

Abstract
This review is focused on proteins with key roles in pathways controlling either reactive oxygen species or DNA damage responses, both of which are essential for preserving the nervous system. An imbalance of reactive oxygen species or inappropriate DNA damage response likely causes mutational or cytotoxic outcomes, which may lead to cancer and/or aging phenotypes. Moreover, individuals with hereditary disorders in proteins of these cellular pathways have significant neurological abnormalities. Mutations in a superoxide dismutase, which removes oxygen free radicals, may cause the neurodegenerative disease amyotrophic lateral sclerosis. Additionally, DNA repair disorders that affect the brain to varying extents include ataxia-telangiectasia-like disorder, Cockayne syndrome or Werner syndrome. Here, we highlight recent advances gained through structural biochemistry studies on enzymes linked to these disorders and other related enzymes acting within the same cellular pathways. We describe the current understanding of how these vital proteins coordinate chemical steps and integrate cellular signaling and response events. Significantly, these structural studies may provide a set of master keys to developing a unified understanding of the survival mechanisms utilized after insults by reactive oxygen species and genotoxic agents, and also provide a basis for developing an informed intervention in brain tumor and neurodegenerative disease progression.
Keywords: Amyotrophic lateral sclerosis, ataxia-telangiectasia-like disorder, Werner syndrome, xeroderma pigmentosum, nitric oxide synthase, superoxide dismutase
Introduction
Proteins that control cellular levels of reactive oxygen species (ROS) or DNA damage responses are essential to the nervous system. This is because an imbalance of ROS may cause significant damage to the cell, which often produces a cytotoxic or mutational outcome. DNA is surprisingly susceptible to both exogenous and endogenous DNA damaging agents, considering that it is the carrier of genetic information. If the DNA damage is not corrected by repair mechanisms, deleterious mutations and genomic instability may arise, leading to aging and cancer. Additionally, since DNA repair mechanisms are imperfect accumulation of both nuclear and mitochondrial DNA damage, in post-mitotic cell may have a central role in neurodegeneration and aging pathologies. Therefore, the defenses against DNA damaging agents merit close attention for their significance to neuropathologies.
Responses to ROS and DNA damage have many critical links to neuroscience. The damage from stroke is likely linked to ROS, with resulting damage to membrane components, proteins, and DNA. The long-term stability of human embryonic stem cells, with their potential for repair of neuronal defects, depends upon their effective DNA repair responses. Damage within actively transcribed genes has to be repaired, otherwise defective proteins are likely to be produced, or the more bulky DNA lesions may block transcription, triggering cell death. DNA damage to regulatory regions can cause the stochastic deregulation of gene expression, and provide a mechanism for age-related degeneration and cell death. Mouse models involving the mutation of key DNA repair enzymes support a role for these proteins in the control of aging and cancer, as well as reveal the etiology of neurological abnormalities in the brain (Hoeijmakers, 2001). Defects in either ROS controlling enzymes or DNA double-strand break (DSB) repair genes are also the cause of several human hereditary diseases. These disorders present clinical phenotypes that are often linked to tumorigenesis and neurological abnormalities. Severe neurodegeneration is clearly apparent in some disorders, such as Cockayne syndrome (Lehmann, 2003), while more aging-related phenotypes are present in others, including Werner syndrome (Goto, 1997). Thus, the emerging knowledge of mutations and polymorphisms in key human ROS and DNA repair genes may provide an informed basis for improved strategies for interventions into brain tumors and neurodegenerative disorders. Yet, to date no unifying theory exists to explain the root causes of aging and neurodegenerative diseases.
Here, we focus on the important gains in our knowledge of the cellular mechanisms controlling ROS and DNA repair events, through insights provided by structural biochemistry studies. We describe the known roles, disease phenotypes and molecular mechanisms of key ROS and DNA repair proteins within the nervous system. We highlight how these proteins coordinate chemical steps within pathways, as well as integrate any signaling and response events, often through dynamic conformational changes in structure. The results of these studies may therefore provide a more unified understanding of molecular survival mechanisms from ROS and DNA damaging insults. Moreover, these findings may provide new templates for rationally based therapeutic design, for the treatment of brain tumors and neurodegenerative diseases.
ROS Removal by Manganese Superoxide Dismutase
The high metabolic activity of post-mitotic neuronal cells typically produces significant quantities of ROS. However, neurons are also relatively sensitive to ROS and neurodegenerative disorders, including amyotrophic lateral sclerosis, Alzheimer disease and Parkinson disease have been linked to damage caused by ROS (Andersen, 2004). 90 % of the ROS in the cell are generated by mitochondria, as the toxic by-products of energy generation. Most of these free radicals are rapidly scavenged in the cell, by the superoxide dismutase (SOD) enzymes. SOD enzymes catalyze the disproportionation of superoxide anion radicals to molecular oxygen and hydrogen peroxide, and are the enzymes that provide the master controls for ROS levels in the cell (Silverman and Nick, 2002).
The important role of SOD in the brain was highlighted by genetic inactivation of the mitochondrial form of SOD, manganese SOD (MnSOD), in mice. This resulted in dilated cardiomyopathy, hepatic lipid accumulation, and early neonatal death (Li et al., 1995). Treatment with an SOD mimetic, MnTBAP, rescued the MnSOD −/− mutant mice from this systemic pathology and dramatically prolonged their survival (Melov et al., 1998). However, surviving animals developed a pronounced movement disorder, which progressed to total debilitation by 3 weeks of age. Neuropathologic evaluation showed a striking spongiform degeneration of the cortex and specific brainstem nuclei. This was associated with gliosis and intramyelinic vacuolization, and was similar to that observed in cytotoxic edema and disorders associated with mitochondrial abnormalities, such as Leigh disease and Canavan disease. It was suggested that because of the failure of MnTBAP to cross the blood-brain barrier, progressive neuropathology is caused by excessive mitochondrial production of ROS (Melov et al., 1998), normally removed by MnSOD.
To define how MnSOD controls ROS levels in the cell, the molecular mechanism of MnSOD has been extensively characterized through combined structural and biochemical studies. The crystal structure of human MnSOD revealed that the enzyme forms a homotetramer (Borgstahl et al., 1992). Each subunit contains a N-terminal helical hairpin and a C-terminal α/β domain. The helical hairpins form two symmetrical 4-helix bundles, which assembles the tetramer. The active site of each subunit is at the junction between the helical hairpin and the α/β domain. The active site contains four amino acid side chains, His26, His74, Asp159 and His163, and one solvent molecule. These coordinate a single manganese ion in a strained trigonal bipyramidal geometry. The active site of the enzyme also shows an apparent hydrogen-bonded network of side chains and water, which extends from the manganese bound solvent molecule at the active site, to solvent-exposed residues and the interface between subunits (Borgstahl et al., 1992, Silverman and Nick, 2002) (Fig. 1). This hydrogen-bonded network is suggested to support proton transfer in catalysis, providing a mechanism for delivering protons to the active site, and a means by which the pKa of the active-site water is regulated (Silverman and Nick, 2002).
Fig. 1.
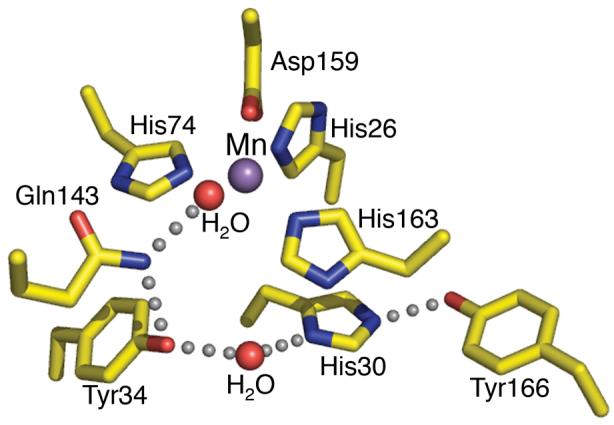
The Human MnSOD active site. In the wild type MnSOD structure (PDB code 1N0J) the His26, His74, His163, and Asp159 side chains and a water molecule chelate a single Mn2+ ion. The active site hydrogen bond network, depicted by grey spheres, starts at the metal bond solvent that forms a hydrogen bond to Gln143 and then follows with a bond to Tyr34. A conserved water molecule then mediates the hydrogen bond between Tyr34 and His30, and His30 also forms a hydrogen bond with Tyr166 from another subunit.
Structure-based mutagenesis (Guan et al., 1998, Leveque et al., 2000, Hearn et al., 2003, Greenleaf et al., 2004, Hearn et al., 2004) or chemical modification (Ayala et al., 2005a) of the hydrogen bonding partners have highlighted how these residues control the enzyme's activity. The network consists of the metal bound solvent forming a hydrogen bond with Gln143 Nε, which subsequently hydrogen bonds to the Tyr34 hydroxyl group. A conserved water molecule then mediates the hydrogen bond between Tyr34 and His30, near the surface, and His30 also forms a hydrogen bond with Tyr166 from another subunit (Fig. 1). Any alterations in these residues significantly affect both the enzyme's catalytic activity and stability, despite minimal structural changes in the active site (Guan et al., 1998, Leveque et al., 2000, Hearn et al., 2003, Hearn et al., 2004, Ayala et al., 2005a, Ren et al., 2006). Structures revealed that substitution with smaller amino acids, allows for water molecules to replace the endogenous side chain, fulfilling the necessary interactions to compliments the hydrogen bond network. Most of these substitutions at Gln143 create an enzyme whose activity is about 2-3 orders of magnitude lower than in the wild-type enzyme (Leveque et al., 2000). The Tyr34Phe mutant has a steady-state turnover resembling that of the wild-type enzyme, but at high superoxide levels the rate decreases by 10-fold (Guan et al., 1998, Ayala et al., 2005a). Changing His30 lowered the Kcat/Km by about one order of magnitude, suggesting that there is some flexibility in the chemical properties of residue 30, but that a histidine in this position creates the most efficient enzyme (Hearn et al., 2003). Similarly, mutation of Tyr166 also shows a decrease in Kcat/Km of about an order of magnitude (Cabelli et al., 1999). Thus, the lower activity of the mutant enzymes suggests that maintenance of the correct hydrogen bond partners in MnSOD is essential for the highly tuned reactivity of the active site, functioning to minimize ROS damage to the brain.
ROS and Copper,Zinc Superoxide Dismutase
One of the most common neurological disorders in humans is amyotrophic lateral sclerosis (ALS or Lou Gehrig's disease), which affects approximately 1 in 200,000 people (Cleveland and Rothstein, 2001). The disorder usually strikes in mid-life and leads to progressive paralysis and death within one to five years of the onset of symptoms, due to the selective killing of motor neurons. The disease is inherited in an autosomal recessive manner in 5–10% of cases, which is referred to as familial ALS (FALS)(Cleveland and Rothstein, 2001). All the genes directly linked to FALS have not been identified. However, mutations in the superoxide dismutase1 (SOD1) gene give rise to approximately 20% of FALS cases (Deng et al., 1993, Rosen et al., 1993, Rakhit et al., 2002) while mutations in several other genes, including ALS2, SETX, or VAPB cause much rarer forms of FALS (Kunst, 2004). SOD1 encodes a cytosolic copper,zinc superoxide dismutase (Cu,Zn SOD), which similar to the mitochondrial MnSOD, is responsible for the disproportionation of harmful superoxide radicals to hydrogen peroxide and oxygen (Fridovich, 1986).
Structural studies on human Cu,Zn SOD have revealed that the enzyme is composed of two identical subunits, which form an exceptionally stable homodimer (Parge et al., 1992). The enzyme uses electrostatic guidance and exquisite substrate specificity, to achieve a faster than diffusion reaction rate (Getzoff et al., 1983, Fridovich, 1986). A multitude of SOD1 mutations have been identified in FALS patients (Gaudette et al., 2000, Andersen, 2001), which are predominantly single amino acid substitutions, along with a few truncations. The mutations are dispersed throughout the 153 amino acid residue SOD polypeptide (Deng et al., 1993). Structural, biochemical and pathological studies have lead to several hypotheses being provided for the molecular mechanisms that lead to the disease. The more recent experimental data support the idea that toxicity of intracellular Cu,Zn SOD aggregates may result from protein misfolding or impaired protein degradation. This is in common with many other neurodegenerative diseases, which are noted to have protein aggregates in brain tissue (Wanker, 2000, Dobson, 2001, Soto, 2001). Aggregates have been observed in both mouse and cell culture models of ALS. These aggregates are found in the cytoplasm, are strongly immunoreactive to Cu,Zn SOD antibodies and cannot be dissociated with strong detergents or reducing agents (Durham et al., 1997, Bruijn et al., 1998). Furthermore, in transgenic mice the ALS aggregates can be detected biochemically months before the onset of symptoms (Shibata et al., 1996, Johnston et al., 2000). One proposed mechanism of FALS mutant SOD-mediated toxicity is the coprecipitation of mutant Cu,Zn SOD with essential cellular components (Bruijn et al., 1998, Johnston et al., 2000, Cleveland and Rothstein, 2001) and this has been demonstrated with the copper chaperone for SOD (CCS)(Kato et al., 2001), nitric oxide synthase (NOS) and phosphorylated neurofilaments (Chou et al., 1996). Yet, it is not entirely clear how the many different Cu,Zn SOD single-site mutations, which are widely dispersed throughout the protein sequence, can give rise to the same FALS pathology. Some clarification was provided by combined structural, biochemical and biophysical characterizations of two FALS mutant SOD proteins (DiDonato et al., 2003). These two FALS proteins represent the two major structural classes of SOD mutations. The first mutant, H43R, disrupts the proteins hydrophobic packing, and the second, A4V, disrupts the dimer interface. To separate possible secondary destabilization and aggregative events due to oxidation of the free cysteine residues (C6 and C111) from changes caused directly by the H43R and A4V mutations, the mutations were examined in the context of the C6A/C111S HSOD (HSOD-AS) parent, which retains the wild-type fold and activity (McRee et al., 1990, Hallewell et al., 1991, Parge et al., 1992). These two mutants retain nearly wild-type structures, but an overall architectural destabilization of these proteins propagates from the point mutations, to promote the formation of fibril-like aggregates (DiDonato et al., 2003). The cysteine-independent aggregation of FALS mutants is enhanced by factors of 7–80 times, compared to the parent HSOD-AS. Aggregation severity is also likely to be enhanced by the presence of the free cysteine residues, which can form disulfide bonds to covalently lock the fibrous aggregates. Visualization of these aggregates by electron microscopy and atomic force microscopy revealed that they resemble those found in post-mortem studies of FALS patients, and bind dyes that detect amyloid-like structure. Thus, the structural biochemistry data provides evidence for a molecular mechanism for initiating FALS toxicity, whereby framework destabilization and loss of dimer assembly specifically promote fibril formation, with consequent implications for mitochondrial damage and motor neuron cytotoxicity (DiDonato et al., 2003).
Hydrogen Peroxide Removal by Catalase
The catalase protein completes the process of eliminating ROS by converting hydrogen peroxide into water and oxygen. In addition to this defense against oxidative damage, human catalase has roles in ethanol metabolism (Zimatkin et al., 1998), inflammation (Halliwell and Gutteridge, 1984), apoptosis (Yabuki et al., 1999), aging and cancer (Miyamoto et al., 1996). Catalases convert two molecules of hydrogen peroxide to two molecules of water and one molecule of oxygen, via a two-step reaction (Deisseroth and Dounce, 1970). In step one, the heme Fe3+ reduces a hydrogen peroxide molecule to water and generates a covalent Fe4+=O oxyferryl species with a porphyrin π-cation radical, referred to as compound I. In step two, compound I oxidizes a second peroxide molecule to molecular oxygen and releases the ferryl oxygen species as water. Several catalase crystal structures have been determined, aiding our understanding of reactive oxygen control by defining the reaction and inhibition mechanisms (Goth, 1997). These studies include the definition of the chemistry of the human catalase, through structures of the resting-state enzyme and complexes of a catalase bound to cyanide and 3AT inhibitors (Putnam et al., 2000).
Human catalase forms a tetrameric assembly and this may be important to ensure that the active site is sequestered and that the enzyme is competent to complete the reaction. Interestingly, an unstable catalase isolated from patients homozygous for a Swiss-type acatalasemia, a hereditary catalase deficiency disorder, rapidly disassociates into inactive dimers with reduced heme content. This suggests that catalase assembly variants may play roles in disease susceptibility (Aebi et al., 1974), in addition to nonsense and splicing mutations (Hirono et al., 1995). From the structural data, a mechanism for the recognition and removal of peroxide by catalase has been proposed (Putnam et al., 2000). Hydrogen peroxide is selected for by a ‘molecular ruler’, a narrow, hydrophobic channel that promotes occupancy of a single peroxide molecule, binding the active site heme and blocks the passage of large molecules. The peroxide forms an iron coordination complex at the bottom of the hydrophobic channel, with one oxygen atom forming hydrogen bonds to two side chains, and the other directly ligating the metal. Catalase uses these two asymmetric interactions with the substrate to prime the otherwise symmetric peroxide bond for heterolytic bond cleavage; good geometry for both iron coordination and hydrogen bond formation to side chains would require stretching of the peroxide bond, furthering the complex towards the cleavage transition state. The reaction then continues by either a concerted hydride transfer or by stepwise transfer of two electrons and a proton. The ligation of the first peroxide to the metal also opens up the channel and this likely promotes binding of a second peroxide by the ruler, to allow the second stage of the reaction and return the enzyme to the resting state for the next catalytic cycle.
ROS and Nitric Oxide Synthase
Another reactive oxygen species, nitric oxide (NO), acts in key physiological processes within the central nervous system. NO may play important roles in neurodevelopment, synaptic plasticity, neurotransmitter release, neurotransmitter reuptake and regulation of gene expression (Guix et al., 2005). Additionally, excessive NO production plays a function in mediating neurotoxicity associated with a variety of neurological disorders, including stroke, Parkinson's Disease, Alzheimer's disease and other neurodegenerative dementias, including HIV dementia (Aliyev et al., 2004, Togo et al., 2004). NO may also be involved in the neurodegeneration observed during normal aging. Therefore, inhibitors of NO production may prove to be attractive compounds for treatment of neurodegenerative disorders and cerebrovascular incidents (Alderton et al., 2001, Iadecola and Alexander, 2001, Rosenfeld et al., 2002).
NO is a small, easily diffusible and transient free radical, whose availability is controlled at the synthesis level, by the nitric oxide synthase (NOS) enzymes. The NOS enzymes produce NO through the conversion of arginine to citrulline, utilizing oxygen and NADPH co-substrates. In mammals there are three NOS isoforms, which have been named after the activity or tissue type in which they were first associated. These NOS isoforms are neuronal NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS). nNOS and eNOS are constitutively expressed isozymes, controlling basal NO levels and synthesizing NO in response to increases in intracellular calcium levels. iNOS is expressed in response to specific cytokines, growth factors or bacterial products, and its NO production is independent from intracellular Ca2+ levels (Mayer and Hemmens, 1997).
Functional NOS isozymes are homodimers and each isozyme subunit contains an N-terminal catalytic oxygenase module (NOSox), a connecting Ca2+/calmodulin (CaM) binding region and a C-terminal electron-supplying reductase module (NOSred) (Abu-Soud et al., 1995) (Fig. 2a). Extensive crystallographic studies on NOSox in complex with substrate, intermediates, and inhibitors of the three isozymes have considerably advanced our understanding of the structural chemistry underlying NOSox catalytic activity (Crane et al., 1997, Alderton et al., 2001, Li et al., 2002, Rosenfeld et al., 2002). The NOSox domain is built from a unique, winged β-sheet fold (Crane et al., 1997). NOSox binds L-arginine substrate, heme, and two tetrahydrobiopterin cofactors and a zinc ion that stabilize the NOS dimer interface (Crane et al., 1998, Raman et al., 1998, Crane et al., 1999, Fischmann et al., 1999). NOSox accepts electrons from NOSred to catalyze the sequential monooxygenation of L-arginine, into N-hydroxyarginine and then into citrulline and NO. The reductase domain binds and transfers electrons from NADPH to the oxygenase domain, with the aid of one flavin adenine dinucleotide (FAD) and one flavin mononucleotide (FMN) moiety. Detailed understanding of the NOSred molecular mechanism was gained from the crystal structure of the NOSred module (Garcin et al., 2004) (Fig. 2). NOSred belongs to a large protein family, including NADPH-dependent cytochrome P450 reductase and sulfite reductase flavoprotein. An α-helical connecting domain (CD) orients flanking FMN- and FAD-binding domains, to align the two flavins (Wang et al., 1997). Electron transfer proceeds from NADPH, to the FAD, to FMN and then to the oxygenase heme (Adak et al., 1999). This last step is rate-limiting (Miller et al., 1999), occurs in trans, from the NOSred of one polypeptide to the NOSox of the other. This step is triggered by conformational changes that are induced by Ca2+/CaM binding (Siddhanta et al., 1996).
Fig. 2.

Proposed holo-NOS. a) The modular structure of nNOS. This schematic representation shows the domain organization of nNOS, indicated above, the regions binding the substrate and cofactors, below, phosphorylation sites are indicated by *, and the regulatory sequence elements are colored in magenta. The N-terminal PDZ domain mediates intracellular localization of nNOS. b) stereoview of the NOSred fold. The different domains of NOSred are labeled and color-coded to match a; from the N to C terminus: yellow, FMN domain; red, hinge; blue, CD; green, β-finger; dark gray, FAD domain; and orange, NADPH domain. NOS regulatory elements include the autoinhibitory helix, CD2A loop, and C-terminal tail (AH, CD2A, and CTN in magenta). Cofactors are depicted as ball-and-stick models and unstructured regions are shown as semitransparent white tubes. c) stereo view of nNOSred dimer. Subunit A-red is related to subunit B-red by the vertical non-crystallographic 2-fold axis. The FMN-binding domains (yellow) protrude in opposite directions (in front for left B-red; in back for right A-red). This figure was adapted from (Garcin et al., 2004)
Structural biochemistry data elucidated from a fully assembled reductase dimer of nNOS (Fig. 2b, c) provided critical insights into this domain's regulation and molecular mechanism (Garcin et al., 2004). An autoinhibitory helix motif, a CD regulatory element, the C-terminal tail and phosphorylation, function to regulate NOS activity, which is exquisitely tuned to control NO production (Roman et al., 2002). In all isozymes, the 21–42-residue C-terminal tail (CT) represses electron transfer, by locking the FMN-binding domain in the electron-accepting position. In addition, eNOS and nNOS contain the 42–45-residue auto-inhibitory helix (AH) within the FMN-binding domain, which interferes with Ca2+/CaM binding and inhibits both intra- and inter-module electron transfers. Also, a regulatory element, the ‘CD2A’ loop of a protruding β-finger present in the CD of eNOS and nNOS, plays an autoinhibitory role in the control of NO by interaction with the CaM-binding peptide (Knudsen et al., 2003). The upregulation of eNOS and nNOS activity is controlled by phosphorylation of both the CT and AH regulatory elements and by reversible Ca2+/CaM-binding.
An experimentally determined structure of full-length NOS remains elusive, perhaps due to the required flexibility of its functional components inhibiting crystal growth. However, the structure and biochemical analysis of the nNOSred dimer provided a template for a model of the holo-nNOS enzyme assembly (Garcin et al., 2004). The NOS model was built by connecting the dimeric NOSox modules and a CaM: NOS-peptide complex (Aoyagi et al., 2003) to the NOSred structure. Model building was constrained by the short CaM binding linker between NOSox to NOSred. Therefore, out of the potential assembly states, a model with the NOSox C-terminus facing the NOSred N-terminus will produce a compatible linker length (Fig. 3a, right). In this model the polypeptide chains cross each other, and the CaM-binding linker brings together opposing NOSox and NOSred modules (Fig. 3a, right). The shortest distance between the FMN and heme cofactors is 70 Å in this model, which is substantially too long for direct intermodule electron transfer (Page et al., 1999). Therefore, the structural, biochemical and modeling results indicate that the entire FMN domain serves as a one-electron shuttle, by swinging back and forth between its two redox partners (Fig. 3b). The flexible hinge region in NOS would serve as the pivot point for this motion (Fig. 2b). This model is consistent with the proposed role of the same region in the structurally related cytochrome P450-BM3 (Sevrioukova et al., 1999) and reminiscent of those found in multiple redox centers containing proteins (Zhang et al., 1998, Lennon et al., 2000, Leys et al., 2003). Additionally, this swinging FMN domain mechanism would account for the slow rate of inter-module electron transfer in NOS. The dimerization NOS would provide a means for fine-tuning this electron transfer mechanism, where the two redox partners of the FMN shuttles are located on adjacent polypeptides. Promotion of inter-molecule transfer would occur through both binding of Ca2+/CaM and phosphorylation of the CT, which would displace the repressive C-terminal tail and thereby unlock the FMN-binding domain for shuttling.
Fig. 3.
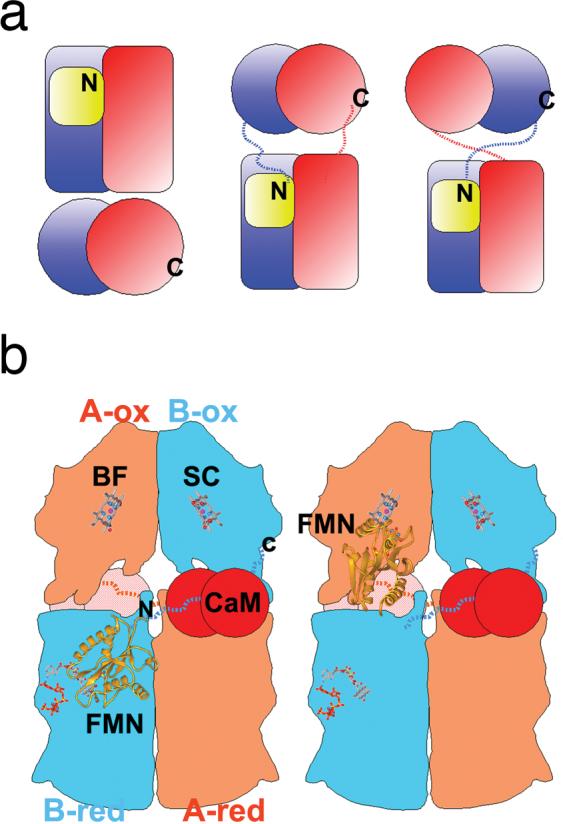
Proposed holo-NOS assembly and domain movements. a) Three possible models for dimeric holo-NOS, dimeric NOSox and NOSred modules are represented by pairs of spheres and rectangles, respectively. The FMN domain is shown as a yellow box. The nNOSox C terminus, labeled C, and nNOSred N terminus, labeled N, are covalently linked in holo-NOS by the intervening CaM-binding regions, indicated by dashed lines. b) Schematic drawing for the proposed holo-NOS assembly and domain rearrangements. The assembly corresponds to the models at the right in a) NOSox and NOSred are colored in orange for chain A and blue for chain B. The CaM: NOS-peptide complex is depicted as two red spheres, the heme, NADPH, FAD, and FMN cofactors as ball-and-stick models and the FMN domain as a yellow ribbon. Binding of CaM frees the FMN-binding domain, and allows trans inter-module electron transfer through alternating electron-accepting, left, and electron-donating, right, positions for the FMN domain. This figure was adapted from (Garcin et al., 2004).
ROS and Pathogens
Pathogens are likely to amplify ROS damage in the CNS during pathogenesis. This includes Neisseria meningitides, which is the major cause of meningitis worldwide. Neisseria pathogenesis is mediated through molecular machines, particularly the type IV pili system (Craig et al., 2004). Recently, structures elucidated through a combination of cryo-electron microscopy and crystallography studies have revealed how type IV pilus fibers function, using surface groves as receptor-like binding sites for adherence and host cell interactions (Craig et al., 2006). This interaction, as part of the invasion process, is able to increase intracellular calcium bursts (Ayala et al., 2005b). Moreover, the pilus of Neisseria gonorrhoeae, which shares substantial sequence conservation with N. meningitides, acts cooperatively with a porin to induce calcium ion transients in infected epithelial cells (Ayala et al., 2005b). Such calcium mediated signaling by pathogen invasion in neuronal cells is expected to increase ROS stress (Koedel and Pfister, 1999), through the calcium-mediated activation of neuronal NOS (Aoyagi et al., 2003). Therefore, a combination of factors may exacerbate ROS production and provide increased risk of damage to the brain. Additionally, apoptosis-mediated pathology may be accentuated post-mitotic neuron cells, which have a high metabolic rate and oxygen utilization. Their high metabolic ROS generation is further accentuated by NO used in signaling and by stimulation of NOS, by calcium burst during invasion. Besides directly increasing cellular apoptosis, these ROS events increase DNA damage, with implications for degenerative diseases.
DNA Repair, Cancer and Aging in the Nervous System
Accumulation of mitochondrial and nuclear DNA damage, which can occur by ROS and a large variety of other genotoxic agents, in post-mitotic cells plays a key role in aging and neurodegeneration. The human DNA repair proteins restore the normal nucleotide sequence and DNA structure after damage (Wood et al., 2001), through an array of different responses that depend on the type of damage. Base excision repair (BER) corrects DNA base alterations, which have not distorted the DNA helix, including oxidative base damage. Evidence for BER in neurons includes the repair of GT and GU mismatches, from studying highly enriched neuronal populations of the cerebellar extracts (Brooks et al., 1996). Nucleotide excision repair (NER) differs from BER by responding to DNA helix distorting damage and by removing short segments of nucleotides in its repair process, rather than single bases. Evidence for NER in the brain also includes studies on cerebellar extracts (Brooks, 1998) and also a few rare hereditary diseases in known NER genes that cause marked neurological pathology, which are discussed later. Mismatch repair (MMR) is another type of excision repair, removing mispaired bases resulting from replication errors. This post-replicative pathway might be expected to be down regulated in the postmitotic neurons, yet several lines of evidence do indicate the presence of MMR proteins in the brain (David et al., 1997, Marietta et al., 1998, Belloni et al., 1999).
In addition to base damage, breaks in the backbone can occur, either as single-strand breaks (SSBs) or as DSBs. SSBs are efficiently repaired by a mechanism that shares many common features with the later steps in BER. DSBs initiate a coordinated response to ensure resolution of the damage and cellular survival. The lack of a complementary strand to act as a template for repair in DSBs means the cell has a particularly dramatic response, where a single unrepaired DSB can be lethal to the cell (Bennett et al., 1993). The repair of DSBs occurs via the homologous recombination repair (HRR) or the nonhomologous end joining (NHEJ) pathways. HR events occur in cells just prior to cell division, with replicated DNA, and strand exchange occurs between the two copies so that a suitable template can be used for correct repair. In the nervous system, HR is particularly important for proliferating cells during development. However, for non-dividing cells NHEJ is the predominant pathway, rejoining two broken ends of DNA without a template and therefore resulting in the potential loss of genetic information. Inactivation of proteins in the DSB repair pathways in mice leads to either embryonic lethality, neurodegeneration or brain tumors, indicating their critical requirement to the brain. Furthermore defects in either HR or NHEJ can lead to certain types of brain tumors. Here, we highlight some of the most recent structural biochemical observations of several key DNA repair genes that when mutated can give rise to severe clinical phenotypes, which include the brain to vary extents.
Mitochondrial DNA Repair by mtPol
Terminally differentiated cells need never replicate their genomes and may therefore dispense with the daunting task of maintaining several DNA repair systems to constantly scan their entire complement of DNA. Yet, the mitochondria of neurons have to replicate as well as repair their DNA. Mutations in mitochondrial DNA (mtDNA) have been linked to several neurodegenerative diseases as well as normal aging (Zeviani and Spinazzola, 2003). The principle mechanisms to avoid mutations in mtDNA are the control of ROS and the repair of DNA damage. Reduced DNA repair or increased ROS are correlated with the induction of apoptosis. Human mitochondria use a single DNA polymerase (pol γ) for both DNA replication and repair (Kaguni, 2004). Mutations in pol γ itself cause autosomal dominant or recessive progressive external ophthalmoplegia (ad/arPEO), a human disorder manifesting exercise intolerance, muscle weakness, peripheral neuropathy, deafness, ataxia, cataracts, and hypogonadism. Additionally, knock-in mice expressing an exonuclease-deficient pol γ display dramatic effects of premature-aging.
The molecular mechanism of DNA synthesis by pol γ has been studied through structure-function analysis. Human pol γ is a two-subunit enzyme (Kaguni, 2004). The catalytic core (pol γ–α) is a member of family A DNA polymerases, the prototypes of which include E. coli DNA polymerase I and T7 DNA polymerase. Pol γ–α contains a polymerase domain and an exonuclease domain required for accurate DNA replication. The high processivity required for mitochondrial DNA replication is ensured by an accessory subunit (pol γ–β). Results from extensive mutagenesis on Drosophila pol γ suggest that the two subunits interact through multiple sites, influencing both DNA binding and activity of the pol γ holo-enzyme (Fan and Kaguni, 2001). The crystal structures of the human and mouse pol γ–β have been determined (Carrodeguas et al., 2001, Fan et al., 2006b), but structural information on the pol γ–α catalytic core has not yet been defined. The two pol γ–β structures are highly homologous, both form a dimer structure and share structural similarity to class IIa aminoacyl-tRNA synthetases. These structures, combined with the structure of the bacteriophage T7 DNA polymerase, a homolog of pol γ–α, have lead to the development of a structural model of the pol γ holo-enzyme ternary complex (Fan et al., 2006b)(Fig. 4). This model also includes primer/template DNA substrate, and an incoming nucleotide (Fig. 4). In keeping with the recent structural, (Carrodeguas et al., 2001, Fan et al., 2006b), biophysical (Carrodeguas et al., 2001, Yakubovskaya et al., 2006) and mutagenesis results (Fan and Kaguni, 2001), the model proposes that the pol γ holo-enzyme consists of two pol γ–β and a single pol γ–α, subunits. The model suggests that multiple regions of subunit interaction between pol γ–β and the γ–α catalytic core allow the holo-enzyme to encircle the newly synthesized double-stranded DNA, and thereby enhance DNA binding affinity and holo-enzyme processivity. The biochemical properties of a set human pol γ–β mutants were also explained by the model, and these helped elucidate the role of the accessory subunit, which acts as a novel type of processivity factor in stimulating pol γ activity and in enhancing processivity (Fan et al., 2006b).
Fig. 4.

Structural model of the pol γ holo-enzyme/template-primer DNA complex. Bacteriophage T7 DNA polymerase in a ternary complex with template–primer DNA and ddGTP was docked onto the surface of the accessory subunit dimer of human pol γ (a) Overview of the pol γ/DNA complex. (b) A partial view of the complex looking down the dsDNA helix, where the dsDNA is represented by the magenta spiral and bases. Pol γ–β is depicted as a backbone structure with monomers colored green and yellow. The human pol γ–β mutants that helped elucidate the role of the accessory subunit, acting as a processivity factor in stimulating pol γ activity and in enhancing processivity (Fan et al., 2006b) are shown in black, magenta, blue and red. T7 pol is in blue ribbon with the exonuclease domain in orange; P, F, T and E designate the palm, fingers and thumb subdomains, and the exonuclease domain, respectively. Thioredoxin in the T7 complex is shown in gray for comparison, and is not part of the pol γ holo-enzyme. This figure was adapted from (Fan et al., 2006b).
Double-Strand Breaks and Mre11/Rad50/Nbs1
The Mre11/Rad50/Nbs1 (MRN) protein complex plays a central role in repairing DNA DSBs during HRR and it may also have a role in NHEJ (Stracker et al., 2004). The essential nature of MRN is highlighted by studies demonstrating that null mutations in any of the three proteins in mice lead to embryonic lethality (Xiao and Weaver, 1997, Luo et al., 1999, Zhu et al., 2001). Moreover, rare defects in the MRN complex cause neurological dysfunction and cancer predisposition in humans. Mutations in the Mre11 component give rise to ataxia-telangiectasia-like disorder (ATLD), with its increased radiosensitivity and an increased level of spontaneously occurring chromosome aberrations, and an ataxia clinical phenotype. Mutations in Nbs1 cause Nijmegen Breakage syndrome, which displays similar symptoms to ATLD and is characterized by microcephaly, radiosensitivity, immunodeficiency, increased cancer risk, particularly lymphoid malignancy and growth retardation.
The MRN complex is among the first set of proteins recruited to sites of DSBs during HRR, where it forms nuclei foci. The role of MRN in these foci is likely connected to DNA end processing events and to cell cycle checkpoint signaling, through both ATM checkpoint kinase (D'Amours and Jackson, 2002, van den Bosch et al., 2003, Assenmacher and Hopfner, 2004) and global genome histone H2AX (Paull et al., 2000). This crucial checkpoint step halts cell cycle progression, until the DSBs breaks are either repaired or the cells are triggered to undergo apoptosis (Khanna and Jackson, 2001). These functions of MRN are being defined at the molecular level, through a combination of biological, biochemical, and structural studies. These indicate that MRN plays multifaceted roles in the following diverse activities. MRN acts as a DNA-damage sensor, as an enzymatic effecter in DNA damage repair and as a transducer of critical damage-response signals to the cell cycle checkpoint apparatus (Stracker et al., 2004). The diverse roles appear to be combined due to MRN functioning in part, as a multi-purpose tether that bridges severed DNA ends (Chen et al., 2001, de Jager et al., 2001, Hopfner et al., 2002b). This finding has been derived through visualization of the MRN molecular machinery by crystallographic studies of the Mre11-Rad50 (MR) core complex and analysis of the overall framework from both electron- and atomic force-microscopic imaging of intact MRN (Chen et al., 2001, de Jager et al., 2001, Hopfner et al., 2002b).
The MR core complex exists as a heterotetrameric assembly (M2R2) whose morphology is divided into distinct head, coil and hook domain regions (Fig. 5). The head of the complex possesses ATP-stimulated nuclease activity, where the Rad50 ATPase controls the Mre11 nuclease. Nbs1 also appears to be part of the head, through its interactions with Mre11 (Zhang et al., 2006). Perhaps the most striking feature of the MRN complex is the long antiparallel coiled-coil tails that are capped by hook-like structures, which that can form an interlocked hook/Zinc/hook bridges joining two Rad50 coiled-coils (Hopfner et al., 2002a). The integrity of this hook is required for Rad50-Rad50 homodimeric interactions and MRN function in vivo (Wiltzius et al., 2005). However, the exact nature of the supramolecular structure of these joined coiled-coils remains to be determined. One intriguing model suggests that they form large loops and that these loops are closed at one end by the global head and linked at the other end by the zinc hooks (Fig. 5). This structure is reminiscent of the proposed ring model for cohesin (Gruber et al., 2003). In the presence of DNA substrates intermolecular joining of two MRN complexes occurs (Hopfner et al., 2001, Hopfner et al., 2002a)(Fig. 5). Two head domains linked by the coiled-coils and zinc hooks may bind to opposing sister chromatids, providing a critical architectural function for early steps of homologous recombination. Thus, it appears that the MRN complex acts as a critical nexus point for both the signaling and effector functions in specific DNA damage responses, through combing regulated enzymatic activities with its fairly unusual architectural features.
Fig. 5.

MRN assembly models. a) Intracomplex Zn-hook minimizes unproductive, intercomplex interactions in the absence of DNA. b) DNA binding straightens the Rad50 coiled coils, which favors inter-complex tethering via Rad50 Zn-hooks with extended and parallel coils. “k” indicates kink regions that intersperse regions with strong coiled coil-forming potential “cc.” The figure was adapted from (Williams and Tainer, 2005).
Double-Strand Breaks, Base Excision Repair and WRN
Hereditary mutations in WRN are associated with Werner syndrome (WS), a rare autosomal recessive disorder that gives rise to multiple progeroid pathologies, including osteoporosis, atherosclerosis and a greatly increased cancer incidence (Goto, 1997). Rapid aging associated symptoms occur post-pubescence and are present in a wide variety of body systems. Increased aging in the central nervous system is still under debate, but analysis of WS individuals revealed brain atrophy in 40% of individuals (Goto, 1997). Schizophrenia was noted in 10% of WS individuals and a few cases of senile dementia, which are not linked to Alzheimer's, were also reported (Goto, 1997). Moreover, sensitive magnetic resonance imaging methods have demonstrated diffuse structural and metabolic tissue damage in the brains of WS individuals (De Stefano et al., 2003).
WRN encodes a 1,432-residue protein that contains a C-terminal nuclear-localization signal (Yu et al., 1996). Mutations in WS individuals are nonsense or frameshift mutations, which leads to a truncated protein that cannot localize to the nucleus (Matsumoto et al., 1997, von Kobbe and Bohr, 2002) and is generally degraded in the cytoplasm (Moser et al., 2000). WRN belongs to the RecQ helicase family that is widely distributed across the domains of life (Hickson, 2003) and is named after the E. coli founder (Nakayama et al., 1984). The human genome also contains four other RecQ helicase family members, RecQ1, BLM, RecQ4L and RecQ5. Mutations in BLM, and RecQ4L cause Bloom syndrome and Rothmund-Thomson syndrome, respectively (Harrigan et al., 2003). Werner, Bloom and Rothmund-Thompson syndromes share a predisposition to cancer, but notable pathological differences suggest that each disease pathway is functionally distinct.
WRN has been implicated to function in multiple DNA metabolism steps, but precise roles remain incompletely defined (Ozgenc and Loeb, 2005). Biochemical characterization of WRN helicase has shown ATPase activity and unwinding of partial-duplex DNA substrates with 3′-5′ polarity (Gray et al., 1997). Alternate DNA conformations are preferred over double-stranded DNA (dsDNA), and substrate specificities suggest roles in DNA replication, recombination and repair events (Shen et al., 1998, von Kobbe et al., 2003b). Significantly, a unique feature of WRN among all the human RecQ helicases is the addition of an exonuclease domain. This domain was first identified by sequence analysis (Moser et al., 1997, Mushegian et al., 1997), and then demonstrated to degrade dsDNA with 3′ recessed termini with a 3′-5′ directionality (Huang et al., 1998). WRN exonuclease functions on a variety of structured DNA substrates that include bubbles, stem-loops, forks and Holliday junctions, as well as on RNA-DNA duplexes, which further implies roles in replication, recombination and repair (von Kobbe et al., 2003b). WRN 3′-5′ exonuclease activity shows substrate specificity similar to that for the helicase, suggesting that the two WRN enzymatic activities may have coordinated functions on several classes of DNA structures (Opresko et al., 2001, Opresko et al., 2003).
WRN has been implicated in certain DNA repair events, as WS cells display hypersensitivity to specific DNA-damaging agents (Bohr, 2005) and show a mild, yet distinct sensitivity to ionizing radiation (Yannone et al., 2001, Cheng et al., 2004, Comai and Li, 2004). Moreover, repair roles have been indicated by physical and functional interactions with proteins in the DNA repair pathways. WRN links to BER include physical and functional interaction with polβ (Harrigan et al., 2003), polδ (Szekely et al., 2000), replication protein A (RPA)(Brosh et al., 1999), flap endonuclease 1 (FEN-1) (Brosh et al., 2001b), PCNA (Lebel et al., 1999) and poly(ADP-ribose)polymerase 1 (PARP-1) (von Kobbe et al., 2003a). Possible roles in the HRR pathway are suggested by interactions with the MRN complex (Cheng et al., 2004) and Rad52 (Baynton et al., 2003) and by colocalization with Rad51 in camptothecin-treated cells (Sakamoto et al., 2001). Similarly, a link to the NHEJ pathway is indicated by in interactions of WRN with the NHEJ-essential protein kinase DNA-PK (Yannone et al., 2001, Karmakar et al., 2002a, Li and Comai, 2002). WRN activity is regulated by holo–DNA-PK (Yannone et al., 2001, Karmakar et al., 2002a), WRN is an in vivo substrate of DNA-PK (Yannone et al., 2001, Karmakar et al., 2002a) and the DNA-PK subunit Ku70/80 stimulates WRN exonuclease activity in vitro (Cooper et al., 2000, Li and Comai, 2000, Li and Comai, 2001, Orren et al., 2001, Karmakar et al., 2002b). Furthermore, WRN has been observed in an endogenous complex with the Ku70/80 subunit and poly(ADP-ribose) polymerase-1 (PARP-1) (Li et al., 2004). Notably, PARP-1 binds sites of SSBs and DSBs and is also implicated in the control of genomic integrity and mammalian life span (Burkle et al., 2005).
WRN has a modular composition (Fig. 6a) and structural studies on the protein's domains and those of homologues are helping to define WRN mediated functions (Killoran and Keck, 2006). The N-terminus of WRN contains the exonuclease domain, the central core contains the helicase region and in the C-terminus two regions have been identified that bind protein partners and/or DNA (Sharma et al., 2006). Crystallographic and structure based mutational studies on the WRN exonuclease domain, have revealed a high degree of structural and mechanistic conservation with the DnaQ family of replicative proofreading exonucleases (Perry et al., 2006) (Fig. 6b). These structural biochemistry studies on WRN exonuclease revealed a two metal ion mediated mechanism of nucleotide substrate degradation, using divalent cations that include Mn2+ and Mg2+. The lanthanide Eu3+ ions inhibit the WRN exonuclease activity, probably due to either a greater charge state or their larger radii or both, causing misalignment of the scissile phosphate of the DNA substrate. Ku70/80 specifically stimulates this WRN exonuclease activity but inhibits the Klenow fragment exonuclease, its closest structural homolog (Perry et al., 2006). This also suggests that the WRN exonuclease domain may help impart functions mediated by WRN–Ku70/80. Additionally, WRN exonuclease activity is required to fully compliment a Werner syndrome DNA-end joining phenotype, in an in vivo plasmid based assay (Perry et al., 2006). The in vivo data did not define a specific cellular pathway, but the elevated microhomology-mediated repair observed in WRN exonuclease deficient cells is similar to the phenotypes associated with essential NHEJ proteins (Melek et al., 1998, Verkaik et al., 2002), possibly linking WRN to this pathway. However, Werner syndrome cells have mild radiation sensitivity, which rules out WRN as an essential DSB repair protein, but WRN exonuclease may nevertheless be used for resolution of a limited class of DSBs.
Fig. 6.

WRN exonuclease structure and hexameric ring model. a) WRN protein is modular, composed of an N-terminal exnuclease domain (blue), helicase lobes (red and yellow), RecQ-Ct region (green, cyan) and C-terminal HRDC domain (purple). b) The WRN exonuclease structure (adapted from (Perry et al., 2006)). WRN exonuclease has an α/β fold belonging the DnaQ family of proofreading exonucleases. The α-helices, β-strands are active site residues, (yellow tubes with red oxygens) are labeled. Two Mn2+ metal ions that bind in the active site and can mediate dsDNA degradation are shown in magenta. c) The WRN exonuclease hexameric ring model, built by structural superimposition with the A. thaliana homlogue (PDB code: 1VK0). The active site of the exonuclease, containing grey spheres denoting metal ions, faces the center of the ring, which is large enough to accommodate dsDNA. d) The E. coli RecQ crystal structure containing helicase lobes I (red) and II (yellow), and the Zn2+ binding domain (green) and winged helix domain (cyan) of the RecQ-Ct domain (Bernstein et al., 2003). e) ATP-γ-S and a Mn2+ ion (purple) are bound in a cleft formed by helicase motifs, I II and motif 0 that forms a pocket for the adenine base. f) A Zn2+ ion (orange) is bound in the RecQ-Ct region by cysteine side chains that are well conserved in RecQ helicases and are critical for function. f) The WRN winged helix domain that binds DNA and protein partners. The side chains that constitute the probable DNA binding interface are depicted (Hu et al., 2005). g) In the E. coli HRDC structure, side chains predominantly on α-helix 1 constitute a ssDNA binding interface (Bernstein and Keck, 2005). However, DNA substrates vary among the RecQ helicases and substrate specificity may be mediated by differences in their HRDC domains.
This substantial functional divergence between WRN exonuclease and its structural homologs, such as Klenow fragment exonuclease, may in part be explained by differences in higher-order structural organization. The WRN exonuclease domain construct that was defined by crystallography studies is monomeric. However, a similar WRN exonuclease construct forms homo-hexamers upon interaction with DNA or PCNA (Xue et al., 2002). Also, a larger WRN N-terminal construct, residues 1-333 and containing the exonuclease domain, forms a stable homomultimer (Huang et al., 2000). Multimerization potentially affects substrate specificities and enzymatic activities of the full-length WRN protein. The multimerization state of WRN homologues is still under debate (Sharma et al., 2006), but the human homolog BLM has been observed to form hexameric and/or tetrameric rings (Karow et al., 1999). A WRN exonuclease hexameric ring model (Fig. 6c) has been built based on a multimeric A. thaliana structural homolog (PDB code: 1VK0) (Perry et al., 2006). This WRN exonuclease ring contains a positively charged central cavity with the exonuclease domain facing the center and it is large enough to accommodate dsDNA. Ring formation would allow for efficient protein handoffs and regulation of DNA-end processing and avoid the risks of release of broken DNA strand.
Important insights into the molecular mechanisms of WRN have also been discovered from the structure of the conserved RecQ helicase core from the E. coli homologue (Bernstein et al., 2003). The RecQ crystal structure revealed the presence of four domains in the helicase core (Fig. 6d). The two N-terminal domains are helicase lobes that share structural similar to superfamily (SF) 1 and 2 helicases, but have some features unique to the RecQ family. The classical seven helicase sequence motifs (I, Ia, II, III, IV, V & VI) which couple nucleotide triophosphate (NTP) binding and hydrolysis with unwinding nucleotide duplexes, are conserved. Motifs I and II (the walker A and B motifs) interact with the non-hydrolyzable ATP analog, ATP-γ-S and Mg2+ in co-crystal structure of E. coli RecQ with these moieties (Bernstein et al., 2003) (Fig. 6e) and this is similar to SF1 & 2 helicases. In addition, a conserved region specific RecQ helicases, ‘motif 0’, forms a pocket to preferentially accommodate the adenine base of ATP, over other NTPs. This is similar to the Q motif in RNA DEAD-box helicases, indicating a possible evolutionary link between the two families. Mutations in the helicase motifs disrupt the function of RecQ helicases (Killoran and Keck, 2006). In WRN, a mutation in Motif I in mice induces a Werner phenotype in mice tail-derived fibroblasts (Wang et al., 2000). However, how DNA binding and unwinding is mediated by the helicase motifs is still undefined, but the lobes likely use strategies similar to SF1 & 2 proteins. The diminished ability to bind DNA substrates with restricted backbone flexibility (Wang et al., 2000) and regions of sequence similarity to SF1 indicates that WRN helicase may function a base-flipping mechanism proposed in SF1, despite overall sequence similarity to SF2 helicases (Bernstein et al., 2003).
Two domains following the helicase lobes in the RecQ conserved core form the RecQ C-terminal region, RecQ-Ct. This region is composed of a Zn2+ binding domain and a winged helix domain (Fig. 6c). Zn2+ is ligated in the E. coli structure via four cysteine side chains from two helices and a connecting loop (Fig. 6f). The cysteine side chains are conserved in the RecQ family and mutations in these residues disrupt RecQ helicase function, and are sufficient to cause Bloom syndrome when mutated in BLM (Ellis et al., 1995). The winged helix domain forms a helix-turn-helix fold in the E. coli crystal structure (Bernstein et al., 2003) and in a more recently determined NMR structure of this domain in WRN (Hu et al., 2005) (Fig. 6g). This domain in WRN binds several alternate DNA substructures, including forks, holding junctions, 3′ recessed dsDNA and binds blunt-ended dsDNA or ssDNA (von Kobbe et al., 2003b). Notably, the WRN winged helix domain facilitates targeting of WRN to the nucleolus (von Kobbe and Bohr, 2002) and interactions of several of the potential protein partners of WRN have also been specifically mapped to this winged helix domain (Lee et al., 2005), indicating the critical and versatile nature of this domain in WRN.
The remaining C-terminal domain of WRN is the HRDC (Helicase RNase D Conserved) domain. The structures of the E. coli and S. cerevisae HRDC domains have been determined, which have a globular all α-helical fold (Liu et al., 1999, Bernstein and Keck, 2005) (Fig. 6h). HRDC domains bind DNA substrates and differences in the composition of HRDC surface residues alters the substrate affinities between the RecQ helicases. In WRN the HRDC domain preferentially binds to forked duplex DNA and holiday junction DNA with high affinity, and 3′ recessed DNA to a lesser extent (von Kobbe et al., 2003b). This suggests that this domain is utilized in replication and recombination functions of WRN. Significantly, the interactions with the WRN C-terminus, containing the winged helix and HRDC domains, have been indicated to regulate the activity of WRN or of the partner protein. The WRN exonuclease domain activity is regulated through the interaction of its C-terminus with p53 (Blander et al., 1999), Ku70/80 (Cooper et al., 2000, Li and Comai, 2000, Brosh et al., 2001a, Karmakar et al., 2002b) and PARP-1 (von Kobbe et al., 2004). While WRN helicase activity is regulated by C-terminal interactions that include TRF2 (Opresko et al., 2002), Rad52 (Baynton et al., 2003) and PARP-1 (von Kobbe et al., 2004). An example of WRN stimulation of partner proteins includes the FEN-1 partner protein, whose structures with DNA and PCNA have been defined (Hosfield et al., 1998, Chapados et al., 2004). WRN interaction that was mapped to the winged helix domain stimulates FEN-1 nucleolytic activity, by more than 80-fold (Brosh et al., 2001b). Therefore, it will be most interesting to define how these interactions are able to regulate WRN catalytic activities, how key DNA and/or protein interactions may potentially allow for controlled handoffs during WRN mediated pathway progression and how the breakdown of this pathway progression, in the absence of functioning WRN, gives rise to the disease phenotype.
NER and the XPB helicase
NER functions to restore short segments of nucleotides containing DNA helix distorting mutations. These typically occur from carcinogenic compounds and covalent linkages between pyrimidine bases caused by UV exposure. Clearly, UV light does not penetrate the skull, but there is evidence that DNA exposed to oxygen radicals generated by ionizing radiation can produce DNA lesions that require NER for repair (Satoh et al., 1993). NER is a particularly versatile DNA repair system that is capable of global genome repair (GG-NER) and also transcription-coupled repair (TC-NER), where the template strand of actively transcribed genes is preferribly repaired. Hereditary mutations in NER genes clearly demonstrate that the inherited DNA repair potential has profound influence on an individual's health and life span (de Laat et al., 1999). Moreover, much of our understanding of NER has been derived from studies on cells from individuals with NER defects that present clinical phenotypes. This includes patients with the rare genetic disorders xeroderma pigmentosum (XP), trichothiodystrophy (TTD) and Cockayne syndrome (CS). These diseases are characterized by high skin and eye photosensitivity, manifested at different levels (Lehmann, 2003). XP individuals are hyperphotosensitive, have marked skin cancer risk and may have neuropathology caused by neuronal degeneration. Some of the XP individuals have a combined XP-CS phenotype, which has the severe neurological and developmental anomalies typical of CS that are caused by demyelination in the brain. The TTD individuals have characteristically brittle hair and nails, and may also present physical and mental retardation.
One key gene that is associated with all three disorders is the XPB helicase (Weeda et al., 1997), which is part of the general transcription factor TFIIH complex (Schaeffer et al., 1993). The XPB ATPase and helicase activities are essential for promoter DNA melting (Tirode et al., 1999) and clearance steps (Tirode et al., 1999) during initiation of transcription by RNA pol II. In addition to these transcriptional functions XPB also plays a role in NER. Recent developments in structural and biochemical characterization of XPB helicase have begun to address some of the key questions on the underlying mechanisms of how XPB and TFIIH function in both transcription and NER (Coin et al., 2004, Coin et al., 2006, Fan et al., 2006a). Structural biochemistry studies have been conducted on a homolog of the human XPB, the archea Archaeoglobus fulgidus XPB (AfXPB) (Fan et al., 2006a). AfXPB shares 42% amino acid sequence similarity with the central region of human XPB, suggesting that the core XPB structure is conserved. As indicated by sequence comparison, the AfXPB structure contains two RecA-like helicase domains (HD1 and HD2) that belong to helicase superfamily 2. However, several other functional regions in XPB were discovered that were not predicted either through sequence analysis or biochemical characterization. A small N-terminal domain is attached to helicase domain HD1 (Fig. 7), which shares structural similarity to the mismatch recognition domain of the DNA mismatch repair protein MutS (Obmolova et al., 2000). This domain in AfXPB has been demonstrated to interact with some types of damaged DNA, and was hence termed the damage recognition domain (DRD) (Fan et al., 2006a). XPB DRD differs from the MutS domain, by lacking a critical Phe residue that is used for interaction with mismatch-specific lesions. Instead, AfXPB DRD likely recognizes distortions in the DNA typically caused by the broad spectrum of NER lesions. Therefore, this region may explain how DNA damage is located and linked to initiation of DNA unwinding during NER steps by XPB/TFIIH. Also present is a highly conserved XPB-family specific RED amino acid motif located in domain HD1, (Fig. 7c). Mutational analysis suggests that this XPB RED motif has a critical role in DNA unwinding function (Fan et al., 2006a). The C-terminal region contains the helicase domain HD2, and a thumb domain (ThM) insert. The ThM domain is predicted to bind DNA in a sequence independent manner, via the phosphodiester backbone. This is based on its similarity with the thumb domain of DNA polymerases and the presence of several conserved positively charged amino acid residues at the interface between the ThM and HD2 domains (Fan et al., 2006a).
Fig. 7.
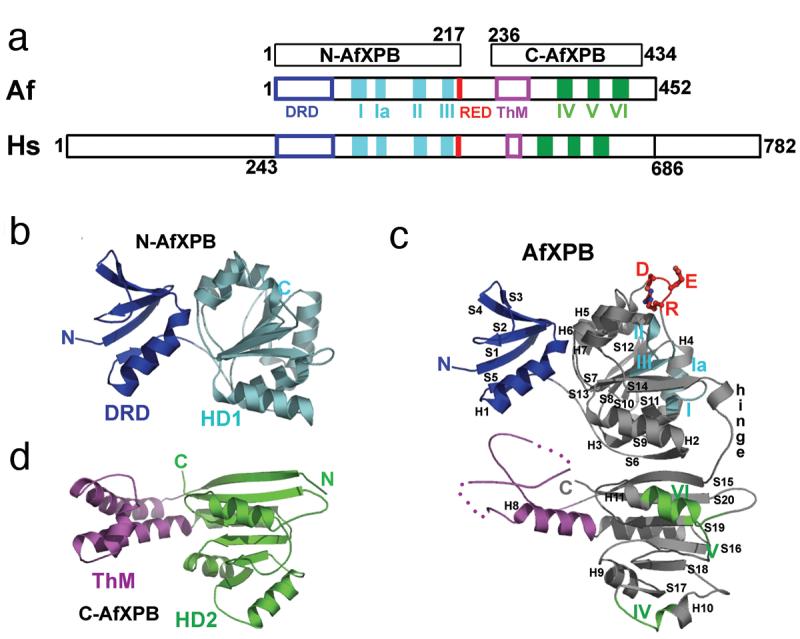
XPB conserved motifs and structural architecture. a) Schematic alignment between AfXPB, Af, and human XPB, Hs. The conserved helicase motifs I-VI and RED are colored by bars, the N-terminal DRD and ThM domains are colored by boxes. b) N-terminal domain of AfXPB showing the MutS-like DRD, blue, joins helicase domain HD1, cyan. c) Full length AfXPB showing the hinge joining HD1 and HD2, the RED motif side chains, plus the architectural arrangement of the domains. d) The C-terminal domain of AfXPB showing HD2, green and protruding helical polymerase-like thumb domain, purple, ThM that is partly disordered in full length AfXPB.
Interestingly, large conformational changes in helicases are known to be required for translocation along the duplex DNA and are coupled by ATP hydrolysis (Soultanas and Wigley, 2000, Soultanas and Wigley, 2001). AfXPB seems to follow this general trend. The relative orientation of the two helicase domains HD1 and HD2 observed in the full-length AfXPB is different than the “closed” conformation observed in crystal structures of nucleotide-bound helicases, suggesting a significant reorientation of the two helicase domains would have to take place to bring the functional helicase motifs to the active cleft (Fig. 8). This could occur through a long flexible loop that connects the N and C terminal helicase domain. These observations have also lead to a proposed mechanism for the involvement of XPB in the unwinding of duplex DNA at sites of DNA repair and transcription (Fig. 8). When XPB is recruited to DNA, the DRD domain is proposed to recognize the distorted and damaged DNA. This interaction induces a reorientation of helicase domain HD2 via a rotation of ∼170°, and allows XPB to wrap around the DNA. During transcription initiation, such a conformational change may result from interaction of the XPB C-terminus (including ThM and HD2 domains) with 3'-overhanging DNA. In both cases, in this new “closed” configuration, the RED motif would be ideally placed at the helicase active site, with the side chains intruding into the distorted DNA duplex. The ThM domain now “grips” one strand of the DNA above helicase domain HD2, whereas the other strand may lie in the groove on the opposite side of the RED motif. In this position, the RED motif would function as a “wedge” to unzip the DNA when ATP hydrolysis drives XPB to move along the duplex DNA during NER. However, it is noticed that DNA melting by XPB during transcription initiation is possibly mediated through an unconventional helicase mechanism (Kim et al., 2000), in which XPB functions as a molecular “wrench”: rotating downstream DNA relative to the fixed upstream protein-DNA interactions. Therefore, the conformation observed in the AfXPB structure may represent a “transcriptional mode” of XPB tuned for this action, whereas the domain reorientation described above is NER-specific and only occurs upon the interactions of the DRD with damaged DNA. If these mechanisms are true, the conformation of XPB will decide whether TFIIH functions as a transcription factor or a DNA repair factor. In other words, XPB acts as a master key, helping TFIIH switch pathway selection for transcription or DNA repair, whenever it is recruited to the DNA.
Fig. 8.

Proposed structure-based mechanism whereby damage verification by XPB promotes unwinding of damaged dsDNA for NER. a) Schematic model shows how XPB DRD, depicted in blue, HD1, cyan, RED motif, red, HD2, green, and ThM, purple, may be activated by dsDNA, yellow, containing a DNA damage site, orange. AfXPB initially binds to damaged DNA in an open conformation. DNA damage verification by N-XPB, DRD and HD1, induces the rotation of the C-terminal XPB domain, HD2/ThM. This forms the closed helicase-DNA complex, facilitated by HD1 mediated ATP hydrolysis that aids dsDNA melting at the lesion, orange, to allow the RED loop residues, red, to intrude between the opened DNA strands. The red arrow indicates the direction in which the RED motif unzips the base pairs. b) The predicted closed AfXPB conformation consisting of structural domains DRD, blue, HD1, cyan, and HD2, green, together with motifs RED, red sphere side chains, and ThM, purple. c) A proposed AfXPB-DNA complex. The AfXPB surface is mapped with the color-coded electrostatic potential, red negative, white neutral, blue positive. The DNA, purple phosphate backbone with bases as gold bars, was taken from the helicase Pcr-DNA complex (PDB code 3PJR) connected to a thymine-dimer-containing DNA from the photolyase-DNA complex (PDB code 1TEZ).
Defining the AfXPB structural biochemistry has uncovered some unexpected structural motifs and functions for XPB. However, AfXPB only correlates to the central region of human XPB. Disease-related mutations exclusively occur in the N- and C-terminal extensions of human XPB, suggesting that mutation to the conserved XPB central region is lethal. AfXPB reflects the basic structure and function of XPB helicases. However, the extensions to the human XPB are likely to contribute to a greater level of complexity and control. Phosphorylation of residue S751 at the C-terminal extension of human XPB was reported to regulate TFIIH activity in NER reactions (Coin et al., 2004). The physical and functional interactions between XPB and other proteins within and outside of the TFIIH complex have been investigated recently (Jawhari et al., 2002, Coin et al., 2006). They occur in the extensions and have profound effects on the TFIIH activities in transcription or DNA repair. Therefore, we expect that future studies will similarly uncover new functions for human XPB. For example, it will be interesting to see how the structural and mechanistic features highlighted above will fit into the ring-structure of human TFIIH complex (Chang and Kornberg, 2000, Schultz et al., 2000) and propagate signals and coordinate pathways in the context of several protein cofactors involved in transcription or DNA repair events.
Perspectives
We have presented the recent structural biochemistry results on proteins that when defective may give rise to severe neurological disorders. These novel studies are considerably aiding our understanding of the molecular basis of the complex disease phenotypes. The structural studies when coupled with in vitro and in vivo biochemistry analysis have provided not only a detailed understanding of catalytic mechanism at atomic resolution, but also provide insights into how the disease phenotypes occur through mutations in specific residues, such as observed in the ROS controlling superoxide dismutases. Some remarkable and unexpected discoveries have also been made into how the proteins and their key complexes are regulating pathway progression, often through conformational changes induced by substrate binding, signaling events or by protein partner binding. Notably, major architectural alterations in the protein assemblies are being suggested, such as those seen with the MRN complex, which likely allows for the bringing together of two distal sister chromatids for the proceeding HRR steps. Another significant example is the fine control of activities of the NOS holo-enzyme, suggested to occur through either promoting or inhibiting a molecular shuttle in the dimeric protein complex. Pathway progession may also be regulated through the mimicry of DNA or protein partners. There are several examples of proteins mimicking DNA interactions to form complexes (Putnam and Tainer, 2005) and these were initially identified in the BER pathway (Mol et al., 1995, Savva and Pearl, 1995, Putnam et al., 1999). A significant example of protein interface mimicry occurs in Rad51 filament formation, which is central to HRR steps. Structural biochemistry studies indicate that filament formation occurs by the sequential binding of adjacent Rad51 monomers mimicking a BRC repeat. This BRC repeat is normally found in the BRCA2 partner that mediates critical functions of Rad51 (Pellegrini et al., 2002, Shin et al., 2003). Thus, it will be interesting to see if interface mimicry and exchange participates in DNA repair events critical to genomic stability in the brain.
Overall, these structure-based studies are producing perhaps the most accurate and highly testable models, which define cellular pathway transitions that occur through the formation of macromolecular assemblies. Such dynamic assemblies are now beginning to be characterized through combined crystallography and small angle x-ray scattering studies to define their architectures (Tsutakawa, 2006). Thus, these advanced structural methods are defining in depth a set of cellular master keys, which control the complex steps in pathway progression that provide genomic stability and protection from insults from ROS and DNA damage agents. Moreover, these novel structural studies have presented possibilities for new therapeutics to be generated through rational structure-based drug design studies. These therapeutics may be geared towards the treatment or the disruption of neurological disease progression. The proteins described may be useful targets for several disease states. This includes MRN complex or the WRN RecQ helicase, which may prove to be suitable targets for therapies against tumorigenesis (O'Malley et al., 2003, Sharma et al., 2005) that includes the brain, this is in addition to their roles in neurological degeneration. The NOS isoforms also have multiple functions that may be targets for therapeutic strategies including the control of blood pressure, wound healing, migraines and Alzheimer's disease (Togo et al., 2004, Thatcher, 2005, Ramadan and Buchanan, 2006, Vignini et al., 2006). This is in addition to the NO mimetic that is currently in clinical trials for Alzheimer's disease (Thatcher et al., 2006).
Acknowledgements
Research results and analyses from the Tainer lab presented in this review come from studies supported by U.S. National Institute of Health grants CA104660 for RecQ studies, CA112093 for XPB analysis, CA92584 for studies on cellular DNA repair machines and AI022160 for pili and reactive oxygen species.
Abreviations
- AfXPB
Archaeoglobus fulgidus XPB
- AH
auto-inhibitory helix
- ATLD
ataxiatelangiectasia-like disorder
- BER
base excision repair
- CS
Cockayne syndrome
- CD
connecting domain
- CT
C-terminal tail
- DRD
damage recognition domain
- DSBs
double-strand breaks
- FAD
flavin adenine dinucleotide
- (F)ALS
(Familial) amyotrophic lateral sclerosis
- FMN
flavin mononucleotide
- GG-NER
global genome nucleotide excision repair
- HD1 and HD2
helicase domains 1 and 2
- HR
homologous recombination
- HRR
homologous recombination repair
- HRDC
helicase RNase D conserved domain
- MMR
mismatch repair
- MRN
Mre11/Rad50/Nbs1
- mtDNA
mitochondrial DNA
- NER
nucleotide excision repair
- NO
nitric oxide
- e, i or nNOS
endothelial, inducible or neuronal nitric oxide synthase
- NOSox
NOS catalytic oxygenase module
- NOSred
NOS reductase module
- NHEJ
nonhomologous end joining
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- SSBs
single-strand breaks
- TC-NER
transcription-coupled nucleotide excision repair
- ThM
thumb domain
- TTD
trichothiodystropy
- WS
Werner syndrome
- XP
xeroderma pigmentosum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Soud HM, Loftus M, Stuehr DJ. Subunit dissociation and unfolding of macrophage NO synthase: relationship between enzyme structure, prosthetic group binding, and catalytic function. Biochemistry. 1995;34:11167–11175. doi: 10.1021/bi00035a023. [DOI] [PubMed] [Google Scholar]
- Adak S, Ghosh S, Abu-Soud HM, Stuehr DJ. Role of reductase domain cluster 1 acidic residues in neuronal nitric-oxide synthase. Characterization of the FMN-FREE enzyme. J Biol Chem. 1999;274:22313–22320. doi: 10.1074/jbc.274.32.22313. [DOI] [PubMed] [Google Scholar]
- Aebi H, Wyss SR, Scherz B, Skvaril F. Heterogeneity of erythrocyte catalase II. Isolation and characterization of normal and variant erythrocyte catalase and their subunits. Eur J Biochem. 1974;48:137–145. doi: 10.1111/j.1432-1033.1974.tb03751.x. [DOI] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliyev A, Seyidova D, Rzayev N, Obrenovich ME, Lamb BT, Chen SG, Smith MA, Perry G, de la Torre JC, Aliev G. Is nitric oxide a key target in the pathogenesis of brain lesions during the development of Alzheimer's disease? Neurol Res. 2004;26:547–553. doi: 10.1179/01610425017613. [DOI] [PubMed] [Google Scholar]
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Andersen PM. Genetics of sporadic ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(Suppl 1):S37–41. doi: 10.1080/14660820152415726. [DOI] [PubMed] [Google Scholar]
- Aoyagi M, Arvai AS, Tainer JA, Getzoff ED. Structural basis for endothelial nitric oxide synthase binding to calmodulin. Embo J. 2003;22:766–775. doi: 10.1093/emboj/cdg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenmacher N, Hopfner KP. MRE11/RAD50/NBS1: complex activities. Chromosoma. 2004;113:157–166. doi: 10.1007/s00412-004-0306-4. [DOI] [PubMed] [Google Scholar]
- Ayala I, Perry JJ, Szczepanski J, Tainer JA, Vala MT, Nick HS, Silverman DN. Hydrogen bonding in human manganese superoxide dismutase containing 3-fluorotyrosine. Biophys J. 2005a;89:4171–4179. doi: 10.1529/biophysj.105.060616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala P, Wilbur JS, Wetzler LM, Tainer JA, Snyder A, So M. The pilus and porin of Neisseria gonorrhoeae cooperatively induce Ca(2+) transients in infected epithelial cells. Cell Microbiol. 2005b;7:1736–1748. doi: 10.1111/j.1462-5822.2005.00586.x. [DOI] [PubMed] [Google Scholar]
- Baynton K, Otterlei M, Bjoras M, von Kobbe C, Bohr VA, Seeberg E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J Biol Chem. 2003;278:36476–36486. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- Belloni M, Uberti D, Rizzini C, Ferrari-Toninelli G, Rizzonelli P, Jiricny J, Spano P, Memo M. Distribution and kainate-mediated induction of the DNA mismatch repair protein MSH2 in rat brain. Neuroscience. 1999;94:1323–1331. doi: 10.1016/s0306-4522(99)00380-2. [DOI] [PubMed] [Google Scholar]
- Bennett CB, Lewis AL, Baldwin KK, Resnick MA. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc Natl Acad Sci U S A. 1993;90:5613–5617. doi: 10.1073/pnas.90.12.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DA, Keck JL. Conferring substrate specificity to DNA helicases: role of the RecQ HRDC domain. Structure (Camb) 2005;13:1173–1182. doi: 10.1016/j.str.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Bernstein DA, Zittel MC, Keck JL. High-resolution structure of the E.coli RecQ helicase catalytic core. Embo J. 2003;22:4910–4921. doi: 10.1093/emboj/cdg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander G, Kipnis J, Leal JF, Yu CE, Schellenberg GD, Oren M. Physical and functional interaction between p53 and the Werner's syndrome protein. J Biol Chem. 1999;274:29463–29469. doi: 10.1074/jbc.274.41.29463. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Deficient DNA repair in the human progeroid disorder, Werner syndrome. Mutat Res. 2005;577:252–259. doi: 10.1016/j.mrfmmm.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Borgstahl GE, Parge HE, Hickey MJ, Beyer WF, Jr., Hallewell RA, Tainer JA. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell. 1992;71:107–118. doi: 10.1016/0092-8674(92)90270-m. [DOI] [PubMed] [Google Scholar]
- Brooks PJ. Detection of excision nuclease in cell-free extracts from the adult mammalian brain. Mutat Res. 1998;408:37–46. doi: 10.1016/s0921-8777(98)00018-4. [DOI] [PubMed] [Google Scholar]
- Brooks PJ, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM, Jr., Karmakar P, Sommers JA, Yang Q, Wang XW, Spillare EA, Harris CC, Bohr VA. p53 Modulates the exonuclease activity of Werner syndrome protein. J Biol Chem. 2001a;276:35093–35102. doi: 10.1074/jbc.M103332200. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Jr., Orren DK, Nehlin JO, Ravn PH, Kenny MK, Machwe A, Bohr VA. Functional and physical interaction between WRN helicase and human replication protein A. J Biol Chem. 1999;274:18341–18350. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Jr., von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. Embo J. 2001b;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Burkle A, Diefenbach J, Brabeck C, Beneke S. Ageing and PARP. Pharmacol Res. 2005;52:93–99. doi: 10.1016/j.phrs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Cabelli DE, Guan Y, Leveque V, Hearn AS, Tainer JA, Nick HS, Silverman DN. Role of tryptophan 161 in catalysis by human manganese superoxide dismutase. Biochemistry. 1999;38:11686–11692. doi: 10.1021/bi9909142. [DOI] [PubMed] [Google Scholar]
- Carrodeguas JA, Theis K, Bogenhagen DF, Kisker C. Crystal structure and deletion analysis show that the accessory subunit of mammalian DNA polymerase gamma, Pol gamma B, functions as a homodimer. Mol Cell. 2001;7:43–54. doi: 10.1016/s1097-2765(01)00153-8. [DOI] [PubMed] [Google Scholar]
- Chang WH, Kornberg RD. Electron crystal structure of the transcription factor and DNA repair complex, core TFIIH. Cell. 2000;102:609–613. doi: 10.1016/s0092-8674(00)00083-0. [DOI] [PubMed] [Google Scholar]
- Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell. 2001;8:1105–1115. doi: 10.1016/s1097-2765(01)00388-4. [DOI] [PubMed] [Google Scholar]
- Cheng WH, von Kobbe C, Opresko PL, Arthur LM, Komatsu K, Seidman MM, Carney JP, Bohr VA. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J Biol Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- Chou SM, Wang HS, Komai K. Colocalization of NOS and SOD1 in neurofilament accumulation within motor neurons of amyotrophic lateral sclerosis: an immunohistochemical study. J Chem Neuroanat. 1996;10:249–258. doi: 10.1016/0891-0618(96)00137-8. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Coin F, Auriol J, Tapias A, Clivio P, Vermeulen W, Egly JM. Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity. Embo J. 2004;23:4835–4846. doi: 10.1038/sj.emboj.7600480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin F, Proietti De Santis L, Nardo T, Zlobinskaya O, Stefanini M, Egly JM. p8/TTD-A as a repair-specific TFIIH subunit. Mol Cell. 2006;21:215–226. doi: 10.1016/j.molcel.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Comai L, Li B. The Werner syndrome protein at the crossroads of DNA repair and apoptosis. Mech Ageing Dev. 2004;125:521–528. doi: 10.1016/j.mad.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912. [PMC free article] [PubMed] [Google Scholar]
- Craig L, Pique ME, Tainer JA. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol. 2004;2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- Craig L, Volkmann N, Arvai AS, Pique ME, Yeager M, Egelman EH, Tainer JA. Type IV Pilus Structure by Cryo-Electron Microscopy and Crystallography: Implications for Pilus Assembly and Functions. Mol Cell. 2006;23:651–662. doi: 10.1016/j.molcel.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, Stuehr DJ, Tainer JA. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- Crane BR, Rosenfeld RJ, Arvai AS, Ghosh DK, Ghosh S, Tainer JA, Stuehr DJ, Getzoff ED. N-terminal domain swapping and metal ion binding in nitric oxide synthase dimerization. Embo J. 1999;18:6271–6281. doi: 10.1093/emboj/18.22.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- David P, Efrati E, Tocco G, Krauss SW, Goodman MF. DNA replication and postreplication mismatch repair in cell-free extracts from cultured human neuroblastoma and fibroblast cells. J Neurosci. 1997;17:8711–8720. doi: 10.1523/JNEUROSCI.17-22-08711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Dotti MT, Battisti C, Sicurelli F, Stromillo ML, Mortilla M, Federico A. MR evidence of structural and metabolic changes in brains of patients with Werner's syndrome. J Neurol. 2003;250:1169–1173. doi: 10.1007/s00415-003-0167-4. [DOI] [PubMed] [Google Scholar]
- Deisseroth A, Dounce AL. Catalase: Physical and chemical properties, mechanism of catalysis, and physiological role. Physiol Rev. 1970;50:319–375. doi: 10.1152/physrev.1970.50.3.319. [DOI] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- DiDonato M, Craig L, Huff ME, Thayer MM, Cardoso RM, Kassmann CJ, Lo TP, Bruns CK, Powers ET, Kelly JW, Getzoff ED, Tainer JA. ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol. 2003;332:601–615. doi: 10.1016/s0022-2836(03)00889-1. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Protein folding and its links with human disease. Biochem Soc Symp. 2001:1–26. [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Fan L, Arvai AS, Cooper PK, Iwai S, Hanaoka F, Tainer JA. Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Mol Cell. 2006a;22:27–37. doi: 10.1016/j.molcel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Fan L, Kaguni LS. Multiple regions of subunit interaction in Drosophila mitochondrial DNA polymerase: three functional domains in the accessory subunit. Biochemistry. 2001;40:4780–4791. doi: 10.1021/bi010102h. [DOI] [PubMed] [Google Scholar]
- Fan L, Kim S, Farr CL, Schaefer KT, Randolph KM, Tainer JA, Kaguni LS. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J Mol Biol. 2006b;358:1229–1243. doi: 10.1016/j.jmb.2006.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat Struct Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide dismutases. Adv Enzymol Relat Areas Mol Biol. 1986;58:61–97. doi: 10.1002/9780470123041.ch2. [DOI] [PubMed] [Google Scholar]
- Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. doi: 10.1080/14660820050515377. [DOI] [PubMed] [Google Scholar]
- Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- Goth L. Further genetic heterogeneity in acatalasemia. Electrophoresis. 1997;18:1942–1943. doi: 10.1002/elps.1150181110. [DOI] [PubMed] [Google Scholar]
- Goto M. Hierarchical deterioration of body systems in Werner's syndrome: implications for normal ageing. Mech Ageing Dev. 1997;98:239–254. doi: 10.1016/s0047-6374(97)00111-5. [DOI] [PubMed] [Google Scholar]
- Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- Greenleaf WB, Perry JJ, Hearn AS, Cabelli DE, Lepock JR, Stroupe ME, Tainer JA, Nick HS, Silverman DN. Role of hydrogen bonding in the active site of human manganese superoxide dismutase. Biochemistry. 2004;43:7038–7045. doi: 10.1021/bi049888k. [DOI] [PubMed] [Google Scholar]
- Gruber S, Haering CH, Nasmyth K. Chromosomal cohesin forms a ring. Cell. 2003;112:765–777. doi: 10.1016/s0092-8674(03)00162-4. [DOI] [PubMed] [Google Scholar]
- Guan Y, Hickey MJ, Borgstahl GE, Hallewell RA, Lepock JR, O'Connor D, Hsieh Y, Nick HS, Silverman DN, Tainer JA. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry. 1998;37:4722–4730. doi: 10.1021/bi972394l. [DOI] [PubMed] [Google Scholar]
- Guix FX, Uribesalgo I, Coma M, Munoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol. 2005;76:126–152. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Hallewell RA, Imlay KC, Lee P, Fong NM, Gallegos C, Getzoff ED, Tainer JA, Cabelli DE, Tekamp-Olson P, Mullenbach GT, et al. Thermostabilization of recombinant human and bovine CuZn superoxide dismutases by replacement of free cysteines. Biochem Biophys Res Commun. 1991;181:474–480. doi: 10.1016/s0006-291x(05)81443-3. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Opresko PL, von Kobbe C, Kedar PS, Prasad R, Wilson SH, Bohr VA. The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activity. J Biol Chem. 2003;278:22686–22695. doi: 10.1074/jbc.M213103200. [DOI] [PubMed] [Google Scholar]
- Hearn AS, Fan L, Lepock JR, Luba JP, Greenleaf WB, Cabelli DE, Tainer JA, Nick HS, Silverman DN. Amino acid substitution at the dimeric interface of human manganese superoxide dismutase. J Biol Chem. 2004;279:5861–5866. doi: 10.1074/jbc.M311310200. [DOI] [PubMed] [Google Scholar]
- Hearn AS, Stroupe ME, Cabelli DE, Ramilo CA, Luba JP, Tainer JA, Nick HS, Silverman DN. Catalytic and structural effects of amino acid substitution at histidine 30 in human manganese superoxide dismutase: insertion of valine C gamma into the substrate access channel. Biochemistry. 2003;42:2781–2789. doi: 10.1021/bi0266481. [DOI] [PubMed] [Google Scholar]
- Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- Hirono A, Sasaya-Hamada F, Kanno H, Fujii H, Yoshida T, Miwa S. A novel human catalase mutation (358 T-->del) causing Japanese-type acatalasemia. Blood Cells Mol Dis. 1995;21:232–234. doi: 10.1006/bcmd.1995.0026. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, Carney JP, Petrini JH, Tainer JA. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002a;418:562–566. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–485. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Putnam CD, Tainer JA. DNA double-strand break repair from head to tail. Curr Opin Struct Biol. 2002b;12:115–122. doi: 10.1016/s0959-440x(02)00297-x. [DOI] [PubMed] [Google Scholar]
- Hosfield DJ, Mol CD, Shen B, Tainer JA. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell. 1998;95:135–146. doi: 10.1016/s0092-8674(00)81789-4. [DOI] [PubMed] [Google Scholar]
- Hu JS, Feng H, Zeng W, Lin GX, Xi XG. Solution structure of a multifunctional DNA- and protein-binding motif of human Werner syndrome protein. Proc Natl Acad Sci U S A. 2005;102:18379–18384. doi: 10.1073/pnas.0509380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Beresten S, Li B, Oshima J, Ellis NA, Campisi J. Characterization of the human and mouse WRN 3′-->5′ exonuclease. Nucleic Acids Res. 2000;28:2396–2405. doi: 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Li B, Gray MD, Oshima J, Mian IS, Campisi J. The premature ageing syndrome protein, WRN, is a 3′-->5′ exonuclease. Nat Genet. 1998;20:114–116. doi: 10.1038/2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr Opin Neurol. 2001;14:89–94. doi: 10.1097/00019052-200102000-00014. [DOI] [PubMed] [Google Scholar]
- Jawhari A, Laine JP, Dubaele S, Lamour V, Poterszman A, Coin F, Moras D, Egly JM. p52 Mediates XPB function within the transcription/repair factor TFIIH. J Biol Chem. 2002;277:31761–31767. doi: 10.1074/jbc.M203792200. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- Karmakar P, Piotrowski J, Brosh RM, Jr., Sommers JA, Miller SP, Cheng WH, Snowden CM, Ramsden DA, Bohr VA. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J Biol Chem. 2002a;277:18291–18302. doi: 10.1074/jbc.M111523200. [DOI] [PubMed] [Google Scholar]
- Karmakar P, Snowden CM, Ramsden DA, Bohr VA. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 2002b;30:3583–3591. doi: 10.1093/nar/gkf482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karow JK, Newman RH, Freemont PS, Hickson ID. Oligomeric ring structure of the Bloom's syndrome helicase. Curr Biol. 1999;9:597–600. doi: 10.1016/s0960-9822(99)80264-4. [DOI] [PubMed] [Google Scholar]
- Kato S, Sumi-Akamaru H, Fujimura H, Sakoda S, Kato M, Hirano A, Takikawa M, Ohama E. Copper chaperone for superoxide dismutase co-aggregates with superoxide dismutase 1 (SOD1) in neuronal Lewy body-like hyaline inclusions: an immunohistochemical study on familial amyotrophic lateral sclerosis with SOD1 gene mutation. Acta Neuropathol (Berl) 2001;102:233–238. doi: 10.1007/s004010000355. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- Killoran MP, Keck JL. Sit down, relax and unwind: structural insights into RecQ helicase mechanisms. Nucleic Acids Res. 2006 doi: 10.1093/nar/gkl538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Ebright RH, Reinberg D. Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science. 2000;288:1418–1422. doi: 10.1126/science.288.5470.1418. [DOI] [PubMed] [Google Scholar]
- Knudsen GM, Nishida CR, Mooney SD, Ortiz de Montellano PR. Nitric-oxide synthase (NOS) reductase domain models suggest a new control element in endothelial NOS that attenuates calmodulin-dependent activity. J Biol Chem. 2003;278:31814–31824. doi: 10.1074/jbc.M303267200. [DOI] [PubMed] [Google Scholar]
- Koedel U, Pfister HW. Oxidative stress in bacterial meningitis. Brain Pathol. 1999;9:57–67. doi: 10.1111/j.1750-3639.1999.tb00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst CB. Complex genetics of amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:933–947. doi: 10.1086/426001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel M, Spillare EA, Harris CC, Leder P. The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J Biol Chem. 1999;274:37795–37799. doi: 10.1074/jbc.274.53.37795. [DOI] [PubMed] [Google Scholar]
- Lee JW, Harrigan J, Opresko PL, Bohr VA. Pathways and functions of the Werner syndrome protein. Mech Ageing Dev. 2005;126:79–86. doi: 10.1016/j.mad.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Lehmann AR. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie. 2003;85:1101–1111. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Lennon BW, Williams CH, Jr., Ludwig ML. Twists in catalysis: alternating conformations of Escherichia coli thioredoxin reductase. Science. 2000;289:1190–1194. doi: 10.1126/science.289.5482.1190. [DOI] [PubMed] [Google Scholar]
- Leveque VJ, Stroupe ME, Lepock JR, Cabelli DE, Tainer JA, Nick HS, Silverman DN. Multiple replacements of glutamine 143 in human manganese superoxide dismutase: effects on structure, stability, and catalysis. Biochemistry. 2000;39:7131–7137. doi: 10.1021/bi9929958. [DOI] [PubMed] [Google Scholar]
- Leys D, Basran J, Talfournier F, Sutcliffe MJ, Scrutton NS. Extensive conformational sampling in a ternary electron transfer complex. Nat Struct Biol. 2003;10:219–225. doi: 10.1038/nsb894. [DOI] [PubMed] [Google Scholar]
- Li B, Comai L. Functional interaction between Ku and the werner syndrome protein in DNA end processing. J Biol Chem. 2000;275:39800. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- Li B, Comai L. Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-Ku70/80 complex. J Biol Chem. 2001;276:9896–9902. doi: 10.1074/jbc.M008575200. [DOI] [PubMed] [Google Scholar]
- Li B, Comai L. Displacement of DNA-PKcs from DNA ends by the Werner syndrome protein. Nucleic Acids Res. 2002;30:3653–3661. doi: 10.1093/nar/gkf488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Navarro S, Kasahara N, Comai L. Identification and biochemical characterization of a Werner's syndrome protein complex with Ku70/80 and poly(ADPribose) polymerase-1. J Biol Chem. 2004;279:13659–13667. doi: 10.1074/jbc.M311606200. [DOI] [PubMed] [Google Scholar]
- Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The novel binding mode of N-alkyl-N'-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Liu Z, Macias MJ, Bottomley MJ, Stier G, Linge JP, Nilges M, Bork P, Sattler M. The three-dimensional structure of the HRDC domain and implications for the Werner and Bloom syndrome proteins. Structure. 1999;7:1557–1566. doi: 10.1016/s0969-2126(00)88346-x. [DOI] [PubMed] [Google Scholar]
- Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci U S A. 1999;96:7376–7381. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marietta C, Palombo F, Gallinari P, Jiricny J, Brooks PJ. Expression of long-patch and short-patch DNA mismatch repair proteins in the embryonic and adult mammalian brain. Brain Res Mol Brain Res. 1998;53:317–320. doi: 10.1016/s0169-328x(97)00311-2. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Shimamoto A, Goto M, Furuichi Y. Impaired nuclear localization of defective DNA helicases in Werner's syndrome. Nat Genet. 1997;16:335–336. doi: 10.1038/ng0897-335. [DOI] [PubMed] [Google Scholar]
- Mayer B, Hemmens B. Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem Sci. 1997;22:477–481. doi: 10.1016/s0968-0004(97)01147-x. [DOI] [PubMed] [Google Scholar]
- McRee DE, Redford SM, Getzoff ED, Lepock JR, Hallewell RA, Tainer JA. Changes in crystallographic structure and thermostability of a Cu,Zn superoxide dismutase mutant resulting from the removal of a buried cysteine. J Biol Chem. 1990;265:14234–14241. doi: 10.2210/pdb3sod/pdb. [DOI] [PubMed] [Google Scholar]
- Melek M, Gellert M, van Gent DC. Rejoining of DNA by the RAG1 and RAG2 proteins. Science. 1998;280:301–303. doi: 10.1126/science.280.5361.301. [DOI] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Miller RT, Martasek P, Omura T, Siler Masters BS. Rapid kinetic studies of electron transfer in the three isoforms of nitric oxide synthase. Biochem Biophys Res Commun. 1999;265:184–188. doi: 10.1006/bbrc.1999.1643. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Hayashi M, Takeuchi A, Okamoto T, Kawashima S, Takii T, Hayashi H, Onozaki K. Identification of a novel growth-promoting factor with a wide target cell spectrum from various tumor cells as catalase. J Biochem (Tokyo) 1996;120:725–730. doi: 10.1093/oxfordjournals.jbchem.a021471. [DOI] [PubMed] [Google Scholar]
- Mol CD, Arvai AS, Sanderson RJ, Slupphaug G, Kavli B, Krokan HE, Mosbaugh DW, Tainer JA. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701–708. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- Moser MJ, Holley WR, Chatterjee A, Mian IS. The proofreading domain of Escherichia coli DNA polymerase I and other DNA and/or RNA exonuclease domains. Nucleic Acids Res. 1997;25:5110–5118. doi: 10.1093/nar/25.24.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MJ, Kamath-Loeb AS, Jacob JE, Bennett SE, Oshima J, Monnat RJ., Jr. WRN helicase expression in Werner syndrome cell lines. Nucleic Acids Res. 2000;28:648–654. doi: 10.1093/nar/28.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian AR, Bassett DE, Jr., Boguski MS, Bork P, Koonin EV. Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc Natl Acad Sci U S A. 1997;94:5831–5836. doi: 10.1073/pnas.94.11.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Nakayama K, Nakayama R, Irino N, Nakayama Y, Hanawalt PC. Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol Gen Genet. 1984;195:474–480. doi: 10.1007/BF00341449. [DOI] [PubMed] [Google Scholar]
- O'Malley BW, Jr., Li D, Carney J, Rhee J, Suntharalingam M. Molecular disruption of the MRN(95) complex induces radiation sensitivity in head and neck cancer. Laryngoscope. 2003;113:1588–1594. doi: 10.1097/00005537-200309000-00034. [DOI] [PubMed] [Google Scholar]
- Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- Opresko PL, Cheng WH, von Kobbe C, Harrigan JA, Bohr VA. Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis. 2003;24:791–802. doi: 10.1093/carcin/bgg034. [DOI] [PubMed] [Google Scholar]
- Opresko PL, Laine JP, Brosh RM, Jr., Seidman MM, Bohr VA. Coordinate action of the helicase and 3′ to 5′ exonuclease of Werner syndrome protein. J Biol Chem. 2001;276:44677–44687. doi: 10.1074/jbc.M107548200. [DOI] [PubMed] [Google Scholar]
- Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem. 2002;277:41110–41119. doi: 10.1074/jbc.M205396200. [DOI] [PubMed] [Google Scholar]
- Orren DK, Machwe A, Karmakar P, Piotrowski J, Cooper MP, Bohr VA. A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 2001;29:1926–1934. doi: 10.1093/nar/29.9.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutat Res. 2005;577:237–251. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Parge HE, Hallewell RA, Tainer JA. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc Natl Acad Sci U S A. 1992;89:6109–6113. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, Cooper PK, Chen DJ, Tainer JA. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Arvai AS, Bourne Y, Tainer JA. Active and inhibited human catalase structures: ligand and NADPH binding and catalytic mechanism. J Mol Biol. 2000;296:295–309. doi: 10.1006/jmbi.1999.3458. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Shroyer MJ, Lundquist AJ, Mol CD, Arvai AS, Mosbaugh DW, Tainer JA. Protein mimicry of DNA from crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with Escherichia coli uracil-DNA glycosylase. J Mol Biol. 1999;287:331–346. doi: 10.1006/jmbi.1999.2605. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Tainer JA. Protein mimicry of DNA and pathway regulation. DNA Repair (Amst) 2005;4:1410–1420. doi: 10.1016/j.dnarep.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- Ramadan NM, Buchanan TM. New and future migraine therapy. Pharmacol Ther. 2006;112:199–212. doi: 10.1016/j.pharmthera.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- Ren X, Tu C, Bhatt D, Perry JJP, Tainer JA, Cabelli DE, Silverman DN. Kinetic and Structural Characterization of Human Manganese Superoxide Dismutase Containing 3-Fluorotyrosines. Journal of Molecular Structure. 2006;1-3:168–173. [Google Scholar]
- Roman LJ, Martasek P, Masters BS. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem Rev. 2002;102:1179–1190. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rosenfeld RJ, Garcin ED, Panda K, Andersson G, Aberg A, Wallace AV, Morris GM, Olson AJ, Stuehr DJ, Tainer JA, Getzoff ED. Conformational changes in nitric oxide synthases induced by chlorzoxazone and nitroindazoles: crystallographic and computational analyses of inhibitor potency. Biochemistry. 2002;41:13915–13925. doi: 10.1021/bi026313j. [DOI] [PubMed] [Google Scholar]
- Sakamoto S, Nishikawa K, Heo SJ, Goto M, Furuichi Y, Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells. 2001;6:421–430. doi: 10.1046/j.1365-2443.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- Satoh MS, Jones CJ, Wood RD, Lindahl T. DNA excision-repair defect of xeroderma pigmentosum prevents removal of a class of oxygen free radical-induced base lesions. Proc Natl Acad Sci U S A. 1993;90:6335–6339. doi: 10.1073/pnas.90.13.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva R, Pearl LH. Nucleotide mimicry in the crystal structure of the uracil-DNA glycosylase-uracil glycosylase inhibitor protein complex. Nat Struct Biol. 1995;2:752–757. doi: 10.1038/nsb0995-752. [DOI] [PubMed] [Google Scholar]
- Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JH, Chambon P, Egly JM. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- Schultz P, Fribourg S, Poterszman A, Mallouh V, Moras D, Egly JM. Molecular structure of human TFIIH. Cell. 2000;102:599–607. doi: 10.1016/s0092-8674(00)00082-9. [DOI] [PubMed] [Google Scholar]
- Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc Natl Acad Sci U S A. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Doherty KM, Brosh RM., Jr. DNA helicases as targets for anti-cancer drugs. Curr Med Chem Anticancer Agents. 2005;5:183–199. doi: 10.2174/1568011053765985. [DOI] [PubMed] [Google Scholar]
- Sharma S, Doherty KM, Brosh RM., Jr. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J. 2006;398:319–337. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JC, Gray MD, Oshima J, Loeb LA. Characterization of Werner syndrome protein DNA helicase activity: directionality, substrate dependence and stimulation by replication protein A. Nucleic Acids Res. 1998;26:2879–2885. doi: 10.1093/nar/26.12.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, Yu DS, Shivji MK, Hitomi C, Arvai AS, Volkmann N, Tsuruta H, Blundell TL, Venkitaraman AR, Tainer JA. Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. Embo J. 2003;22:4566–4576. doi: 10.1093/emboj/cdg429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddhanta U, Wu C, Abu-Soud HM, Zhang J, Ghosh DK, Stuehr DJ. Heme iron reduction and catalysis by a nitric oxide synthase heterodimer containing one reductase and two oxygenase domains. J Biol Chem. 1996;271:7309–7312. doi: 10.1074/jbc.271.13.7309. [DOI] [PubMed] [Google Scholar]
- Silverman DN, Nick HS. Catalytic pathway of manganese superoxide dismutase by direct observation of superoxide. Methods Enzymol. 2002;349:61–74. doi: 10.1016/s0076-6879(02)49321-4. [DOI] [PubMed] [Google Scholar]
- Soto C. Protein misfolding and disease; protein refolding and therapy. FEBS Lett. 2001;498:204–207. doi: 10.1016/s0014-5793(01)02486-3. [DOI] [PubMed] [Google Scholar]
- Soultanas P, Wigley DB. DNA helicases: ‘inching forward’. Curr Opin Struct Biol. 2000;10:124–128. doi: 10.1016/s0959-440x(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Soultanas P, Wigley DB. Unwinding the ‘Gordian knot’ of helicase action. Trends Biochem Sci. 2001;26:47–54. doi: 10.1016/s0968-0004(00)01734-5. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Theunissen JW, Morales M, Petrini JH. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst) 2004;3:845–854. doi: 10.1016/j.dnarep.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Szekely AM, Chen YH, Zhang C, Oshima J, Weissman SM. Werner protein recruits DNA polymerase delta to the nucleolus. Proc Natl Acad Sci U S A. 2000;97:11365–11370. doi: 10.1073/pnas.97.21.11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher GR. An introduction to NO-related therapeutic agents. Curr Top Med Chem. 2005;5:597–601. doi: 10.2174/1568026054679281. [DOI] [PubMed] [Google Scholar]
- Thatcher GR, Bennett BM, Reynolds JN. NO chimeras as therapeutic agents in Alzheimer's disease. Curr Alzheimer Res. 2006;3:237–245. doi: 10.2174/156720506777632925. [DOI] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3:87–95. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- Togo T, Katsuse O, Iseki E. Nitric oxide pathways in Alzheimer's disease and other neurodegenerative dementias. Neurol Res. 2004;26:563–566. doi: 10.1179/016164104225016236. [DOI] [PubMed] [Google Scholar]
- Tsutakawa SEH, Frankel KA, Cooper PK, Tainer JA. Structural analysis of flexible proteins in solution by small angle X-ray scattering combined with crystallography. Journal of Structural Biology. 2006 doi: 10.1016/j.jsb.2006.09.008. G.L. [DOI] [PubMed] [Google Scholar]
- van den Bosch M, Bree RT, Lowndes NF. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003;4:844–849. doi: 10.1038/sj.embor.embor925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkaik NS, Esveldt-van Lange RE, van Heemst D, Bruggenwirth HT, Hoeijmakers JH, Zdzienicka MZ, van Gent DC. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur J Immunol. 2002;32:701–709. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Vignini A, Nanetti L, Moroni C, Tanase L, Bartolini M, Luzzi S, Provinciali L, Mazzanti L. Modifications of platelet from Alzheimer disease patients: A possible relation between membrane properties and NO metabolites. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.05.010. [DOI] [PubMed] [Google Scholar]
- von Kobbe C, Bohr VA. A nucleolar targeting sequence in the Werner syndrome protein resides within residues 949-1092. J Cell Sci. 2002;115:3901–3907. doi: 10.1242/jcs.00076. [DOI] [PubMed] [Google Scholar]
- von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, Cheng WH, Bohr VA. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol. 2003a;23:8601–8613. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, Harrigan JA, Schreiber V, Stiegler P, Piotrowski J, Dawut L, Bohr VA. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004;32:4003–4014. doi: 10.1093/nar/gkh721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, Thoma NH, Czyzewski BK, Pavletich NP, Bohr VA. Werner syndrome protein contains three structure-specific DNA binding domains. J Biol Chem. 2003b;278:52997–53006. doi: 10.1074/jbc.M308338200. [DOI] [PubMed] [Google Scholar]
- Wang L, Ogburn CE, Ware CB, Ladiges WC, Youssoufian H, Martin GM, Oshima J. Cellular Werner phenotypes in mice expressing a putative dominant-negative human WRN gene. Genetics. 2000;154:357–362. doi: 10.1093/genetics/154.1.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci U S A. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker EE. Protein aggregation in Huntington's and Parkinson's disease: implications for therapy. Mol Med Today. 2000;6:387–391. doi: 10.1016/s1357-4310(00)01761-5. [DOI] [PubMed] [Google Scholar]
- Weeda G, Eveno E, Donker I, Vermeulen W, Chevallier-Lagente O, Taieb A, Stary A, Hoeijmakers JH, Mezzina M, Sarasin A. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet. 1997;60:320–329. [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Tainer JA. A nanomachine for making ends meet: MRN is a flexing scaffold for the repair of DNA double-strand breaks. Mol Cell. 2005;19:724–726. doi: 10.1016/j.molcel.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Wiltzius JJ, Hohl M, Fleming JC, Petrini JH. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat Struct Mol Biol. 2005;12:403–407. doi: 10.1038/nsmb928. [DOI] [PubMed] [Google Scholar]
- Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997;25:2985–2991. doi: 10.1093/nar/25.15.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ratcliff GC, Wang H, Davis-Searles PR, Gray MD, Erie DA, Redinbo MR. A minimal exonuclease domain of WRN forms a hexamer on DNA and possesses both 3′- 5′ exonuclease and 5′-protruding strand endonuclease activities. Biochemistry. 2002;41:2901–2912. doi: 10.1021/bi0157161. [DOI] [PubMed] [Google Scholar]
- Yabuki M, Kariya S, Ishisaka R, Yasuda T, Yoshioka T, Horton AA, Utsumi K. Resistance to nitric oxide-mediated apoptosis in HL-60 variant cells is associated with increased activities of Cu,Zn-superoxide dismutase and catalase. Free Radic Biol Med. 1999;26:325–332. doi: 10.1016/s0891-5849(98)00203-2. [DOI] [PubMed] [Google Scholar]
- Yakubovskaya E, Chen Z, Carrodeguas JA, Kisker C, Bogenhagen DF. Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J Biol Chem. 2006;281:374–382. doi: 10.1074/jbc.M509730200. [DOI] [PubMed] [Google Scholar]
- Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, Campisi J, Chen DJ. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem. 2001;276:38242–38248. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- Zeviani M, Spinazzola A. Mitochondrial disorders. Curr Neurol Neurosci Rep. 2003;3:423–432. doi: 10.1007/s11910-003-0026-9. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhou J, Lim CU. The role of NBS1 in DNA double strand break repair, telomere stability, and cell cycle checkpoint control. Cell Res. 2006;16:45–54. doi: 10.1038/sj.cr.7310007. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Electron transfer by domain movement in cytochrome bc1. Nature. 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001;11:105–109. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]
- Zimatkin SM, Liopo AV, Deitrich RA. Distribution and kinetics of ethanol metabolism in rat brain. Alcohol Clin Exp Res. 1998;22:1623–1627. [PubMed] [Google Scholar]