

Abstract
Estimating the number of ancestral lineages of a sample of DNA sequences at time t in the past can be viewed as a variation on the problem of estimating the time to the most recent common ancestor. To estimate the number of ancestral lineages, we develop a maximum-likelihood approach that takes advantage of a prior model of population demography, in addition to the molecular data summarized by the pattern of polymorphic sites. The method relies on a rejection sampling algorithm that is introduced for simulating conditional coalescent trees given a fixed number of ancestral lineages at time t. Computer simulations show that the number of ancestral lineages can be estimated accurately, provided that the number of mutations that occurred since time t is sufficiently large. The method is applied to 986 present-day human sequences located in hypervariable region 1 of the mitochondrion to estimate the number of ancestral lineages of modern humans at the time of potential admixture with the Neanderthal population. Our estimates support a view that the proportion of the modern population consisting of Neanderthal contributions must be relatively small, less than ∼5%, if the admixture happened as recently as 30,000 years ago.
MUCH attention has been paid in population genetics to the estimation of the time since the most recent common ancestor (TMRCA) of a sample of homologous genes (Tavaré et al. 1997; Wilson and Balding 1998; Thomson et al. 2000; Tang et al. 2002). This has been particularly true for the TMRCA of human mitochondrial DNA and of the nonrecombining portion of the Y chromosome. Arguments based on TMRCA estimates have been used in evaluating historical models for human evolution, as smaller estimates are thought to be more compatible with a recent departure of anatomically modern humans from Africa followed by replacement of all other existing hominids (Cann et al. 1987; Vigilant et al. 1991), whereas larger estimates are potentially suggestive of ancient admixture with Eurasian Homo erectus (see Garrigan and Hammer 2006).
In this article, we consider a variation of the TMRCA estimation problem. Rather than estimating the TMRCA, i.e., the first time (measured backward from the present) when the genealogical tree contains only one lineage, we aim at estimating the number of ancestral lineages at a fixed point of time in the past. To ensure that there are a unique number of ancestral lineages at time t, we assume that the sequences under consideration are nonrecombining. The population of interest is assumed to be panmictic, but possibly of varying size, so that a simple coalescent approximation holds.
The problem of estimating the number of ancestral lineages is of particular relevance in testing for admixture between pairs of ancient populations. When the first Neanderthal mtDNA sequence was published, Krings et al. (1997) concluded that admixture between Neanderthals and modern humans was unlikely because the Neanderthal sequence coalesced with the modern human sequences much further in the past than the time of the most recent common ancestor of the modern human sample. However, Nordborg (1998) argued that the evidence against admixture was considerably weaker than was suggested by Krings et al. (1997), as the 986 modern human sequences studied by Krings et al. coalesced to a much smaller number of sequences contemporaneous with the Neanderthal sequence. Thus, the likelihood of admixture should have been evaluated by assessing the chance that this smaller number of ancestral sequences would coalesce separately from the Neanderthal.
Because at the time of the potential admixture, the number of sequences ancestral to modern humans may have been rather small, the ability to reject ancient gene flow between Neanderthals and modern humans may be very low. Using a coalescent model as an approximation to the genealogy of the sequences, Nordborg (1998) argued that the number of ancestral sequences is likely to have been small, except if the beginning of the human population expansion was further in the past than the potential contact between the two populations. Whereas Nordborg was making theoretical predictions concerning the number of ancestral lineages of modern humans, we propose to estimate this number using molecular data. The coalescent-based statistical method that we present in this article uses the pattern of polymorphic sites contained in a mitochondrial DNA data set similar to that used by Krings et al. (1997) to estimate the number of ancestral mtDNA sequences of modern humans at the time of potential admixture.
A variety of statistical methods have been proposed for inferential problems in a coalescent setting. The oldest methods used moment estimators, which rely on a simple formula connecting the expected value under the coalescent model of some summary statistic, such as the number of segregating sites, and a parameter of interest. For example, a widely used moment estimator is Watterson's estimator (Watterson 1975), which relies on the number of segregating sites to estimate θ, the product of the effective population size N and the mutation rate per generation. A disadvantage of moment estimators, however, is that confidence intervals may be hard to derive, and the estimators usually do not apply as the model becomes more complex.
Maximum-likelihood methods that use all of the data available are a second class of methods. These methods must contend with the fact that computing the probability of the data requires knowledge of the unknown genealogy on which sequences have evolved. Importance sampling methods (Griffiths and Tavaré 1994a) and Markov chain Monte Carlo (MCMC) approaches (Kuhner et al. 1995) have been proposed for integrating over possible genealogies. Although in principle these methods give the most accurate estimates, in practice they are quite computationally intensive.
A third type of approach that is becoming increasingly popular is rejection sampling (see, e.g., Fu and Li 1997; Tavaré et al. 1997; Weiss and von Haeseler 1998; Pritchard et al. 1999; Beaumont et al. 2002; Jakobsson et al. 2006). These methods typically consist of accepting the values of model parameters that produce summaries of simulated data that match summaries of the observed data and rejecting values for which the simulations do not match the observations. Rejection methods can combine a reasonable level of accuracy with more rapid execution compared to maximum likelihood.
For the problem of estimating the number of ancestral lineages from a set of sequences, we propose a maximum-likelihood method that is based on summaries of the observed data. The likelihood of a given value for the number of ancestral lineages is computed using an algorithm that simulates coalescent trees conditional on a given number of lineages at a given point of time in the past. We propose a new rejection algorithm for these simulations. The statistical properties of the maximum-likelihood estimator are then investigated using simulated genetic data, and the estimator is applied to data on 986 human sequences of hypervariable region 1 of mtDNA. Our method provides estimates of the number of ancestral mtDNA lineages of modern humans that were contemporary to the Neanderthals, with consequent implications for the possible levels of admixture between the Neanderthal population and early modern humans.
THEORY AND METHODS
The pattern of segregating sites:
Segregating sites are sites that are variable in a sample of DNA sequences. They can be classified according to their sizes or their types. The size of a mutation is defined as the number of individuals that carry the mutation. Because we assume that the sequences evolve according to the infinitely many sites model (Watterson 1975), there are exactly two alleles for each segregating site, and the type of the mutation is defined as the smaller of the counts of the two alleles. Because a mutation that affects the whole sample does not generate a polymorphic site, there are only n − 1 possible sizes of segregating sites in a sample of n sequences. We denote by ζi the number of segregating sites that are of size i and by 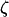 the vector of these counts:
the vector of these counts:
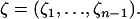 |
The computation of the size of a site assumes that the ancestral nucleotide at this site is known, for example, using an outgroup sequence. When the ancestral nucleotide is unknown, the type of a mutation—rather than its size—should be determined. We denote by τi the number of segregating sites that are of type i and by 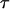 the vector of these counts:
the vector of these counts:
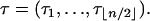 |
The vectors 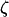 and
and 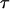 denote the site frequency spectrum and the folded site frequency spectrum, respectively. We choose the site frequency spectrum and the folded site frequency spectrum as our summary statistics for two reasons. First, the pattern of segregating sites (i.e., the site frequency spectrum or the folded site frequency spectrum) is highly informative about underlying population-genetic parameters such as θ, the product of the effective population size and the mutation rate (Fu 1994). Second, once the coalescent tree is known, the probability of a pattern of segregating sites can be computed using an explicit formula (Fu 1998). By contrast, the probability distributions of most summary statistics [e.g., mean pairwise differences, Tajima's D (Tajima 1989)] do not have known explicit formulas, even when the coalescent tree is known. For most summary statistics, the probability of a particular set of values is computed using a Monte Carlo approximation by jointly simulating coalescent trees and mutations. When computing the probability of a particular pattern of segregating sites, however, because of the existence of an exact formula for the probability of the pattern given a coalescent tree, only coalescent trees need to be simulated, but not mutations.
denote the site frequency spectrum and the folded site frequency spectrum, respectively. We choose the site frequency spectrum and the folded site frequency spectrum as our summary statistics for two reasons. First, the pattern of segregating sites (i.e., the site frequency spectrum or the folded site frequency spectrum) is highly informative about underlying population-genetic parameters such as θ, the product of the effective population size and the mutation rate (Fu 1994). Second, once the coalescent tree is known, the probability of a pattern of segregating sites can be computed using an explicit formula (Fu 1998). By contrast, the probability distributions of most summary statistics [e.g., mean pairwise differences, Tajima's D (Tajima 1989)] do not have known explicit formulas, even when the coalescent tree is known. For most summary statistics, the probability of a particular set of values is computed using a Monte Carlo approximation by jointly simulating coalescent trees and mutations. When computing the probability of a particular pattern of segregating sites, however, because of the existence of an exact formula for the probability of the pattern given a coalescent tree, only coalescent trees need to be simulated, but not mutations.
The probability of a particular pattern of segregating sites can be computed conditional on the coalescent tree as follows. First, the number of segregating sites S is a Poisson random variable of rate θℓ, where ℓ is the total length of the coalescent tree. Second, conditional on S = s, the vector of the sizes of the segregating sites 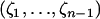 has a multinomial
has a multinomial 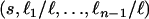 distribution, where ℓi denotes the total length of the branches ancestral to exactly i individuals. Therefore, conditional on the coalescent tree
distribution, where ℓi denotes the total length of the branches ancestral to exactly i individuals. Therefore, conditional on the coalescent tree 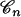 , the probability of the vector
, the probability of the vector 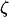 that counts the number of mutations of each size is
that counts the number of mutations of each size is
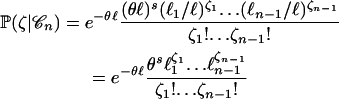 |
(1) |
(Fu 1998). Similarly, the probability of the vector  that counts the mutations of each type is
that counts the mutations of each type is
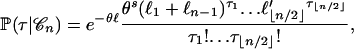 |
(2) |
where 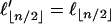 if n is even and
if n is even and 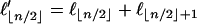 if n is odd.
if n is odd.
Because the coalescent tree 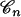 is not known, Equations 1 and 2 must be integrated over the space of coalescent trees. This is accomplished using Monte Carlo integration and is the subject of the next section. More precisely, the computation of the likelihood of the parameter j that denotes the number of ancestors is performed as follows:
is not known, Equations 1 and 2 must be integrated over the space of coalescent trees. This is accomplished using Monte Carlo integration and is the subject of the next section. More precisely, the computation of the likelihood of the parameter j that denotes the number of ancestors is performed as follows:
Simulate M coalescent trees
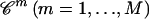 , given that the number of ancestors at time t = j.
, given that the number of ancestors at time t = j.Compute the likelihood of j using a Monte Carlo estimator
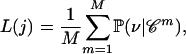 |
(3) |
where 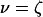 or
or 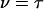 .
.
The second step of the algorithm is performed using Equation 1 or 2. The next section proposes a method for performing the first step, that is, for simulating coalescent trees given that the number of ancestors at time t = j. Nevertheless it is important to point out that using the complete vector of the sizes or the types of the segregating sites may be too time consuming. If a sample with 1000 sequences, for example, contains one mutation of size 400, it is unlikely that the simulated coalescent trees will contain a branch leading to 400 individuals. Thus, most of the time, the likelihood computed in step 2 will be zero. To avoid this problem, the segregating sites are binned into different categories that may contain one or more possible sizes (or types). The likelihood is then computed as in Equations 1 and 2, but the parameter of the multinomial distribution that corresponds to the binned category containing mutations of sizes (or types) s1 to s2 is equal to the sum of the branch lengths ancestral to 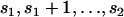 individuals divided by the total branch length of the tree. Because coalescent branches ancestral to a small number of individuals are more likely to occur than coalescent branches ancestral to a large number (see Blum and François 2005, Theorem 1, and Rosenberg 2006, Theorem 4.4), the general binning strategy that we adopted was to bin the mutations with large sizes (or types) into large clusters and to bin mutations with small sizes (or types) into small clusters. Note that binning all the mutations into a single category is equivalent to using the number of segregating sites as the only summary statistic in the estimation framework.
individuals divided by the total branch length of the tree. Because coalescent branches ancestral to a small number of individuals are more likely to occur than coalescent branches ancestral to a large number (see Blum and François 2005, Theorem 1, and Rosenberg 2006, Theorem 4.4), the general binning strategy that we adopted was to bin the mutations with large sizes (or types) into large clusters and to bin mutations with small sizes (or types) into small clusters. Note that binning all the mutations into a single category is equivalent to using the number of segregating sites as the only summary statistic in the estimation framework.
Simulating coalescent trees conditional on the number of ancestors at time t:
The simplest approach for simulating coalescent trees conditional on a fixed number of ancestors j at time t is basic rejection sampling. This method consists of simulating standard coalescent trees and accepting the trees whose number of ancestors at time t = j. However, the number of simulated coalescent trees that is required to simulate only one conditional coalescent tree may be prohibitively large, especially when having j lineages at time t is unlikely under the coalescent model. Therefore, we propose an alternative method for simulating conditional coalescent trees. This method is based on the conditional distribution of the intercoalescence times given that there is a fixed number of lineages j at time t.
For a sample of size n lineages, we denote by An(x) the random number of lineages in the coalescent process at time x and by qn,i(x) the probabilities 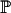 (An(x) = i) (Tavaré 1984; Takahata and Nei 1985). The intercoalescence times are denoted by Ti (
(An(x) = i) (Tavaré 1984; Takahata and Nei 1985). The intercoalescence times are denoted by Ti (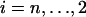 ), where Ti corresponds to the time during which there are exactly i lineages. Initially, we assume that the population size is constant, so that the distribution of the time Ti is exponential with parameter λi = i(i − 1)/2. To lighten the notation, we denote by ui+1 the time elapsed since the (n − i)th coalescence event and time t
), where Ti corresponds to the time during which there are exactly i lineages. Initially, we assume that the population size is constant, so that the distribution of the time Ti is exponential with parameter λi = i(i − 1)/2. To lighten the notation, we denote by ui+1 the time elapsed since the (n − i)th coalescence event and time t
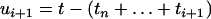 |
(see Figure 1). By convention, we set un+1 = t. The number of lineages An(x) starts at n at time x = 0 and eventually reaches 1 for x = TMRCA. The remaining part of this section is devoted to simulating coalescent trees given that An(t) = j.
Figure 1.—

A coalescent tree with n = 5 sequences conditioned on having j = 3 lineages at time t. The Ti's correspond to the intercoalescence times and ui corresponds to the time elapsed between the (n − i)th coalescence event and time t.
Because the random process An(x) is independent of the topology of the coalescent process (see, e.g., Tavaré 2004, pp. 44–46), the distribution of the topology of the coalescent conditional on An(t) = j remains the same as in the unconditional case. Using the Markov property for coalescences more ancient than t, it is clear that the distribution of the intercoalescence times 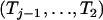 is the same in the conditional and the unconditional cases and that the intercoalescence time Tj during which there are j lineages is the sum of uj+1 and an exponential random variable of rate λj. Thus, the only difficulty when simulating a coalescent tree conditional on having j ancestors at time t resides in the simulation of the joint intercoalescence times
is the same in the conditional and the unconditional cases and that the intercoalescence time Tj during which there are j lineages is the sum of uj+1 and an exponential random variable of rate λj. Thus, the only difficulty when simulating a coalescent tree conditional on having j ancestors at time t resides in the simulation of the joint intercoalescence times 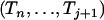 given that An(t) = j. In appendix a, we show that the conditional distribution of the intercoalescence time Ti (
given that An(t) = j. In appendix a, we show that the conditional distribution of the intercoalescence time Ti (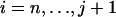 ) given
) given 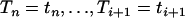 and An(t) = j is a mixture of truncated exponential distributions with positive and negative coefficients
and An(t) = j is a mixture of truncated exponential distributions with positive and negative coefficients
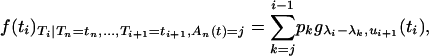 |
(4) |
where gγ,τ denotes the probability density function (p.d.f.) of the exponential distribution of parameter γ truncated at time τ and the coefficients of the mixture pk are given in appendix a (Equation A3). Mixtures with positive and negative coefficients can be simulated using a simple rejection algorithm of Bignami and De Matteis (1971) (see also Devroye 1986, p. 74). The Bignami and De Matteis method uses the fact that the mixture is less than or equal to the sum of only its positive components, so that
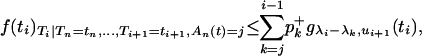 |
where 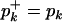 if pk > 0 and
if pk > 0 and 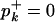 otherwise. The algorithm for simulating Ti given the intercoalescence times
otherwise. The algorithm for simulating Ti given the intercoalescence times 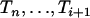 and An(t) = j is as follows.
and An(t) = j is as follows.
Algorithm 1—Bignami and De Matteis rejection sampling for simulating Ti given the intercoalescence times Tn, … , Ti+1 and An(t) = j, where j < i ≤ n:
Generate a random variate X with density
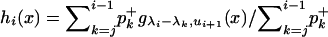 .
.Generate a uniform-[0, 1] random variate U.
If
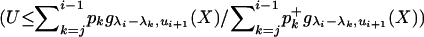 return X; otherwise go back to step 1.
return X; otherwise go back to step 1.
Step 1 is performed by simulating a random variate according to hi. This is straightforward, because hi is a finite mixture of truncated exponential distributions with positive coefficients (see Devroye 1986, p. 66).
The expected number of iterations to get one acceptance in the Bignami and De Matteis algorithm is 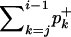 (Bignami and De Matteis 1971). Using Equation 4 and the expressions for the coefficients of the mixture, we find in our setting that the expected number of iterations to get one acceptance is proportional to 1/qi,j(ui+1). This means that for some values of n, i, and t, the algorithmic cost of the rejection algorithm will be prohibitive. This happens when qi,j(ui+1) is very small, that is, when it is extremely unlikely under the coalescent model that i lineages will be reduced to j lineages during ui+1 units of time. Using simulations, we found that for values of the intercoalescence time <0.05, the rejection method is not tractable because of its prohibitive algorithmic cost (results not shown). Therefore, when ui+1 < 0.05, our simulation method instead relies on asymptotic results obtained by Griffiths (1984, Theorem 6) for the distribution of the number of lineages under the coalescent.
(Bignami and De Matteis 1971). Using Equation 4 and the expressions for the coefficients of the mixture, we find in our setting that the expected number of iterations to get one acceptance is proportional to 1/qi,j(ui+1). This means that for some values of n, i, and t, the algorithmic cost of the rejection algorithm will be prohibitive. This happens when qi,j(ui+1) is very small, that is, when it is extremely unlikely under the coalescent model that i lineages will be reduced to j lineages during ui+1 units of time. Using simulations, we found that for values of the intercoalescence time <0.05, the rejection method is not tractable because of its prohibitive algorithmic cost (results not shown). Therefore, when ui+1 < 0.05, our simulation method instead relies on asymptotic results obtained by Griffiths (1984, Theorem 6) for the distribution of the number of lineages under the coalescent.
Griffiths showed that the number of coalescences that occur during x units of time when x is small can be approximated by a Poisson distribution with parameter λix, where i is the number of initial lineages
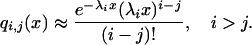 |
(5) |
Using the Markov property and the definition of conditional probabilities, we have for i > j
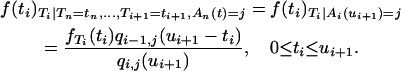 |
Thus, approximating qi−1,j(ui+1) by a Poisson distribution (Equation 5) provides an approximate p.d.f. for the conditional intercoalescence time Ti,
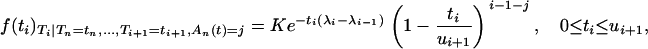 |
(6) |
where K is the normalizing constant. Simulating variates according to this p.d.f. is performed using a simple rejection algorithm, and the details of the simulation procedure are given in appendix b. The approximation of Equation 4 by Equation 6 is excellent for t < 0.05. For instance, Figure 2 displays the exact p.d.f. and the approximate p.d.f. for the conditional intercoalescence time when t = 0.01. The approximation appears to be quite accurate.
Figure 2.—

For different values of i and j, the exact (Equation 4) and approximate p.d.f. (Equation 6) of the intercoalescence time Ti given that An(t) = j and 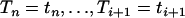 . The dashed lines correspond to the approximate p.d.f. and the points correspond to the exact p.d.f. The time elapsed between
. The dashed lines correspond to the approximate p.d.f. and the points correspond to the exact p.d.f. The time elapsed between 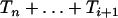 and t is fixed at ui+1 = 0.01.
and t is fixed at ui+1 = 0.01.
We can now write an algorithm for generating coalescent trees given that there are j lineages at time t. Two example trees simulated by the following algorithm are displayed in Figure 3.
Figure 3.—

Two coalescent trees with n = 10 individuals conditional on having, at time t = 2, (a) j = 2 lineages or (b) j = 8 lineages. Both trees were simulated using Algorithm 2.
Algorithm 2—algorithm to generate a coalescent tree given that there are j lineages at time t:
Simulate the topology of a standard coalescent tree and set u = t.
For i = n to 2 do
If i > j
If (u > 0.05) use Equation 4 and Algorithm 1 to simulate Ti conditional on
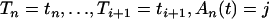 .
.If (u ≤ 0.05) use Equation 6 and the rejection method given in appendix b to simulate Ti conditional on
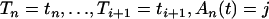 .
.Set u = u − Ti.
If i = j, simulate Ti = u + Exp(λi).
If i < j, simulate Ti = Exp(λi).
So far, we have assumed that the population size is constant so that the intercoalescence times are exponentially distributed. The framework above can easily be extended using the method of Griffiths and Tavaré (1994b) (see also Tavaré 2004, pp. 23–29) that describes the coalescent process when the population size evolves deterministically. We denote by N(r) the population size r generations before the present. Time is measured in units of 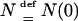 generations. The relative size function sN(x) is defined by
generations. The relative size function sN(x) is defined by
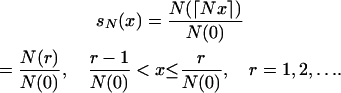 |
We suppose that the limit of sN(x) as N goes to infinity exists and is denoted by s(x). We also denote by {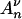 (x)}x≥0 the process that counts at time x the number of ancestors of a sample with initial size n, when the population size is not constant. The result obtained by Griffiths and Tavaré (1994b) (see also Tavaré 2004, pp. 27–28) is that the process
(x)}x≥0 the process that counts at time x the number of ancestors of a sample with initial size n, when the population size is not constant. The result obtained by Griffiths and Tavaré (1994b) (see also Tavaré 2004, pp. 27–28) is that the process 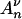 (·) may be constructed using the equality
(·) may be constructed using the equality
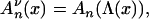 |
(7) |
where An(·) is the ancestral process when the population size is constant and
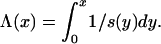 |
When population size varies, it follows from Equation 7 that generating coalescent trees given that there are j lineages at time t can be performed using Algorithm 2. More precisely, a coalescent tree given that there are j lineages at time Λ(t) can be generated using Algorithm 2. The number of lineages in the simulated coalescent jumps at times Tn, 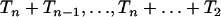 . Thus, the Griffiths and Tavaré equality ensures that
. Thus, the Griffiths and Tavaré equality ensures that 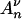 (·) jumps at times Λ−1(Tn), Λ−1(Tn + Tn−1), … ,
(·) jumps at times Λ−1(Tn), Λ−1(Tn + Tn−1), … , 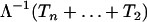 . If we denote by
. If we denote by 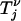 the time during which
the time during which 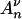 (·) has j ancestors, we have
(·) has j ancestors, we have
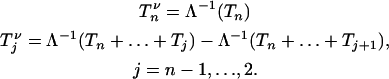 |
SIMULATIONS
We performed computer simulations to study the statistical properties of the maximum-likelihood estimator of the number of ancestral lineages. The coalescent trees were simulated using Algorithm 2, assuming a population of constant size, and mutations were propagated along the trees assuming an infinitely many sites model. The rate at which mutation occurs, θ = Nμ (μ is the mutation rate per generation and N is the effective population size, that is, the number of female individuals when considering mtDNA) was set to 1, 5, or 10 to mimic the scaled mutation rate in the mtDNA control region in humans [note that our definition of θ is half of the value used in the usual definition (Tavaré 2004)]. The number of simulated trees that were used for the estimation of the likelihood at each value of the parameter j was fixed at 1000 and the location  with the highest likelihood was found by simply choosing the value of j that maximized the estimated likelihood. All estimates were obtained using the site frequency and folded site frequency spectra. The number of sequences was set to 50 except where otherwise specified. The different binning schemes that were used are shown in Table 1.
with the highest likelihood was found by simply choosing the value of j that maximized the estimated likelihood. All estimates were obtained using the site frequency and folded site frequency spectra. The number of sequences was set to 50 except where otherwise specified. The different binning schemes that were used are shown in Table 1.
TABLE 1.
The binning schemes used when the simulated site frequency spectrum and the simulated folded site frequency spectrum were analyzed
| No. of sequences (n) | Binning scheme | |||||
|---|---|---|---|---|---|---|
| n = 50 | Sizes of mutations | 1 | 2–5 | 6–10 | 11–25 | 26–49 |
| Types of mutations | 1 | 2–5 | 6–10 | 11–17 | 18–25 | |
| n = 100 | Sizes of mutations | 1 | 2–5 | 6–25 | 26–50 | 51–99 |
| Types of mutations | 1 | 2–5 | 6–14 | 15–24 | 25–50 | |
To compute confidence intervals, we used a parametric bootstrap percentile method (see, e.g., Carpenter and Bithell 2000). Parametric bootstrapping proceeds by approximating the null distribution of an estimator by the distribution of the estimator applied to samples simulated under the null hypothesis. The simulations are performed by assuming that the true value of the parameter is equal to the estimated value. The number of bootstrap replicates is denoted by B. In our setting, the parametric bootstrap consists of simulating genetic data on simulated coalescent trees given that the number of lineages at time t is equal to the maximum-likelihood estimate 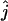 . For each bootstrap replicate, a maximum-likelihood estimate (
. For each bootstrap replicate, a maximum-likelihood estimate (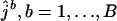 ) of the number of ancestral lineages is found. The lower and upper endpoints of the 95% confidence interval are estimated simply by the 2.5 and 97.5% quantiles of the set
) of the number of ancestral lineages is found. The lower and upper endpoints of the 95% confidence interval are estimated simply by the 2.5 and 97.5% quantiles of the set 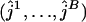 . In our analysis, the number of bootstrap replicates is set at B = 1000.
. In our analysis, the number of bootstrap replicates is set at B = 1000.
To provide an example of our estimation procedure, we computed the log-likelihood of the parameter j using a simulated data set. The simulated data set contained n = 100 sequences, and the number of ancestors j one coalescent time unit before the present was set to 25. The mutation parameter θ was fixed at 5. On a 1.6 GHz Centrino Duo processor, evaluating the likelihood for all values of the parameter j took ∼81 sec. The profile of the log-likelihood is displayed in Figure 4. When using either the site frequency spectrum or the folded site frequency spectrum, the maximum-likelihood estimate was 26, and the 95% confidence interval ranged from 19 to 35. When using only the number of segregating sites as the summary statistic, we found that the maximum-likelihood estimate was 33 and the confidence interval was much wider, ranging from 18 to 49.
Figure 4.—

The profile of the log-likelihood of the number of ancestors estimated from a simulated data set summarized in each of three ways. The number of sequences was set at n = 100 and the number of ancestors 1 coalescent time unit before the present was set at 25. The mutation rate was fixed at θ = 5. The log-likelihood functions have been shifted so that their maximum values are 0.
To investigate the statistical properties of the maximum-likelihood estimator 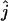 , we estimated its bias
, we estimated its bias 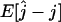 and its root mean square error (RMSE)
and its root mean square error (RMSE) 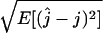 . The bias and the RMSE were both estimated using Monte Carlo approximation with 1000 simulated genetic data sets. For both the site frequency spectrum and the folded site frequency spectrum, the bias of the estimator is displayed in Figure 5 for different values of θ and t, as is the difference between the biases of the estimator using the site frequency and folded site frequency spectra. Provided that the value of the mutation parameter is large enough (θ ≥ 5) and the time at which the number of lineages has been fixed is also large enough (t ≥ 1), the estimator based on the site frequency spectrum is almost unbiased. In other words, the estimator is unbiased if enough mutations have occurred since the time at which we want to estimate the number of lineages. When using the folded site frequency spectrum, the bias is slightly larger, but it remains small (<0.5). When the true number of ancestral lineages is sufficiently large, the estimator is always almost unbiased. However, for t = 0.5 and j = 5 or 10, the bias of the estimator is substantially larger when using the folded site frequency spectrum (see Figure 5). Also, the estimator overestimates the number of ancestral lineages when the true value is 1. This result is expected because the estimator necessarily finds a value ≥1.
. The bias and the RMSE were both estimated using Monte Carlo approximation with 1000 simulated genetic data sets. For both the site frequency spectrum and the folded site frequency spectrum, the bias of the estimator is displayed in Figure 5 for different values of θ and t, as is the difference between the biases of the estimator using the site frequency and folded site frequency spectra. Provided that the value of the mutation parameter is large enough (θ ≥ 5) and the time at which the number of lineages has been fixed is also large enough (t ≥ 1), the estimator based on the site frequency spectrum is almost unbiased. In other words, the estimator is unbiased if enough mutations have occurred since the time at which we want to estimate the number of lineages. When using the folded site frequency spectrum, the bias is slightly larger, but it remains small (<0.5). When the true number of ancestral lineages is sufficiently large, the estimator is always almost unbiased. However, for t = 0.5 and j = 5 or 10, the bias of the estimator is substantially larger when using the folded site frequency spectrum (see Figure 5). Also, the estimator overestimates the number of ancestral lineages when the true value is 1. This result is expected because the estimator necessarily finds a value ≥1.
Figure 5.—

The bias of the estimator, 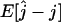 . At each of several values for the number of ancestors at time t, the bias was estimated using 1000 simulated genetic data sets with a sample size of n = 50. (A) Bias for the estimator based on the site frequency spectrum. (B) Bias for the estimator based on the folded site frequency spectrum. (C) Bias in B minus bias in A.
. At each of several values for the number of ancestors at time t, the bias was estimated using 1000 simulated genetic data sets with a sample size of n = 50. (A) Bias for the estimator based on the site frequency spectrum. (B) Bias for the estimator based on the folded site frequency spectrum. (C) Bias in B minus bias in A.
Similarly to the bias, the RMSE of the estimator is large when t is small (t = 0.5) and θ is small (θ = 1) (see Figure 6). When the number of ancestral lineages j = 1, the RMSE is rather small. As j increases, the RMSE increases, reaching a plateau around j = 10 and decreasing slightly for j > 25. To give a sense of the quality of our estimator, we assume that the maximum-likelihood estimator is approximately unbiased and Gaussian. This means that we assume the estimate ranges with a probability of 95% from 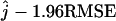 to
to 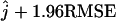 . When j = 25, for example, the worst situation corresponds to t = 0.5 and θ = 1. Using the site frequency spectrum, we find that the estimate ranges between 13 and 36. When the mutation parameter increases 10-fold, the interval where the estimator is likely to be found is reduced, ranging from 19 to 31. The most favorable case for estimating j = 25 corresponds to t = 2 and θ = 10. In that scenario, the probability is 95% that the estimate ranges between 22 and 28. The difference of RMSEs obtained when using the site frequency spectrum and the folded site frequency spectrum is small except when t = 0.5 and j = 5 or 10, where the difference is >1.
. When j = 25, for example, the worst situation corresponds to t = 0.5 and θ = 1. Using the site frequency spectrum, we find that the estimate ranges between 13 and 36. When the mutation parameter increases 10-fold, the interval where the estimator is likely to be found is reduced, ranging from 19 to 31. The most favorable case for estimating j = 25 corresponds to t = 2 and θ = 10. In that scenario, the probability is 95% that the estimate ranges between 22 and 28. The difference of RMSEs obtained when using the site frequency spectrum and the folded site frequency spectrum is small except when t = 0.5 and j = 5 or 10, where the difference is >1.
Figure 6.—

The root mean square error (RMSE) of the estimator, 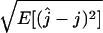 . At each of several values for the number of ancestors at time t, the RMSE was estimated using 1000 simulated genetic data sets with a sample size of n = 50. (A) RMSE for the estimator based on the site frequency spectrum. (B) RMSE for the estimator based on the folded site frequency spectrum. (C) RMSE in B minus RMSE in A.
. At each of several values for the number of ancestors at time t, the RMSE was estimated using 1000 simulated genetic data sets with a sample size of n = 50. (A) RMSE for the estimator based on the site frequency spectrum. (B) RMSE for the estimator based on the folded site frequency spectrum. (C) RMSE in B minus RMSE in A.
We investigated the gain obtained by using the site frequency spectrum or the folded site frequency spectrum rather than the number of segregating sites. The relative difference of RMSEs—the difference between the RMSE using the number of segregating sites and the RMSE using one of the spectra, divided by the RMSE using the number of segregating sites—is displayed in Figure 7. This difference is always positive, as the site frequency spectrum contains more information than the number of segregating sites. The relative decrease of the RMSE when using one of the spectra ranges between 0 and 60%. When the number of ancestral lineages is large (40) there is a clear increase of the relative difference of RMSE except when t = 0.5. When the number of ancestors ranges from 5 to 25, the gain in accuracy of the estimator is consistently larger when using the site frequency spectrum than when using the folded site frequency spectrum, as is expected from Figure 6. For these values of the number of ancestral lineages, the relative decrease of RMSE is small when using the folded site frequency spectrum, ranging from 0 to 25%. In contrast, the relative decrease of RMSE when using the site frequency spectrum ranges from 10 to 50%. When the number of ancestral lineages is 1, there is no major decrease of RMSE, except mainly when θ = 10.
Figure 7.—

The relative difference between the RMSE of the estimator computed from the number of segregating sites and the RMSE of the estimator computed from the (A) site frequency spectrum or the (B) folded site frequency spectrum [the relative difference between two variables A and B is defined by (A − B)/A]. At each of several values for the number of ancestors at time t, the RMSEs were estimated using 1000 simulated genetic data sets with a sample size of n = 50.
Last, we investigated if the maximum-likelihood estimator is robust to violations of the infinitely many sites assumption. Using the software Seq-Gen (Rambaut and Grassly 1997), we simulated 422 bp according to the Hasegawa–Kishino–Yano (HKY85) substitution model (Hasegawa et al. 1985). We assumed equal base frequencies and a transition–transversion ratio of 4. The sites containing three or four distinct nucleotides were removed so that the frequency spectrum could be computed. We considered the folded site frequency spectrum as the summary statistic. The scaled mutation rate was set at θ = 1 and θ = 3.58. A value of θ at 3.58 corresponds to a mutation rate of 2.5 × 10−6/site/generation, which is consistent with estimates of the mutation rate for the control region of mtDNA (Tamura and Nei 1993; Jazin et al. 1998) and an effective population size of 3400 female individuals (Nordborg 1998). As can be seen from Figure 8, the estimate is biased downward when the number of ancestors at time t is large. The bias also increases with the time t at which the number of ancestral lineages has been fixed. Essentially, the bias increases as the number of sites at which multiple mutations occur increases. Despite this slight bias for large values of the number of ancestors, the RMSE of the estimator remains moderate.
Figure 8.—

The (A) bias and the (B) RMSE of the estimator when the genetic data are simulated according to a finite-sites model. At each of several values for the number of ancestors at time t, the RMSEs were estimated using 1000 simulated genetic data sets with a sample size of n = 50.
APPLICATION TO HUMAN mtDNA DIVERSITY
We estimated the number of ancestral lineages of mtDNA in modern humans, both 30,000 years ago and 100,000 years ago. These estimates are of particular interest when investigating the level of admixture between Neanderthals and modern humans using mtDNA data. Indeed, Nordborg (1998) showed that the finding of a Neanderthal mtDNA fragment with a large number of sequence differences from 986 sequences of modern humans (Krings et al. 1997) was not sufficient proof for the absence of admixture. Nordborg introduced a simple model of admixture under which Neanderthals formed an isolated population until the time of admixture tm, when a fraction of the Neanderthal population merged with modern humans to form a single panmictic population. From now on, we refer to modern humans who lived at the time of the admixture as “early modern humans.” We denote by c the proportion of the early modern human population consisting of Neanderthals, at the time tm of admixture (and just after the admixture occurred). A model with no admixture corresponds to c = 0 and a model where all present-day humans descend from Neanderthals corresponds to c = 1. Assuming that the sampled Neanderthal comes from the Neanderthal population at a time that is more ancient than the time when Neanderthals mixed with modern humans, the probability that the Neanderthal sequence differs strikingly from the sequences of modern humans is equal to the probability that none of the ancestors at the time of the admixture came from the Neanderthal fraction. The probability that none of the early modern humans are Neanderthal descendants is simply (1 − c)j, where j is the number of ancestral sequences of modern humans (Nordborg 1998). Because all the ancestors are not descendants of Neanderthals, there is no more extreme scenario for rejecting admixture. Thus, the probability that none of the ancestors at the time of the admixture came from the Neanderthal fraction can be viewed as a P-value for a null model in which admixture occurred with parameter c. The P-value is estimated simply by 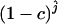 , where
, where 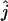 is the maximum-likelihood estimate of the number of lineages at the time of the potential admixture.
is the maximum-likelihood estimate of the number of lineages at the time of the potential admixture.
For the sake of comparison with Nordborg's results, we chose 986 worldwide mtDNA HV1 sequences (422 bp long) contained in the database MOUSE (Burckhardt 2002). We removed 102 sites that were missing in more than one-fourth of the individuals. Because we assumed the infinitely many sites model, we removed 30 sites that contained 3 or 4 distinct nucleotides. These excess mutations can be explained by an actual violation of the infinitely many sites model or by the occurrence of laboratory artifacts (Bandelt et al. 2002). The remaining sequences were 290 bp long and still contained some missing data. At each nucleotide position, missing nucleotides were artificially replaced by nucleotides simulated according to the nucleotide frequencies at that position. Thus, in the end, we analyzed 986 sequences each containing 290 nucleotides. Two values of the mutation rate were used in the analysis, a value of 5 × 10−5/site/generation as estimated from human pedigree studies (Parsons et al. 1997) and a value of 2.5 × 10−6/site/generation as estimated from the divergence time between humans and chimpanzees (Tamura and Nei 1993; Jazin et al. 1998). The generation time of the human female population was set at 20 years/generation.
To apply the maximum-likelihood method, the sizes or the types of the mutations must be computed. As mitochondrial sequences from chimps are available, it is in principle possible to determine the state of the ancestral sequence. However, because the control region of the mtDNA sequence evolves relatively fast, the chimp sequence is likely to differ from the chimp–human ancestral sequence. As a result, we used the folded site frequency spectrum, because its use does not require knowledge of the ancestral sequence. To check that our results were not too dependent on the binning scheme, we considered two different ways of binning the folded site frequency spectrum of the human mtDNA data (see Table 2).
TABLE 2.
Two different ways of binning the folded site frequency spectrum of the 986 human mtDNA sequences
| Binning scheme 1 | ||||||||||
| Types of mutations | 1 | 2–5 | 6–10 | 11–50 | 51–100 | 101–250 | 251–493 | |||
| No. of sites | 36 | 45 | 20 | 30 | 8 | 5 | 1 | |||
| Binning scheme 2 | ||||||||||
| Types of mutations | 1 | 2 | 3–5 | 6–10 | 11–25 | 26–50 | 51–75 | 76–100 | 101–201 | 201–493 |
| No. of sites | 36 | 20 | 25 | 20 | 21 | 9 | 4 | 4 | 4 | 2 |
For the demographic model of the human population, we used a model where the effective population size is constant and equals 3400 (Nordborg 1998) and two models of population expansion. The first of these models was considered by Nordborg and assumes that the population was constant before the date of the expansion, 50,000 years ago, and then grew exponentially to 5 × 108 individuals. The second model of expansion is better supported by demographic estimates for the human population (Biraben 1979; Cohen 1995). It assumes that the population was constant before the date of the first expansion, 50,000 years ago, grew exponentially to attain 2.5 × 106 female individuals 10,000 years ago, and then grew at a faster rate to reach 3 × 109 female individuals today. A mathematical description of the population growth models can be found in appendix c.
Figure 9 displays the profile of the log-likelihood function when the larger mutation rate is assumed. For all models of population demography, the maximum-likelihood estimate of the number of ancestors at tm = 30,000 years and tm = 100,000 years is always 1. Moreover, the likelihood function decreases very fast: the confidence interval of the number of ancestors is restricted to 1 when a model of constant population size is assumed and ranges from 1 to 2 or 3 when population growth models are assumed. In contrast, when the smaller mutation rate is assumed, the maximum-likelihood estimate of the number of ancestral lineages is sensitive to the model of human demography and to the time tm at which the number of lineages is estimated (Figure 10). The estimated number of ancestral lineages is always larger than the expectation of the number of lineages that was computed by Nordborg (1998), using a coalescent model without any data (Table 3). The number of ancestral lineages of the 986 sequences of modern humans 30,000 years ago was estimated at 111 for the model with constant population size and at 50 and 52 for the two models of expansion. One hundred thousand years ago, the number of ancestral lineages was estimated at 41 for the model of constant population size, at 20 for the model with a single stage of expansion, and at 1 for the model with two stages of expansion. The confidence intervals of the number of ancestral lineages are much wider when the smaller mutation rate is assumed. These results were almost unaffected by the choice of binning scheme (see Figures 9 and 10).
Figure 9.—



The log-likelihood of the number of ancestral lineages of 986 human HV1 sequences, 30,000 years ago and 100,000 years ago. The likelihood was computed using a mutation rate of 5 × 10−5/site/generation. Scheme 1 and scheme 2 correspond to the two binning schemes (see Table 2). The log-likelihood functions have been shifted so that their maximum values are 0. (A) Constant population size, (B) one stage of population expansion, (C) two stages of population expansion.
Figure 10.—



The log-likelihood of the number of ancestral lineages of 986 human HV1 sequences 30,000 years and 100,000 years ago. The likelihood was computed using a mutation rate of 2.5 × 10−6/site/generation. Scheme 1 and scheme 2 correspond to the two binning schemes (see Table 2). The log-likelihood functions have been shifted so that their maximum values are 0. (A) Constant population size, (B) one stage of population expansion, (C) two stages of population expansion.
TABLE 3.
Number of ancestors contemporary to the Neanderthal sequence and the minimum value cmin of the admixture coefficient c such that the admixture model can be rejected with a type I error rate of 0.05
| Constant population size: tm (yr)
|
One stage of population growth: tm (yr)
|
Two stages of population growth: tm (yr)
|
|||||
|---|---|---|---|---|---|---|---|
| Mutation rate | Quantity | 30,000 | 100,000 | 30,000 | 100,000 | 30,000 | 100,000 |
| 2.5 × 10−6/site/generation | 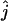 |
111 (4.86) | 41 (1.75) | 52 (782) | 20 (2.86) | 50 | 1 |
| 95% confidence interval | 94–144 | 35–50 | 32–77 | 1–40 | 32–74 | 1–5 | |
| cmin | 0.03 | 0.07 | 0.06 | 0.14 | 0.06 | 0.95 | |
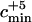 |
0.03 | 0.06 | 0.05 | 0.11 | 0.05 | 0.39 | |
| 5 × 10−5/site/generation | 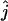 |
1 (4.86) | 1 (1.75) | 1 (782) | 1 (2.86) | 1 | 1 |
| 95% confidence interval | 1–1 | 1–1 | 1–2 | 1–3 | 1–2 | 1–2 | |
| cmin | 0.95 | 0.95 | 0.95 | 0.95 | 0.95 | 0.95 | |
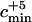 |
0.39 | 0.39 | 0.39 | 0.39 | 0.39 | 0.39 | |
The parameter 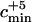 corresponds to the minimum value of the admixture coefficient c such that the admixture model can be rejected with a type I error rate of 0.05 when five sequenced early modern humans (Serre et al. 2004) were added to the estimated number of ancestral lineages. Numbers in parentheses represent values from the study of Nordborg (1998).
corresponds to the minimum value of the admixture coefficient c such that the admixture model can be rejected with a type I error rate of 0.05 when five sequenced early modern humans (Serre et al. 2004) were added to the estimated number of ancestral lineages. Numbers in parentheses represent values from the study of Nordborg (1998).
In Table 3, we display the minimum value of c, cmin, such that the admixture model can be rejected with a type I error rate of 0.05. The value cmin was computed by finding the value of c such that the P-value 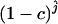 = 0.05. By definition of cmin, all the models of admixture with a value of c > cmin can be rejected with a 0.05 type I error rate. When the larger mutation rate is assumed, cmin is always 0.95. Therefore, when assuming the larger mutation rate, the finding of a Neanderthal sequence that coalesces deeper than the MRCA of the modern human sequences is not sufficient evidence for the rejection of admixture, except if the admixture hypothesis is that the Neanderthal population constituted almost the entire population of early modern humans (c > 0.95). When the smaller mutation rate is assumed, the conclusions depend on the demographic model considered. In the following, the results using the smaller mutation rate are described. If the admixture happened 30,000 years ago, the admixture model can be rejected unless the Neanderthal proportion of the admixed population was quite small (c < 0.06). If the admixture happened 100,000 years ago, the choice of demographic model matters. When the constant population size model or the expansion model with one stage is assumed, the admixture model can be rejected unless the Neanderthal proportion of the admixed population was small (c < 0.07 and c < 0.14). However, when assuming the expansion model with two stages, we cannot reject the admixture model except if the admixture was almost complete (c > 0.95).
= 0.05. By definition of cmin, all the models of admixture with a value of c > cmin can be rejected with a 0.05 type I error rate. When the larger mutation rate is assumed, cmin is always 0.95. Therefore, when assuming the larger mutation rate, the finding of a Neanderthal sequence that coalesces deeper than the MRCA of the modern human sequences is not sufficient evidence for the rejection of admixture, except if the admixture hypothesis is that the Neanderthal population constituted almost the entire population of early modern humans (c > 0.95). When the smaller mutation rate is assumed, the conclusions depend on the demographic model considered. In the following, the results using the smaller mutation rate are described. If the admixture happened 30,000 years ago, the admixture model can be rejected unless the Neanderthal proportion of the admixed population was quite small (c < 0.06). If the admixture happened 100,000 years ago, the choice of demographic model matters. When the constant population size model or the expansion model with one stage is assumed, the admixture model can be rejected unless the Neanderthal proportion of the admixed population was small (c < 0.07 and c < 0.14). However, when assuming the expansion model with two stages, we cannot reject the admixture model except if the admixture was almost complete (c > 0.95).
Last, we took into account the fact that Serre et al. (2004) sequenced mtDNA from five early humans from the Upper Pleistocene. Because their mtDNA sequences indicated that these individuals are not likely to be direct ancestors to modern human mtDNA sequences, we add five ancestors to the estimated number of early modern human ancestors. When we take into account the five extra ancestors, the minimum value of the admixture parameter such that the admixture model can be rejected is denoted 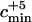 . When the number of ancestors estimated before incorporating these five lineages was larger than one, the results were not qualitatively modified by the inclusion of these lineages. However, when the estimated number of ancestors was equal to one prior to the incorporation of the five extra early modern human lineages, the minimum value of the admixture parameter that enabled rejection of the admixture model changed from cmin = 0.95 to
. When the number of ancestors estimated before incorporating these five lineages was larger than one, the results were not qualitatively modified by the inclusion of these lineages. However, when the estimated number of ancestors was equal to one prior to the incorporation of the five extra early modern human lineages, the minimum value of the admixture parameter that enabled rejection of the admixture model changed from cmin = 0.95 to 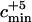 = 0.39.
= 0.39.
DISCUSSION
On the basis of a partition of polymorphic sites in a sample of DNA sequences—the site frequency spectrum or the folded site frequency spectrum—we constructed a maximum-likelihood framework for estimating the number of ancestral lineages j at a given point of time t in the past. The computation of the likelihood of the parameter j relies on an algorithm that we have devised for simulating coalescent trees conditional on having j ancestral lineages at time t. When analyzing mtDNA data, we estimated the number of ancestral lineages assuming the mutation rate θ was known. Nevertheless, our framework can be extended to jointly estimate the mutation rate and the number of ancestral lineages by searching for maximum-likelihood estimates over a grid of values for θ and j.
The accuracy of the maximum-likelihood estimate increases with the mutation rate θ and the time t at which the number of ancestral lineages has to be estimated. However, by analyzing simulated replicates with 50 sequences, we observed that the RMSE of the estimator is always >1 when the number of ancestral lineages is at least five. This lack of precision may be due partly to the randomness of the mutation process and partly to the randomness of the genealogical process (see Joyce 1999 for a mathematical description of the effects of these two sources of randomness in another context). Because the parameter j that has to be estimated is by definition a one-locus parameter—the numbers of ancestral lineages at time t of two different loci might be different—it is not possible to reduce the variance of the estimator by averaging the estimates across loci. The TMRCA, for example, is also a one-locus parameter and the TMRCAs for autosomal and uniparental markers may differ by a factor of 10 or more (Takahata et al. 2001; Tishkoff and Verrelli 2003). Note, however, that multilocus data could potentially be used to infer the genomewide distribution of the number of ancestors at time t.
Using our maximum-likelihood framework, we estimated the number of ancestral lineages of a worldwide sample containing 986 human sequences of the mitochondrial gene HV1. The number of ancestral lineages of modern humans that are contemporary to the sequenced Neanderthal individuals is of particular importance when testing for admixture between Neanderthals and modern humans (Nordborg 1998). When we utilized a rather high mtDNA mutation rate that had been inferred using pedigree analysis (Parsons et al. 1997), the number of ancestors 30,000 years ago was estimated at 1. Because it is very unlikely that the TMRCA of human mtDNA lineages is younger than 30,000 years, it is unlikely that the number j of ancestral lineages 30,000 years ago was 1. This suggests that the mutation rate that always produces j = 1 may be too high to be accurate. When using a smaller mtDNA mutation rate based on the date of divergence between humans and chimps (Tamura and Nei 1993), we obtained that the estimate of the number of ancestral lineages 30,000 years ago is >50. This has the consequence that scenarios of recent admixture between modern humans and Neanderthals (30,000 years ago) can be rejected, except if the proportion of the modern human population that consisted of Neanderthals at the time of the admixture was small (c < 0.06). Note that because we assumed the infinitely many sites model, we might have underestimated the number of ancestral lineages (see simulations). By relaxing the infinitely many sites assumption to allow recurrent mutations, the maximum possible level of admixture would be consequently reduced. A maximum admixture of ∼5% is five times smaller than the previous estimate of Serre et al. (2004), which was also based on the current absence of Neanderthal lineages in the modern human genealogy, but which did not make use of the data to estimate the number of ancestral lineages of modern humans. However, using a spatial range expansion for modeling the process by which Neanderthals were replaced by humans and not using the model of instantaneous admixture that we considered, Currat and Excoffier (2004) found that the absence of mtDNA lineages in the modern human sample was compatible only with much smaller admixture rates of the order of 0.1%.
When aiming to detect ancient admixture, considerable power can be gained by using multilocus data rather than single-locus data (Nordborg 2001). Recent technological advances have made possible the sequencing of multilocus data from Neanderthal fossils (Green et al. 2006; Noonan et al. 2006). Thus, the number of ancestors of modern humans at the time of the potential admixture with Neanderthals could potentially be estimated across the genome to identify the loci that may be the most informative about the issue of ancient admixture. Multilocus data from contemporary human DNA sequences have already been analyzed, with a different approach, for testing ancient admixture in humans (Plagnol and Wall 2006). Plagnol and Wall suggested that ancient admixture may explain an elevated level of linkage disequilibrium observed in the genomes of modern humans.
It is initially surprising that the estimate of the number of ancestral lineages was larger in the constant population size model than in the models of population expansion. This trend is the opposite of what Nordborg (1998) found using a coalescent model for computing the number of ancestral lineages. The result of Nordborg is expected when the onset of the expansion is more ancient than time t, so that most coalescence events occur more anciently than time t. To explain why we observed an opposite trend, consider one conditional coalescent tree with j lineages at time t in a constant-sized population and one conditional coalescent tree with j lineages at time t in an expanding population. If the time since the beginning of the expansion is greater than t, the coalescent tree is likely to have a larger total length in the expanding population than in the stationary population, because in the expanding population, lineages do not have a high rate of coalescence between time t and the beginning of the expansion (see Figure 11). Thus, to explain the same level of genetic variation as in a constant-sized population, the estimate of the number of lineages at time t needs to be smaller in the expanding population.
Figure 11.—

A genealogical tree of n = 5 individuals conditioned on having three lineages at time t, in a population of constant size and in an expanding population for which the beginning of the expansion is more ancient than t. The initial population size in the expanding population is the same as the present-day population size in the constant-population-size model. Because the coalescent tree in the expanding population is likely to have a longer total length, the number of mutations that occur along the genealogy from the expanding population is likely to be larger. Thus, the same number of ancestral lineages at time t will produce a larger genetic diversity in the expanding population. When analyzing the same amount of genetic diversity, this explains why the maximum-likelihood estimates of the number of ancestral lineages are smaller when assuming an expanding population rather than a constant-size population.
The mutation model has an influence on the estimated number of ancestral lineages. Our method of estimation assumes that sequences evolve according to the infinitely many sites model and that each site evolves at the same rate. However, these assumptions may be violated for two principal reasons: first, the rate of mutation may vary across sites, as has been shown for human mtDNA (Meyer et al. 1999); second, recurrent mutations may occur, invalidating the infinitely many sites model. Overcoming the first issue is straightforward by assuming a gamma distribution for the mutation rate θi at site i,
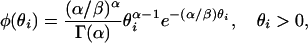 |
where α measures the level of rate heterogeneity and β denotes the mean rate of mutation per site (measured in units of events per N generations). Because a negative binomial distribution is generated from a Poisson distribution where the rate of the Poisson distribution is random and gamma distributed, the number of segregating sites has a negative binomial distribution when the mutation parameter across sites is gamma distributed:
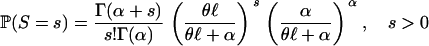 |
(8) |
(Tamura and Nei 1993). Equation 8 is a simple extension of the classical result that states that the number of mutations per site has a negative binomial distribution when the mutation parameter across sites is gamma distributed (Tamura and Nei 1993; Zhang and Gu 1998, Equation 8). Taking rate heterogeneity into account amounts to using a negative binomial distribution rather than a Poisson distribution for the number of segregating sites. As a result, the probability of the site frequency spectrum given a coalescent tree  is no longer given by Equation 1 but rather, by the following equation:
is no longer given by Equation 1 but rather, by the following equation:
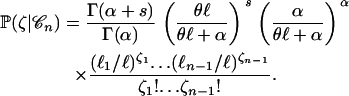 |
(9) |
In contrast, dealing with the second issue, namely the fact that recurrent mutation can happen, is more difficult. Our approach relies on the computation of the probability of the site frequency spectrum. In the infinitely many sites model, the site frequency spectrum is a convenient summary of the data. Indeed, the probability of the site frequency spectrum given a coalescent tree and the total number of mutations is given by the multinomial distribution. This property does not hold for more complex models of DNA substitution such as the Jukes–Cantor model (Jukes and Cantor 1969), and for such models we would need to resort to joint simulations of the mutation process and the coalescent process when computing the probability of the site frequency spectrum. However, the infinitely many sites assumption can still be used as an approximation, as we found through simulations that the maximum-likelihood estimator still behaves well even when the simulated data follow a finite-sites model.
We also note that Algorithm 2 for the simulation of coalescent trees given that the number of ancestors at time t is equal to j may be useful for purposes other than the estimation of the number of ancestral lineages. Using Algorithm 2, prior information about the number of ancestral lineages can be added when estimating demographic parameters from genetic data. Rejection methods simulate genetic data using coalescent replicates and accept the parameters that produce genetic data close to the observed genetic data. When taking prior information about the number of ancestral lineages into account, coalescent replicates could be generated according to Algorithm 2 instead of using a standard coalescent. This estimation procedure may be appropriate, for example, when analyzing mtDNA of Native Americans. Genetic studies have suggested that modern Native Americans are represented by five distinct haplogroups (e.g., Schurr 2004). If we assume that Native American lineages carrying distinct haplogroups coalesced longer ago than the time of the migration through the Bering strait—a view supported by the presence of these haplogroups outside of the Americas (e.g., Kolman et al. 1996)—estimation of demographic parameters such as the size of the founding group might be performed conditional on an assumption of five ancestral mtDNA lineages at the time of the migration.
Acknowledgments
We thank two anonymous reviewers for comments on the manuscript. The research of N.A.R. is supported by a Burroughs Wellcome Fund Career Award in the Biomedical Sciences and by an Alfred P. Sloan Research Fellowship.
APPENDIX A
We show in the following that the distribution of the intercoalescence time Ti given that An(t) = j and 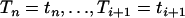 is a mixture of truncated exponential distributions
is a mixture of truncated exponential distributions
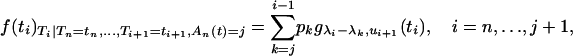 |
(A1) |
where gγ,τ denotes the p.d.f. of the exponential distribution of parameter γ truncated at time τ and the coefficients of mixture pk are given in the proof (Equation A3). When i = n, Equation A1 corresponds to the conditional distribution of Tn given that An(t) = j. Before proving (A1), we first recall some basic properties of truncated exponential distributions.
The truncated exponential p.d.f. gγ,τ is given by
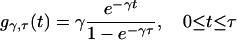 |
and is 0 everywhere else. Its cumulative probability distribution is given by
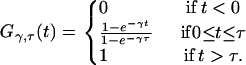 |
The simulation of truncated exponential random variables is easily performed by the inversion method. The inversion method consists of simulating a random variable with cumulative probability distribution G by simulating a uniform random variable U over the interval (0, 1) and returning G−1(U) or (1 − G)−1(U) (Devroye 1986, pp. 27–28). For truncated exponential random variables, it consists of computing −ln(U + (1 − U)e−γτ)/γ, where U is uniform over the interval (0, 1).
Proof of (A1). The derivation of (A1) relies on the distribution of An(t), which can be found in Tavaré (2004, p. 19). We have
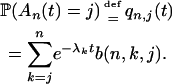 |
The values b(n, k, j) are defined as
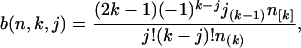 |
where
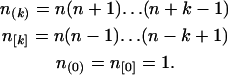 |
The cumulative probability distribution of Ti given An(t) = j and 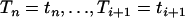 is
is
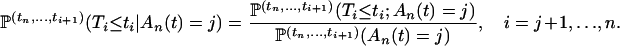 |
(A2) |
In Equation A2, the conditional probability 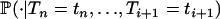 is denoted by
is denoted by 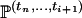 . When i > j, the fact that An(·) is a Markov process leads to
. When i > j, the fact that An(·) is a Markov process leads to
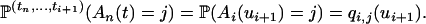 |
By conditioning on Ti, we compute the numerator of (A2):
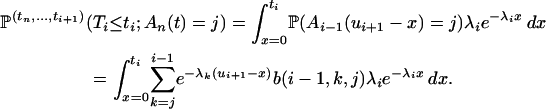 |
Some computations lead to
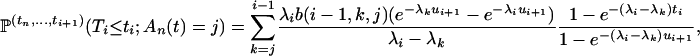 |
which completes the proof and gives the coefficients of the mixture
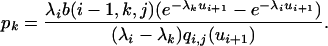 |
(A3) |
▪
APPENDIX B
This section is devoted to the construction of a rejection sampling algorithm for simulating random variates ranging from 0 to ui+1, with p.d.f. given by
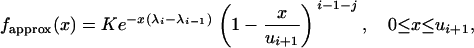 |
where K denotes the normalizing constant. We are interested in the p.d.f. fapprox because it is a good approximation, when t is small, of the conditional p.d.f. of the intercoalescence time Ti given that An(t) = j and 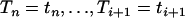 . Because λi − λi−1 > 0, we have
. Because λi − λi−1 > 0, we have
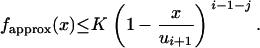 |
We denote by gapprox the following p.d.f.:
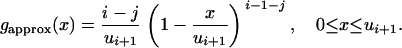 |
Then the standard rejection method (Devroye 1986, pp. 40–42) for simulating a random variable with p.d.f. given by fapprox can simply be written as follows:
Generate a random variate X with density gapprox.
Generate a uniform-[0, 1] random variate U.
If
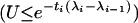 return X; otherwise go back to step 1.
return X; otherwise go back to step 1.
Generating a random variate X with density gapprox is performed using the inversion method (Devroye 1986, pp. 27–28). In that context, it consists of computing 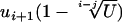 , where U is uniform over the interval (0, 1).
, where U is uniform over the interval (0, 1).
APPENDIX C
We used two models of population growth: a model with one stage of expansion and a model with two stages of expansion. Both models have geometric growth when time is discrete and exponential growth when time is continuous and scaled in units of 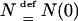 generations. The model with one stage of expansion assumes that the population has constant size prior to generation V and geometric growth from then to the present time. Thus, for some δ ∈ (0, 1), the population size r generations before the present is given by
generations. The model with one stage of expansion assumes that the population has constant size prior to generation V and geometric growth from then to the present time. Thus, for some δ ∈ (0, 1), the population size r generations before the present is given by
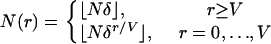 |
(Tavaré 2004, pp. 23–24). We suppose that V = ⌊Nν⌋ for some ν > 0, so that the expansion started ν time units ago. Then
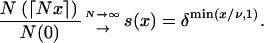 |
The model with two stages of expansion is a simple extension of the model with one stage of expansion. For some δ1 and δ2 ∈ (0, 1), we have
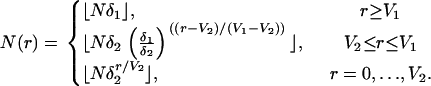 |
We suppose that V1 = ⌊Nν1⌋ and V2 = ⌊Nν2⌋ for some ν1, ν2 > 0. Then
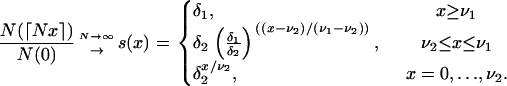 |
When analyzing the human mtDNA data, we set N = 5 × 108, δ = 6.8 × 10−6, and ν = 5 × 10−6 for the expansion model with one growth rate. When using the model of expansion with two growth rates, we set N = 3 × 109, δ1 = 1.13 × 10−6, δ2 = 8.33 × 10−4, ν1 = 8.33 × 10−7, and ν2 = 1.66 × 10−7. The first model of expansion assumes that the initial population containing 3400 individuals was constant before the date of the expansion 50,000 years ago and then grew exponentially to 5 × 108 individuals. The second model assumes that the initial population of 3400 individuals was constant before the date of the first expansion 50,000 years ago, grew exponentially to 2.5 × 106 individuals 10,000 years ago, and then grew at a faster rate to reach 3 × 109 female individuals today.
References
- Bandelt, H.-J., L. Quintana-Murci, A. Salas and V. Macaulay, 2002. The fingerprint of phantom mutations in mitochondrial DNA data. Am. J. Hum. Genet. 71: 1150–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont, M. A., W. Zhang and D. J. Balding, 2002. Approximate Bayesian computation in population genetics. Genetics 162: 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignami, A., and A. De Matteis, 1971. A note on sampling from combinations of distributions. IMA J. Appl. Math. 8: 80–81. [Google Scholar]
- Biraben, J.-N., 1979. Essai sur l'évolution du nombre des hommes. Population 1: 13–25. [Google Scholar]
- Blum, M. G. B., and O. François, 2005. On statistical tests of phylogenetic tree imbalance: the Sackin and other indices revisited. Math. Biosci. 195: 141–153. [DOI] [PubMed] [Google Scholar]
- Burckhardt, F., 2002. MOUSE (Mitochondrial and Other Useful SEquences) a compilation of population genetic markers. Bioinformatics 18: 890–891. [DOI] [PubMed] [Google Scholar]
- Cann, R. L., M. Stoneking and A. C. Wilson, 1987. Mitochondrial DNA and human evolution. Nature 325: 31–36. [DOI] [PubMed] [Google Scholar]
- Carpenter, J., and J. Bithell, 2000. Bootstrap confidence intervals: When, which, what? A practical guide for medical statisticians. Stat. Med. 19: 1141–1164. [DOI] [PubMed] [Google Scholar]
- Cohen, J. E., 1995. How Many People Can the Earth Support? W. W. Norton, New York.
- Currat, M., and L. Excoffier, 2004. Modern humans did not admix with Neanderthals during their range expansion into Europe. PLoS Biol. 2: 2264–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devroye, L., 1986. Non-Uniform Random Variate Generation. Springer-Verlag, New York.
- Fu, Y.-X., 1994. Estimating effective population size or mutation rate using the frequencies of mutations of various classes in a sample of DNA sequences. Genetics 138: 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., 1998. Probability of a segregating pattern in a sample of DNA sequences. Theor. Popul. Biol. 54: 1–10. [DOI] [PubMed] [Google Scholar]
- Fu, Y.-X., and W.-H. Li, 1997. Estimating the age of the common ancestor of a sample of DNA sequences. Mol. Biol. Evol. 14: 195–199. [DOI] [PubMed] [Google Scholar]
- Garrigan, D., and M. F. Hammer, 2006. Reconstructing human origins in the genomic era. Nat. Rev. Genet. 7: 669–680. [DOI] [PubMed] [Google Scholar]
- Green, R. E., J. Krause, S. E. Ptak, A. W. Briggs, M. T. Ronan et al., 2006. Analysis of one million base pairs of Neanderthal DNA. Nature 444: 330–336. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., 1984. Asymptotic line-of-descent distributions. J. Math. Biol. 21: 67–75. [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1994. a Sampling probability distributions in the coalescent. Theor. Popul. Biol. 46: 131–159. [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1994. b Sampling theory for neutral alleles in a varying environment. Philos. Trans. R. Soc. Lond. B 344: 403–410. [DOI] [PubMed] [Google Scholar]
- Hasegawa, M., H. Kishino and T. Yano, 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22: 160–174. [DOI] [PubMed] [Google Scholar]
- Jakobsson, M., J. Hagenblad, S. Tavaré, T. Säll, C. Halldén et al., 2006. A unique recent origin of the allotetraploid species Arabidopsis suecica: evidence from nuclear DNA markers. Mol. Biol. Evol. 23: 1217–1231. [DOI] [PubMed] [Google Scholar]
- Jazin, E., H. Soodyall, P. Jalonen, E. Lindholm, M. Stoneking et al., 1998. Mitochondrial mutation rate revisited: hot spots and polymorphism. Nat. Genet. 18: 109–110. [DOI] [PubMed] [Google Scholar]
- Joyce, P., 1999. No BLUE among phylogenetic estimators. J. Math. Biol. 39: 421–438. [DOI] [PubMed] [Google Scholar]
- Jukes, T. H., and C. R. Cantor, 1969. Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, edited by H. N. Munro. Academic Press, New York.
- Kolman, C. J., N. Sambuughin and E. Bermingham, 1996. Mitochondrial DNA analysis of Mongolian populations and implications for the origin of New World founders. Genetics 142: 1321–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krings, M., A. Stone, R. W. Schmitz, H. Krainitzki, M. Stoneking et al., 1997. Neandertal DNA sequences and the origin of modern humans. Cell 90: 19–30. [DOI] [PubMed] [Google Scholar]
- Kuhner, M. K., J. Yamato and J. Felsenstein, 1995. Estimating effective population size and mutation rate from sequence data using Metropolis-Hastings sampling. Genetics 140: 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, S., G. Weiss and A. von Haeseler, 1999. Pattern of nucleotide substitution and rate heterogeneity in the hypervariable regions I and II of human mtDNA. Genetics 152: 1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan, J. P., G. Coop, S. Kudaravalli, D. Smith, J. Krause et al., 2006. Sequencing and analysis of Neanderthal genomic DNA. Science 314: 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., 1998. On the probability of Neanderthal ancestry. Am. J. Hum. Genet. 63: 1237–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., 2001. On detecting ancient admixture, pp. 123–136 in Genes, Fossils, and Behaviour: An Integrated Approach to Human Evolution, Vol. 310, edited by P. Donnelly and R. Foley. IOS Press, Amsterdam.
- Parsons, T. J., D. S. Muniec, K. Sullivan, N. Woodyatt, R. Alliston-Greiner et al., 1997. A high observed substitution rate in the human mitochondrial DNA control region. Nat. Genet. 15: 363–368. [DOI] [PubMed] [Google Scholar]
- Plagnol, V., and J. D. Wall, 2006. Possible ancestral structure in human populations. PLoS Genet. 2: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. T. Seielstad, A. Pérez-Lezaun and M. W. Feldman, 1999. Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol. Biol. Evol. 16: 1791–1798. [DOI] [PubMed] [Google Scholar]
- Rambaut, A., and N. C. Grassly, 1997. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 13: 235–238. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., 2006. The mean and variance of the numbers of r-pronged nodes and r-caterpillars in Yule-generated genealogical trees. Ann. Comb. 10: 129–146. [Google Scholar]
- Schurr, T. G., 2004. The peopling of the new world: perspectives from molecular anthropology. Annu. Rev. Anthropol. 33: 551–583. [Google Scholar]
- Serre, D., A. Langaney, M. Chech, M. Teschler-Nicola, M. Paunovic et al., 2004. No evidence of Neanderthal mtDNA contribution to early modern humans. PLoS Biol. 2: 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., and M. Nei, 1985. Gene genealogy and variance of interpopulational nucleotide differences. Genetics 110: 325–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., S.-H. Lee and Y. Satta, 2001. Testing multiregionality of modern human origins. Mol. Biol. Evol. 18: 172–183. [DOI] [PubMed] [Google Scholar]
- Tamura, K., and M. Nei, 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10: 512–526. [DOI] [PubMed] [Google Scholar]
- Tang, H., D. O. Siegmund, P. Shen, P. J. Oefner and M. W. Feldman, 2002. Frequentist estimation of coalescence times from nucleotide sequence data using a tree-based partition. Genetics 161: 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S., 1984. Line-of-descent and genealogical processes, and their applications in population genetics models. Theor. Popul. Biol. 26: 119–164. [DOI] [PubMed] [Google Scholar]
- Tavaré, S., 2004. Ancestral inference in population genetics, pp. 1–188 in Lectures on Probability Theory and Statistics. Ecole d'Eté de Probabilités de Saint-Flour XXXI-2001, Vol. 1837, edited by J. Picard. Springer-Verlag, Berlin.
- Tavaré, S., D. J. Balding, R. C. Griffiths and P. Donnelly, 1997. Inferring coalescence times from DNA sequence data. Genetics 145: 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, R., J. K. Pritchard, P. Shen, P. J. Oefner and M. W. Feldman, 2000. Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc. Natl. Acad. Sci. USA 97: 7360–7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff, S. A., and B. C. Verrelli, 2003. Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu. Rev. Genomics Hum. Genet. 4: 293–340. [DOI] [PubMed] [Google Scholar]
- Vigilant, L., M. Stoneking, H. Harpending, K. Hawkes and A. C. Wilson, 1991. African populations and the evolution of human mitochondrial DNA. Science 253: 1503–1507. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Weiss, G., and A. von Haeseler, 1998. Inference of population history using a likelihood approach. Genetics 149: 1539–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, I. J., and D. J. Balding, 1998. Genealogical inference from microsatellite data. Genetics 150: 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J., and X. Gu, 1998. Correlation between the substitution rate and rate variation among sites in protein evolution. Genetics 149: 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]