

Abstract
A novel gene detected in mouse embryonic sites of hematopoiesis was cloned and shown to be a eukaryotic analog of the Escherichia coli selenophosphate synthetase gene. Unlike the E. coli enzyme, which is not a selenoprotein, the presence of selenocysteine in the mouse enzyme is indicated by a TGA codon in the open reading frame of the gene in a position corresponding to the essential cysteine of the E. coli enzyme. An ionized selenol group in place of a cysteine sulfhydryl group could render this mammalian selenocysteine-containing enzyme a more active catalyst. The native cDNA clone and also a mutant form containing a TGC (cysteine) codon in place of TGA were expressed in a baculovirus–insect cell system. Based on recovery of purified proteins, expression of the mutant enzyme was about 40 times higher than wild-type enzyme. The cysteine mutant enzyme exhibited selenophosphate synthetase activity in the assay that measures selenide-dependent AMP formation from ATP. Although expression of wild-type enzyme has not been optimized, the mutant form of the fetal mouse enzyme can be produced in amounts sufficient for isolation in homogeneous form and precise physicochemical and mechanistic studies allowing direct comparison with the analogous cysteine-containing prokaryotic enzyme.
Keywords: eukaryotic selenophosphate synthetase
It is well established that the UGA codon can direct the specific incorporation of selenocysteine into proteins. In prokaryotes at least four genes, selA, selB, selC, and selD, are required for synthesis and specific insertion of selenocysteine into selenoenzymes (1, 2). The 37-kDa selD gene product catalyzes the formation of a reactive selenium donor compound from ATP and selenide (3). Using 31P NMR spectroscopy, it was established that this labile selenium donor compound is a selenophosphate (4), and by comparison with an authentic compound, the labile compound was identified as monoselenophosphate (5). Selenophosphate synthetase, the selD gene product, has been isolated from an overproducing Escherichia coli strain, and analysis of mutant enzymes produced by site-specific mutagenesis showed that Cys-17 and Lys-20, located in a glycine-rich region near the N terminus, are essential for catalytic activity (6–8).
With antibodies elicited to the overproduced E. coli enzyme, selenophosphate synthetase was detected in extracts of various mammalian tissues and an archaean, Methanococcus vannielii (9). Low et al. (10) cloned and characterized human and mouse genes designated sps1 that revealed regions of similarity with the E. coli selenophosphate synthetase gene. The deduced sps1 sequences exhibited an amino acid motif GTGCK (residues 28–32) that resembles the E. coli selenophosphate synthetase 16- to 20-residue segment with the exception that Cys-17 was replaced with threonine. Expression of this human gene was shown to regulate the synthesis of 75Se-labeled 5′ deiodinase, providing evidence of the production of selenophosphate in the system.
A related gene, sps2, was detected in the mouse embryo at early stages of development (11, 12). However, from the deduced sequence, the comparable amino acid motif encoded by this mouse gene, as well as its human counterpart, contains a selenocysteine in place of the essential Cys-17 residue in the E. coli enzyme (Fig. 1). Recently, selenophosphate synthetase genes that contain a TGA codon at the position corresponding to Cys-17 in the E. coli enzyme also have been detected in the complete genomic sequences of Haemophilus influenzae (13) and an extreme thermophilic archae, Methanococcus jannaschii (14). The occurrence of selenocysteine in a catalytically important conserved position of an enzyme that synthesizes an essential precursor of selenocysteine-dependent selenoenzymes suggests that a bypass mechanism should exist in these cases. The possibility that these selenocysteine-containing selenophosphate synthetases may be much better catalysts than the E. coli enzyme is also of particular interest. In the present studies with the mouse selenophosphate synthetase, we expressed the native cDNA clone sps2 and also a mutant form containing a TGC (cysteine) codon in place of TGA in a baculovirus–insect cell system and determined the catalytic activity and some properties of the cysteine-containing mutant enzyme.
Figure 1.

Partial amino acid sequences of eukaryotic and prokaryotic selenophosphate synthetase. The dashed box indicates the active site glycine-rich region of E. coli selenophosphate synthetase (eSPS). Selenocysteine residues indicated by circled U in mouse SPS2 and M. jannaschii (mSPS) and threonine residue of human SPS1 indicated by squared T occur in place of Cys-17 in eSPS, which is essential for catalytic activity.
MATERIALS AND METHODS
Materials.
[8-14C]ATP (44.9 mCi/mmol; 1 Ci = 37 Gbq) was purchased from DuPont/NEN. Cellulose polyethylenimine plastic-backed thin layer chromatography sheets were from J.T. Baker. Polyacrylamide (12%) gel and an immunoblot module were obtained from NOVEX, San Diego. Anti-FLAG M2 monoclonal antibody and anti-FLAG M2 affinity gel were purchased from IBI. Goat anti-mouse immunoglobin G(H+L) conjugated to alkaline phosphatase and a 5-bromo-4-chloro-3-indoylphosphate/nitroblue tetrazolium phosphatase system were obtained from Kirkegaard & Perry Laboratories.
Expression of Mouse sps2 cDNAs in Insect Cell, sf9.
The native sps2 cDNA without its 3′ untranslated region (3′-UTR) was modified by introduction of a nucleotide sequence encoding a FLAG peptide (N-Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys-C, IBI) at the N terminus of the SPS2 protein and was cloned into the plasmid vector pME18X as described (12), using following primers: 5′-CATGAAGGGCTGCGGCTGCAAGGTC-3′ and 5′-GACCTTGCAGCCTGCGCCCTTCATG-3′. The resulting plasmid is designated pME18X-SPS2-N-FLAG. Also, a mutant cDNA containing a TGC (cysteine) codon in place of TGA (selenocysteine) was obtained by PCR (12). The recombinant plasmid containing the mutant cDNA is pME18X-CYS-N-FLAG. To express the sps2 gene in an insect cell, sf9, the cDNA was subcloned into a baculovirus expression vector, pAcSG2 (PharMingen). The recombinant plasmids, pME18X-SPS2-N-FLAG and pME18X-CYS-N-FLAG, were digested with two restriction enzymes, KpnI and XhoI. The resulting DNA fragments containing either the sps2 gene or the mutant gene were eluted from 1.0% agarose gel and then ligated with KpnI/XhoI-digested pAcSG2. The resulting recombinant plasmids were designated pAcSPS2-N-FLAG and pAcCYS-N-FLAG, respectively. Maintenance of sf9 cells, transfection of the cells with recombinant plasmids, and expression of the genes, native and cysteine mutant, were done according to the procedure provided by PharMingen. The expressed proteins were identified by immunoblot assay using anti-FLAG M2 monoclonal antibody, goat anti-mouse immunoglobin G(H+L) conjugated to alkaline phosphatase, and a 5-bromo-4-chloro-3-indoyl phosphate/nitroblue tetrazolium phosphatase system according to established procedures.
Production and Purification of SPS2-N-FLAG and CYS-N-FLAG Proteins.
The insect cells infected by recombinant baculovirus were cultured in spinner culture bottles, harvested after 3 days, and lysed in 1 × PBS (pH 7.4) by homogenization. The cell lysates were immunopurified using anti-FLAG M2 affinity gel according to the procedure provided by IBI. The purified protein preparation was analyzed by SDS/PAGE, and the FLAG-conjugated protein band was identified by immunoblotting using anti-FLAG M2 monoclonal antibody. Protein concentrations were measured by the Bio-Rad protein assay.
Selenophosphate Synthetase Activity Assay.
Reaction mixtures (100 μl) containing 100 mM Tricine-KOH (pH 7.2), 1.5 mM [8-14C]ATP (0.25 μCi), 3 mM MgCl2, 10 mM DTT, 1.5 mM NaSeH, and an appropriate amount of enzyme were incubated at 37°C under argon. The NaSeH was prepared as described by Veres et al. (6). The reactions were terminated by the addition of HClO4, and aliquots of the supernatant solutions, after neutralization with KOH, were chromatographed on polyethyleneimine-cellulose thin layer sheets developed in 1.0 M LiCl. Radioactivity in the AMP spots was measured by liquid scintillation spectroscopy.
RESULTS AND DISCUSSION
Transient Expression of Mouse sps2 Gene in Mammalian Cells.
For transient expression of SPS2-N-FLAG and CYS-N-FLAG in mammalian cells, the recombinant plasmids were introduced into COS-1 or HeLa cells by Lipofectin-mediated transfection using LipofectAmine reagent (Life Technologies, Grand Island, NY). Expressed proteins were detected by immunoblotting with anti-FLAG M2 antibody (IBI). CYS-N-FLAG proteins were expressed after a 48-hr incubation, whereas the native form, SPS2-N-FLAG, which should contain selenocysteine, was not detected even at 64 hr after transfection (data not shown). Because expression levels of the wild-type and mutant proteins were very low in both cell systems, protein bands were not detectable on gels by Coomassie brilliant blue staining. In these experiments, we used constructs containing the cDNA without the 3′-UTR. As shown by Guimarães et al. (12), inclusion of the 3′-UTR in a construct containing the sps2 increased protein expression by more than 20 times. Incorporation of 75Se into the SPS2 protein was observed with this 3′-UTR containing construct indicating the existence of a selenocysteine insertion sequence in the 3′-UTR. Selenocysteine insertion sequence elements present in 3′-UTR regions of other eukaryotic genes have been shown to be required for incorporation of selenocysteine into selenoproteins (15, 16).
Expression of Mouse sps2 Gene in Insect Cells, sf9.
To increase the expression level of wild-type and mutant SPS2 proteins, SPS2-N-FLAG and CYS-N-FLAG cDNAs were subcloned into a baculovirus transfer vector, pAcSG2 (PharMingen). The resulting plasmids, designated pAcSPS2-N-FLAG and pAcCYS-N-FLAG, respectively, were introduced into an insect cell line, sf9. The expressed proteins were identified by immunoblotting using an anti-FLAG M2 monoclonal antibody (Fig. 2). As compared with the transient expression systems, levels of the gene products were considerably higher in the baculovirus–insect cell system. Since full-length SPS2-N-FLAG protein was expressed, it is clear that readthrough of the UGA codon in the message occurred in the insect cell system even in the absence of the 3′-UTR. The amount of protein, however, was lower than the amount of the cysteine mutant, CYS-N-FLAG, product. In immunoblot assays using polyclonal antibodies elicited to overproduced E. coli selenophosphate synthetase, neither the SPS2-N-FLAG protein nor the CYS-N-FLAG protein produced in the baculovirus system was recognized (data not shown). These same antibodies, however, reacted strongly with selenophosphate synthetase in immunoblot assays of adult rat tissue extracts (9). Likewise the enzyme was readily detected in crude extracts of an archae, M. vannielii (9). In view of the crossreactivity of the antibodies to the prokaryotic enzyme with antigens of eukaryotic and archael sources under the same assay conditions, it is possible that the acidic FLAG peptide sequence at the N termini of SPS2-N-FLAG and CYS-N-FLAG prevented recognition of these mammalian proteins produced in the baculovirus system. Since the CYS-N-FLAG protein was active catalytically (see below) it must have been folded correctly in the native state.
Figure 2.

Expression of mouse sps2 cDNAs in insect cell, sf9. Recombinant plasmids, pAcSPS2-N-FLAG and pAcCYS-N-FLAG, were introduced into sf9 cells by the baculovirus system. Crude extracts of the sf9 cells were prepared by homogenization and analyzed by 12% SDS/PAGE. Expressed proteins were detected by Coomassie blue staining (A) and immunoblotting with anti-FLAG M2 antibody (B). Lane 1, molecular weight markers; lane 2, crude extract of sf9 cells without plasmid; lane 3, crude extract of cells containing CYS-N-FLAG; lane 4, crude extract of cells containing SPS2-N-FLAG. Arrowheads indicate expressed proteins. The faint band detected in control cells (lane 2) by the anti-FLAG antibody is due to contamination from the heavy protein band in the adjacent lane.
Purification of the Expressed Proteins.
Harvested baculovirus-infected insect cells were homogenized, and extracts were clarified by centrifugation. The extracts were applied to anti-FLAG M2 gel affinity columns (IBI), and the adsorbed proteins were eluted with 0.1 M glycine buffer (pH 3.0). From 68-mg samples of crude lysate protein applied to the columns, the amounts of purified enzyme recovered were 1.05 mg of CYS-N-FLAG protein and 0.028 mg of SPS2-N-FLAG protein, indicating enzyme levels of 1.54 and 0.04%, respectively (Fig. 3). The low wild-type gene product level obtained is consistent with the problems encountered generally in expression of selenocysteine-containing proteins in heterologous systems.
Figure 3.

Purification of SPS2-N-FLAG and CYS-N-FLAG. Extracts (68 mg of protein) prepared from sf9 cells containing SPS2-N-FLAG or CYS-N-FLAG were fractioned on an anti-FLAG M2 affinity gel column (3 ml bed volume) equilibrated with PBS buffer (pH 7.4). After washing the column with three sequential column volumes of PBS (pH 7.4), the bound FLAG fusion proteins were eluted with 6 × 1-ml aliquots of 0.1 M glycine-HCl (pH 3.0). The fractions were neutralized immediately with 1 M Tris·HCl (pH 8.0). The enzymes in each fraction were detected by immunoblotting with anti-FLAG M2 antibody. After combining fractions containing the protein, comparable aliquots of each preparation were analyzed by 12% SDS/PAGE. The relative amounts of enzyme stained with Coomassie blue in the two extracts are shown. Lane 1, molecular weight markers; lane 2, CYS-N-FLAG; lane 3, SPS2-N-FLAG.
Selenophosphate Synthetase Activity of the Expressed Protein.
Selenophosphate synthetase activity of the CYS-N-FLAG protein purified by immunoaffinity chromatography was monitored by measuring the selenide-dependent formation of [14C]AMP from [14C]ATP (6). As shown in Fig. 4, AMP formation from ATP was a linear function of enzyme concentration (2.5–7 μg per 100 μl reaction mixture) in the presence of sodium selenide, whereas, no AMP was formed in the absence of selenide. An apparent specific activity of 0.3 nmol·min−1·mg−1 CYS-N-FLAG is to be contrasted to that of 49.9 nmol·min−1·mg−1 protein for the E. coli selenophosphate synthetase under the same assay conditions. Since the lower activity of the isolated eukaryotic CYS-N-FLAG enzyme could be due to the presence of the N-terminal FLAG peptide, an enzyme preparation was treated with enterokinase under conditions that have been used to selectively cleave the Lys—Met peptide bond to release the FLAG peptide. This proved to be unsatisfactory for the CYS-N-FLAG construct because extensive nonspecific cleavage of the enzyme was observed. Alternatively, the low catalytic activity of the CYS-N-FLAG could be due to a bound inhibitory substance on the enzyme, to differing pH or metal ion requirements for maximal catalytic activity or other reasons. The E. coli enzyme, for example, is markedly inhibited by zinc ion.
Figure 4.

Selenophosphate synthetase activity of CYS-N-FLAG. Reaction mixtures (100 μl) containing 100 mM Tricine-KOH (pH 7.2), 1.5 mM [14C]ATP (0.25 μCi), 3 mM MgCl2, 10 mM DTT, with (▪) or without (□) 1.5 mM NaSeH, and an appropriate amount of CYS-N-FLAG protein as indicated were incubated at 37°C under argon. [14C]AMP produced was measured as described in Materials and Methods.
Use of Ap5A to Inhibit Adenylate Kinase Contaminant.
Adenylate kinase, a common contaminant of apparently homogenous E. coli selenophosphate synthetase preparations, can interfere in enzyme assays by catalyzing the conversion of the AMP product to ADP according to reaction 1 (6).
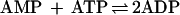 |
1 |
Although P1,P5-di(adenosine 5′)pentaphosphate (Ap5A) is an effective inhibitor of mammalian adenylate kinase, the bacterial enzymes are much less sensitive (17). However, levels of 100–200 μM Ap5A were found to inhibit the contaminating adenylate kinase activity present in an E. coli selenophosphate synthetase preparation effectively (6). In the present study, addition of Ap5A to reaction mixtures containing E. coli selenophosphate synthetase that had not been additionally purified by phenyl-Sepharose chromatography (6), increased the amount of AMP detected about 2.3-fold to a specific activity of about 115 nmol·min−1·mg−1 protein (Fig. 5). Addition of similar concentrations (0.1–0.2 mM) of Ap5A to reaction mixtures containing affinity FLAG-purified CYS-N-FLAG also increased AMP levels about 4-fold in the presence of selenide to a specific activity of about 2.0 nmol·min−1·mg−1 protein (Fig. 6). In the absence of Ap5A, labeled ADP, formed from AMP and ATP by adenylate kinase also was detected on the cellulose thin layer chromatograms. Thus mammalian selenophosphate synthetase preparations purified by FLAG-affinity chromatography may contain significant levels of contaminating adenylate kinase.
Figure 5.

Effects of Ap5A on E. coli selenophosphate synthetase activity estimated by AMP formation. Reaction mixtures as shown in the legend to Fig. 4 were incubated with 2 μM (7 μg) E. coli selenophosphate synthetase with or without 1.5 mM NaSeH and with or without 0.1 mM Ap5A and analyzed as described in Materials and Methods.
Figure 6.

Effects of Ap5A on selenophosphate synthetase activity of CYS-N-FLAG estimated by AMP formation. Reaction mixtures as shown in the legend to Fig. 4 were incubated with 15 μg of CYS-N-FLAG protein with (▪) or without (□) 1.5 mM NaSeH and the indicated amounts of Ap5A and were analyzed as described in Materials and Methods.
Effects of Cations on Selenophosphate Synthetase Activity.
Mg2+, which is required for E. coli selenophosphate synthetase activity, also is required for the CYS-N-FLAG-catalyzed reaction, and the effects of various concentrations of Mg2+ on AMP formation at a fixed concentration of ATP are shown in Fig. 7. Under these conditions a molar ratio of Mg2+ to ATP in the range 2:1 or 4:1 appears to be optimal. A molar ratio of Mg2+ to ATP between 1:1 and 2:1 is the optimal range for E. coli selenophosphate synthetase (4). A monovalent cation, K+, NH4+, or Rb+ is required for E. coli selenophosphate activity and Li+ or Na+ is ineffective as an activator. Sodium ion is slightly inhibitory in the presence of K+. Thus, addition of 40 mM NaCl to the reaction mixtures containing 20 mM KCl caused a decrease in reaction velocity of 16%, whereas with KCl alone the concentration could be increased to 80 mM without a significant change in the reaction velocity (6). However, high concentrations (5–20 mM) of Na+ ion had little effect on CYS-N-FLAG enzyme activity in the presence or absence of 1.5 mM K+ ion in the reaction mixture (data not shown) indicating that Na+ is an effective monovalent cation. Binding of Mn-ATP to the E. coli selenophosphate synthetase required K+, and substitution of K+ with Na+ abolished Mn-ATP binding to the enzyme indicating monovalent cation induced conformational effects (18). From these observations, it appears that the CYS-N-FLAG enzyme differs from the E. coli selenophosphate synthetase in availability and/or specificity of monovalent cation binding sites.
Figure 7.

Effects of MgCl2 concentration on selenophosphate synthetase activity of CYS-N-FLAG. Reaction mixtures as shown in the legend to Fig. 4 were incubated with 15 μg of CYS-N-FLAG protein with (□) or without (▪) 0.1 mM Ap5A and the indicated amounts of MgCl2. [14C]AMP produced was measured as described in Materials and Methods.
Other Properties.
Supplementation of ATP-containing reaction mixtures with equal concentrations of other nucleoside triphosphates had no effect on the formation of [14C]AMP from [8-14C]ATP (data not shown), indicating that like E. coli selenophosphate synthetase, the CYS-N-FLAG is an ATP-specific enzyme and that the other common nucleoside triphosphates have little affinity for the protein. Without the N-terminal FLAG sequence, the predicted molecular weight of the cysteine mutant of SPS2 is 47,786 (12) as contrasted to 36,661 for the E. coli enzyme (1) and 42,267 for SPS1 (10). The molecular weights of the proteins were calculated by the peptidesort program in the GCG package. The amino acid composition of SPS2 is qualitatively similar to that of E. coli selenophosphate synthetase except that there are six tryptophan residues in SPS2,and only one tryptophan residue in the E. coli enzyme.
Acknowledgments
MJG is supported by Junta Nacional de Investigacão Cietifica e Tecnológica, Portugal (CI NCIA/BD/2685/93). DNAX is supported by Schering-Plough.
Footnotes
Abbreviations: 3′-UTR, 3′ untranslated region; Ap5A, P1,P5-di(adenosine 5′)pentaphosphate.
References
- 1.Leinfelder W, Forchhammer K, Zinoni F, Sawer G, Mandrand-Berthelot M-A, Böck A. J Bacteriol. 1988;170:540–546. doi: 10.1128/jb.170.2.540-546.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Böck A, Stadtman T C. BioFactors. 1988;1:245–249. [PubMed] [Google Scholar]
- 3.Leinfelder W, Forchhammer K, Veprek B, Zehelein E, Böck A. Proc Natl Acad Sci USA. 1990;87:543–547. doi: 10.1073/pnas.87.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veres Z, Tsai L, Scholz T D, Politino M, Balaban R S, Stadtman T C. Proc Natl Acad Sci USA. 1992;89:2975–2979. doi: 10.1073/pnas.89.7.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glass R S, Singh W P, Jung W, Veres Z, Tsai L, Scholz T D, Stadtman T C. Biochemistry. 1993;32:12555–12559. doi: 10.1021/bi00210a001. [DOI] [PubMed] [Google Scholar]
- 6.Veres Z, Kim I Y, Scholz T D, Stadtman T C. J Biol Chem. 1994;269:10597–10603. [PubMed] [Google Scholar]
- 7.Kim I Y, Veres Z, Stadtman T C. J Biol Chem. 1992;267:19650–19654. [PubMed] [Google Scholar]
- 8.Kim I Y, Veres Z, Stadtman T C. J Biol Chem. 1993;268:27020–27025. [PubMed] [Google Scholar]
- 9.Kim I Y, Stadtman T C. Proc Natl Acad Sci USA. 1995;92:7710–7713. doi: 10.1073/pnas.92.17.7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Low S C, Harney J W, Berry M J. J Biol Chem. 1995;270:21659–21664. doi: 10.1074/jbc.270.37.21659. [DOI] [PubMed] [Google Scholar]
- 11.Guimarães M J, Bazan J F, Zlotnik A, Wiles M V, Grimaldi J C, Lee F, McClanahan T. Development (Cambridge, UK) 1995;121:3335–3346. doi: 10.1242/dev.121.10.3335. [DOI] [PubMed] [Google Scholar]
- 12.Guimarães M J, Peterson D, Vicari A, Cocks B G, Copeland N G, Gilbert D J, Jenkins N C, Ferrick D A, Kastelein R A, Bazan J F, Zlotnik A. Proc Natl Acad Sci USA. 1996;93:15086–15091. doi: 10.1073/pnas.93.26.15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleishmann R D, Adams R D, White O, Clayton R A, Kirkness E F, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 14.Bult C J, White O, Olsen G J, Zhou L, Fleishmann R D, et al. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 15.Berry M J, Banu L, Chen Y, Mandel S J, Kieffer J D, Harney J W, Larsen P R. Nature (London) 1991;353:273–276. doi: 10.1038/353273a0. [DOI] [PubMed] [Google Scholar]
- 16.Berry M J, Banu L, Harney J W, Larsen P R. EMBO J. 1993;12:3315–3322. doi: 10.1002/j.1460-2075.1993.tb06001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Girons I S, Gilles A-M, Margarita D, Michelson S, Monnot M, Fermandjian S, Danchin A, Barzu O. J Biol Chem. 1987;262:622–629. [PubMed] [Google Scholar]
- 18.Kim I Y, Stadtman T C. Proc Natl Acad Sci USA. 1994;91:7326–7329. doi: 10.1073/pnas.91.15.7326. [DOI] [PMC free article] [PubMed] [Google Scholar]