

Abstract
The promyelocytic leukemia (PML) gene, a tumor suppressor inactivated in acute promyelocytic leukemia (APL), regulates apoptosis induced by DNA damage. However, the molecular mechanisms by which PML modulates apoptosis following genotoxic stress are only partially elucidated. PML is essential for p53-dependent induction of programmed cell death upon γ-irradiation through PML-nuclear body (NB)–mediated control of p53 acetylation. Here, we show that PML selectively regulates proapoptotic transcription factors upon different types of DNA damage. We find that Pml inactivation protects fibroblasts from UV-induced apoptosis in a p53-independent manner. We demonstrate that c-Jun is required for UV-induced apoptosis and that PML is essential for both c-Jun transcriptional activation and DNA binding upon UV radiation. We find that PML physically interacts with c-Jun and that upon UV radiation the PML-NBs reorganize into novel nuclear microspeckled structures (UV-NBs), where PML and c-Jun dynamically accumulate. These data identify a novel PML-dependent pathway for c-Jun transcriptional activation and induction of apoptosis in response to DNA damage and shed new light on the role of PML in tumor suppression.
Introduction
The promyelocytic leukemia (Pml) gene encodes a tumor suppressor involved in the t(15:17) chromosomal translocation associated with acute promyelocytic leukemia (APL).1-5 PML is a Really Interesting New Gene (RING) finger protein found localized in subnuclear structures known as PML-nuclear bodies (PML-NBs), which have been implicated in transcriptional regulation.6,7 PML is essential for the proper formation and stability of these subnuclear structures, since in Pml-/- primary cells the PML-NBs are disrupted and their components acquire an aberrant nuclear localization pattern.7-9 Upon cellular stresses such as γ-irradiation and acute oncogene exposure, PML acts as a p53 transcriptional coactivator, at least in part through the recruitment of p53 into the PML-NB and its consequent CREB (cAMP [cyclic adenosine monophosphate] response element-binding protein)–binding protein (CBP)–mediated acetylation.5,10,11 As a consequence, Pml-/- thymocytes are resistant to γ-ray–induced apoptosis.10 Remarkably, Pml-/- mice and cells are also protected from several p53-independent apoptotic stimuli, although the molecular mechanisms underlying the role of PML in these pathways are currently unknown.12
The apoptotic response to short wavelength ultraviolet (UV) light is not mediated by p53 in primary fibroblasts, since both p53 and p21 null primary fibroblasts are more sensitive to UV-induced apoptosis.13-17 By contrast, primary embryonic fibroblasts devoid of c-Jun-N-terminal kinases (JNKs), the upstream activators of c-Jun, are resistant to UV-induced apoptosis.18 Phosphorylation of c-Jun was shown to be required for UV-triggered apoptosis.19 Nevertheless, the role of c-Jun in UV-induced cell death is still controversial and a matter of debate, because of conflicting reports.19-21
In this study, we demonstrate that c-Jun regulates UV-induced apoptosis in primary cells, and that PML controls c-Jun function specifically in response to UV irradiation. Remarkably, upon UV radiation, PML redistributes to multiple microspeckles where it colocalizes with c-Jun. On the contrary, in unirradiated cells, PML and c-Jun do not colocalize in the PML-NB, and this correlates with the inability of PML to regulate c-Jun function in the absence of cellular stress.
Materials and methods
Apoptosis analysis
Mouse embryo fibroblasts (MEFs) were fixed in acetone-methanol (1:4) and stained with propidium iodide (10 μg/mL). Subdiploid peak analysis was performed to evaluate the percentage of apoptotic cells. Alternatively, cell death was evaluated by trypan blue uptake. Mitochondrial transmembrane potential (ΔΨm) was measured by JC-1 staining following manufacture's instruction (Molecular Probes, Eugene, OR).
Western blotting and immunoprecipitation
MEFs were lysed in buffer A (50 mM Tris (tris(hydroxymethyl)aminomethane), pH 7.6, 200 mM NaCl, 1 mM EDTA (ethylenediaminetetraacetic acid),10 mM MgCl2, 10 mM MnCl2, 1% Triton-X100, 50 mM NaF, 0.5 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/mL leupeptin, aprotinin, and pepstatin). Antibodies used were anti–c-Jun-Ser 63 or -Ser 73 (Cell Signaling Technology, Beverly, MA), anti–c-Jun (BD Transduction Laboratories, San Diego, CA), anti-actin (Sigma, St Louis, MO), anti-HSP90 (BD Transduction Laboratories), p53 (Oncogene Science, Cambridge, MA), p53 Ser 18 (Cell Signaling Technology), p21 (Santa Cruz Biotechnology, Santa Cruz, CA), Bax (Santa Cruz), and HSP90 (Transduction Laboratories). For immunoprecipitation, WI-38 cells were lysed in 10 mM Tris, pH 7.6, 150 mM NaCl, 0.2% Triton-X100, 1 mM EDTA, 50 mM NaF, 0.5 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 10 μg/mL leupeptin, aprotinin, and pepstatin. PML was immunoprecipitated by using anti–human PML (Santa Cruz Biotechnology). Purified glutathione S transferase (GST)–c-Jun was incubated with in vitro–translated 35S-PML and -PML mutants in binding buffer (20 mM Tris, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1 mM DTT [dithiothreitol], 1 mM PMSF, 0.2% NP-40 [(Octylphenoxy)polyethoxyethanol]) for 1 hour. Beads were washed 5 times in binding buffer containing 200 mM NaCl.
Cell infection
Pml+/+ and Pml-/- MEFs were infected with high-titer pBabe or pBabe–c-Jun retroviral supernatants and selected in puromycin (Sigma) for 48 hours. Cells were then replated and UV-irradiated (60 J/m2).
Transcriptional assay
MEFs were transfected by using Effectene (Qiagen, Valencia, CA). The following plasmids were used: galactosidase-4 (GAL4)–c-Jun and S63A/S73A GAL4–c-Jun (GAL4–c-JunAA) (kind gifts of Dr L. Zon, Harvard University), GAL4-Luc (Promega, Madison, WI), jun2CRE-Luc (4 jun2 CRE [cAMP-responsive element] elements from the c-jun promoter cloned upstream the SV40 [simian virus 40] minimal promoter), collTRE-Luc (5 TRE [12-O-tetradecanoyl-phorbol-13-acetate (TPA)–responsive element] elements from the collagenase promoter cloned upstream the SV40 minimal promoter), thymidine kinase (TK)–Renilla-Luc (Promega), pSG5 (Stratagene, La Jolla, CA), pSG5-PML3 and pSG5-PML-RARα (retinoic acid receptor α). Luciferase activity was measured by using the Luciferase Assay System (Promega).
Gel shift and chromatin immunoprecipitation
Nuclear extracts from UV-treated MEFs were incubated with either 32P-labeled jun2 CRE element-like (AGCTCCCGTGACGTCACCCG) for 20 minutes at room temperature. For supershift analysis, extracts were preincubated with antibodies against c-Jun (Santa Cruz) or activating transcription factor-2 (ATF-2; Santa Cruz) for 30 minutes on ice before adding the labeled probe. DNA-protein complexes were resolved on a 4% polyacrilamide gel and exposed. For chromatin immunoprecipitation, we processed cells as previously described.22 After O/N immunoprecipitation, DNA-protein complexes were analyzed by polymerase chain reaction ATGTAAGCATGTTTACCTTC and CATGGTGCCCAGCAGTCC (collagenase TRE 5′ and 3′ primers, respectively), ACAAGCCGAAGCTGCGCGC and TTGGCTTGCGTCGTTCTCAG (c-jun jun2 5′ and 3′ primers, respectively), and AGCCTTCGCGGGCCCAG and CAACTCTGAGTCCTTATCCA (c-jun jun1 5′ and 3′ primers, respectively).
Immunofluorescence and confocal microscopy
Briefly, WI-38 cells or MEFs grown on glass coverslips were fixed and permeabilized. Cells were then incubated with combinations of the following antibodies: mouse anti–human PML (Santa Cruz), rabbit anti–mouse Pml (kind gift of Dr Freemont, Imperial College, London, United Kingdom), anti–small ubiquitin-related modifier 1 (SUMO1; Santa Cruz), anti-DAXX (Santa Cruz), anti-CBP (Santa Cruz), and anti–phosphorylated Ser 63–c-Jun (Cell Signaling Technology). Fluorescein isothiocyanate (FITC)- and phycoerythrin (PE)-onjugated secondary antibodies were purchased from Molecular Probes. The imaging medium was Prolong Antifade kit (Molecular Probes). Images were taken with 40×/1.3 numerical aperture Plan NeoFluor or 63×/1.2 numerical aperture Plan Apochromat lenses. A Zeiss Axiovert 100 M, PMT detector, and LSM 510 acquisition software (all from Carl Zeiss, Thornwood, NY).
Results
PML is required for UV-induced apoptosis
Since PML has been involved in several apoptotic pathways elicited by different cellular stresses,12 we set to determine whether PML also regulates apoptosis induced by UV irradiation. To this end, we exposed Pml+/+ and Pml-/- MEFs to 60 J/m2 UV light and found that the percentage of sub-G1 hypodiploid cells was markedly impaired in Pml-/- MEFs (Figure 1A) and was dependent on neosynthesis (not shown). Reduction of apoptosis in Pml-/- cells was accompanied by a decrease in mitochondrial membrane potential depolarization (not shown). By contrast, similarly to previous reports,16,17 p53-/- cells were not protected from UV-induced apoptosis at 60 J/m2 (not shown), but they were more susceptible to UV-induced cell death at lower doses (not shown). Since p53 induction and activation by UV radiation trigger cell growth arrest through the up-regulation of p21,21 we analyzed the UV-induced cell cycle arrest in Pml-/- cells. Pml-/- MEFs properly underwent cell cycle arrest (24 hours after 20 J/m2; not shown). Furthermore, Pml inactivation did not affect p53 induction or p53 Ser 18 phosphorylation (Figure 1B). p53 Target genes such as p21 and Bax were also normally induced at both mRNA and protein levels in Pml-/- cells (Figure 1B-C). Taken together these data demonstrate that PML regulates apoptosis upon UV radiation in a p53-independent manner.
Figure 1.

PML modulates the apoptotic response to UV radiation in a p53-independent manner. (A) Pml+/+ ( ) and Pml-/- (▪) MEFs were UV-irradiated with 60 J/m2, and apoptosis was measured at 0, 12, and 24 hours by propidium iodide staining and subdiploid peak analysis. Average of 3 independent experiments ± standard deviation (SD) is shown. (B) p53 Function upon UV radiation is unaltered in Pml-/- MEFs. Pml+/+ and Pml-/- MEFs were UV-treated (60 J/m2) and lysed at 0, 1, 3, 6, 12, and 24 hours. Total extracts were probed with antibodies against p53, phospho (P)–p53 (Ser 18), p21, and Bax. HSP90 levels were measured as loading control. Levels of p53 expression and phosphorylation are represented as fold of induction over untreated controls. (C) Bax and p21 mRNA levels in Pml+/+ and Pml-/- MEFs were analyzed by Northern blot at 0, 6, 12, and 18 hours upon UV irradiation (60 J/m2). Ethidium bromide staining of 28S rRNA is shown as loading control.
) and Pml-/- (▪) MEFs were UV-irradiated with 60 J/m2, and apoptosis was measured at 0, 12, and 24 hours by propidium iodide staining and subdiploid peak analysis. Average of 3 independent experiments ± standard deviation (SD) is shown. (B) p53 Function upon UV radiation is unaltered in Pml-/- MEFs. Pml+/+ and Pml-/- MEFs were UV-treated (60 J/m2) and lysed at 0, 1, 3, 6, 12, and 24 hours. Total extracts were probed with antibodies against p53, phospho (P)–p53 (Ser 18), p21, and Bax. HSP90 levels were measured as loading control. Levels of p53 expression and phosphorylation are represented as fold of induction over untreated controls. (C) Bax and p21 mRNA levels in Pml+/+ and Pml-/- MEFs were analyzed by Northern blot at 0, 6, 12, and 18 hours upon UV irradiation (60 J/m2). Ethidium bromide staining of 28S rRNA is shown as loading control.
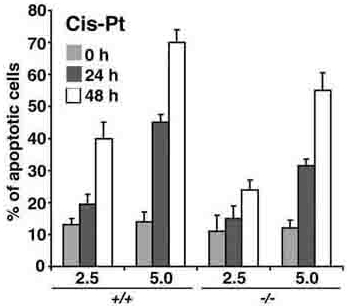
Apoptosis triggered by cisplatin (cis-pt), a chemotherapy agent that, similarly to UV radiation, induces the formation of DNAcross-links and can cause cell death in a p53-independent manner,23 was also impaired in Pml-/- MEFs at all doses tested, although at a milder extent (Supplemental Figure S1, available at the Blood website; see the Supplemental Figures link at the top of the online article). These results indicate that PML can also regulate p53-independent apoptotic pathways induced by both UV radiation and cis-pt.
Functional cross-talk between PML and c-Jun upon UV radiation
The c-Jun/JNK pathway has been involved in the regulation of UV-induced apoptosis.18,19,21 To better clarify the role of c-Jun in UV-induced cell death and to test whether PML and c-jun would cooperate in this function, we, at first, used a dominant-negative mutant of c-Jun (DN–c-Jun), which inhibits c-Jun transcriptional activity.24 Fibroblasts infected with a DN–c-Jun retroviral vector displayed reduction in cell death upon UV radiation (Figure 2A). Importantly, the dominant-negative effect of DN–c-Jun on UV-induced cell death was significantly reduced in Pml-/- cells, suggesting a possible functional cooperation between PML and c-Jun (Figure 2A). Thus, UV-induced apoptosis requires intact PML and c-Jun functions. In this respect, it is worth noting that cis-pt–induced apoptosis also depends on functional c-Jun.25
Figure 2.

Role of PML and c-Jun in UV-induced apoptosis. (A) Inhibition of c-Jun transcriptional function results in reduction of cell death upon UV radiation. Pml+/+ and Pml-/- MEFs were infected with pBabe () or with a DN–c-Jun (▪). After 2 days of selection, cells were left untreated or UV-irradiated (60 J/m2). Cell death was measured by trypan blue uptake at 24 hours after the irradiation. Bars indicate fold induction of cell death over unirradiated controls. DN–c-Jun levels were measured in pBabe (B)- and DN–c-Jun (DN)–infected cells by using an anti–c-Jun antibody directed against the c-Jun C-terminus. Actin is shown as loading control. Data shown are representative of 3 independent experiments performed in duplicate. (B) c-Jun overexpression potentiates UV-triggered cell death. Pml+/+ and Pml-/- MEFs were infected with pBabe () or c-Jun (▪) retroviruses and UV-treated as described earlier. Cell death was measured by trypan blue uptake at 24 hours after irradiation. Bars indicate fold induction of cell death over unirradiated controls. c-Jun levels were measured in pBabe (B)- and c-Jun (J)–infected cells by using an anti–c-Jun antibody directed against the c-Jun N-terminus. Actin is shown as control of loading. Data shown are representative of 3 independent experiments performed in duplicate. Error bars indicate standard deviation.
Because DN–c-Jun can in principle inhibit the transcriptional activity of other c-Jun family members, we analyzed whether c-Jun itself could potentiate UV-induced cell death. In unirradiated cells, c-Jun overexpression caused increased proliferation and reduced basal cell death in both wild-type and Pml-/- MEFs (not shown). By contrast, UV-induced cell death was further increased by c-Jun in wild-type MEFs, while c-Jun was ineffective in Pml-/- cells (Figure 2B). Taken together, these data demonstrate that c-Jun exerts a proapoptotic function upon UV radiation and that PML plays an important role in executing this function.
c-Jun transcriptional activity is impaired by Pml inactivation
We then studied the molecular mechanisms by which PML regulates this pathway. c-Jun transcriptional activity is enhanced following UV exposure.25 Therefore, we tested the effect of PML overexpression on c-Jun transcriptional activity in MEFs upon UV radiation. Upon UV irradiation, transactivation by a GAL4–c-Jun construct was induced in MEFs (Figure 3A). Overexpression of PML strongly potentiated UV-triggered c-Jun transcriptional activation (Figure 3A) in a dosedependent manner (not shown). Importantly, coactivation of c-Jun by PML was only observed in irradiated cells, thus demonstrating that PML-mediated regulation of c-Jun transcriptional activity is UV radiation dependent (Figure 3A). We next compared GAL4–c-Jun activity in Pml+/+ and Pml-/- MEFs and found that the UV-dependent transcriptional activation of c-Jun was significantly impaired in the absence of PML (Figure 3B). This defect was directly due to the absence of PML, since reintroduction of PML into Pml-/- MEFs almost completely rescued c-Jun–regulated transcription (Figure 3B) in a dose-dependent fashion (not shown). We next studied the transcriptional activity of endogenous c-Jun upon UV radiation. To this end, we tested 2 types of c-Jun responsive elements: multimerized TPA-responsive elements (TREs), which bind c-Jun/c-Fos heterodimers, and CRE-like sequences, which bind c-Jun/ATF-2 dimers.26,27 TRE activity was not significantly induced upon UV radiation in both wild-type and Pml-/- MEFs (not shown). By contrast, CRE basal activity was clearly enhanced in Pml+/+ MEFs upon UV radiation, while, once again, in Pml-/- MEFs its transactivation was completely impaired (Figure 3C).
Figure 3.

PML regulates c-Jun transcriptional activation upon UV irradiation. (A) PML coactivates c-Jun upon UV irradiation. Activation of GAL4–c-Jun by PML in MEFs. Pml+/+ MEFs were transfected with the GAL4-Luciferase reporter construct and the indicated combinations of GAL4–c-Jun and PML. After 36 hours, cells were left untreated () or UV-irradiated with 40 J/m2 (▪). Reporter activity was measured 3 hours after irradiation. Luciferase is expressed as arbitrary light units and normalized to an internal Renilla luciferase control. Data shown are the mean ± SD of 3 independent experiments performed in triplicate. (B) PML is required for UV-dependent activation of GAL4–c-Jun. Pml+/+ () and Pml-/- (▪) MEFs were transfected with the GAL4-Luciferase reporter construct and the indicated combinations of GAL4–c-Jun and PML and subsequently UV-treated (40 J/m2) for 3 hours. Data shown are the mean ± SD of 3 independent experiments performed in triplicate. (C) The transcriptional activity of endogenous c-Jun is impaired in Pml-/- cells. Pml+/+ and Pml-/- MEFs were transfected with a jun2CRE-Luc reporter plasmid. After 24 hours, transfected cells were left untreated () or UV-treated with 60 J/m2 (▪). Reporter activity was measured 3 hours after irradiation and normalized to an internal Renilla luciferase control. UV-triggered jun2CRE-Luc transcription is expressed as fold induction over luciferase activity in untreated cells. Luciferase activity in untreated cells is arbitrarily shown as 1.0. Data shown are the mean ± SD of 3 independent experiments performed in duplicate. (D) In vivo c-Jun binding to CRE sites is induced by UV irradiation and is defective in the absence of PML. Pml+/+ and Pml-/- MEFs (indicated as +/+ and-/-) were left untreated or UV-irradiated (40 J/m2) and then cultured for 2 hours. Cross-linked chromatin from untreated or UV-treated Pml+/+ and Pml-/- cells was incubated with anti–c-Jun antibody or with control rabbit immunoglobulin G (IgG). Immunoprecipitates were analyzed by PCR with primers specific for jun1 and jun2 regions of the c-jun promoter and for the collagenase (coll) promoter. A sample representing 0.5% of total input chromatin (input) was included in the PCR reactions. Data shown are representative of 3 independent experiments.
We next investigated whether PML influences c-Jun transcriptional activity by regulating its DNA binding ability and found that, upon UV irradiation, the c-Jun/ATF-2 binding activity was clearly induced in wild-type MEFs, while, remarkably, it was impaired in Pml-/- cells (Figure S2). Supershift analysis using anti–c-Jun and anti–ATF-2 antibodies confirmed the presence of both c-Jun and ATF-2 in the CRE complex (Figure S2). Similar results were obtained in UV radiation cross-linking experiments (not shown).
We next set to determine whether c-Jun DNA binding activity was influenced by Pml inactivation in vivo by chromatin immunoprecipitation experiments (ChIP) using an anti–c-Jun antibody. We studied the binding of c-Jun to its own promoter, which contains 2 CRE sequences, termed jun1 and jun2.27 The binding of c-jun to both jun1 and jun2 promoter regions was induced by UV radiation in Pml+/+ cells (Figure 3D). By contrast, we observed a reproducible and consistent reduction of c-Jun binding to both the jun1 and jun2 in Pml-/- cells (Figure 3D). ChIP pull-downs from untreated or UV-treated Pml+/+ cells contained almost undetectable amounts of collagenase promoter fragments containing a TRE site (Figure 3D). All together these data demonstrate that PML is important for c-Jun transcriptional activity and in vivo DNA binding upon UV irradiation.
PML localization is disrupted upon UV irradiation
We then analyzed the effects of UV irradiation on the PML-NB. In unirradiated MEFs, PML was found typically concentrated in 10 to 15 PML-NBs/nucleus (Figure 4A). Strikingly, PML relocalized into multiple microspeckles upon UV radiation in both MEFs and primary human fibroblasts (Figure 4A, and not shown). This phenomenon affected more than 60% of cells. By contrast, treatment of MEFs with γ-rays did not result in such changes (data not shown). A recent study reported that PML relocalization upon UV exposure is p53 dependent in tumor cell lines.28 Nevertheless, PML is able to relocalize to microspeckles in p53-/- MEFs, thus indicating that this phenomenon is not p53 dependent in primary cells (not shown). Similar to UV radiation, cis-pt treatment resulted in PML delocalization (Figure 4B). The UV-induced microspeckles partitioned with the nuclear insoluble fraction and the effect was not accompanied by an increase in PML protein levels (not shown). We next studied the localization of other PML-NB components. SUMO1 and DAXX were found to relocalize along with PML into the UV-induced microspeckles (Figure 4C-D). While CBPand p53 accumulate into the PML-NB upon γ-rays,10 they were not found to accumulate in the UV-induced microspeckles (Figure 4E, and not shown). Thus, the regulation of c-Jun transcriptional function by PML occurs along with its delocalization from the PML-NB into novel microspeckled nuclear structures, hereafter referred to as UV-nuclear bodies (UV-NBs). A recent report demonstrated that PML deSUMOylation results in the reorganization of the PML-NB and in c-Jun activation.29 However, we were unable to detect changes in PML SUMOylation upon UV radiation (not shown). Moreover, SUMO1 localizes to the UV-NB (Figure 4C), suggesting that PML delocalization upon UV radiation might be triggered by different signals or posttranslational modifications. Overall, this evidence suggests that the function of PML upon UV radiation is PML-NB independent.
Figure 4.
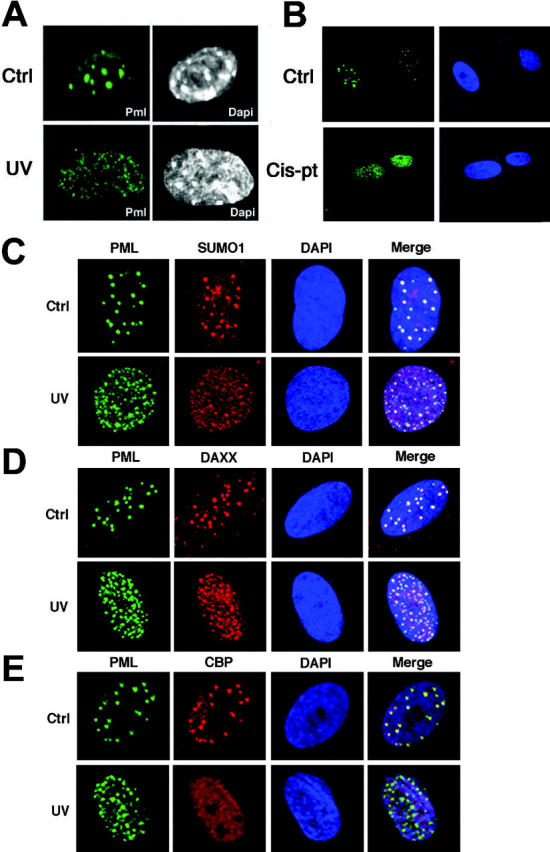
PML relocalizes into novel microspeckles upon UV radiation. (A) MEFs were UV-irradiated (60 J/m2) and then stained with an antibody against mouse PML. Nuclei were visualized by DAPI (4′,6-diamidino-2-phenylindole). (B) PML is delocalized upon cisplatin treatment. WI-38 cells were treated with cis-pt at 10 μg/mL for 6 hours and subsequently fixed and stained with an antibody against human PML (green). Nuclei were visualized by DAPI. (C-E) Analysis of PML-NB components in UV-irradiated fibroblasts. (C) WI-38 cells grown on coverslip were UV-irradiated (60 J/m2), stained with antibodies anti-PML (green) and anti-SUMO1 (red), and analyzed by confocal fluorescence microscopy. Nuclei were visualized by DAPI (blue). Colocalization of PML and SUMO1 is shown (yellow). (D) WI-38 cells were stained with antibodies against PML (green) and DAXX (red). Colocalization of PML and DAXX is shown (yellow). (E) Cells were stained with antibodies against PML (green) and CBP (red). Colocalization of PML and CBP is shown (yellow).
PML and UV-activated c-Jun colocalize and physically interact
As PML regulates p53 function in the PML-NBs upon γ-ray irradiation,10 we next set to determine whether UV light would cause c-Jun to colocalize with PML in the UV-NBs. While an antibody against total c-Jun showed a rather diffuse staining upon UV radiation (not shown), an antibody recognizing phosphorylated c-Jun (P–c-Jun) detected nuclear microspeckles in more than 80% of irradiated cells, in both primary human fibroblasts and MEFs (Figure 5A, and not shown). Remarkably, a statistically significant colocalization of P–c-Jun and PML in the UV-NB was found in primary fibroblasts at the endogenous level (Figure 5A; Figure S3). These nuclear speckles might potentially represent sites of active c-Jun–dependent transcription. In agreement with the fact that UV induces c-Jun phosphorylation, we did not observe P–c-Jun staining in the majority of unirradiated cells, and P–c-Jun was never found in the PML-NB in untreated cells (not shown). Since c-Jun and PML colocalize upon UV radiation, we tested whether they physically interact in vivo and observed that endogenous PML and c-Jun proteins were readily coimmunoprecipitated from UV-irradiated fibroblasts (Figure 5B). To test whether the c-Jun/PML interaction was direct, we performed GST pull-down experiments by using GST–c-Jun and in vitro–translated PML. GST–c-Jun and PML directly interacted (Figure 5C). We next identified the RBCC N-terminal domain as the PML region responsible for the PML/c-Jun interaction, since a PML mutant lacking the RBCC did not interact with GST–c-Jun (Figure 5C).
Figure 5.

PML and c-Jun colocalize and interact. (A) PML colocalizes with phosphorylated c-Jun into the UV-NB. Primary human fibroblasts were UV-treated (60 J/m2) and stained with antibodies against PML (green) and phosphorylated c-Jun (Ser 63) (P–c-Jun; red). Arrows indicate sites of PML and P–c-Jun colocalization (yellow). (B) PML interacts with c-Jun. WI-38 cells were left untreated or UV-treated (60 J/m2) and lysed after 3 hours. Anti-PML immunoprecipitates were probed with antibodies against c-Jun or PML. An isotypic IgG antibody (ISO) was used as negative control of coimmunoprecipitation. (C) c-Jun and PML directly interact. GST–c-Jun glutathione beads were incubated with in vitro–translated PML, PML RBCC (RING finger-B box-coiled coil), or PML ΔRBCC and then extensively washed. The input is 20% of the in vitro–translated PML used in the pull-down experiment. Data shown are representative of 3 independent experiments. (D) A model for PML function upon distinct genotoxic stresses. While PML regulates p53 upon γ-irradiation, it selectively modulates c-Jun proapoptotic function upon UV irradiation.
Discussion
Accumulating evidence indicates that PML exerts its tumor suppressive function by regulating cellular senescence and promoting apoptosis.5 PML is part of a p53-controlled tumor suppressive pathway for the induction of senescence upon oncogenic transformation.5 Furthermore, PML and p53 functionally interact during apoptosis induced by γ-irradiation in thymocytes.10 However, PML also controls apoptotic pathways that do not rely on p53.5 The mechanisms regulating the induction of apoptosis upon UV irradiation has been the object of intense research efforts in the past 2 decades. Several cellular pathways can modulate this process. The p53 tumor suppressor has been shown to mainly modulate cell cycle arrest in UV-irradiated primary cells, while its role in apoptosis is unclear.13-17 Disruption of the c-Jun/JNK pathway results in alteration of apoptosis upon UV radiation.18,21 However, although the c-Jun/JNK pathway has been implicated in the regulation of apoptosis upon UV radiation,18,19,21 the precise molecular mechanisms underlying its activation upon cellular stress remain largely unknown. In the present report, we proved that c-Jun indeed mediates UV-triggered cell death and provide direct evidence that PML selectively regulates distinct transcription factors upon different DNA-damaging agents: p53 upon γ-irradiation and c-Jun upon UV irradiation (Figure 5D). Importantly, we demonstrate that PML modulates the proapoptotic function of c-Jun upon UV radiation by potentiating its transcriptional activity.
Strikingly, UV light induces the disruption of the PML-NB and the formation of novel microspeckled structures, the UV-NB, in which PML and phosphorylated c-Jun dynamically accumulate. The formation of these structures does not rely on p53, since it normally occurs in UV-irradiated p53-/- MEFs (not shown). By contrast, upon γ-rays the PML-NB is not disrupted and serves a critical role as a center for p53 modification and transcriptional activation.10 On the basis of our data, we, therefore, propose a model by which the nuclear dynamics of PML upon distinct apoptotic stimuli dictate the selective activation of different proapoptotic transcription factors.
Interestingly, while the PML tumor suppressor regulates c-Jun activity in a strict UV-dependent fashion, we find that the PML-RARα oncoprotein of APL acts as a constitutive and UV-independent c-Jun transcriptional coactivator (data not shown). Thus, it could be envisioned that PML-RARα constitutively triggers the oncogenic potential of c-Jun, while PML would specifically regulate c-Jun proapoptotic function modulating its UV-dependent role. As point mutations of the PML gene have been recently discovered in aggressive cases of APL,30 it would be intriguing to test whether these mutants are defective in activating c-Jun upon DNA damage, thus further protecting APL cells from cell death induced upon DNA damage. Furthermore, this pathway could be altered in solid tumors, as cancers of various histologic origins have been found to lack expression of the PML protein.31
Supplementary Material
Acknowledgments
We thank Maria Barna, Francesca Bernassola, Davide Ruggero, Stefano Cairo, Len Zon, Jennifer Best, and John Petrini for useful discussion. We also thank Scott Lowe for mouse anti-PML antibodies and Katia Manova for confocal studies.
Prepublished online as Blood First Edition Paper, December 30, 2004; DOI 10.1182/blood-2004-09-3782.
Supported by the National Institutes of Health (NIH) (CA-71692 to P.P.P.). R.B. was the recipient of an NIH T32 Training Grant.
The online version of the article contains a data supplement.
An Inside Blood analysis of this article appears in the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Pandolfi PP, Grignani F, Alcalay M, et al. Structure and origin of the acute promyelocytic leukemia myl/RAR alpha cDNA and characterization of its retinoid-binding and transactivation properties. Oncogene. 1991;6: 1285-1292. [PubMed] [Google Scholar]
- 2.de The H, Lavau C, Marchio A, et al. The PML-RAR alpha fusion mRNA generated by the t(15; 17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66: 675-684. [DOI] [PubMed] [Google Scholar]
- 3.Kakizuka A, Miller WH Jr, Umesono K, et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell. 1991;66: 663-674. [DOI] [PubMed] [Google Scholar]
- 4.Goddard AD, Borrow PS, Freemont PS, Solomon E. Characterization of a zinc finger gene disrupted by the t(15;17) in acute promyelocytic leukemia. Science. 1991;254: 1371-1374. [DOI] [PubMed] [Google Scholar]
- 5.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108: 165-170. [DOI] [PubMed] [Google Scholar]
- 6.Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene. 2001;20: 7223-7233. [DOI] [PubMed] [Google Scholar]
- 7.Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of the PML-NB. Nat Cell Biol. 2000;2: E85-E90. [DOI] [PubMed] [Google Scholar]
- 8.Wang ZG, Delva L, Gaboli M, et al. Role of PML in cell growth and the retinoic acid pathway. Science. 1998;279: 1547-1551. [DOI] [PubMed] [Google Scholar]
- 9.Zhong S, Muller S, Ronchetti S, et al. Role of SUMO-1-modified PML in nuclear body formation. Blood. 2000;95: 2748-2752. [PubMed] [Google Scholar]
- 10.Guo A, Salomoni P, Luo J, et al. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2: 730-736. [DOI] [PubMed] [Google Scholar]
- 11.Pearson M, Carbone R, Sebastiani C, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406: 207-210. [DOI] [PubMed] [Google Scholar]
- 12.Wang ZG, Ruggero D, Ronchetti S, et al. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20: 266-272. [DOI] [PubMed] [Google Scholar]
- 13.Brugarolas J, Chandrasekaran C, Gordon JI, et al. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377: 552-557. [DOI] [PubMed] [Google Scholar]
- 14.Waldman T, Lengauer C, Kinzler KW, Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature. 1996;381: 713-716. [DOI] [PubMed] [Google Scholar]
- 15.Bissonnette N, Hunting DJ. p21-induced cycle arrest in G1 protects cells from apoptosis induced by UV-irradiation or RNA polymerase II blockage. Oncogene. 1998;16: 3461-3469. [DOI] [PubMed] [Google Scholar]
- 16.Smith ML, Fornace AJ Jr. p53-mediated protective responses to UV irradiation. Proc Natl Acad Sci U S A. 1997;94: 12255-12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lackinger D, Kaina B. Primary mouse fibroblasts deficient for c-Fos, p53 or for both proteins are hypersensitive to UV light and alkylating agent-induced chromosomal breakage and apoptosis. Mut Res. 2000;457: 113-123. [DOI] [PubMed] [Google Scholar]
- 18.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288: 870-874. [DOI] [PubMed] [Google Scholar]
- 19.Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21: 326-329. [DOI] [PubMed] [Google Scholar]
- 20.Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999;18: 188-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaulian E, Schreiber M, Piu F, et al. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 2000;103: 897-907. [DOI] [PubMed] [Google Scholar]
- 22.Boyd KE, Wells J, Gutman J, Bartley SM, Farnham PJ. c-Myc target gene specificity is determined by a post-DNA binding mechanism. Proc Natl Acad Sci U S A. 1998;95: 13887-13892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci. 2000;57: 1229-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young MR, Li JJ, Rincon M, et al. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci U S A. 1999;96: 9827-9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103: 239-252. [DOI] [PubMed] [Google Scholar]
- 26.van Dam H, Duyndam M, Rottier R, et al. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12: 479-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dam H, Castellazzi M. Distinct roles of Jun: Fos:ATF dimmers in oncogenesis. Oncogene. 2001;20: 2453-2464. [DOI] [PubMed] [Google Scholar]
- 28.Seker H, Rubbi C, Linke SP, et al. UV–c-induced DNA damage leads to p53-dependent nuclear trafficking of PML. Oncogene. 2003;22: 1620-1628. [DOI] [PubMed] [Google Scholar]
- 29.Best JL, Ganiatsas S, Agarwal S, et al. SUMO-1 protease-1 regulates gene transcription through PML. Mol Cell. 2002;10: 843-855. [DOI] [PubMed] [Google Scholar]
- 30.Gurrieri C, Nafa K, Merghoub T, et al. Mutations of the PML tumor suppressor gene in acute promyelocytic leukemia. Blood. 2004;103: 2358-2362. [DOI] [PubMed] [Google Scholar]
- 31.Gurrieri C, Capodieci P, Bernardi R, et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst. 2004;96: 269-279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}