

Abstract
An assembly of two receptor ligand bonds in series will typically break at the weaker complex upon application of an external force. The rupture site depends highly on the binding potentials of both bonds and on the loading rate of the applied force. A model is presented that allows simulations of force-induced rupture of bonds in series at a given force and loading rate based on the natural dissociation rates kR0,S0 and the potential width ΔxR,S of the reference and sample bonds. The model is especially useful for the analysis of differential force assay experiments. This is illustrated by experiments on molecular force balances consisting of two 30-bp oligonucleotide duplexes where kR0,S0 and ΔxR,S have been determined for different single nucleotide mismatches. Furthermore, prediction of the rupture site of two bonds in series is demonstrated for DNA duplexes in combination with streptavidin/biotin and anti-digoxigenin/digoxigenin, respectively.
INTRODUCTION
Being a pivotal parameter in classical physics for centuries, force was not accessible on a molecular level in the biological sciences for a long time. Just recently single molecule experiments by means of force probe instruments such as atomic force microscope (AFM) (1–5), optical trap (6,7) magnetic tweezers (8,9), and biomolecular force probe (10,11) made it possible to investigate the mechanical properties of biological macromolecules (12,13). Receptor ligand bonds, which are commonly characterized by affinity measurements, have also been subject to single molecule force measurements. While affinities solely reflect the binding energy corresponding to the depth of the binding potential and therefore to the natural dissociation rate k0, force measurements furthermore reveal an additional parameter, which is dependent on the unbinding pathway, the potential width Δx (14–16).
To derive Δx, a variety of force transducers like cantilevers (16–18), optical tweezers (19,20), and even biological membranes (11,15) have been employed to record force extension curves of receptor ligand bonds. Recently a fundamentally different approach to probe molecular forces has been introduced by our group (Fig. 1) (21–25). Here the microscopic force transducer is substituted by a single molecular bond, which is linked to the sample bond in series, thereby forming a molecular force balance. Upon application of a force at the ends of the balance, both bonds are probed simultaneously until the weaker one fails (21,22).
FIGURE 1.

Experimental setup of the molecular force balance. A molecular force balance consisting of a reference bond (blue) and a sample bond (red) is immobilized between the chip surfaces in contact (left). Upon separation of the surfaces, force builds up in the balance until one of the bonds fails (right). The location of the fluorescent label after the separation indicates whether the reference or the sample bond was ruptured. The bond survival probability ΦR,S depends on the rates of the reference bond kR and the sample bond kS.
For highly symmetric combinations of sample (S) and reference (R) bonds, excellent sensitivity in terms of force differences is achieved, as demonstrated by detection of single nucleotide mismatches in DNA duplexes (21,22). However, due to the lack of a theoretical description, only ratios of discrimination have been assessed instead of quantitative differences in the natural dissociation rate of the reference kR0 and the sample kS0 bond as well as the potential width ΔxR and ΔxS.
Here we present a new theoretical approach for the analysis of force-induced unbinding of two bonds in series. By means of a reference bond where the binding potential is well characterized (known kR0 and ΔxR), we are able to calculate the dissociation rate kS0 and potential width ΔxS for an arbitrary sample bond based on the well-established Bell-Evans model for a given force and loading rate (14,26–28).
We apply this new analysis to experiments where small mutations have been introduced into a 30-bp oligonucleotide duplex. The results are compared to previous measurements on DNA oligonucleotides (17) and single chain antibodies performed by other groups (18). In general, we corroborate the finding of those earlier studies, namely that differences in kR0 of a receptor ligand bond are closely coupled to the potential width ΔxR (17,18).
Moreover, the theoretical model allows for predictions on alternative unbinding of any biological system where two bonds are probed in series. The model is particularly useful for simulations of force experiments (AFM, biomolecular force probe, and magnetic- and optical tweezers) where the sample is immobilized by affinity tags (7,20,29–34). Our approach allows one to predict whether a certain immobilization tag will survive the forces applied during the experiment or not.
MATERIALS AND METHODS
The principle of the molecular force balance
The technology of the molecular force balance has been described elsewhere in detail (22). Briefly, amino-labeled oligonucleotides are immobilized on an activated slide at their 3′-termini. Cy3 labeled oligonucleotides are now hybridized to the receptors, thereby forming a sample duplex. Subsequently biotin labeled oligonucleotides are hybridized to the sample, thereby forming a reference duplex. This results in a molecular balance as it is depicted in Fig. 1. A stamp covered with streptavidin having an elevated microstructure is pressed onto the slide at this time. When the elevated area of the stamp makes contact with the slide, the biotinylated oligonucleotides will bind to the streptavidin. After the formation of biotin-streptavidin bond, the stamp is pulled away from the slide. The applied force will gradually increase until the reference duplex or the sample duplex ruptures, depending on which one is the weaker link. The Cy3-label of the middle oligonucleotide will therefore end up on the stamp when the sample duplex fails or on the slide when the reference duplex fails. Reading out the slide using a fluorescent scanner determines how much of the Cy3-oligonucleotide remained on the slide (Cy3REM) in relation to the starting intensity Cy3START. Thus, the normalized Cy3REM intensity reflects the survival probability of the sample duplex ΦS when probed against a certain reference duplex ΦR. In principle, ΦS could also be deduced from the distribution of the Cy3-label between the stamp and the slide as described in Albrecht et al. (22); however, it is easier and more accurate to accomplish the readout just from the information on the slide. Therefore, in the following the stamp images are not used for the analysis.
The structure of the molecular balances is shown in Table 1.
TABLE 1.
Four molecular force balances are depicted each comprising an amino-oligo (top), a Cy3-oligo (middle), and a biotin-oligo (bottom)
| Dir. | Duplex | No. | Sequence |
|---|---|---|---|
| 3′-5′ | 30PM | #66 | NH2-20t-CTGCAGGAATTCGATATCAAGCTTATCGAT |
| 5′-3′ | #74 | GACGTCCTTAAGCTATAGTTCGAATAGCTAc-17t-cATCGATAAGCTTGATATCGAATTCCTGCAGttttt-Cy3 | |
| 3′-5′ | 30PM | #62 | TAGCTATTCGAACTATAGCTTAAGGACGTC-20*t-Biotin |
| 3′-5′ | 30GG | #66 | NH2-20t-CTGCAGGAATTCGATATCAAGCTTATCGAT |
| 5′-3′ | #75 | GACGTCCTTAAGGTATAGTTCGAATAGCTAc-17t-cATCGATAAGCTTGATATCGAATTCCTGCAGttttt-Cy3 | |
| 3′-5′ | 30PM | #62 | TAGCTATTCGAACTATAGCTTAAGGACGTC-20*t-Biotin |
| 3′-5′ | 30CC | #66 | NH2-20t-CTGCAGGAATTCGATATCAAGCTTATCGAT |
| 5′-3′ | #80 | GACGTCCTTAACCTATAGTTCGAATAGCTAc-17t-cATCGATAAGCTTGATATCGAATTCCTGCAGttttt-Cy3 | |
| 3′-5′ | 30PM | #62 | TAGCTATTCGAACTATAGCTTAAGGACGTC-20*t-Biotin |
| 3′-5′ | 29CC | #66 | NH2-20t-CTGCAGGAATTCGATATCAAGCTTATCGAT |
| 5′-3′ | #86 | _ACGTCCTTAACCTATAGTTCGAATAGCTAc-17t-cATCGATAAGCTTGATATCGAATTCCTGCAGttttt-Cy3 | |
| 3′-5′ | 30PM | #62 | TAGCTATTCGAACTATAGCTTAAGGACGTC-20*t-Biotin |
Amino- and biotin-oligos are identical in all balances. Mutations are only introduced by single nucleotide exchanges (30GG, 30CC, 29CC) and a single nucleotide deletion (29CC) in the Cy3-oligonucleotides (underlined bases).
Surfaces and instrumentation
The silicone stamp had 16 protruding contact areas which themselves were microstructured by 100 × 100 μm squares elevated by 5 μm. It was functionalized by treatment with epoxy-silane, subsequently coated with bifunctional amino-biotin-PEG and finally covered with streptavidin. The amino-oligonucleotides were labeled at the 3′-termini and attached to aldehyde-functionalized glass slides. After the hybridization of the other two oligonucleotides, stamp and slide were brought in contact by a piezoelectric element. The piezo allowed for precisely defined pulling velocities during surface separation (see (22) for details).
Analysis of fluorescence images
The image analysis procedure is shown in Fig. 2. The light-shaded DNA spot (Fig. 2 A) on a slide is depicted, where the fluorescence intensity of the Cy3-labeled oligo was measured. The area of contact between slide and stamp is equivalent to the darker squares, where the balances were probed and Cy3-labeled oligonucleotides were removed from the slide. The intensity of the darker squares corresponds to the remaining signal Cy3REM. The light grid area is equivalent to the start intensity Cy3START, where no contact was established between stamp and slide. The solid light-shaded line in Fig. 2 A indicates a region of interest (ROI-1) corresponding to the Cy3REM signal. Another two dashed black lines are enclosing the area of ROI-2, which corresponds to Cy3START. The fluorescence intensity of the dark-shaded spot in Fig. 2 B represents biotin residues that have not coupled to the stamp, and which have been labeled by soluble AlexaFluor647-streptavidin after the Cy3-scan was performed. Accordingly, the AlexaFluor647 intensity of the dark squares reflects the coupling efficiency, as discussed in Albrecht et al. (22). Here (ROI-3) and (ROI-4) are enclosing the remaining biotin density AF647REM and the starting biotin density AF647START.
FIGURE 2.

Fluorescent images are shown for the slide after separation of the stamp. (A) Image of the Cy3 fluorescent signal of a spot after separation. (B) The same spot after binding of fluorescent-streptavidin to the free biotin. Dark squares correspond to the contact area where the balances have ruptured. Cy3START intensity and AF647START intensity have been derived from ROI-1 and ROI-3, respectively. Cy3REM intensity and AF647REM intensity have been derived from ROI-3 and ROI-4 and correspond to the grid around one contact area.
For each of the 25 contact areas the mean intensities Cy3REM, Cy3START, AF647REM, and AF647START have been determined. The degree of coupling was obtained by AF647REM/AF647START. To correct the Cy3-signal for the coupling efficiency, an offset of Cy3 was calculated from
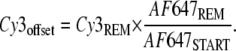 |
(1) |
Finally, the survival probability of the sample duplex ΦS was calculated from Cy3REM, corrected for the coupling efficiency, and normalized to Cy3START, according to Eq. 2:
 |
(2) |
Since not all of the contact areas showed a high homogeneity in terms of intensity and a good coupling efficiency, those ΦS1-25 values were selected that resulted in the minimal standard deviation (SD) for all of the measurement spots. This was the case for the following criteria: Coupling efficiency > 85%; SD(Cy3START) < 14%; SD(Cy3REM) < 12%; SD(AF647START) < 15%; and SD(AF647REM) < 49%. According to those criteria, the SD for every kind of measurement spot (30PM, 30CC, 30GG, and 29CC) was <5%, as indicated in Table 2.
TABLE 2.
Summary of the input parameters for the simulations and the resulting potential width ΔxR,S values
| Duplex | ΦS | ΔGR,S [kBT] | kR0,S0 [s−1] | ΔxR,S [nm] |
|---|---|---|---|---|
| Reference | — | −45.02 | 2.82 × 10−14 | 2.800 |
| 30PM | 0.406 ± 0.016 | −45.02 | 2.82 × 10−14 | 2.834 ± 0.006 |
| 30GG | 0.271 ± 0.010 | −41.88 | 6.49 × 10−13 | 2.612 ± 0.005 |
| 30CC | 0.199 ± 0.010 | −37.24 | 6.76 × 10−11 | 2.228 ± 0.005 |
| 29CC | 0.179 ± 0.009 | −35.39 | 4.26 × 10−10 | 2.072 ± 0.005 |
ΔGR,S is the Gibbs free energy difference, kR0,S0 is the natural dissociation rate, and ΦS is the measured survival probability.
Calculation of dissociation rates kR0,S0 and the Gibb's free energy differences ΔGR,S
The Gibb's free energy difference ΔGR,S for the DNA perfect match and mismatch duplexes was calculated by the computer program HyTher (Wayne State University, Detroit, MI; http://ozone3.chem.wayne.edu/cgi-bin/login/login/showLoginPage.cgi), which is based on the nearest-neighbor algorithm refined for mismatches by SantaLucia (36,37), for 150 mM Na+, 25°C, and 100 nM of each oligonucleotide. The dissociation rate kR0,S0 of the duplexes were then calculated from the equilibrium constant of dissociation
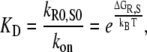 |
(3) |
and with kon = 106 M−1 s−1, based on the assumption that kon is diffusion-limited and identical for all DNA duplexes (38).
Determination of loading rate and rupture force
The loading rate r and the corresponding rupture force f of the balance were estimated from the applied velocity v = 5 nm/s, the polymer spacer length L0 = 30 nm, dissociation rate kR0 = 2.81 × 10−14 s−1, and potential width ΔxR = 2.8 nm of the reference duplex, according to the model from Friedsam et al. (28).
Simulations
Each survival probability for the reference ΦR and the sample duplex ΦS was simulated by increasing the force f and keeping the loading rate r0,1,2,3,4 fixed at the same time. Simulations and data fits were performed with Mathematica (Wolfram Research, Champaign, IL) and IGOR (WaveMetrics, Portland, OR).
RESULTS
Theoretical model
Despite the fact that a differential force assay is carried out on a large ensemble of molecular balances at the same time, each of the balances is probed individually without cooperative effects from neighboring molecules. Therefore, a single molecule approach is adequate for the theoretical description of the assay.
According to the experiments presented later, where the molecular balance comprises a constant reference duplex and a variable sample duplex, the goal of the simulation is to derive the survival probability ΦS of the sample from the potential width ΔxS and the dissociation rate kS0 in a combination with a certain reference bond with ΔxR and kR0.
To calculate ΦS, the molecular force balance is modeled based on reaction rates, as shown in Fig. 1, where ΦR and ΦS indicate survival probabilities for the reference and sample duplex, respectively. According to Fig. 1, the time-dependent rupture probabilities can be expressed as a system of coupled ordinary differential equations, depending only on the dissociation rates kR and kS:
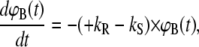 |
(4A) |
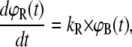 |
(4B) |
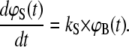 |
(4C) |
To solve Eqs. 4A–4C, the probabilities of ϕB (t = 0) = 1, ϕR (t = 0) = 0, and ϕS (t = 0) = 0 are used as boundary conditions at t = 0 s when the bonds are just about to rupture. The association reaction is neglected based on the assumption that forced unbinding happens much faster than the association reaction. In contrast to the force balance experiment where intact bonds are detected by means of the fluorophore, the solution of the differential equations equals probabilities (ϕR(t)) and (ϕS(t)) corresponding to ruptured bonds. Therefore, the survival probabilities ΦR(t) and ΦS(t) of the intact bonds are calculated from
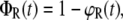 |
(5A) |
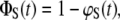 |
(5B) |
and plotted as a function of time in Fig. 3 A. Please note that all probability diagrams represent experiments on very large ensembles (n ∼ 109) of molecular balances and not single molecule data.
FIGURE 3.

Solutions of the Eqs. 7A–7C are plotted for a symmetric balance with tR0 = 1/kR0 = 3.55 × 1013 s, kR0,S0 = 2.82 × 10−14 s−1, and ΔxR,S = 2.8 nm. (A) Survival probabilities of the reference bonds (blue) and sample bonds (red) are plotted as function of the logarithm of the normalized dissociation time for rupture forces of f0 = 0 pN, f1 = 10 × fR, f2 = 20 × fR, f3 = 30 × fR, and f4 = 40 × fR with fR = kBT/ΔxR = 1.48 pN. (B) The same survival probabilities are plotted as shown in panel A as a function of normalized force f/fR for loading rates r0 = 0 pN/s, r1 = 4 × 10−9 pN/s, r2 = 1.5 × 10−4 pN/s, r3 = 4 pN/s, and r4 = 1 × 105 pN/s. Black circles indicate the point where all balances are ruptured for different rupture forces f0, 1, 2, 3, 4.
To convert kR and kS into force-dependent rates, the Bell-Evans model for force-induced unbinding was applied to obtain
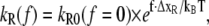 |
(6A) |
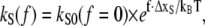 |
(6B) |
where ΔxR,S is the distance between the bound and the transition state in a triangular binding potential and f is the external applied force. The constants kR0 and kS0 correspond to the natural dissociation rates in equilibrium, which were calculated from the free energy equilibrium constants KD, as discussed above. The applied force tilts the binding potential and thus lowers the transition state energy by f × ΔxR,S, causing the reference and sample bond to dissociate faster. The rate constants in Eq. 1 are therefore substituted by the force-dependent rates kR = kR(f) and kS = kS(f). An example for survival probability functions ΦR(t, f) and ΦS(t, f) for different external applied forces was plotted in Fig. 3 A over the logarithm of time. The timescale was normalized with respect to the time of dissociation of the reference bond at zero force 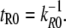 Since the experimentally accessible variables are the separation velocity v and force f, the loading rate r and the time were substituted by r = df/dt, as shown in Fig. 3 B. With this substitution, the final form of the differential equations is given in Eqs. 7A–7C and the solutions are plotted in Fig. 3 B:
Since the experimentally accessible variables are the separation velocity v and force f, the loading rate r and the time were substituted by r = df/dt, as shown in Fig. 3 B. With this substitution, the final form of the differential equations is given in Eqs. 7A–7C and the solutions are plotted in Fig. 3 B:
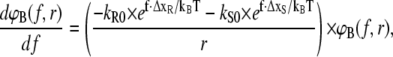 |
(7A) |
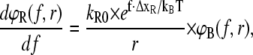 |
(7B) |
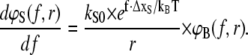 |
(7C) |
Here, the survival probability of the sample ΦS and the reference bond ΦR were plotted as a function of normalized force f/fR, with respect to the reference bond, where fR = kBT/ΔxR is the characteristic force (10).
The experimentally acquired survival probability of the sample duplex ΦS corresponds to the point where the dotted red line converges toward the solid red line in the following diagrams. In Fig. 3 B and the following diagrams, this point is highlighted by a black circle.
Simulations
In Fig. 4, A–D, the binding potentials and survival probabilities for different types of molecular balances are depicted in dependence of loading rates and rupture forces. Depending on the grade of asymmetry, the external force will bend the binding potential of the reference bond (blue lines) and the sample bond (red lines) to a different degree according to Eqs. 7A–7C. In particular, the reference bond is characterized by a Gibbs free energy difference of −45.02 kBT and a potential width of 2.8 nm. With these two values, the binding potential and the potential width in Fig. 4 were normalized. Furthermore, the natural dissociation rate was calculated to kR0 = 2.82 × 10−14 s−1, using Eq. 3. The potential width of the reference bond ΔxR sets the characteristic force for the reference bond according to fR = 4.14 pN/nm/2.8 nm = 1.48 pN.
FIGURE 4.

Influence of the shape of triangular binding potentials for two bonds in series. The two binding potentials of the reference (blue) and the sample (red) bond are plotted on left. The differences in potential width and depth are magnified in the inserts. The resulting survival probabilities of the two bonds are plotted on the right side. The loading rates are r0 = 0 pN/s, r1 = 4 × 10−9 pN/s, r2 = 1.5 × 10−4 pN/s, r3 = 4 pN/s, and r4 = 1 × 105 pN/s. The applied forces are f0 = 0 pN, f1 = 10 × fR, f2 = 20 × fR, f3 = 30 × fR, and f4 = 40 × fR with fR = kBT/ΔxR = 1.48 pN. (A) Perfectly symmetric molecular balance with identical potential width ΔxR,S = 2.8 nm and natural dissociation rates kR0,S0 = 2.82 × 10−14 s−1 . Black circles indicate the survival probability for the sample bond (red curves) for a given loading rate r0,1,2,3,4 and rupture force f0,1,2,3,4. (B) Asymmetry in potential depth, which corresponds to an asymmetry in kR0,S0 for a molecular balance with identical ΔxR,S but different kS0 = 8.46 × 10−14 s−1. The survival probability of the sample bonds indicated by the black circles is the same for different loading rates r0,1,2,3,4 and rupture forces f0,1,2,3,4. (C) Asymmetry in ΔxR,S with identical kR0,S0 but different ΔxS = 2.66 nm. The survival probability of the sample bonds, indicated by the black circles, rises with higher loading rates and rupture forces. (D) Mixed asymmetry with differences in kR0,S0 (kR0 = 2.82 × 10−14 s−1, kS0 = 8.46 × 10−14 s−1) and ΔxR,S (ΔxR = 2.8 nm, ΔxS = 2.66 nm). The sample bonds (red curve) have a lower survival probability than the reference bonds (blue curve) for low loading rates but rises with higher loading rates and rupture forces. The crossover occurs at a rate of rc = 9 × 10−4 pN/s.
In Fig. 4, A–D, left, the binding potential was tilted by f × ΔxR. Here we applied five different forces of multiples of fR: f0 = 0 pN, f1 = 10 × fR, f2 = 20 × fR, f3 = 30 × fR, and f4 = 40 × fR. The simulations for the survival probability ΦS as a function of the normalized force f/fR were then performed at five different loading rates of r0 = 0 pN/s, r1 = 4 × 10−9 pN/s, r2 = 1.5 × 10−4 pN/s, r3 = 4 pN/s, and r4 = 1 × 105 pN/s (Fig. 4, A–D, right).
Perfectly symmetric balances
A balance where the reference and the sample bond are equal in terms of ΔxR,S and kR0,S0 could be considered as perfectly symmetric. In Fig. 4 A two diagrams are shown for a perfectly symmetric molecular balance, where the binding potential for the reference bond and the sample bond is identical along the pulling direction (kR0 = kS0 = 2.82 × 10−14 s−1 and ΔxR = ΔxS = 2.8 nm). For simplicity, a triangular potential was assumed based on the Bell-Evans model. On the left, the normalized binding potential ΔE/ΔGR is depicted for different normalized unbinding forces f/fR. The potentials of the reference bond (hidden blue line) and the sample bond (red line) are deformed to the same degree (left) and no shift in the survival probability ΦS is observed in the probability diagram (right).
Asymmetry in kR0,S0
In this case, a difference in kR0,S0 was introduced into the sample bond by lowering the transition state by 1 kBT (kS0 = 8.46 × 10−14 s−1) while the reference bond was not changed at all (kR0 < kS0), but leaving the potential width equal (ΔxR = ΔxS). As shown for the potentials in Fig. 4 B (left), the red and the blue lines are shifted to each other, indicating the asymmetry between the two bonds (right). As for the symmetric case in Fig. 4 A, the system is not affected in terms of the rupture probabilities by a change in loading rate, since both binding potentials are tilted by the same energy f × ΔxR = f × ΔxS.
Asymmetry in ΔxR,S
Here the potential width of the reference bond was chosen to be wider than the sample bond (ΔxR > ΔxS, ΔxS = 2.66 nm) while the potential depths were kept equal (kR0 = kS0). As demonstrated in Fig. 4 C (right), the asymmetry in ΔxR,S results in a pronounced dependence of the unbinding process on the force rate. Since the sample bond has a smaller ΔxS, larger rupture probabilities than for the reference bond are observed. As a consequence, the survival probability of the sample bond ΦS (red line, right) converges toward 1 for high loading rates.
Combined asymmetry
Finally in Fig. 4 D an example for the combination of asymmetries in potential widths (ΔxS = 2.66 nm, ΔxR > ΔxS) and depths (kS0 = 8.46 × 10−14 s−1, kR0 < kS0) is presented. As evidenced in the probability diagram, the sample bond is weaker then the reference bond at zero force (f = 0) and for small loading rates. However, at higher loading rates the disadvantage due to the larger kS0 is successively compensated by the advantage of a shorter ΔxS. After the red line in the right graph has crossed the 0.5 line (between rupture forces f2 and f3) again, an asymptotic increase toward 1 is observed.
Experiments
The theoretical concept presented above was corroborated with experiments on short oligonucleotide duplexes as previously published (22) with the goal to derive ΔxS values for several mutations in the sample duplex. With the data available in the literature (17), the assumption of triangular potentials for the reference and the sample complex are made as shown in Fig. 4, A–D.
For the simulation, the following input parameters are required:
The survival probabilities of the sample bond ΦS, which are calculated from the fluorescence intensities as described in Materials and Methods and listed in Table 2.
The velocity v by which the molecules are probed is assumed to be identical to the pulling velocity.
The loading rate and the corresponding rupture force were simulated from kR0, ΔxR, and the velocity v as described in Materials and Methods. Based on the pulling velocity of v = 5 nm/s, the loading rate equals 37 pN/s at a rupture force of 48 pN.
Natural dissociation rates kR0,S0 at zero force for the reference duplex and the sample duplex were calculated from ΔGR,S values as described in Materials and Methods.
The potential width of the reference bond ΔxR was derived from Strunz et al. (17), which is 2.8 nm.
For the simulation, the above-mentioned variables were kept constant and only ΔxS was varied until the minima of the curves matched the measured ΦS values (compare to Fig. 4 and Fig. 5 A). Then the corresponding potential width ΔxS was extracted.
FIGURE 5.

Combined data from different experiments (light shaded bars) and simulations (solid line) on a 30-mer PM, 30-mer GG MM, 30-mer CC MM, and a 29-mer CC MM. (A) Survival property of the sample duplex ΦS plotted as a function of normalized dissociation rate kS0/kR0 on a log scale. (B) Calculated normalized potential width of the sample duplex ΔxS/ΔxR is plotted as a function of the corresponding normalized dissociation rate of the sample duplex kS0/kR0 that matches the survival probabilities in panel A. The pulling velocity was v = 5 nm/s, resulting in a loading rate of 37 pN/s at a rupture force of 48 pN.
The probability diagrams for all sample duplexes are plotted in Fig. 5 A, showing experimental data (light shaded bars) for the perfect match as well as the 30GG, 30CC, and the 29CC mismatches. To get an impression about the correlation between the rupture probability and the dissociation rate kS0, the measured survival probability of the sample bond ΦS was plotted in Fig. 5 A over the normalized natural dissociation rate kS0/kR0. To calculate survival probabilities of the sample bond (shown as solid line), only the potential width of the sample bond was varied. The resulting normalized potential width ΔxS/ΔxR is plotted in Fig. 5 B as a function of the normalized natural dissociation rate of the sample complex kS0/kR0.
Table 2 summarizes the input parameters for all sample duplexes and the reference duplex. The ΔxR,S values are also listed, calculated by using the differential equation Eqs. 7A–7C (with exception of ΔxR,S for the 30-bp perfect match, which was taken from the data provided in (17)).
Besides the analysis of force-balance measurements, our simulations are useful for designing force experiments where the sample is bound to a surface by a receptor ligand immobilization tag. In Fig. 6, two simulations of AFM experiments are depicted, where streptavidin/biotin (Fig. 6 A) and anti–digoxigenin/digoxigenin (Fig. 6 B), respectively, are used to immobilize a 15-bp DNA duplex. Again, survival probabilities for the DNA (blue) and for the immobilization tag (red) are plotted as function of the normalized force for different loading rates. Here, the reference duplex is a 15-bp DNA duplex with a natural dissociation rate of kR0 = 3.16 × 10−5 s−1, a potential width of ΔxR = 1.75 nm, and a characteristic force fR = 2.36 pN. For a loading rate of 37 pN/s, the survival probability of the immobilization tag (red) is highlighted by a black circle. It is evident from Fig. 6 A, which the streptavidin survives over the whole force range with a likelihood of >99%. In contrast, the antibody bond is not sufficiently strong for the experiment and would fail with a probability of ∼90%.
FIGURE 6.

Simulation of possible AFM experiments using two bonds in series. The reference bond is a 15-bp DNA duplex (blue), which was immobilized by (A) streptavidin/biotin (red) and (B) anti-digoxigenin/digoxigenin (red). Black circles indicate survival probability of the immobilization tag at f1 = 23 pN and r1 = 37 pN/s (dotted curves). The black circle on the solid line indicates survival probabilities at zero force (f0) and loading rate (r0). Reference: kR0 = 3.16 × 10−5 s−1 and ΔxR = 1.75 nm for 15 bp DNA (17). Sample: (A) kS0 = 3.71 × 10−6 s−1 and ΔxS = 1.31 nm for streptavidin/biotin (41). (B) kS0 = 0.015 s−1 and ΔxS = 1.15 nm for anti-digoxigenin/digoxigenin (16).
DISCUSSION
We can proceed from the assumption that for a molecular force balance, which is perfectly symmetric in terms of kR0,S0 and ΔxR,S, the relative rupture probability is independent of any changes in pulling velocity and loading rate (Fig. 4 A). However, even small variations of the sample duplex binding potential breaks the symmetry and gives rise to significant differences in survival probability as illustrated by Fig. 4, B and C. Compared to the fully symmetric balance in 4 A, where both the reference and the sample bond survival probabilities converge toward 0.5, a huge shift is observed when ΔGS and ΔxS of the sample bond are changed just between 2 and 5% compared to the reference bond. Interestingly, for changes in kS0, ΦS stays constant over the entire loading range, while for changes in ΔxS, ΦS varies with an increase of the applied loading rate.
As shown by Strunz et al. (17), deletions of peripheral basepairs from a 30-bp DNA duplex give rise to decreasing kS0 and increasing ΔxS. Since the deletion affects both variables in the opposite direction, it is evident that a substantial part of the lowering in binding energy is compensated by a decrease in ΔxS when we consider the resulting survival probabilities. This is illustrated by Fig. 4 D where a case with differences in kR0,S0 and ΔxR,S was simulated. It is evident from the red line that the drop in the survival probability of the sample complex ΦS becomes smaller for increasing loading rates, since the difference in ΔxR,S causes an asymptotic rise, which compensates the differences between kR0 and kS0.
In contrast to DNA duplexes, no such correlation was generally expected for an antibody antigen system like that investigated by Schwesinger et al. (18). There the effect of single amino-acid exchanges in the binding pocket of anti-hapten single-chain fragment antibodies was analyzed by AFM force spectroscopy. Since the mutations should not have altered the geometry of the binding pocket, ΔxR,S was assumed to be constant despite changes in the binding energy. Surprisingly, the antibody system shows a qualitatively similar behavior as the DNA duplexes studied by Strunz. Again, a linear correlation between the logarithm of the natural dissociation rate kS0 and the potential width ΔxS was found as shown in Fig. 5 A.
For the experiments presented here, we have introduced internal basepair mismatches to the DNA duplex, because such mutations have about the half effect on ΔGS compared to a 10-bp deletion (17), but without affecting the contour length of the duplex. Therefore, we expected the effect of the mutations on ΔxS to be rather small compared to the 10-bp deletions.
To facilitate a quantitative comparison of our data to that of Strunz et al. (17), we calculated ΔGR,S values based on the nearest-neighbor algorithm as explained above for both experiments. In Fig. 5 B, the normalized potential width ΔxS/ΔxR over normalized dissociation rate kS0/kR0 is plotted, and in accordance to the procedure from Schwesinger et al. (18), we obtained a slope for the Strunz data of 0.53 (17) close to the value measured by Schwesinger for the single-chain fragment antibodies of 0.3. In contrast to that, our point mutations resulted in an even higher slope of 0.8. In other words, a single-point mutation like the CC homoduplex accounts for a difference in ΔGS comparable to an 8-bp deletion (7.8 kBT) but equals a shift in ΔxS comparable to a 10-bp deletion according to the equation from Strunz. Hence, the compensation of ΔGS (kS0) by ΔxS in terms of the resulting survival probability ΦS is more pronounced for internal mismatches than for peripheral deletions in DNA. This may be an explanation for the fact that discrimination of mismatches has not been reported for the AFM so far, in contrast to energetically comparable peripheral deletions (17).
As seen in Fig. 5, the experimental data are reproduced very well using the approximation of triangular potentials for the reference and the sample complex (17). Furthermore, this framework of comparing two bonds in series is also extendible to other potential shapes. If the reference potential is known in better detail, a wide range of loading rates can be measured, because only the differences between the potentials of the reference and the sample complexes are measured. And even if these differences are small, this method has the potential to resolve them.
There are some potential challenges in applying this technique directly to molecular interactions when the spontaneous dissociation rates become comparable to the timescale of the experiment. If the sample and the reference complex are too weak, they start to dissociate even during assembly of the assay. The consequence will be a change of concentrations of reference and sample complex during the experiment without applying a force. But if one uses the experimental setup above to measure the interactions of molecules that bind to just the sample DNA, in principle it is also possible to measure weaker interactions. These interactions act only on the sample complex in the time frame of the contact of the stamp with the slide and not during the assembly of the assay.
In addition to analyzing force-balance experiments, the theoretical model also was applied to prove the suitability of immobilization tags for force experiments, as illustrated in Fig. 6. The fact that streptavidin/biotin (Fig. 6 A) is very well suited for immobilization of samples under force is again corroborated by the simulation for forces at least up to 100 pN. In contrast to that, it is critical to rely on a single digoxigenin/antibody bond, which fails for low loading rates in comparison to a 15-mer DNA (Fig. 6 B).
Furthermore, it is evident that DNA duplexes may be used as reference bonds to determine the kS0 and ΔxS values of protein receptors like the anti-digoxigenin antibody. However, for the streptavidin/biotin bond it would be hard to find a matching DNA duplex, since the rupture force of 15 bp is much too low and even much longer DNA double strands will not exceed the 65 pN barrier of the BS-transition (39,40).
Acknowledgments
We thank Robert Lugmaier, Julia Morfill, Kay Gottschalk, Dominik Ho, Katja Falter, and Erich Sackmann for helpful discussions.
This work was supported by the Deutsche Forschungsgemeinschaft.
Editor: Jane Clarke.
References
- 1.Rief, M., M. Gautel, F. Oesterhelt, J. M. Fernandez, and H. E. Gaub. 1997. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 276:1109–1112. [DOI] [PubMed] [Google Scholar]
- 2.Rief, M., F. Oesterhelt, B. Heymann, and H. E. Gaub. 1997. Single molecule force spectroscopy on polysaccharides by atomic force microscopy. Science. 275:1295–1297. [DOI] [PubMed] [Google Scholar]
- 3.Grandbois, M., M. Beyer, M. Rief, H. Clausen-Schaumann, and H. E. Gaub. 1999. How strong is a covalent bond? Science. 283:1727–1730. [DOI] [PubMed] [Google Scholar]
- 4.Hugel, T., and M. Seitz. 2001. The study of molecular interactions by AFM force spectroscopy. Macromol. Rapid Commun. 22:989–1016. [Google Scholar]
- 5.Hugel, T., N. B. Holland, A. Cattani, L. Moroder, M. Seitz, and H. E. Gaub. 2002. Single-molecule optomechanical cycle. Science. 296:1103–1106. [DOI] [PubMed] [Google Scholar]
- 6.Bustamante, C. 2005. Unfolding single RNA molecules: bridging the gap between equilibrium and non-equilibrium statistical thermodynamics. Q. Rev. Biophys. 38:291–301. [DOI] [PubMed] [Google Scholar]
- 7.Bockelmann, U. 2004. Single-molecule manipulation of nucleic acids. Curr. Opin. Struct. Biol. 14:368–373. [DOI] [PubMed] [Google Scholar]
- 8.Charvin, G., T. R. Strick, D. Bensimon, and V. Croquette. 2005. Tracking topoisomerase activity at the single-molecule level. Annu. Rev. Biophys. Biomol. Struct. 34:201–219. [DOI] [PubMed] [Google Scholar]
- 9.Gore, J., Z. Bryant, M. Nollmann, M. U. Le, N. R. Cozzarelli, and C. Bustamante. 2006. DNA overwinds when stretched. Nature. 442:836–839. [DOI] [PubMed] [Google Scholar]
- 10.Evans, E. 1998. Energy landscapes of biomolecular adhesion and receptor anchoring at interfaces explored with dynamic force spectroscopy. Faraday Discuss. 111:1–16. [DOI] [PubMed] [Google Scholar]
- 11.Merkel, R. 2001. Force spectroscopy on single passive biomolecules and single biomolecular bonds. Phys. Rep. Rev. Phys. Lett. 346:344–385. [Google Scholar]
- 12.Holland, N. B., T. Hugel, G. Neuert, A. Cattani-Scholz, C. Renner, D. Oesterhelt, L. Moroder, M. Seitz, and H. E. Gaub. 2003. Single molecule force spectroscopy of azobenzene polymers: switching elasticity of single photochromic macromolecules. Macromolecules. 36:2015–2023. [Google Scholar]
- 13.Neuert, G., T. Hugel, R. R. Netz, and H. E. Gaub. 2006. Elasticity of poly(azobenzene-peptides). Macromolecules. 39:789–797. [Google Scholar]
- 14.Bell, G. I. 1978. Models for the specific adhesion of cells to cells. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- 15.Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- 16.Neuert, G., C. Albrecht, E. Pamir, and H. E. Gaub. 2006. Dynamic force spectroscopy of the digoxigenin-antibody complex. FEBS Lett. 580:505–509. [DOI] [PubMed] [Google Scholar]
- 17.Strunz, T., K. Oroszlan, R. Schafer, and H. J. Guntherodt. 1999. Dynamic force spectroscopy of single DNA molecules. Proc. Natl. Acad. Sci. USA. 96:11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwesinger, F., R. Ros, T. Strunz, D. Anselmetti, H. J. Guntherodt, A. Honegger, L. Jermutus, L. Tiefenauer, and A. Pluckthun. 2000. Unbinding forces of single antibody-antigen complexes correlate with their thermal dissociation rates. Proc. Natl. Acad. Sci. USA. 97:9972–9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishizaka, T., H. Miyata, H. Yoshikawa, S. Ishiwata, and K. Kinosita. 1995. Unbinding force of a single motor molecule of muscle measured using optical tweezers. Nature. 377:251–254. [DOI] [PubMed] [Google Scholar]
- 20.Lang, M. J., P. M. Fordyce, A. M. Engh, K. C. Neuman, and S. M. Block. 2004. Simultaneous, coincident optical trapping and single-molecule fluorescence. Nat. Methods. 1:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albrecht, C., K. Blank, M. Lalic-Multhaler, S. Hirler, T. Mai, I. Gilbert, S. Schiffmann, T. Bayer, H. Clausen-Schaumann, and H. E. Gaub. 2003. DNA: a programmable force sensor. Science. 301:367–370. [DOI] [PubMed] [Google Scholar]
- 22.Albrecht, C. H., H. Clausen-Schaumann, and H. E. Gaub. 2006. Differential analysis of biomolecular rupture forces. J. Phys. Condens. Matter. 18:S581–S599. [Google Scholar]
- 23.Blank, K., A. Lankenau, T. Mai, S. Schiffmann, I. Gilbert, S. Hirler, C. Albrecht, M. Benoit, H. E. Gaub, and H. Clausen-Schaumann. 2004. Double-chip protein arrays: force-based multiplex sandwich immunoassays with increased specificity. Anal. Bioanal. Chem. 379:974–981. [DOI] [PubMed] [Google Scholar]
- 24.Blank, K., T. Mai, I. Gilbert, S. Schiffmann, J. Rankl, R. Zivin, C. Tackney, T. Nicolaus, K. Spinnler, F. Oesterhelt, M. Benoit, H. Clausen-Schaumann, and H. E. Gaub. 2003. A force-based protein biochip. Proc. Natl. Acad. Sci. USA. 100:11356–11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert, L., S. Schiffmann, S. Rubenwolf, K. Jensen, T. Mail, C. Albrecht, A. Lankenau, G. Beste, K. Blank, H. E. Gaub, and H. Clausen-Schaumann. 2004. Double chip protein arrays using recombinant single-chain Fv antibody fragments. Proteomics. 4:1417–1420. [DOI] [PubMed] [Google Scholar]
- 26.Evans, E., and K. Ritchie. 1997. Dynamic strength of molecular adhesion bonds. Biophys. J. 72:1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans, E., and K. Ritchie. 1999. Strength of a weak bond connecting flexible polymer chains. Biophys. J. 76:2439–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedsam, C., A. K. Wehle, F. Kühner, and H. E. Gaub. 2003. Dynamic single-molecule force spectroscopy: bond rupture analysis with variable spacer length. J. Phys. Condens. Matter. 15:S1709–S1723. [Google Scholar]
- 29.Allemand, J. F., D. Bensimon, and V. Croquette. 2003. Stretching DNA and RNA to probe their interactions with proteins. Curr. Opin. Struct. Biol. 13:266–274. [DOI] [PubMed] [Google Scholar]
- 30.Berquand, A., N. Xia, D. G. Castner, B. H. Clare, N. L. Abbott, V. Dupres, Y. Adriaensen, and Y. F. Dufrene. 2005. Antigen binding forces of single antilysozyme Fv fragments explored by atomic force microscopy. Langmuir. 21:5517–5523. [DOI] [PubMed] [Google Scholar]
- 31.Evans, E. 2001. Probing the relation between force lifetime and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 30:105–128. [DOI] [PubMed] [Google Scholar]
- 32.Gore, J., Z. Bryant, M. D. Stone, M. Nollmann, N. R. Cozzarelli, and C. Bustamante. 2006. Mechanochemical analysis of DNA gyrase using rotor bead tracking. Nature. 439:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinterdorfer, P., and Y. F. Dufrene. 2006. Detection and localization of single molecular recognition events using atomic force microscopy. Nat. Methods. 3:347–355. [DOI] [PubMed] [Google Scholar]
- 34.Williams, M. C., and I. Rouzina. 2002. Force spectroscopy of single DNA and RNA molecules. Curr. Opin. Struct. Biol. 12:330–336. [DOI] [PubMed] [Google Scholar]
- 35.Reference deleted in proof.
- 36.SantaLucia, J., Jr. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 95:1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peyret, N., P. A. Seneviratne, H. T. Allawi, and J. SantaLucia, Jr. 1999. Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A·A, C·C, G·G, and T·T mismatches. Biochemistry. 38:3468–3477. [DOI] [PubMed] [Google Scholar]
- 38.Northrup, S. H., and H. P. Erickson. 1992. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. USA. 89:3338–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith, S. B., Y. J. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 271:795–799. [DOI] [PubMed] [Google Scholar]
- 40.Cluzel, P., A. Lebrun, C. Heller, R. Lavery, J. L. Viovy, D. Chatenay, and F. Caron. 1996. DNA: an extensible molecule. Science. 271:792–794. [DOI] [PubMed] [Google Scholar]
- 41.Pincet, F., and J. Husson. 2005. The solution to the streptavidin-biotin paradox: the influence of history on the strength of single molecular bonds. Biophys. J. 89:4374–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]