 integrase and 14 generated molecules
Table 4. Energy values from AutoDock.
| Molecule | RMSDA | Estimated free energy kcal/mol | Final docked energy Kcal/mol |
|---|---|---|---|
| 1 | 0.360 | -6.73 | -7.94 |
| 2 | 0.669 | -8.10 | -10.44 |
| 3 | 0.361 | -8.65 | -7.51 |
| 4 | 0.525 | -6.96 | -8.31 |
| 5 | 0.815 | -8.49 | -10.33 |
| 6 | 0.335 | -7.46 | -8.64 |
| 7 | 0.565 | -8.03 | -10.20 |
| 9 | 0.627 | -6.35 | -7.79 |
| 10 | 0.417 | -7.64 | -10.05 |
| 11 | 0.448 | -7.56 | -9.18 |
| 12 | 0.524 | -7.14 | -7.77 |
| 13 | 0.496 | -7.28 | -8.63 |
| 14 | 0.551 | -7.76 | -9.81 |
Conclusion
The purpose of this study is to generate novel compounds as HIV-1 integrase inhibitors. A receptor based pharmacophore model was developed for HIV-1 integrase with a variety of pharmacophore features (hydrogen-bond acceptor and donor, hydrophobic, and excluded volumes). In this study, fourteen molecules were identified as potential integrase inhibitors. Docking results from using AutoDock illustrated the molecular interactions between the 14 compounds and the HIV-1 integrase. Further studies on their binding properties will provide useful information towards the rational design of effective HIV-1 integase inhibitors.
Acknowledgments
The authors are grateful to Dr. K. N. Somasekharan, Dr. S. Swaminathan & Mr. N. Vijayaraj for their support of this work
Footnotes
Citation:Jaganatharaja & Gowthaman, Bioinformation 1(4): 112-117 (2006)
References
- 1.Andrake MD, Skalka AM. J Biol Chem. 1996;271:19633. doi: 10.1074/jbc.271.33.19633. [DOI] [PubMed] [Google Scholar]
- 2.Thomas M, Brady L. Trends Biotechnol. 1997;15:167. doi: 10.1016/S0167-7799(97)01016-0. [DOI] [PubMed] [Google Scholar]
- 3.Lee SP, Han MK. Biochemistry. 1996;35:3837. doi: 10.1021/bi952056p. [DOI] [PubMed] [Google Scholar]
- 4.Zheng R, et al. Proc Natl Acad Sci. 1996;93:13659. doi: 10.1073/pnas.93.24.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SP, et al. Biochemistry. 1997;36:173. doi: 10.1021/bi961849o. [DOI] [PubMed] [Google Scholar]
- 6.Khan E, et al. Nucleic Acids Res. 1991;19:851. doi: 10.1093/nar/19.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lodi P, et al. Biochemistry. 1995;34:9826. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- 8.Cai M, et al. Nat Struct Biol. 1997;4:567. doi: 10.1038/nsb0797-567. [DOI] [PubMed] [Google Scholar]
- 9.Eijkelenboom AP, et al. Nat Struct Biol. 1995;2:807. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
- 10.Dyda F, et al. Science. 1994;266:1981. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 11.Bujacz G, et al. J Mol Biol. 1995;253:333. doi: 10.1006/jmbi.1995.0556. [DOI] [PubMed] [Google Scholar]
- 12.Deng J, et al. J Med Chem. 2005;48:1496. doi: 10.1021/jm049410e. [DOI] [PubMed] [Google Scholar]
- 13. http://mcl1.ncifcrf.gov/integrase/asv_y3.html.
- 14.Lubkowski J, et al. App Biol Sci. 1998;95:4831. doi: 10.1073/pnas.95.9.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berman HM, et al. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brady GP, et al. J Comp Aided Mol Design. 2000;14:383. doi: 10.1023/a:1008124202956. [DOI] [PubMed] [Google Scholar]
- 17.Wang R, et al. J Mol Model. 2000;6:498. [Google Scholar]
- 18.Barreca ML, et al. J Med Chem. 2005;48:7084. doi: 10.1021/jm050549e. [DOI] [PubMed] [Google Scholar]
- 19.Nicklaus MC, et al. J Med Chem. 1997;40:920. doi: 10.1021/jm960596u. [DOI] [PubMed] [Google Scholar]
- 20.Hong H, et al. J Med Chem. 1997;40:930. doi: 10.1021/jm960754h. [DOI] [PubMed] [Google Scholar]
- 21.Neamati N, et al. Mol Pharmacol. 1997;52:1041. doi: 10.1124/mol.52.6.1041. [DOI] [PubMed] [Google Scholar]
- 22.Dayam R, et al. J Med Chem. 2005;48:8009. doi: 10.1021/jm050837a. [DOI] [PubMed] [Google Scholar]
- 23.Deng J, et al. J Med Chem. 2005;48:1496. doi: 10.1021/jm049410e. [DOI] [PubMed] [Google Scholar]
- 24.Carlson HA, et al. J Med Chem. 2000;43:2100. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 25.Deng J, et al. J Med Chem. 2006;49:1684. doi: 10.1021/jm0510629. [DOI] [PubMed] [Google Scholar]
- 26.Wang R, et al. Perspectives in Drug Design and Discovery. 2000;19:47. [Google Scholar]
- 27.Wang R, et al. J Mol Model. 1998;4:379. [Google Scholar]
- 28. http://www.molinspiration.com/
- 29.Goodsell DS, et al. J of Mol Recog. 1996;9:1. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]