

Summary
The 3′ processing of most bacterial precursor tRNAs involves exonucleolytic trimming to yield a mature CCA end. This step is carried out by RNase T, a member of the large DEDD family of exonucleases. We report the crystal structures of RNase T from Escherichia coli and Pseudomonas aeruginosa, which show that this enzyme adopts an opposing dimeric arrangement, with the catalytic DEDD residues from one monomer closely juxtaposed with a large basic patch on the other monomer. This arrangement suggests that RNase T has to be dimeric for substrate specificity, and agrees very well with prior site-directed mutagenesis studies. The dimeric architecture of RNase T is very similar to the arrangement seen in oligoribonuclease, another bacterial DEDD family exoribonuclease. The catalytic residues in these two enzymes are organized very similarly to the catalytic domain of the third DEDD family exoribonuclease in E. coli, RNase D, which is monomeric.
Introduction
All stable RNAs are synthesized as precursors. Maturation of stable RNAs often involves an initial cleavage by endoribonucleases followed by exonucleolytic trimming. In E. coli, seven distinct exoribonucleases have been identified (Ezraty et al., 2005; Zuo and Deutscher, 2001). Although many of them, including RNase D, RNase PH, RNase II and RNase T, contribute to this trimming process, RNase T (RNT) appears to be the most important for the final step of maturation of many stable RNAs (Kelly and Deutscher, 1992; Li and Deutscher, 1995; Li and Deutscher, 1996; Li et al., 1998; Li et al., 1999). In fact, RNase T is essential for generating the mature 3′-ends of 5S and 23S rRNAs (Li and Deutscher, 1995; Li et al., 1999). RNT is also involved in tRNA end turnover, which consists of the removal and regeneration of the –CCA end of tRNA (Deutscher et al., 1985). The latter half of this process involves a universally conserved –CCA adding enzyme, tRNA nucleotidyl transferase. Despite its many important roles, RNT orthologs have been found in only a small group of bacteria, the γ division of Proteobacteria (Zuo and Deutscher, 2001).
RNase T is a 3′ to 5′ hydrolytic exoribonuclease (Deutscher and Marlor, 1985). It is one of several E. coli proteins belonging to the DEDD superfamily, a large family of 3′-5′ exonucleases which also includes the proof-reading domain of many DNA polymerases (Zuo and Deutscher, 2001). Members of this superfamily contain four highly conserved acidic residues distributed in three conserved motifs (Figure 1). In the Klenow fragment, the four acidic residues were shown to bind two divalent metal ions and form the catalytic center (Beese and Steitz, 1991). It is believed that all DEDD family members share a common catalytic mechanism involving two divalent metal ions coordinated by the conserved acidic residues (Steitz and Steitz, 1993). The DEDD family can be divided into two subfamilies, DEDDh and DEDDy, depending on whether a histidine (e.g., in ε subunit of DNA polymerase III) or a tyrosine residue (e.g., in Klenow fragment) is conserved in the ExoIII motif (Zuo and Deutscher, 2001). Other than a slight difference in local folding surrounding this residue, the DEDDh and DEDDy exonucleases share a nearly identical fold with the His/Tyr residues occupying an essentially equivalent position. It is believed that a His in DEDDh functions the same way as a Tyr in DEDDy (Breyer and Matthews, 2000; Hamdan et al., 2002). The significance of the difference between a histidine and a tyrosine residue is still unclear. However, it cannot be responsible for the difference between DNases and RNases since both classes of enzymes are found in the DEDDh and DEDDy groups. RNase T is a member of the DEDDh subgroup. Sequence analysis suggests that RNase T is closely related to DP3E, the proof-reading ε subunit of DNA polymerase III, which is another DEDDh member (Koonin and Deutscher, 1993; Zuo and Deutscher, 2001). Interestingly, RNase T also displays strong DNA exonucleases activity (Viswanathan et al., 1998; Zuo and Deutscher, 1999).
Figure 1.
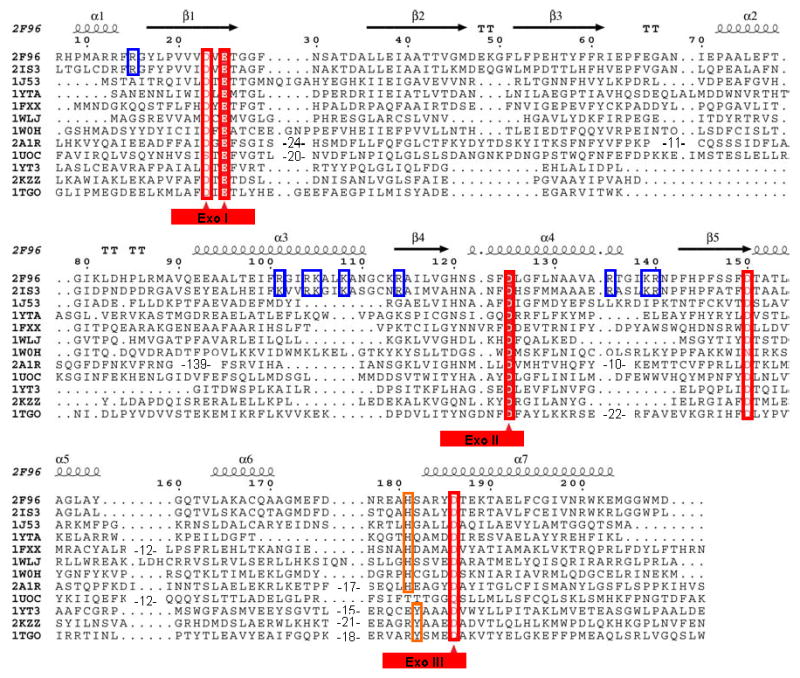
Structure based multiple alignment of E. coli and P. aeruginosa RNase T and related nucleases. Structures included here are: E. coli RNase T (PDB ID: 2IS3); P. aeruginosa putative RNase T (PDB ID: 2F96); E. coli DNA polymerase III ε subunit (PDB ID: 1J53); E. coli Oligoribonuclease (PDB ID: 1YTA); E. coli exonuclease I (PDB ID: 1FXX); three human 3′-5′ exoribonucleases, PARN (PDB ID: 2A1R), 3′hExo (PDB ID: 1W0H) and ISG20 (PDB ID: 1WLJ); yeast POP2 protein exonuclease domain (PDB ID: 1UOC); E. coli RNase D (PDB ID: 1YT3); Klenow fragment (PDB ID: 2KZZ); and T. gorgonarius DNA polymerase (PDB ID: 1TGO). The last three belong to the DEDDy subgroup, while the others have the DEDDh folds. Sequence alignments were initially generated using Tcoffee (http://www.tcoffee.org) (Notredame et al., 2000), followed by some manual adjustment. The three conserved Exo motifs are labeled at bottom with the DEDD residues marked by red triangles. The NBS basic residues conserved in RNase T orthologs are highlighted using blue rectangles. The numbering on the top is based on the E. coli RNase T sequence, with the secondary structure of P. aeruginosa RNase T.
Like all DEDD exonucleases, RNase T is a single-strand specific exonuclease and requires divalent metal ions, such as Mg2+ or Mn2+, for its activity (Deutscher and Marlor, 1985; Zuo and Deutscher, 2002c). RNase T is distributive, and displays an unusual base specificity, discriminating against pyrimidines and, particularly, C residues (Zuo and Deutscher, 2002c). Although RNase T appears to bind up to about 10 nucleotides, its substrate specificity is defined largely by the last 4 residues. Another major reason for the specific involvement of RNase T in stable RNA maturation and tRNA end turnover is that RNase T appears to be the only enzyme capable of efficiently removing residues close to a duplex structure without unwinding the double helix. RNase T action slows down significantly as it approaches a duplex, however, it can digest RNA up to the first base pair (Li and Deutscher, 1995) and generate blunt-ended DNA duplex (Zuo and Deutscher, 1999).
RNase T forms a homodimer in vitro and in vivo, and formation of the dimer is required for it to function (Li et al., 1996a). In addition to confirming that the conserved DEDDh residues are essential for RNase T activity, mutagenesis also identified three short conserved nucleic acid binding sequence (NBS) segments important for substrate binding (Zuo and Deutscher, 2002a). These three NBS segments are rich in positively charged Arg/Lys residues. Homology modeling using oligoribonuclease, a related DEDD family exoribonuclease as a template, suggested that the NBS segments cluster and form a positively charged surface patch on the face of the RNase T monomer opposite the DEDDh catalytic center (Zuo and Deutscher, 2002b). It was proposed that the two subunits of an RNase T dimer complement each other with substrate-binding and catalysis to form fully functional RNase T active sites (Zuo and Deutscher, 2002b).
Here we report two independently solved structures of RNase T, a 2.1 Å resolution RNT structure from Pseudomonas aeruginosa (H. Zheng, M. Chruszcz, M. Cymborowski, T. Skarina, A. Savchenko, and W. Minor), and a 3.1 Å resolution RNT structure from Escherichia coli (Y. Zuo, Y. Wang and A. Malhotra). Although RNase T is mostly related to the proof-reading ε subunit of DNA polymerase III in sequence, our structures reveal that RNase T adopts an oligoribonuclease-like homodimer architecture with the two monomers arranged in an opposing orientation as proposed previously (Zuo and Deutscher, 2002b). This arrangement juxtaposes the NBS patch from one monomer in the vicinity of the DEDD active center pocket of the other monomer, explaining the requirement of homodimer formation for RNase T activity.
Results
Structure Determination of RNase T
RNase T from P. aeruginosa (Midwest Center for Structural Genomics target APC5754) crystallized in the P21 space group with one RNT dimer in the asymmetric unit. Phasing was performed using single wavelength anomalous diffraction (SAD), and the structure was refined to a resolution of 2.1 Å with a final R factor of 16% (Table 1); RNase T from E. coli crystallized in the I422 space group with four monomers (two dimers) in the asymmetric unit. Phases were obtained using MAD to 3.1 Å, and the structure was refined to an R factor of 19.6% (Table 1).
Table 1.
Summary of Data Collection and Refinement Statistics.
| Data Collection | P. aeruginosa RNaseT | E. coli RNase T | |
|---|---|---|---|
| Wavelength (Å) | 0.9794 | 0.9786 | 0.9788 |
| Space group | P21 | I422 | |
| Unit cell dimensions (Å) | a=49.9, b=76.6, c=61.7, 90.0, 93.55, 90.0 | a=213.1, b=213.1, c=149.2, 90.0, 90.0, 90.0 | |
| Resolution Range (Å)* | 19.97–2.09 (2.15–2.09) | 30.0–3.1 (3.21–3.1) | 30.0–3.1 (3.21–3.1) |
| Rmerge ** | 0.125 (0.441) | 0.092 (0.696) | 0.085 (0.845) |
| I/σ(I) | 17.9 (2.6) | 28.9 (2.1) | 27.3 (1.4) |
| Reflections | |||
| Measured | 117,164 | 406,651 | 396,810 |
| Unique | 27,229 | 31,325 | 31,269 |
| Completeness (%) | 99.5 (96.5) | 99.5 (95.1) | 98.9 (89.3) |
| Refinement Statistics | |||
| R factors | |||
| Rwork | 0.158 | 0.196 | |
| Rfree (5% data) | 0.204 | 0.231 | |
| Number of Atoms | |||
| Protein | 3099 | 6039 | |
| Solvent (H2O/Sulfate) | 233 | 65 | |
| Average B-factor (Å2) | 28.6 | 77.4 | |
| r.m.s.d. from ideality | |||
| bond length (Å) | 0.014 | 0.013 | |
| bond angle (°) | 1.345 | 1.369 | |
| Ramachandran Plot Statistics (%) *** | |||
| Residues in most favored regions | 94.0 | 87.9 | |
| Residues in additional allowed regions | 5.7 | 11.1 | |
| Residues in generously allowed regions | 0.3 | 1.0 | |
| Residues in disallowed regions | 0.0 | 0.0 | |
Data in parentheses are for the highest resolution shell.
Rmerge=Σ(|Ij−<I>|Σ<I>, where Ij is the observed intensity of reflection j and <I> is the average intensity of multiple observations. The calculation is performed for merged Bijovet pairs.
Ramachandran Plot statistics are taken from PROCHECK (Laskowski et al., 1993).
RNT forms a compact alpha/beta fold with the N- and C- termini coming together at the base of the molecule (Figure 2A). This part of the molecule appears flexible, with the N-terminal ends disordered in both structures (residues 1–18 in P. aeruginosa; 1–6 in E. coli could not be seen in the electron density.) Electron density for several residues at the C-terminal end is missing in both structures. Two internal loop regions in molecule B of E. coli RNT (residues 27–36 and 65–84) are invisible in the density map, and appear to be disordered in the crystal. The E. coli and P. aeruginosa RNT sequences share more than 60% identical residues over the entire sequence. Other than 12 additional residues at the N-terminal end of P. aeruginosa RNT, pairwise alignment between P. aeruginosa RNT and E. coli RNT shows no gaps (Figure 1). Unless otherwise mentioned, the residue numbers in the entire paper will be based on E. coli RNT. The use of the E. coli RNT numbering facilitates correlations to earlier biochemical studies, and the residue numbers for P. aeruginosa RNT can be readily determined by adding 12. The structures of P. aeruginosa RNT and E. coli RNT have essentially the same fold with pairwise r.m.s.d. between monomer Cα atoms from 0.9–1.6 Å, not much higher than pairwise r.m.s.d. between monomers within each structure (0.6 Å between P. aeruginosa RNT monomers, and 0.6–1.2 Å among E. coli RNT monomers). Though several loop regions are flexible, the biggest movement appears to be in the DEDDh-specific α2 region above the DEDD cavity (Figure 2C). All analyses apply to both P. aeruginosa and E. coli structures unless otherwise specified.
Figure 2.
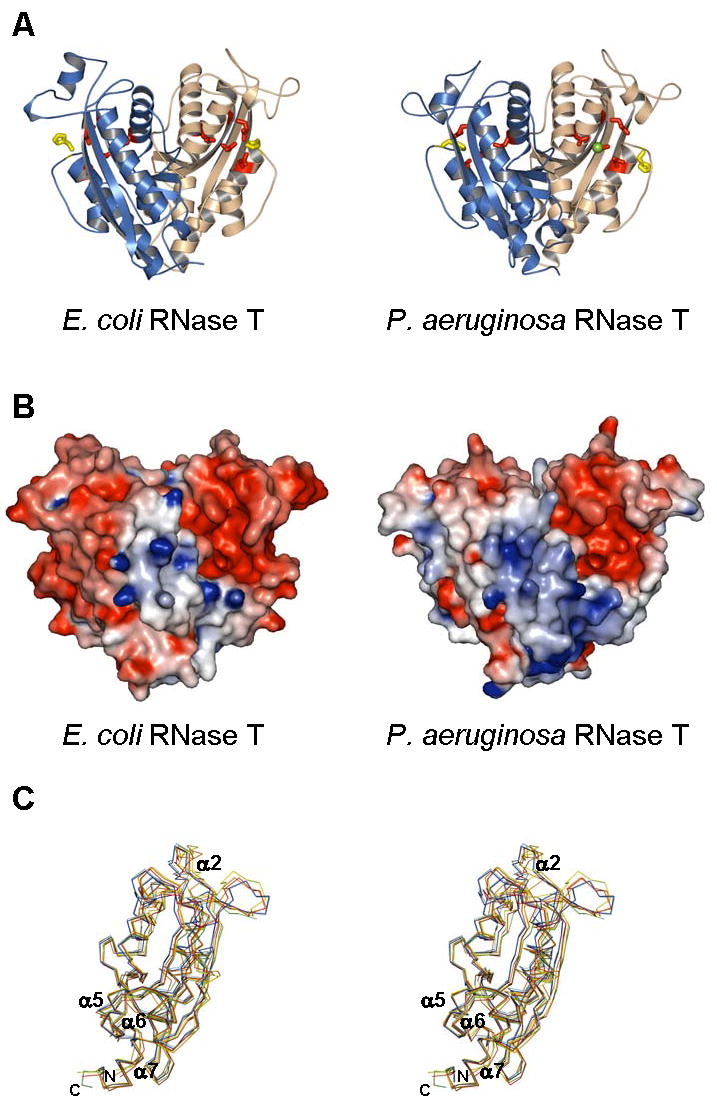
Crystal structure of E. coli and P. aeruginosa RNase T. (A). RNase T in a ribbon representation, with the two subunits in each dimer colored differently. Also shown are conserved DEDDh residues (sticks) and bound metal ions (balls). (B). Molecular surface of RNase T, colored by local electrostatic potential using GRASP (Nicholls et al., 1991). Red color indicates negative potential, blue color for positive potential, and both panels use similar scales for electrostatic shading. The larger “blue” patch in the P. aeruginosa RNase T reflects a larger number of basic residues in the NBS patch. (C) Comparison of RNT monomers. The two monomers of P. aeruginosa RNase T (PDB ID: 2F96) and the four monomers of E. coli RNase T (PDB ID: 2IS3) are superimposed, with only the Cα backbone shown. Each monomer is colored differently: cyan – 2F96 chain A, blue – 2F96 chain B, yellow – 2IS3 chain A, green – 2IS3 chain B, red – 2IS3 chain C, and orange – 2IS3 chain D.
Overall Fold of RNase T
RNase T is a dimer with monomers facing opposite ends (Figure 2). As identified using the DALI server (Holm and Sander, 1996), each monomer has an alpha/beta fold very similar to the catalytic domains of other DEDD family exonucleases, such as the ε subunit of DNA polymerase III (when compared to an E. coli RNase T monomer, Z-score is 17.7; pdb ID: 1J53) (Hamdan et al., 2002), the nuclease domain of exonuclease I (Z-score: 16.1; pdb ID: 1FXX) (Breyer and Matthews, 2000), E. coli oligoribonuclease (Z-score: 13.4; pdb ID: 1YTA) (Fiedler, Zuo and Malhotra, unpublished data), the nuclease domain of POP2 (Z-score: 11.5; pdb ID: 1UOC) (Thore et al., 2003) and several human DEDDh exoribonucleases, 3′hExo (Z-score: 16.1; pdb ID: 1W0H) (Cheng and Patel, 2004), ISG20 (Z-score: 13.5; pdb ID: 1WLJ) (Horio et al., 2004) and PARN (Z-score: 12.5; pdb ID: 2A1R) (Wu et al., 2005). The top Z-scores are all against DEDDh proteins. For the DEDDy subfamily, the protein with highest structural similarity to RNT is Thermococcus DNA polymerase (pdb ID: 1TGO) (Hopfner et al., 1999) with a Z-score of 11.2.
Oligoribonuclease is another homodimeric DEDD exonuclease whose structure has been solved (Fiedler, Zuo and Malhotra, unpublished data; Gilliland et al., 2002). RNase T and oligoribonuclease share low sequence identity (~15%), yet adopt a very similar homodimeric conformation. The opposing arrangement of two monomers in the RNT homodimer allows putative RNA-binding sites from one monomer to lie very close to the active site of the other monomer (Figure 2B) (Zuo and Deutscher, 2002b). Another DEDD exonuclease, human poly(A) specific exoribonuclease PARN, has also been shown to be a homodimer (Wu et al., 2005). However, PARN is a large multidomain protein, and uses a different dimerization interface when compared to RNT (Wu et al., 2005).
RNT Dimeric Interface
Approximately 1,300 Å2 are buried for each monomer at the RNase T dimer interface (more than 12% of total surface area of the monomer). The interface features complementary protuberances and cavities (Figure 3A) that come together to allow RNT to form a very stable dimer, as verified by size exclusion chromatography at various salt concentrations (Li et al., 1996a). At the center of the highly hydrophobic interface are residues Leu157 and Trp201 from one monomer protruding deep into the other monomer (Figure 3A), contributing about 20% of the buried interface. These two residues are surrounded by conserved hydrophobic residues, including Phe14, Phe149, Leu154, Phe194, Ile197, and Leu204 (Met in P. aeruginosa RNT), which form a hydrophobic core at the dimer interface.
Figure 3.
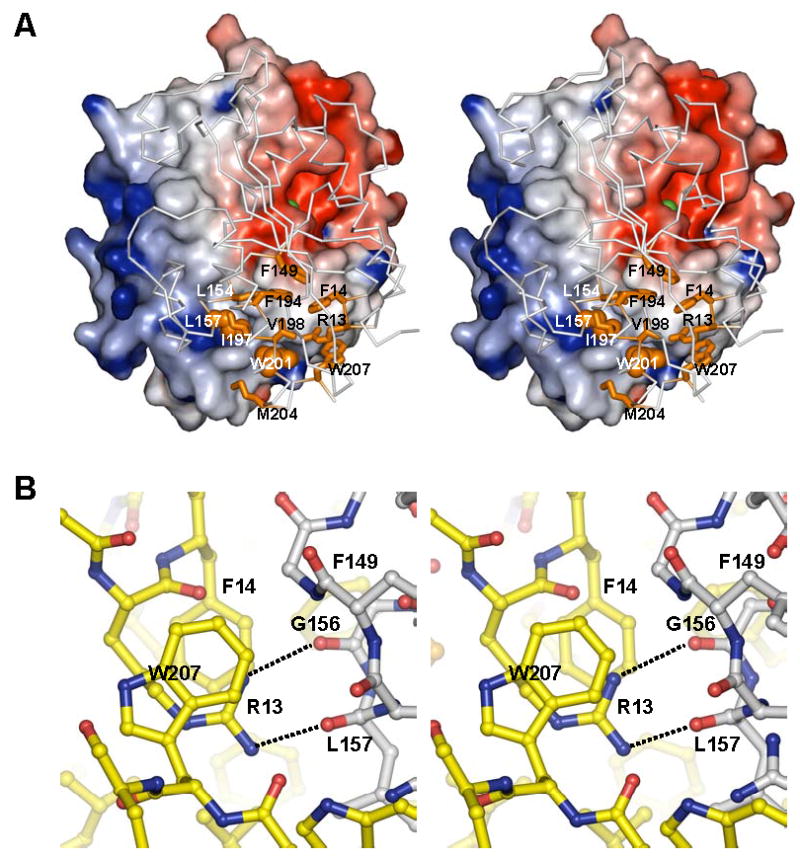
Dimer interface of RNase T. (A) View of the P. aeruginosa RNase T monomer from the dimer interface. One monomer is shown with its molecular surface colored by electrostatic potential as in Figure 2, while the other is shown as Cα trace (grey) with major interface contributors shown in orange as sticks (Arg13, Phe14, Phe149, Leu154, Phe194, Ile197, Val198, Met204 and Trp207) and balls (Leu157 and Trp201). Both Leu157 and Trp201 side chains protrude deep into the partner subunit. The metal ion at the active center of the monomer is shown as a green sphere. The residue numbers shown here are that of corresponding E. coli RNase T residues. Met204 corresponds to Met216 in P. aeruginosa RNase T, and is Leu204 in E. coli RNase T. (B) Details of P. aeruginosa RNase T dimer interface, showing the position of Arg13 and its interactions with Phe14 and Trp207, and the backbone of the partner subunit. The carbon atoms from the two subunits are colored differently.
Several charged residues also contribute to RNT dimerization. The long aliphatic side chain of Arg15 is part of the hydrophobic dimer interface. Asp150, a residue conserved in almost all DEDD proteins, is also buried at the interface. Replacing Asp150 with alanine leads to a significant loss of RNase T activity (Zuo and Deutscher, 2002a). However, Asp150 appears to be more important for maintaining the local fold rather than contributing to dimerization. A surprising observation is that Arg13 is buried at the interface, forming two hydrogen bonds with the backbone of the other subunit. In a previous study, Arg13 was predicted to be involved in substrate-binding, which does not explain the dramatic effect on RNase T activity by an Arg13 to alanine mutation, as this single mutation led to a large increase in Km (~15 fold) and a more than 100 fold decrease in Vmax (Zuo and Deutscher, 2002a). It is evident from the crystal structure that the Arg13 to alanine mutation likely affects RNase T dimerization. Previous experiments have also shown that mutation of Trp207 makes RNase T temperature-sensitive due to a destabilized dimer conformation (Zuo and Deutscher, 2002a). The crystal structure suggests that Trp207 serves to sequester Arg13 from the bulk solvent. Therefore, the temperature sensitivity of Trp207 mutants might be more of an indirect effect of an exposed Arg13 residue, which would weaken the hydrogen bonds at the dimer interface (Figure 3B).
RNase T Active Center
All DEDD family exonucleases share a common active site geometry, with four acidic side chains that coordinate two divalent metal ions: a site A metal ion coordinated by the three conserved acidic residues from ExoI and ExoIII sequence motifs (Figure 1), and a site B metal ion coordinated by the conserved aspartate residue in ExoI and indirectly by the conserved aspartate in ExoII. Both metal ions interact with the terminal phosphate in the presence of a substrate and a general catalytic mechanism was proposed involving the two bound metal ions and a conserved tyrosine residue in ExoIII motif in the case of a DEDDy protein (Beese and Steitz, 1991; Steitz and Steitz, 1993). RNT, a DEDDh member, has all the four conserved acidic residues (Asp23 and Glu25 in ExoI, Asp125 in ExoII and Asp186 in ExoIII) and a conserved histidine in ExoIII (His181). The geometry of the four conserved acidic residues in RNT agrees very well with other DEDD family members. Of the 203 Cα atoms in one P. aeruginosa RNT monomer, 140 can be superimposed on E. coli exonuclease I Cα traces with an r.m.s.d. of 1.60 Å, 109 can be superimposed on Klenow fragment exonuclease domain Cα traces with an r.m.s.d. of 1.58 Å.
No metals were observed in the E. coli structure, but a metal ion was observed at the B site of each monomer of the P. aeruginosa RNT structure (Figure 4). This B site metal ion has an octahedral coordination typical for a magnesium ion. It is interesting to point out that, in contrast to DEDDy proteins, in which the B site is usually a weaker metal-binding site than the A site (Zuo et al., 2005), DEDDh proteins appear to have stronger B site metal binding in the absence of a substrate. A single metal ion at B site has also been observed in E. coli exonuclease I (Breyer and Matthews, 2000). The absence of a metal ion at A site of P. aeruginosa RNT could also be due to preference for a non-magnesium metal ion at this site, even though Mg2+ by itself supports RNase T activity (Deutscher and Marlor, 1985). In fact, a water molecule (Water-164) occupying the A site in P. aeruginosa RNT (chain A) might mimic a non-magnesium metal ion with 5 potential coordination ligands.
Figure 4.
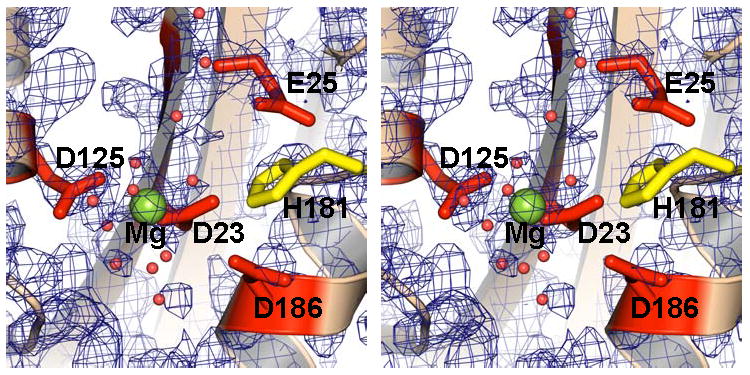
Close-up of the DEDDh active site in P. aeruginosa RNase T (monomer B in stereoview). The conserved DEDDh residues are shown as sticks (red and yellow). The Mg ion located at the “B” site is shown as a green sphere. Also shown are a few water molecules (small red spheres) at the active center. Experimental electron density map after solvent flattening is shown contoured at 2σ.
Like other DEDDh proteins, the conserved histidine in ExoIII motif is part of a highly flexible loop. This conserved histidine residue in DEDDh can occupy a position spatially equivalent to DEDDy tyrosine and has been proposed to function in a manner similar to the conserved tyrosine residue in the DEDDy exonucleases (Breyer and Matthews, 2000; Hamdan et al., 2002). Mutating His181 of E. coli RNase T abolishes its exonuclease activity (Zuo and Deutscher, 2002a), suggesting an important role of His181 for RNase T catalysis. Interestingly, the two DEDDh histidine residues in each RNT homodimer (two homodimers in E. coli RNT structure and one homodimer in P. aeruginosa RNT structure) adopt different conformations; one is partially directed toward the active center (Figure 4), while the other one is facing away from the active center. It is not clear whether this is just a crystal packing artifact as the histidine directed toward the active center is close to the symmetry related molecule or an indication of flexibility in absence of substrate in the RNT active site.
RNase T Substrate Binding Site
Both E. coli RNT and P. aeruginosa RNT are highly negatively-charged proteins with theoretical pIs of 5.2 and 5.0, respectively. However, surface charge distribution is strongly uneven in both proteins (Figure 2B). Nine out of the 18 Arg/Lys residues in E. coli RNT (11 out of 23 in P. aeruginosa RNT) are found in three short conserved sequence segments, named NBS segments for their likely roles in nucleic acid-binding (Zuo and Deutscher, 2002a). The greater number of Arg/Lys residues in P. aeruginosa RNT NBS segments is reflected in a larger positive patch (Figure 2B). Except for Arg13, which is buried at the dimer interface, all the NBS basic residues cluster on one side of the monomer surface to form a basic patch, which contains no acidic residues. This basic patch is on a side opposite to the DEDD catalytic center cavity. Another basic residue, Lys166, on the edge of the DEDD cavity, extends the basic patch. Mutating the basic residues of the NBS patch, individually or in combination, leads to significantly increased Km with little effect on Vmax of RNase T catalysis (Table 2) (Zuo and Deutscher, 2002a). This is consistent with the suggested role of substrate-binding for the NBS patch. The involvement of NBS patch in substrate binding is also supported by the observation that several SO42− anions are bound to this region in the E. coli RNase T structure.
Table 2.
Effect of Mutations on E. coli RNase T Activity.
| RNase T Variant | Km (μM) | Relative Activity* |
|---|---|---|
| Wild-type RNT | 10 | 100 |
| D23A RNT | 23 | 1.4 |
| E25A RNT | ND** | 1.3 |
| H120A RNT | ND | 5.4 |
| D125A RNT | 33 | 34 |
| H181A RNT | ND | 1.4 |
| D186A RNT | 25 | 0.7 |
| D150A RNT | ND | 1.4 |
| R13A RNT | ~150 | 0.04 |
| R15A RNT | ~100 | 14 |
| K108A | ~150 | 26 |
| R114A | ~120 | 24 |
| K108A/R114A | ~250 | 6 |
| K139A | ~80 | 35 |
| K108A/K139A | ~200 | 10 |
| K108A/R114A/K139A | >300 | 2 |
| K108A/R114A/K139A/R140A | >300 | 0.6 |
Data from Zuo and Deutscher, (2002a)
Relative Activity was estimated with 20 μg tRNA substrate in a 50 μl reaction (~ 16 μM).
ND, not determined.
Discussion
Requirement of Dimerization for RNase T Action
Prior mutagenesis studies have revealed that the conserved DEDDh residues are essential for RNase T activity, indicating that they likely form the RNase T catalytic center in a manner similar to that found in other DEDD exonucleases (Zuo and Deutscher, 2002a). Our structures confirm that RNase T shares the same spatial arrangement of active center residues as seen in other DEDD enzymes. Mutagenesis studies also identified three short sequence segments (NBS segments) important for RNase T substrate-binding. These three NBS segments are highly conserved in RNase T orthologs and are rich in positively charged Arg/Lys residues. Like oligoribonuclease, RNase T forms a homodimer and the homodimeric form is required for function (Li et al., 1996a). An oligoribonuclease-like homodimer architecture was thus proposed for RNase T (Zuo and Deutscher, 2002b). According to this homodimer model, the NBS substrate binding patch and the DEDD center from different subunits are brought together to form fully active sites. Our results confirm this model, and both the E. coli and the P. aeruginosa RNase T structures show the basic patch from one monomer positioned right next to the DEDD catalytic center cavity of the other monomer in the homodimer (Figure 2B).
While there is good agreement between the homology model and the crystal structure, there is a surprising observation that the conserved basic residue Arg13 is buried in the otherwise highly hydrophobic dimer interface and forms two hydrogen bonds with the backbone of the other monomer. Arg13 had been predicted to be part of the NBS patch, though its mutation showed a different effect on RNase T activity when compared to mutations of other NBS basic residues (Table 2). In addition to an increased Km value as seen with other NBS mutations, the Arg13 mutant also displayed a dramatically reduced Kcat, which is inconsistent with a residue involved only in substrate-binding. The observation that Arg13 is buried at the dimerization interface in the crystal structures suggests that mutating Arg13 might disrupt or significantly weaken the RNase T dimer. The observation of a buried Arg13 also suggests that the temperature-sensitivity seen in Trp207 mutants might be more of an indirect effect. In the crystal structure, Trp207 serves to sequester Arg13 from the bulk solvent; Arg13 is sandwiched between Trp207 and Phe14 and participates in cation-pi like interactions with these sidechains (Figure 3B). Mutating Trp207 to an Ala or a stop codon would expose Arg13, and thus weaken the hydrogen bonding with the partner subunit. The same could also be true for another C-terminal mutation, Gly206Ser (Li et al., 1996a), which displays an effect consistent with weaker dimerization of RNase T. A Gly206 to Ser mutation would disrupt the C-terminal folding and thus expose Arg13, even if it does not directly interfere with dimerization.
In contrast to the unexpected involvement of Arg13 in RNT dimerization, the hydrophobic residue Cys168, which was implied in RNT dimerization (Li et al., 1996a), is not at the dimer interface. Instead, Cys168 is part of helix α6 and is buried at the α6/α7 interface. Both α6 and α7 helices are important for shaping the DEDD active center cavity. Mutating Cys168 to less hydrophobic residues (Li et al., 1996a) would likely weaken the anchoring of α6 to α7. It is expected that a displaced α6 would compromise RNase T activity by disrupting the DEDD cavity and the basic patch as this helix also harbors the conserved basic residue Lys166. It is also possible that a displacement of this helix will interfere with RNT dimerization as α6 is close to, both in sequence and in space, α5 and the C-terminus of α7, which are major contributors to the dimer interface.
Making Sense of RNase T Substrate Binding
RNase T has been shown to be important for the 3′ maturation of many stable RNAs as well as tRNA end turnover in vivo (Deutscher et al., 1985; Kelly and Deutscher, 1992; Li and Deutscher, 1995; Li and Deutscher, 1996; Li et al., 1998; Li et al., 1999). All RNase T substrates share a common feature, i.e., their 5′- and 3′-ends pair with each other to form a stable, double-stranded stem followed by a few unpaired 3′-nucleotides (Li et al., 1998). Later studies with model substrates demonstrated that RNase T is a single strand specific exonuclease that can act on both DNA and RNA substrates (Zuo and Deutscher, 1999; Zuo and Deutscher, 2002c). The important role of RNase T in stable RNA maturation was thus attributed to its unique ability to efficiently remove residues near a stable duplex without substrate unwinding.
Docking studies (data not shown) indicate that RNase T can make favorable interactions with up to 11–12 nucleotides upstream of the 3′ end for a flexible single-stranded substrate. Other than the 3′-terminal 3–4 nucleotides, which are mostly inside the DEDD cavity, contacts are expected between the nucleic acid backbone and the positively charged side chains on the NBS basic patch.
Interactions of duplex substrates with RNase T are likely to be more restricted due to the need for accommodating the helical stem on the NBS and for proper presentation of the 3′ overhang into the DEDD active center. For duplex containing RNA substrates, such as tRNA or tRNA precursors, a 3′-overhang of 5 nucleotides or longer appears to be preferred. Due to potential steric hindrance by the complementing 5′-end residues, a substrate with shorter 3′-overhang would either not be able to interact with the NBS patch at the far end or have to disrupt the stacking on the 3′ end (Figure 5A). This explains the impeding effect of a duplex on RNase T activity. In fact, the active center cavity can well accommodate a DNA/RNA duplex with a one nucleotide 3′ overhang, suitable for generating blunt-ended duplex (Zuo and Deutscher, 1999). Docking shows that a DNA duplex substrate with a 3′ overhang as short as a single nucleotide should be able to nicely interact with the entire RNase T NBS patch due to its wide major groove; on the other hand, an A-form RNA duplex with a short 3′ overhang is likely to make fewer contacts with the NBS patch due to steric clashes (Figure 5B).
Figure 5.
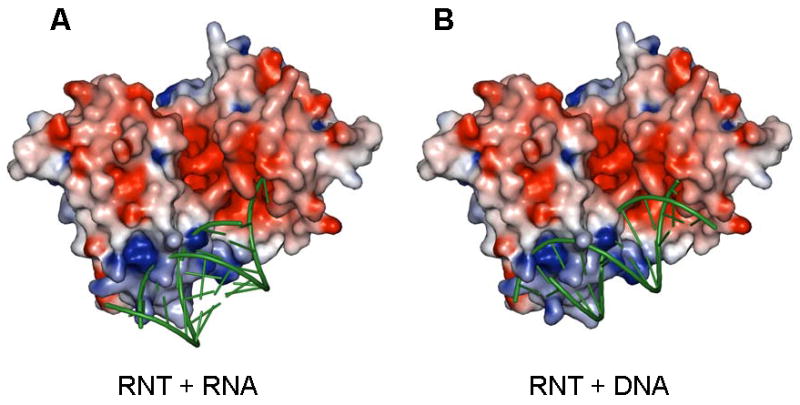
Possible binding modes for duplex containing substrates on P. aeruginosa RNase T. (A) Docking of an ideal A form RNA duplex with a two nucleotide 3′ overhang. While the unpaired 3′ overhang is shown stacked here, this region is likely to be distorted for hydrolytic cleavage. (B) Docking of an ideal B form DNA helix with a single nucleotide 3′ overhang. RNase T is shown as a surface potential diagram colored as in Figure 2, and substrates are shown as green ribbons. Only one substrate molecule was docked; the active pocket/RNA binding surface on the opposite side of RNase T is shown empty, and could presumably accommodate another substrate molecule in an analogous manner. Clashes in the docking models are mostly due to the long side chains of a few solvent exposed basic residues (Arg101, Arg114, Lys139 and Arg140). These sidechains are likely to adopt alternative conformations upon substrate binding.
The physiological role of RNase T is also defined by its unusual sequence specificity, discriminating against pyrimidine and particularly, C residues at the 3′ end. This sequence specificity is defined largely by the last 4 residues (Zuo and Deutscher, 2002c). A single 3′-terminal C residue can reduce RNase T action by more than 100-fold, and two terminal C residues essentially stop the enzyme. RNase T prefers a substrate with a CCAN, especially CCAA, sequence at the 3′ end (Zuo and Deutscher, 2002c). This makes RNase T well suited for tRNA maturation while preventing over-digestion of the CCA end. All E. coli tRNA genes encode the CCA ends, and 43% (37/86) of them have an additional A residue immediately following the CCA sequence. Due to the high flexibility of the α2 region on top of the DEDD cavity, it is difficult to predict with high confidence the specific interaction between RNase T and the terminal residues of the substrate based on docking. However, it is reasonable to speculate that the two terminal bases would stack between phenylalanine side chains, namely, between Phe77 and Phe29 and between Phe29 and Phe146, by analogy to the stacking of the terminal substrate base between Leu361 and Phe473 in the Klenow Fragment (Beese and Steitz, 1991). Similar stacking has also been observed in structures of other DEDD exonucleases with substrates, such as the stacking of the terminal base between Ile34 and Phe115 in human PARN (Wu et al., 2005). In protein-RNA interactions, phenylalanine is typically disfavored in interactions with pyrimidine bases, especially cytidines (Jones et al., 2001). The “sandwiching” of terminal bases by phenylalanine side chains was speculated to account for RNase T discrimination of pyrimidine residues (Zuo and Deutscher, 2002c). However, a Phe29Tyr/Phe77Tyr double mutant did not significantly alter RNase T specificity (data not shown), suggesting a more complicated mechanism for its unusual base specificity.
Comparison to Other DEDD Exonucleases
E. coli has three DEDD family exoribonucleases – RNase T, oligoribonuclease (ORN) and RNase D (RND). RNase D is a monomeric enzyme of the DEDDy subfamily. It has two additional HRDC domains and forms a ring-like arrangement (Zuo et al., 2005). RNase T and oligoribonuclease are homodimeric DEDDh enzymes. Both RNase T and oligoribonuclease display strong RNase and DNase activity. ORN favors RNA oligonucleotides 5 nucleotides or shorter with highest activity on dinucleotide substrates. In contrast, RNase T acts on longer substrates with essentially no activity on RNA dinucleotides. Albeit its specificity is not well defined, RNase D is highly active on tRNA-like substrates: tRNA precursors and denatured tRNAs. Corresponding to these differences in substrate specificity, we also observe differences in the architecture of these enzymes (Figure 6). RNT and ORN share very similar homodimeric arrangement with major contribution of substrate binding from the complementing subunit. However, RNT has a large basic patch suitable for binding of longer substrates, while there is only a small basic patch on ORN surface for favorable binding of short oligonucleotides. In favor of shorter substrates, ORN also has a unique elongated DEDD cavity long enough for 3–4 nucleotides (Fiedler, Zuo and Malhotra, unpublished data). Both RNT and ORN have deep DEDD cavities shaped by dimerization, whereas monomeric RND has a narrower DEDD cavity. RND also has extended basic patch, on the HRDC domains, for potential substrate binding, though its ring structure may also play an important role in binding substrates (Zuo et al., 2005).
Figure 6.
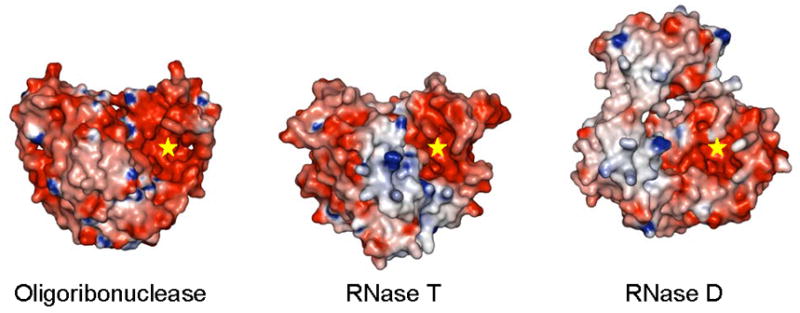
Comparison of the structures of the three DEDD family exoribonucleases from E. coli. Molecular surfaces colored by electrostatic potential are shown for these enzymes with one DEDD domain from each aligned in the same orientation. The active centers of these enzymes are highlighted (“star”).
The use of basic patches for substrate binding appears to be a common feature of the three E. coli DEDD exoribonucleases. While the DEDD domain contributes the active site, another domain or subunit contributes to the basic patch. Unlike other DEDD exonucleases, RNT and ORN form homodimers. Interestingly, both RNT and ORN use equivalent regions for dimerization. For non-homodimeric DEDD family members, this same interface region is involved in inter-domain or inter-subunit interactions.
Experimental Procedures
Cloning, Expression, Purification and Crystallization
P. aeruginosa RNase T
The P. aeruginosa RNase T open reading frame was amplified by PCR from genomic DNA (American Type Culture Collection) as previously described (Zhang et al., 2001). The gene was cloned into the NdeI and BamHI sites of a modified pET15b expression vector p11-toronto1 (Novagen Inc.). Modifications include a 6xHis tag in a 22-aa N-terminal fusion peptide, with a TEV protease cleavage site, followed by a double stop codon downstream from the BamHI site. The resulting construct is an N-terminal 6xHis tag separated from the gene by a TEV protease recognition site sequence (ENLYFQG). The modified pET15b vector containing the cloned P. aeruginosa RNT was then transformed into E. coli BL21(DE3) gold magic. Fresh transformants were inoculated into 25-ml culture grown in a 250-ml flask with ampicillin, and then inoculated into 2 liters of LB in a custom-baffled 4-liter flask. The sample was induced at an optical density A600 of 0.6–0.8 with 0.4mM isopropyl-1-thio-D-galactopyranoside (IPTG) after growth at 37°C and grown overnight at 15°C. The cells were harvested by centrifugation (10 min at 8000 rpm; Beckman Coulter Avanti J-20 centrifuge). The cell pellet was resuspended in 40 ml binding buffer, supplemented with 1 mM protease inhibitors phenylmethylsulfonyl fluoride (PMSF) and 1 mM benzamidine, flash-frozen in liquid nitrogen, and stored at −70°C.
The purification procedure used buffers containing 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, and 5, 30, and 250 mM imidazole for the binding, wash, and elution buffers, respectively. The harvested cells were lysed by adding 0.5% Nonidet P-40 to the thawed sample before sonication. Fresh protease inhibitors were added before the sample was clarified by centrifugation (30 min at 17000 rpm; Beckman Coulter Avanti J-25 centrifuge). The clarified lysate was passed by gravity through a DE52 column in series with a Ni2+ column. Contaminating proteins were removed by washing the Ni2+ column with 50 column volumes of wash buffer. The bound protein was removed with elution buffer as qualitatively determined by the Bradford assay (Bio-Rad Protein Assay) (Bradford, 1976). The sample was then supplemented with 0.5 mM EDTA and 0.5 mM dithiothreitol (DTT) (final concentration). The His tag was removed by cleavage with recombinant His-tagged TEV protease (60 μg TEV/mg recombinant protein). The cleavage step was done concurrently with dialysis in binding buffer without imidazole at 4°C overnight. The cut His tag and His-tagged TEV protease were removed from the purified recombinant protein by passage through a second Ni2+ column. The sample was prepared for crystallization screening by a second dialysis in 10 mM HEPES, pH 7.5, 500 mM NaCl, followed by concentration to 10 mg/ml using a BioMax concentrator (Millipore Inc.). Finally, any particulate matter was removed from the sample by passage through a 0.2μm Ultrafree-MC centrifugal filtration device (Millipore Inc.) (Zhang et al., 2001).
The protein was crystallized at 20°C using vapor diffusion method in sitting drops. Cryschem Plate was used in the crystallization setup. An initial hit was obtained with Hampton Research Index Screen, conditions #82 (200 mM magnesium chloride hexahydrate, 25% w/v PEG3350, 100 mM Bis-Tris pH 5.5). After optimization, single, well-formed, diffraction-quality crystals were grown from a condition containing 25% w/v PEG3350, 200 mM magnesium chloride, 0.4% w/v NDSB256, and 300 mM Bis-Tris pH 5.5.
E. coli RNase T
RNase T was initially overexpressed and purified using a pUT18-based overexpression system with the wild-type rnt gene cloned into the pUC18 vector (Zuo and Deutscher, 2002a). To achieve higher overexpression, E. coli RNase T gene was cloned into the NcoI and BamHI sites of the pET15b expression vector (Novagen Inc.). The resulting pETT plasmid for wild-type RNase T production was confirmed by DNA sequencing. Plasmid pETT was then transformed into the E. coli BL21(DE3) strain for RNase T overexpression. RNase T was overexpressed by adding 1 mM IPTG (final concentration) to 2 L of cells grown in Luria Broth (Invitrogen Inc.) with ampicillin to an A600 of ~ 1.0 at 37°C; the induction proceeded for about 2 hours. Cells were pelleted by centrifugation and then flash-frozen using liquid nitrogen and stored at −80°C.
RNase T was purified chromatographically using established procedures (Deutscher and Marlor, 1985; Li et al., 1996b) with some modifications. Briefly, the cell extract was applied on an Affi-Gel Blue column (Bio-Rad) followed by a hydroxyapatite column. RNase T was further purified and concentrated using a MonoQ anion exchange column (GE Healthcare Inc.). The RNase T-containing peak fractions were then applied on a Superdex S200 (GE Healthcare Inc.) gel filtration column equilibrated and eluted with a Tris buffer (20 mM Tris-Cl pH 7.5, 250 mM NaCl, 1 mM DTT and 10% glycerol). RNase T was then concentrated to 50 mg/ml. The concentrated protein was kept at −80°C after flash-freezing in liquid nitrogen. All centrifugation and chromatographic steps were carried out at 4°C. SeMet-RNT was produced from the same expression vector under conditions of methionine pathway inhibition as described previously (Fiedler et al., 2004). SeMet-RNT was purified in the same manner as the wild type RNT.
E. coli RNase T protein crystallizes at room temperature in the presence of 2.3–2.9 M ammonium sulfate and various buffer conditions. The hanging-drop technique was used. RNase T crystals appear overnight and grow to more than 0.5 mm in size. These crystals are typically round-shaped and diffract very poorly. The best diffracting crystals were grown from a condition containing 2.4 M ammonium sulfate and 100 mM MES pH 6.0.
Data Collection, Processing and Structure Determination
P. aeruginosa RNase T
Prior to the data collection, P. aeruginosa RNT crystals were transferred to a cryosolution prepared by addition of 9% glycerol, 9% ethylene glycol, 9% sucrose, 0.2M NaCl to well solution, and frozen by plunging into liquid nitrogen. Data collection for P. aeruginosa RNT was performed on the 19-ID beamline of Structural Biology Center (Rosenbaum et al., 2006) at the Advanced Photon Source (APS) at 100K. Data collection, structure determination, and refinement statistics are summarized in Table 1. Data from Se-Met labeled samples were processed with HKL-2000 (Otwinowski and Minor, 1997). Structures were solved using SAD data, and initial models were built with HKL-3000 (Minor et al., 2006) coupled with SHELXD (Schneider and Sheldrick, 2002), MLPHARE (Otwinowski, 1991), DM (Cowtan, 1994), O (Jones et al., 1991), SOLVE (Terwilliger and Berendzen, 1999) and RESOLVE (Terwilliger, 2000). Initial models were refined with multiple runs of REFMAC5 (Murshudov et al., 1997) and manual model adjustments in COOT (Emsley and Cowtan, 2004). In the crystal structure of P. aeruginosa RNT, the first eighteen residues (MSEDNFDDEFDGSLPSGP) and the last four residues (DDDD) could not be observed in the final electron density map.
E. coli RNase T
Prior to data collection, E. coli RNT crystals were transferred to reservoir solution and then transferred in steps to increase glycerol to about 20% for cryoprotection. Data collection was performed at 100K on the X-12C beamline, National Synchrotron Light Source (NSLS), Brookhaven National Laboratory (BNL). Data were reduced using HKL-2000 (Otwinowski and Minor, 1997); Phases were determined from a two-wavelength MAD experiment on SeMet-RNT. Twenty-four selenium sites (six per molecule) were found and refined in BnP (Xu et al., 2005). Albeit at 3.1 Å resolution, the initial map after solvent-flattening (~ 70% solvent content) clearly displays ORN-like heart-shaped density for the RNase T dimer. The model was first roughly fitted with the homology model (Zuo and Deutscher, 2002b) and then manually built using O (Jones et al., 1991) and refined in CNS (Brunger et al., 1998). After several rounds of iterative model building and refinement, the final structure was refined at 3.1 Å with a simulated annealing procedure using CNS followed by REFMAC5 (Murshudov et al., 1997). In the crystal structure of E. coli RNT, two loop regions in molecule B (residues 27–36 and 65–84), as well as the first 6 residues and the last 6 residues in each monomer (last 4 residues of molecule B) were not built due to poor electron density.
Structure validation
MOLPROBITY (Lovell et al., 2003) and PROCHECK (Laskowski et al., 1993) were used for structure validation. All model quality indicators were either within the normal range or better. Data collection, structure determination, and refinement statistics are summarized in Table 1. Molecular interface interactions were analyzed by using the Protein-Protein Interaction Server http://www.biochem.ucl.ac.uk/bsm/PP/server (Jones and Thornton, 1996). All molecular figures were generated using PYMOL (DeLano, 2002) with surface potentials calculated using GRASP (Nicholls et al., 1991).
Accession Numbers
Atomic coordinates and experimental structure factors have been deposited with the RCSB Protein Data Bank - accession code 2F96 for Pseudomonas aeruginosa RNase T, and 2IS3 for Escherichia coli RNase T.
Acknowledgments
We thank Professor Murray Deutscher for assistance and valuable discussions. We thank Anand Saxena at the X12-C beamline, National Synchrotron Light Source, Brookhaven National Laboratory, for assistance with data collection. NSLS financial support comes principally from the Offices of Biological and Environment Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the National Institutes of Health. We thank all of the members of Midwest Center of Structural Genomics and the Structural Biology Center at Argonne National Laboratory, especially Andrzej Joachimiak and Aled Edwards for discussions and assistance. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02–06CH11357.
Footnotes
The work toward E. coli RNT structure was supported in part by a National Institutes of Health Grant GM69972 (A.M.). Y.Z. was supported in part by an American Heart Association Florida/Puerto Rico Affiliate Postdoctoral Fellowship (0525530B). The work toward Pseudomonas aeruginosa RNT structure was supported by a National Institutes of Health Grant GM62414.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Breyer WA, Matthews BW. Structure of Escherichia coli exonuclease I suggests how processivity is achieved. Nat Struct Biol. 2000;7:1125–1128. doi: 10.1038/81978. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Patel DJ. Crystallographic structure of the nuclease domain of 3′hExo, a DEDDh family member, bound to rAMP. J Mol Biol. 2004;343:305–312. doi: 10.1016/j.jmb.2004.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994;31:34–38. [Google Scholar]
- DeLano WL. The PYMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- Deutscher MP, Marlor CW. Purification and characterization of Escherichia coli RNase T. J Biol Chem. 1985;260:7067–7071. [PubMed] [Google Scholar]
- Deutscher MP, Marlor CW, Zaniewski R. RNase T is responsible for the end-turnover of tRNA in Escherichia coli. Proc Natl Acad Sci USA. 1985;82:6427–6430. doi: 10.1073/pnas.82.19.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ezraty B, Dahlgren B, Deutscher MP. The RNase Z homologue encoded by Escherichia coli elaC gene is RNase BN. J Biol Chem. 2005;280:16542–16545. doi: 10.1074/jbc.C500098200. [DOI] [PubMed] [Google Scholar]
- Fiedler TJ, Vincent HA, Zuo Y, Gavrialov O, Malhotra A. Purification and crystallization of Escherichia coli oligoribonuclease. Acta Crystallogr D Biol Crystallogr. 2004;60:736–739. doi: 10.1107/S0907444904002252. [DOI] [PubMed] [Google Scholar]
- Gilliland GL, Teplyakov A, Obmolova G, Tordova M, Thanki N, Ladner J, Herzberg O, Lim K, Zhang H, Huang K, et al. Assisting functional assignment for hypothetical Heamophilus influenzae gene products through structural genomics. Curr Drug Targets Infect Disord. 2002;2:339–353. doi: 10.2174/1568005023342281. [DOI] [PubMed] [Google Scholar]
- Hamdan S, Carr PD, Brown SE, Ollis DL, Dixon NE. Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure. 2002;10:535–546. doi: 10.1016/s0969-2126(02)00738-4. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Eichinger A, Engh RA, Laue F, Ankenbauer W, Huber R, Angerer B. Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc Natl Acad Sci USA. 1999;96:3600–3605. doi: 10.1073/pnas.96.7.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horio T, Murai M, Inoue T, Hamasaki T, Tanaka T, Ohgi T. Crystal structure of human ISG20, an interferon-induced antiviral ribonuclease. FEBS Lett. 2004;577:111–116. doi: 10.1016/j.febslet.2004.09.074. [DOI] [PubMed] [Google Scholar]
- Jones S, Daley DT, Luscombe NM, Berman HM, Thornton JM. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kelly KO, Deutscher MP. The presence of only one of five exoribonucleases is sufficient to support the growth of Escherichia coli. J Bacteriol. 1992;174:6682–6684. doi: 10.1128/jb.174.20.6682-6684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Deutscher MP. RNase T shares conserved sequence motifs with DNA proofreading exonucleases. Nucleic Acids Res. 1993;21:2521–2522. doi: 10.1093/nar/21.10.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MV, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Li Z, Deutscher MP. The tRNA processing enzyme RNase T is essential for maturation of 5S RNA. Proc Natl Acad Sci USA. 1995;92:6883–6886. doi: 10.1073/pnas.92.15.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Deutscher MP. Maturation pathways for E. coli tRNA precursors: a random multienzyme process in vivo. Cell. 1996;86:503–512. doi: 10.1016/s0092-8674(00)80123-3. [DOI] [PubMed] [Google Scholar]
- Li Z, Pandit S, Deutscher MP. 3′ exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc Natl Acad Sci USA. 1998;95:2856–2861. doi: 10.1073/pnas.95.6.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Pandit S, Deutscher MP. Maturation of 23S ribosomal RNA requires the exoribonuclease RNase T. RNA . 1999;5:139–146. doi: 10.1017/s1355838299981669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhan L, Deutscher MP. Escherichia coli RNase T functions in vivo as a dimer dependent on cysteine 168. J Biol Chem. 1996a;271:1133–1137. doi: 10.1074/jbc.271.2.1133. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhan L, Deutscher MP. The role of individual cysteine residues in the activity of Escherichia coli RNase T. J Biol Chem. 1996b;271:1127–1132. doi: 10.1074/jbc.271.2.1127. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. Daresbury Study Weekend Proceedings. 1991. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, et al. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thore S, Mauxion F, Seraphin B, Suck D. X-ray structure and activity of the yeast Pop2 protein: a nuclease subunit of the mRNA deadenylase complex. EMBO Rep. 2003;4:1150–1155. doi: 10.1038/sj.embor.7400020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan M, Dower KW, Lovett ST. Identification of a potent DNase activity associated with RNase T of Escherichia coli. J Biol Chem. 1998;273:35126–35131. doi: 10.1074/jbc.273.52.35126. [DOI] [PubMed] [Google Scholar]
- Wu M, Reuter M, Lilie H, Liu Y, Wahle E, Song H. Structural insight into poly(A) binding and catalytic mechanism of human PARN. EMBO J. 2005;24:4082–4093. doi: 10.1038/sj.emboj.7600869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Weeks CM, Hauptman HA. Optimizing statistical Shake-and-Bake for Se-atom substructure determination. Acta Crystallogr D Biol Crystallogr. 2005;61:976–981. doi: 10.1107/S0907444905011558. [DOI] [PubMed] [Google Scholar]
- Zhang RG, Skarina T, Katz JE, Beasley S, Khachatryan A, Vyas S, Arrowsmith CH, Clarke S, Edwards A, Joachimiak A, Savchenko A. Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure. 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Deutscher MP. The DNase activity of RNase T and its application to DNA cloning. Nucleic Acids Res. 1999;27:4077–4082. doi: 10.1093/nar/27.20.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Deutscher MP. Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res. 2001;29:1017–1026. doi: 10.1093/nar/29.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Deutscher MP. Mechanism of action of RNase T. I. Identification of residues required for catalysis, substrate binding, and dimerization. J Biol Chem. 2002a;277:50155–50159. doi: 10.1074/jbc.M207706200. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Deutscher MP. Mechanism of action of RNase T. II. A structural and functional model of the enzyme. J Biol Chem. 2002b;277:50160–50164. doi: 10.1074/jbc.M207707200. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Deutscher MP. The physiological role of RNase T can be explained by its unusual substrate specificity. J Biol Chem. 2002c;277:29654–29661. doi: 10.1074/jbc.M204252200. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Wang Y, Malhotra A. Crystal structure of Escherichia coli RNase D, an exoribonuclease involved in structured RNA processing. Structure. 2005;13:973–984. doi: 10.1016/j.str.2005.04.015. [DOI] [PubMed] [Google Scholar]