

Abstract
We demonstrate the use of a DNA minicircle competition binding assay, together with DNA cyclization kinetics and gel-phasing methods, to show that the DNA-binding domains (dbd) of the heterodimeric leucine zipper protein Fos–Jun do not bend the AP-1 target site. Our DNA constructs contain an AP-1 site phased by 1–4 helical turns against an A-tract-directed bend. Competition binding experiments reveal that (dbd)Fos–Jun has a slight preference for binding to linear over circular AP-1 DNAs, independent of whether the site faces in or out on the circle. This result suggests that (dbd)Fos–Jun slightly stiffens rather than bends its DNA target site. A single A-tract bend replacing the AP-1 site is readily detected by its effect on cyclization kinetics, in contrast to the observations for Fos–Jun bound at the AP-1 locus. In contrast, comparative electrophoresis reveals that Fos–Jun–DNA complexes, in which the A-tract bend is positioned close (1–2 helical turns) to the AP-1 site, show phase-dependent variations in gel mobilities that are comparable with those observed when a single A-tract bend replaces the AP-1 site. Whereas gel mobility variations of Fos–Jun–DNA complexes decrease linearly with increasing Mg2+ contained in the gel, the solution binding preference of (dbd)Fos–Jun for linear over circular DNAs is independent of Mg2+ concentration. Hence, gel mobility variations of Fos–Jun–DNA complexes are not indicative of (dbd)Fos–Jun-induced DNA bending (upper limit 5°) in the low salt conditions of gel electrophoresis. Instead, we propose that the gel anomalies depend on the steric relationship of the leucine zipper region with respect to a DNA bend.
DNA bending by proteins has been studied by x-ray crystallography (1, 2), NMR spectroscopy (3), gel-based circular permutation (4) and phasing assays (5), and more recently by solution-based DNA cyclization kinetics (6, 7). Circular permutation of a protein-binding site within a DNA fragment and phasing of a binding site against a sequence-directed bend have reliably indicated bend location and direction in many protein–DNA systems. These gel methods to study DNA bending are based on predictions from electrophoretic theory that bent DNA helices will migrate slower than straight helices because of their shorter average end-to-end distances (8). In the application of these methods to study protein-induced DNA bending, DNA conformation is assumed to dominate the gel mobilities of protein–DNA complexes. However, recent studies have indicated that in specific cases, the conformations of proteins may also affect the migration of protein–DNA complexes through a gel matrix, giving rise to anomalous results (7, 9).
Recent work has suggested that the elongated and helical structures of leucine zipper proteins dominate the gel mobilities of their complexes with DNA. Gel-based assays have yielded contradictory results in the case of leucine zipper proteins including eukaryotic oncoproteins Fos and Jun (9–11) and the yeast transcriptional factor GCN4 (12). Results obtained from a sensitive gel-phasing assay (10, 13) have been interpreted to show that full-length Fos–Jun and several peptide fragments of Fos–Jun bend DNA away from the protein surface and toward the minor groove by ≈10–25° (13–18). However, our previously reported observations (9) based on gel phasing and solution-based DNA cyclization experiments indicate that both full-length Fos–Jun and DNA-binding domains of Fos and Jun [(dbd)Fos–Jun] do not bend DNA (upper limit 5°), in agreement with the x-ray crystal structure of the Fos–Jun–DNA complex (19). Similarly, whereas gel-based circular permutation experiments (12) suggest that GCN4 bends DNA significantly, gel phasing (12, 20) and DNA cyclization experiments (21) reveal that GCN4 does not induce curvature at the AP-1 recognition site, again in agreement with x-ray crystallography (22–24). Furthermore, more recently it was shown that gel mobility anomalies induced by the Myc/Max basic helix–loop–helix/leucine zipper proteins are a function of the presence and length of the leucine zipper protein motif and probably not an indication of structural distortions of the DNA target site (7). These studies have highlighted the importance of the development and use of solution-based methods, which do not depend on gel migrations, to study DNA bending by proteins with elongated conformations.
In this report we use a combination of solution and gel methods to reinvestigate DNA bending by (dbd)Fos–Jun, a heterodimer of Fos(118–211) and Jun(199–334). This heterodimer contains not only the minimal bZIP domains of Fos and Jun but also additional regions at the amino-terminal ends that are believed to promote DNA bending by an electrostatic mechanism (16, 18, 25). We use an independent minicircle competition binding assay to study DNA bending by proteins in solution. This assay is based on the thermodynamic premise that a protein that bends DNA will bind preferentially to a properly phased bent rather than linear DNA substrate. Based on the previously established quasi thermodynamic cycle between cyclization and protein binding (6), this assay provides an independent means of checking DNA cyclization kinetics results. The minicircle binding assay reveals that (dbd)Fos–Jun has similar binding affinities for both circular and linear AP-1 DNA, confirming our previously reported DNA cyclization results, which show that Fos–Jun does not bend DNA in solution (9).
We have also carried out studies aimed at understanding the origin of gel mobility anomalies detected in studies of Fos–Jun with DNA (10, 13, 15–18). We observe that gel mobility differences of (dbd)Fos–Jun bound to differently phased DNA constructs is a function of the ionic-strength of the gel-running buffer. Our results suggest that gel-buffer conditions may affect the conformational flexibility of the leucine-zipper region and therefore the extent of anomalous gel mobility patterns induced by these zipper proteins. We propose that gel mobility anomalies of Fos–Jun–DNA complexes are a function of the conformation and orientation of the helical leucine-zipper region with respect to a DNA helix, and not a reflection of DNA bending by Fos–Jun.
MATERIALS AND METHODS
Preparation of DNA Constructs.
Previously reported 158-bp DNA constructs 17A9, 13A13, and 9A17 containing AP-1 DNA from the his3–189 supersite or the human collagenase gene (hcg) were used in these studies (9). These constructs contain phasing linkers (defined as the distance in bp from the center of the closest A-tract to the center of the AP-1 site) of 35, 39, and 43 bp and are therefore also referred to as hcg/yeastDNA35, hcg/yeastDNA39, and hcg/yeastDNA43, respectively. In addition, a set of DNA constructs hcgDNA11, hcgDNA15, and hcgDNA19 (Fig. 1) that contain phasing linkers of 11, 15, and 19 bp, respectively, were prepared as follows. Mutagenic PCR methods were used to construct a 122-bp fragment from template molecule 17A9 with primers (JA13) 5′-CGCCATGGAATCGATGAATTC and mutagenic primer 5′-CCTAGGTCTAGAATTCGGAACCGGTTT (containing a unique AgeI site). The 122-bp PCR product was then restricted with AgeI to yield a 96-bp fragment containing 6 A-tracts from 17A9 with a 4-bp overhang. Synthetic DNA oligonucleotides 5′-CCGGTGACTCATTGTCGATTGCTCGAATCCGAATTCTAGACCTAGGATGCGCTACAACGTGCTGCC-3′ (66 bp) and 5′-GGCAGCACGTTGTAGCGCATCCTAGGTCTAGAATTCGGATTCGAGCAATCGACAATGAGTCA-3′ (62 bp) containing the 7-bp AP-1 site from hcg (underlined) were hybridized to form a duplex with a 4-bp overhang. The hybridized duplex was ligated to the above 96-bp fragment to create a 160-bp final template molecule for PCR. As previously described (9) primers JA13 and 5′-GCAGATATCGATTCCATGGCAGCACGTTGTAGC were used in PCR to synthesize internally radiolabeled AP-1 DNA followed by restriction with ClaI to yield the product 158-bp molecule hcgDNA11 (Fig. 1). Molecules hcgDNA15 and hcgDNA19 were prepared by using a similar strategy, with the use of appropriate pairs of synthetic duplexes containing the AP-1 site moved internally by 4 and 8 bp, respectively, with respect to hcgDNA11. Similarly, a set of control molecules AtractDNA11, AtractDNA15, and AtractDNA19 (Fig. 1) containing a single 6-bp A-tract in place of the AP-1 site was prepared by using the above procedure.
Figure 1.

DNA constructs (158 bp) with short phasing linkers. The top three DNA constructs contain the AP-1 site (in bold) from the hcg. The center of the AP-1 site is positioned 11, 15, and 19 bp, respectively, from the center of the closest A-tract sequence. The bottom three DNAs are control molecules that contain a single A-tract site in place of the AP-1 site.
Minicircle Binding Assay.
DNA minicircles were prepared by overnight ligation of a 300-μl reaction mixture containing 4 nM ClaI-restricted radiolabeled PCR product and 5000 units/ml T4 DNA ligase at 21°C. The reaction mixtures were phenol/chloroform-extracted, and the minicircles subsequently were purified on a 5% polyacrylamide gel.
The relative affinities of (dbd)Fos–Jun for circular vs. linear AP-1 DNAs were determined by a competitive binding experiment. N-terminal hexahistidine fusion forms of (dbd)Fos–Jun, corresponding to amino acids 118–211 of Fos and 199–334 of Jun, were a generous gift from T. K. Kerppola and D. A. Leonard (University of Michigan School of Medicine). The specific conditions used to prepare (dbd)Fos–Jun heterodimer and to assay its DNA binding were identical to those previously reported (9, 26, 27). A reaction mixture containing 1–4 nM linear DNA was incubated with ≈5 nM (dbd)Fos–Jun for 2 min, followed by the addition of 1–3 nM circular DNA. In a parallel experiment, circular DNA was incubated with (dbd)Fos–Jun followed by the addition of linear DNA. Limiting concentrations of protein were used in these experiments to ensure a competitive binding condition in which approximately 50% of the DNA substrates bound to protein. The two order of addition experiments were essential to establish whether the system had reached equilibrium. The reaction mixtures were then incubated for 30–60 min and analyzed on 8% native gels (75:1, acrylamide/N,N′-methylenebisacrylamide, 50 mM TBE, 1.5 mm thick, 250 V) at 21°C. The gels were phosphoroimaged and quantitated with a FUJIX Bio-imaging Analyzer System (BAS 1000) with MacBas.
The relative binding affinity (Krel) of (dbd)Fos–Jun for linear vs. circular DNA was calculated based on the following analyses.
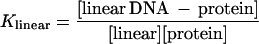 |
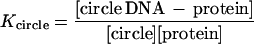 |
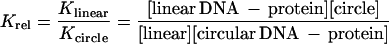 |
DNA Cyclization and Gel Phasing.
DNA cyclization of molecules AtractDNA11, AtractDNA15, and AtractDNA19 was exactly carried out according to previously reported procedures (9). (dbd)Fos–Jun bound to differently phased constructs hcgDNA11, hcgDNA15, and hcgDNA19 were analyzed on 8% native gels (75:1, acrylamide/N,N′-methylenebisacrylamide, 50 mM TBE, 0.75 mm thick, 200 V) at 21°C by constant temperature gel electrophoresis in the presence of increasing amounts of Mg2+ (0–3 mM). The Mg2+ ion concentration in the gel and buffer was maintained by constant recirculation of the buffer between the top and bottom chambers.
RESULTS
Competition Binding Assay.
If a protein bends DNA, it will bind preferentially to properly phased circular DNAs as opposed to linear DNAs (6). Based on this thermodynamic premise, we investigated whether (dbd)Fos–Jun would preferentially bind to appropriately phased minicircle DNAs containing the AP-1 target site. Competition binding was carried out with sets of DNAs that successively differ in their phasings by 4 bp (Fig. 2 and Table 1). For all three DNA isomers, (dbd)Fos–Jun binds preferentially to linear over circular DNA, an effect opposite to that predicted for a protein that bends DNA. This binding preference is independent of the nature of the sequences flanking the AP-1 site, as observed in the comparison of AP-1 DNAs from yeast and humans. Furthermore, the Krel values are independent of the lengths of the phasing linkers; (dbd)Fos–Jun shows a similar preference in binding AP-1 molecules with short (11–19 bp) and long (35–43 bp) phasing linkers (Table 1). Importantly, in binding buffers containing 5 mM Mg2+, the measured apparent relative binding affinity (Krel) of (dbd)Fos–Jun for DNA is independent of the order in which circular and linear DNA is added to the individual reaction mixtures (Fig. 2). Thus in buffers containing 5 mM Mg2+, the relative amounts of (dbd)Fos–Jun–DNA adducts measured are true representations of equilibrium binding and not reflective of reaction kinetics.
Figure 2.

Competition binding of (dbd)Fos–Jun to differently phased linear and circular DNA isomers. Lanes 1a and 1b contain isomer hcgDNA11, 2a and 2b contain isomer hcgDNA15, and 3a and 3b contain isomer hcgDNA19. The measured Krel (Klinear/Kcircle) values are relatively constant across the differently phased DNAs and the different orders of addition.
Table 1.
Relative binding affinities (Krel) of (dbd)Fos–Jun for linear and circular DNAs: Effect of DNA length and DNA phasing
| DNA | DNA length, bp | Phasing* | Krel (Klinear/Kcircle) | [J(−fj)/J(+fj)]† |
|---|---|---|---|---|
| hcgDNA11 | 158 | 11 | 1.3 | |
| hcgDNA15 | 158 | 15 | 1.4 | |
| hcgDNA19 | 158 | 19 | 1.2 | |
| hcgDNA35 | 158 | 35 | 1.7 | |
| hcgDNA39 | 158 | 39 | 1.6 | |
| hcgDNA43 | 158 | 43 | 1.5 | |
| hcgDNA35 | 154 | 35 | 1.4 | |
| hcgDNA39 | 154 | 39 | 1.3 | |
| hcgDNA43 | 154 | 43 | 1.4 | |
| yeastDNA35 | 158 | 35 | 1.4 | 0.80 |
| yeastDNA39 | 158 | 39 | 1.3 | 1.10 |
| yeastDNA43 | 158 | 43 | 1.2 | 0.71 |
The Krel values are obtained in buffers containing 5 mM Mg2+ and represent averages of at least three experiments.
The phasing represents the distance between the centers of the AP-1 site and the closest A-tract sequence.
The term [J(−fj)/J(+fj)] is the ratio of the J factors (cyclization probabilities) in the absence and presence of Fos–Jun, calculated from the data in ref. 9.
To further ensure the accuracy of the binding preferences observed, competition binding was carried out with DNAs of different lengths. Competition binding experiments used to detect protein-induced DNA bending will yield consistent results only for proteins that do not affect the extent of DNA twisting at their target site. Therefore, irrespective of protein-induced DNA bending, a protein that unwinds or overwinds DNA will have different Krel values for minicircles of different lengths. Specifically a protein that unwinds DNA will prefer to bind to an underwound circle as opposed to an overwound circle. For a protein that overwinds DNA, the effects will be reversed. Therefore, competition binding was carried out with DNAs that are 2 bp longer (158 bp) and shorter in length (154 bp) than the previously determined (9) optimum 156-bp length for these DNA circles. We observe that the relative binding affinities of (dbd)Fos–Jun for linear vs. circular DNAs are independent of DNA circle length (Table 1), consistent with our earlier observation that Fos–Jun does not unwind or overwind the AP-1 target site (9). This result further validates our conclusion that Fos–Jun does not bend the AP-1 recognition site.
Competition Binding and DNA Cyclization.
A thermodynamic relationship between binding and DNA cyclization was previously demonstrated in the studies of DNA bending by the catabolite activator protein (6). In this way, competition binding provides a check and confirmation of the results obtained from DNA cyclization. The relative binding affinities of (dbd)Fos–Jun for linear and circular DNA substrates are equivalent, within a factor of 2, to the previously reported relative J factors for DNA cyclization in the absence and presence of Fos–Jun (Table 1) (9).
To determine the sensitivity of the DNA cyclization assay to small DNA bends, we carried out cyclization of control molecules AtractDNA11, AtractDNA15, and AtractDNA19 (Fig. 1) containing a variably phased single A-tract reference bend (≈18°) in place of the AP-1 site. The results indicate (Fig. 3) that a bend of this magnitude induces a significant 15–17-fold effect on the J factors of in-phase as compared with out-of-phase DNAs. This indicates that previously proposed Fos–Jun-induced DNA bends of 10–25° (10, 13, 15–18) should be easily detectable by DNA cyclization kinetics. Because of its equivalence to the cyclization assay, the competition binding assay should also easily detect bends of this magnitude. However, the lack of any detectable effect of Fos–Jun on cyclization and binding clearly indicates the absence of Fos–Jun-induced DNA curvature. We estimate from theoretical simulations (21) that a protein-induced DNA bend of 5° should produce roughly a 5-fold variation in the J factors and Krel values for differently phased DNA constructs.
Figure 3.

Cyclization kinetic measurements on out-of-phase AtractDNA15 (Left) and in-phase AtractDNA19 (Right). (Lower) Determination of cyclization and bimolecular ligation rate constants and J factors for each isomer.
Effect of Mg2+ on Gel Electrophoretic Phasing Analyses.
Distinct gel mobility differences were observed for (dbd)Fos–Jun bound to oppositely phased DNAs (hcgDNA11, hcgDNA15, and hcgDNA19) with short phasing linkers (Figs. 2 and 4). The magnitude and direction of the observed gel mobility amplitudes are in agreement with previously reported gel-phasing experiments carried out with DNAs containing short phasing linkers (10, 13, 15, 16, 18). By using the standard interpretation, the data suggest that (dbd)Fos–Jun bends DNA toward the minor groove by a magnitude that is comparable with an A-tract-directed bend (Fig. 4) of ≈18°. In contrast to these observations, our earlier report indicates that (dbd)Fos–Jun and full-length protein Fos–Jun bound to DNAs with long phasing linkers (35–43 bp) show no significant gel mobility differences.
Figure 4.

(A) Effect of Mg2+ on the gel mobility shifts of (dbd)Fos–Jun bound to differently phased linear DNA isomers. The Mg2+ concentration refers to that contained in the gel and running buffer. The phasing numbers 11, 15, and 19 refer to AP-1 molecules hcgDNA11, hcgDNA15, and hcgDNA19, except in the A-tract site DNAs, where the phasing refers to molecules AtractDNA11, AtractDNA15, and AtractDNA19. The mobilities of the protein–DNA complexes relative to free DNA are normalized relative to each other. The amplitude is the difference between the maximum and minimum relative mobility values. (B) Linear relationship between relative gel mobility amplitude and Mg2+: comparison of Fos–Jun–DNA complexes and A-tract site DNA. The error bars represent the standard deviations for an average of at least 3 data points.
Interestingly, gel mobility differences decrease linearly with increasing Mg2+ concentrations contained in the gel and running buffer (Fig. 4). Furthermore, the stability of (dbd)Fos–Jun–DNA complexes decreases dramatically in gels containing Mg2+, possibly from an increase in the dissociation of (dbd)Fos–Jun from DNA in high ionic strength conditions. Specifically, (dbd)Fos–Jun–DNA complexes electrophoresed in gels containing ≥3 mM Mg2+ are not clearly detectable (data not shown). Whereas the relative amplitude of phase-dependent variations in the gel mobility of (dbd)Fos–Jun–DNA complexes decreases with increasing Mg2+, relative gel mobility amplitudes of A-tract site-containing DNAs increases with increasing Mg2+ (Fig. 4B). Previous work has shown a similar effect of increasing concentrations of Mg2+, up to about 10 mM, on gel migrations of A-tract containing curved DNAs (28). These data suggest that low ionic strength buffer conditions, in a gel containing no Mg2+, may promote (dbd)Fos–Jun-induced DNA bending. Alternatively, gel mobility variations may arise from altered flexibilities and conformations of (dbd)Fos–Jun in low salt-containing gels.
Effect of Mg2+ on Competition Binding.
To address whether low salt promotes (dbd)Fos–Jun-induced DNA bending, competition binding with circular and linear substrates was carried out in buffers containing varying concentrations of Mg2+. The measured relative binding affinities of (dbd)Fos–Jun for differently phased DNAs remain constant in buffers containing 0–5 mM Mg2+, with (dbd)Fos–Jun reproducibly showing a slight preference for linear over circular substrates (Table 1 and Figs. 2 and 5). However, the dissociation kinetics of (dbd)Fos–Jun bound to DNA is significantly slower in buffers containing ≤2 mM Mg2+ as evidenced from the significant order of addition effect observed (compare Figs. 2 and 5). Specifically, the amount of protein–DNA complex formed with the substrate added first decreases very slowly in the presence of the competitor DNA added second (Fig. 5). The measured Krel values for each order of addition experiment converge over a period of hours to the equilibrium value of ≈1.2–1.4, and the geometric mean of the measured Krel values for each time point remains approximately constant, as simulation of kinetic models for the process suggests should be the case (unpublished results). We therefore conclude that low salt conditions do not give rise to a (dbd)Fos–Jun-induced DNA bend.
Figure 5.

Competition binding (dbd)Fos–Jun to oppositely phased hcgDNA15 and hcgDNA19 isomers in buffers containing 2 mM Mg2+: kinetic order of addition effect. The DNA substrate added first was incubated with limiting amounts of (dbd)Fos–Jun for 2 min, followed by incubation with the DNA substrate added second, for the times indicated in the figure. Note that the geometric means of the measured Krel values, which are constant at the different time points, represent the equilibrium values. The Krel values for both isomers are similar in magnitude indicating that low salt does not promote DNA bending by (dbd)Fos–Jun.
DISCUSSION
The competition binding assay used in this work is an independent method to study protein-induced DNA bending in solution. Our studies show that the heterodimer (dbd)Fos–Jun does not bind preferentially to circular over linear DNAs, confirming our previously reported conclusions based on cyclization kinetics (9) that these proteins do not bend AP-1 DNA in solution. In contrast, (dbd)Fos–Jun prefers to bind to unbent DNAs by a small but reproducible extent, suggesting that it stiffens rather than bends DNA. The relative binding affinities of (dbd)Fos–Jun for linear vs. circular AP-1 DNA substrates are consistently >1, which reflects the energetic cost for (dbd)Fos–Jun to straighten the prebent AP-1 site in minicircular DNAs. Similarly, the yeast transcriptional leucine zipper protein GCN4 (21) and the Myc/Max basic helix–loop–helix/leucine zipper family of proteins (7) also prefer to bind to linear DNAs over circular DNA templates, implying that this may be a general phenomenon associated with leucine zipper proteins. DNA stiffening induced by leucine zipper proteins, which will decrease the inherent flexibility of the protein-binding site, may prevent sliding of the zipper’s two α-helices bound in the major groove of DNA.
The minicircle binding assay is a complementary assay to the previously described method of DNA cyclization (6, 9, 29) and can be carried out under a variety of ionic conditions. The relative binding affinities measured are equivalent within a factor of 2 to the previously reported effects of Fos–Jun on DNA cyclization rates (J factors). Thus both methods can be used interchangeably to study protein-induced DNA bending in solution, and each assay provides an independent verification of the other. The competition binding assay has some advantages in that it is generally easier and more accurate, it can be applied under a variety of ionic conditions, and it can be carried out at low protein concentrations where nonspecific binding does not interfere with the measurements. We verified specific binding by control experiments in which no electrophoretic mobility shifts were observed when Fos–Jun was added to the molecules in which the AP-1 site is replaced by a single A-tract (data not shown).
The observation that Fos–Jun has no binding preference for minicircles or no measurable effect on DNA cyclization (9) does not reflect the limitations of the competition binding and cyclization kinetics methods to detect small DNA bends. It has been suggested (30) that our previously reported cyclization kinetics experiments (9) were not sensitive to small DNA bends of ≈5–20° because of interference from additional A–T-rich regions of flexibility included in our DNA constructs. However, DNA cyclization kinetics of comparable DNA constructs have been shown to detect both a large 90° DNA bend (6) induced by the catabolite activator protein, as well as a small 8° bend intrinsic to the ATF/CREB site recognized by GCN4 (21). Similarly, in this work we have demonstrated that a single A-tract bend of ≈18° has a 15–17-fold effect on DNA cyclization. Thus, our experimental results clearly demonstrate that in solution (dbd)Fos–Jun does not induce appreciable curvature at the AP-1 recognition site. This result does not depend on the nature of the sequences flanking the AP-1 site or on the distance separating the AP-1 site and A-tract-directed bend in these molecules (Table 1). Moreover, our 158-bp DNA constructs, which include a large standard 108° A-tract bend, are optimally designed to measure the phasing effects of small bends on DNA cyclization. These effects should be comparable with the energetic contributions of small DNA bends on DNA looping in multiprotein transcription complexes. We therefore conclude that any small (dbd)Fos–Jun-induced DNA bends that are undetected in our current and previously reported work (9) will have no thermodynamic significance for gene regulatory phenomena.
In comparison, results obtained from gel phasing are clearly dependent on the distance between the AP-1 site and the A-tract-directed bend center. Our previously reported gel-phasing experiments carried out with DNAs containing long (35–43 bp) phasing linkers show no gel mobility differences for the complexes of Fos–Jun bound to differently phased DNA constructs. However in this work, (dbd)Fos–Jun bound to DNAs with short (11–19 bp) phasing linkers (Fig. 4A) demonstrates clear phase-dependent mobility shifts that are comparable in magnitude with previously reported gel mobility variations, also obtained with DNAs containing short phasing linkers (10, 13–18). We propose that differences in the results obtained with short and long phasing linkers reflect the gel mobilities of differently shaped three-armed species formed between an elongated zipper arm and a DNA helix (Fig. 6).
Figure 6.

Model to explain gel mobility anomalies of Fos–Jun–DNA complexes. The two rectangular shaped boxes represent the heterodimer Fos–Jun, bound to the AP-1 site in DNA containing a curved A-tract region. (A) Fos–Jun bound to oppositely phased DNA isomers with short (11–19 bp) phasing linkers. Y-shaped, slower moving complexes are formed between Fos–Jun and hcgDNA11 and hcgDNA19; however F-shaped, faster moving complexes are formed between Fos–Jun and hcgDNA15. The gel-phasing amplitude observed is significant. (B) The effect of Mg2+, incorporated into the gel and running buffer, on the gel mobilities of Fos–Jun–DNA complexes. We propose that Mg2+ competes with stabilizing electrostatic interactions between Fos–Jun and DNA, which results in an increase in the flexibility of the orientation of the leucine zipper arm with respect to the DNA helix and a decrease in the gel-phasing amplitude. (C) Fos–Jun bound to oppositely phased DNA isomers with long (35–43 bp) phasing linkers. The local structure at the Fos–Jun–DNA junction is T-shaped for oppositely phased isomers, which is reflected in their similar gel mobilities and lack of gel-phasing amplitude.
Gel mobility anomalies induced by leucine zipper motifs of Fos–Jun are enhanced in molecules with short phasing linkers (11–19 bp) between the A-tract and AP-1 site. Molecules hcgDNA11 and hcgDNA19 with a major groove-bound zipper arm positioned out of phase with respect to the A-tract-directed minor groove bend will resemble “Y”-shaped species. In comparison, the molecule hcgDNA15 with a leucine zipper arm that is positioned in phase with the A-tract bend will resemble an “F”-shaped species (Fig. 6). We predict and experimentally observe that an F-shaped molecule, with two of its three arms pointed in the same direction, migrates faster than Y-shaped molecules, with all its three arms pointed in different directions (Figs. 2 and 4A). Similarly, bulged three-way DNA junctions show variations in gel mobilities that are dependent on the angle between the three DNA arms (31). In comparison, the local shapes of complexes formed between Fos–Jun and DNAs containing long phasing linkers for both the in-phase (35- and 43-bp phasing linkers) and out-of-phase (39-bp phasing linker) molecules resemble “T”-shaped species and thus have similar gel mobilities. Furthermore, DNAs with long phasing linkers have the AP-1 site located closer to the end of the molecules than those with the short phasing linkers. It is likely that complexes with a zipper arm located toward the center of the DNA molecule will impose greater frictional drag than complexes with an elongated zipper arm located at the DNA ends.
Gel mobility shift variations of (dbd)Fos–Jun bound to DNAs containing short phasing linkers decrease linearly with increasing concentrations of Mg2+. However, the relative binding affinity of (dbd)Fos–Jun for linear vs. circular DNA is independent of the Mg2+ concentration in the binding buffer. Clearly, low salt conditions do not promote Fos–Jun-induced DNA curvature. We propose that in low ionic strength gels lacking Mg2+, additional electrostatic interactions between the basic N-terminal region of Fos–Jun and DNA help to orient the leucine zipper motif more rigidly with respect to the DNA helix (Fig. 6). Therefore, in gels lacking Mg2+, complexes of (dbd)Fos–Jun bound to differently phased DNA isomers show distinct gel mobility variations. In comparison, high concentrations of Mg2+ compete out weak electrostatic interactions between the zipper and the DNA helix, which results in increased flexibility of the zipper arm and a decrease in the gel anomalies of Fos–Jun–DNA complexes.
A decrease in gel mobility anomalies in the presence of multivalent cations has been recently reported for DNA complexes formed with full-length Fos–Jun and various chimeric and mutant forms of Fos–Jun (16). Similarly, protein-DNA complexes formed with Fos–Jun mutants, containing an aspartate and alanine in place of arginines at the N-terminal basic region, induce lesser gel mobility anomalies than the wild-type heterodimer (18). Whereas these observations have been interpreted to show that the N-terminal basic residues and the transcription activation domains of Fos–Jun promote DNA bending (16, 18, 25) by an electrostatic mechanism, we caution that electrostatic interactions can affect the conformational flexibility of the leucine zipper with respect to a DNA helix.
Acknowledgments
We thank T. K. Kerppola and D. A. Leonard for providing peptides Fos(118–211) and Jun(199–334). This work was supported by National Institutes of Health Grant GM21966.
ABBREVIATIONS
- hcg
human collagenase gene
- dbd
DNA-binding domains
References
- 1.Kim J L, Nikolov D B, Burley S K. Nature (London) 1993;365:520–527. doi: 10.1038/365520a0. [DOI] [PubMed] [Google Scholar]
- 2.Schultz S C, Shields G C, Steitz T A. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 3.Werner M H, Huth J R, Gronenborn A M, Clore G M. Cell. 1995;81:705–714. doi: 10.1016/0092-8674(95)90532-4. [DOI] [PubMed] [Google Scholar]
- 4.Wu H-M, Crothers D M. Nature (London) 1984;308:509–513. doi: 10.1038/308509a0. [DOI] [PubMed] [Google Scholar]
- 5.Zinkel S S, Crothers D M. Nature (London) 1987;328:178–181. doi: 10.1038/328178a0. [DOI] [PubMed] [Google Scholar]
- 6.Kahn J D, Crothers D M. Proc Natl Acad Sci USA. 1992;89:6343–6347. doi: 10.1073/pnas.89.14.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCormick R J, Badalian T, Fisher D E. Proc Natl Acad Sci USA. 1996;93:14434–14439. doi: 10.1073/pnas.93.25.14434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deutsch J M. Science. 1988;240:922–924. doi: 10.1126/science.3363374. [DOI] [PubMed] [Google Scholar]
- 9.Sitlani A, Crothers D M. Proc Natl Acad Sci USA. 1996;93:3248–3252. doi: 10.1073/pnas.93.8.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerppola T K, Curran T. Cell. 1991;66:317–326. doi: 10.1016/0092-8674(91)90621-5. [DOI] [PubMed] [Google Scholar]
- 11.Kerppola T, Curran T. Nature (London) 1995;373:199–200. doi: 10.1038/373199a0. [DOI] [PubMed] [Google Scholar]
- 12.Gartenberg M R, Ampe C, Steitz T A, Crothers D M. Proc Natl Acad Sci USA. 1990;87:6034–6038. doi: 10.1073/pnas.87.16.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerppola T K, Curran T. Science. 1991;254:1210–1214. doi: 10.1126/science.1957173. [DOI] [PubMed] [Google Scholar]
- 14.Kerppola T K, Curran T. Mol Cell Biol. 1993;13:5479–5489. doi: 10.1128/mcb.13.9.5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerppola T K. Proc Natl Acad Sci USA. 1996;93:10117–10122. doi: 10.1073/pnas.93.19.10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerppola T K, Curran T. EMBO J. 1997;16:2907–2916. doi: 10.1093/emboj/16.10.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajaram N, Kerppola T K. EMBO J. 1997;16:2917–2925. doi: 10.1093/emboj/16.10.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leonard D A, Rajaram N, Kerppola T K. Proc Natl Acad Sci USA. 1997;94:4913–4918. doi: 10.1073/pnas.94.10.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glover J N M, Harrison S C. Nature (London) 1995;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- 20.Paolella D N, Palmer C R, Schepartz A. Science. 1994;264:1130–1133. doi: 10.1126/science.8178171. [DOI] [PubMed] [Google Scholar]
- 21.Hockings S C, Kahn J D, Crothers D M. Proc Natl Acad Sci USA. 1998;95:1410–1415. doi: 10.1073/pnas.95.4.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller W, Konig P, Richmond T J. J Mol Biol. 1995;254:657–667. doi: 10.1006/jmbi.1995.0645. [DOI] [PubMed] [Google Scholar]
- 23.Konig P, Richmond T J. J Mol Biol. 1993;233:139–154. doi: 10.1006/jmbi.1993.1490. [DOI] [PubMed] [Google Scholar]
- 24.Ellenberger T E, Brandl C J, Struhl K, Harrison S C. Cell. 1992;71:1223–1237. doi: 10.1016/s0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]
- 25.Strauss-Soukup J K, Maher L J., III Biochemistry. 1997;36:10026–10032. doi: 10.1021/bi970215u. [DOI] [PubMed] [Google Scholar]
- 26.Abate C, Luk D, Gentz R, Rauscher F J, III, Curran T. Proc Natl Acad Sci USA. 1990;87:1032–1036. doi: 10.1073/pnas.87.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abate C, Luk D, Curran T. Mol Cell Biol. 1991;11:3624–3632. doi: 10.1128/mcb.11.7.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diekmann S. Nucleic Acids Res. 1987;15:247–265. doi: 10.1093/nar/15.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kahn J D, Yun E, Crothers D M. Nature (London) 1994;368:163–166. doi: 10.1038/368163a0. [DOI] [PubMed] [Google Scholar]
- 30.Hagerman P J. Proc Natl Acad Sci USA. 1996;93:9993–9996. doi: 10.1073/pnas.93.19.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welch J B, Duckett D R, Lilley D M J. Nucleic Acids Res. 1993;21:4548–4555. doi: 10.1093/nar/21.19.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]