

Abstract
The neurodegenerative diseases of adulthood, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), pose an enormous and growing public health burden. While effective symptomatic treatments exist for PD, and, to a lesser extent, for AD, there is no therapy for these disorders which will forestall their progression. With the rise of the concept of programmed cell death (PCD) came the realization that even in the absence of complete knowledge of proximate causes, neuroprotection may nevertheless be possible by targeting the pathways of PCD. One set of signaling pathways that have been implicated in cell death are the mitogen-activated protein kinase (MAPK) pathways. The possibility of blocking these pathways and thereby providing neuroprotection has recently been put to the test in a clinical trial of a mixed lineage kinase inhibitor in the treatment of PD. Unfortunately, this trial failed to demonstrate a protective effect. Based on considerations related to the implementation of the trial, it would be premature to conclude that inhibition of MAPK signaling is a failed strategy. In spite of these negative results, the MAPK and related kinase pathways retain their importance as potential targets in PD. In relation to pathogenesis, the discovery of mutations in the MLK-like kinase LRRK2 suggests a role for these kinases in regulating the viability of dopamine neurons. In relation to treatment, the survival signaling kinase Akt has been demonstrated in vivo to mediate striking neurotrophic and anti-apoptotic effects. Thus, it is likely that therapeutic targets related to these kinase signaling pathways will emerge.
Keywords: Alzheimer’s disease, Parkinson’s disease, c-jun, JNK, mixed lineage kinases, Akt, LRRK2
1. Introduction
1.1 Epidemiology and current treatment of neurodegenerative diseases
Neurodegenerative diseases of adulthood, including Alzheimer’s, Parkinson’s, and motor neuron disease as three principal examples, represent an enormous and rapidly growing public health burden globally. These disorders are estimated to affect 20 million individuals worldwide (Mayeux, 2003). Each of these diseases increases in its incidence with age, so with the steady increase in the average age of populations, particularly in developed nations, the morbidity and economic burdens which they impose will grow. Alzheimer’s disease (AD), which manifests principally as a steady decline in cognitive abilities, increases virtually exponentially in its prevalence with age. Less than 1% of individuals aged 60–64 years are affected, whereas 24–33% of individuals 85 years or older are (Blennow, de Leon, & Zetterberg, 2006). Medical costs for patients with AD are predicted to increase from $91 billion in 2005 to $160 billion by 2010 (Alzheimer’s Association Fact Sheet). Parkinson’s disease (PD) manifests initially mainly with impairments of motor function, including tremor, rigidity, slowness of movement and postural instability, but with time, these patients develop non-motor impairments, including dementia, depression and autonomic failure. Approximately one million Americans are estimated to be affected with PD (Mayeux, 2003). Like AD, its incidence increases exponentially with age (Bower, Maraganore, McDonnell, & Rocca, 2000). These figures do not reflect, of course, the emotional and personal devastation which these diseases visit upon patients and their families. Nor do they reflect the health burdens imposed by common, but often unmentioned, related disorders; the cognitive decline caused by the fronto-temporal dementias or diffuse Lewy body disease, or the parkinson-like impairments caused by progressive supranuclear palsy, the multiple system atrophies, cortico-basal ganglionic degeneration, and many others.
In view of the increasing magnitude of these problems, there is desperate need to develop therapies which will halt or hopefully even reverse their inexorable progression. At present there are none, for any of them. On the basis of experimental evidence that cognitive impairment in AD is due, at least in part, to the degeneration of cholinergic neurons of the basal forebrain, pharmacologic approaches have been directed towards enhancement of neurotransmission by acetylcholine. A number of anticholinesterases are now available that are effective in providing some cognitive improvement in mild to moderate AD, but they, of course, do not modify the progression of the disease (Blennow et al., 2006).
There are many effective medical and surgical therapies for the symptomatic treatment of the motor signs of Parkinson’s disease (reviewed in (Lang & Lozano, 1998)), but they often lose efficacy and develop complications in the long term. They do not benefit, and in some instances exacerbate, disabling non-motor features of the disease. As is the case for AD, there is no therapy currently available which prevents the inexorable worsening of the disease due to progressive neurodegeneration.
For both AD and PD, great strides have been made in recent years in our understanding of the molecular basis of neurodegeneration, and these advances have illuminated many possible molecular targets for the development of neuroprotective approaches. For both disorders, the clearest advances have been made possible by the discovery of the genetic causes of the relatively less common familial cases. While the familial forms of these diseases may be less common than the sporadic forms, the molecular insights into possible disease mechanisms provided by the identification of specific disease-causing mutations provide powerful tools. In PD, for example, in the past 10 years disease-causing mutations have been identified in six different genes (reviewed in (Cookson, 2005; Moore, West, Dawson, & Dawson, 2005; Jain, Wood, & Healy, 2005). Importantly, many of these discoveries have suggested common themes in pathogenesis, including protein aggregation and the mis-handling of cellular proteins, and abnormalities in mitochondrial function. These important advances notwithstanding, it is important, in the experimental development of neuroprotective strategies, to recognize that what we clinically diagnose as PD may have diverse proximate causes, especially among the sporadic forms, so neuroprotective approaches based on select “upstream” mechanisms of disease may not benefit all patients. Furthermore, we may find that, in spite of our best efforts, strategies directed at proximate causes may not yield to attempts at therapeutic intervention. Therefore, in the development of neuroprotective approaches, it is worthwhile to target molecular mechanisms of neuron death which appear to be shared by diverse forms of neurologic disease: the pathways of programmed cell death (PCD).
1.2 Programmed cell death
The concept that the molecular pathways of PCD may participate in the processes of chronic neurodegeneration emerged about 15 years ago (Oppenheim, 1991; Ellis, Yuan, & Horvitz, 1991; Johnson & Deckwerth, 1993; Thompson, 1995) and this hypothesis remains under consideration for many of the major neurodegenerative diseases including AD and PD. While the precise role of any specific pathway of PCD remains to be known for these diseases, the concept of PCD has provided a new way of thinking about neuroprotection for them. Even in the absence of exact knowledge of the proximate causes of neurodegeneration, and even in the presence of diverse possible causes, it may be feasible to abrogate neuron death by inhibition of these widely shared pathways.
Several theoretical concerns are often raised about the efficacy of targeting the pathways of PCD. There has been concern that they may be too far “downstream” to preserve sufficient cellular functionality if they are blocked. There has also been concern that these pathways are so redundant and diverse that any attempt, either pharmacologic or genetic, to block them will be futile. And finally, there is the consideration that blocking PCD may protect the neuron cell body, but do little to protect axons, which are likely to degenerate due to molecular mechanisms distinct from those which play a role in PCD (Raff, Whitmore, & Finn, 2002). While all of these concerns are valid, they must be put to empirical test, and there is no a priori reason to believe that they are insurmountable.
Among the diverse and numerous pathways of PCD (reviewed in (Bredesen, Rao, & Mehlen, 2006), there is now an abundance of evidence that the c-jun N-terminal kinase (JNK) signaling cascade may be a therapeutic target in neuron death, including that of dopamine neurons, one of the populations of neurons which degenerate in PD. This pre-clinical evidence has led to the first trial of a drug which blocks PCD to attempt to provide neuroprotection in a neurodegenerative disease; the PRECEPT trial of the mixed linage kinase (MLK) inhibitor CEP1347 in the treatment of PD. In spite of pre-clinical data reviewed and summarized in Sections 2.2 and 2.3 below, this trial has unfortunately failed to find benefit, and it has brought this strategy to a crossroads (Waldmeier, Bozyczko-Coyne, Williams, & Vaught, 2006). We will comment on the trial, (unpublished at the time of this review), and consider its implications for future approaches based on inhibition of the JNK signaling cascade.
Independent of this theme relating the JNK cascade to regulation of the viability of dopamine neurons, an additional theme has emerged to suggest the importance of kinase signaling in the control of the viability of dopamine neurons, and possibly other neuronal populations as well. We have recently found that the survival signaling kinase Akt/PKB has striking trophic effects in vivo on dopamine neuron viability and functionality. These results demonstrate that the many trophic effects of Akt observed in vitro generalize to the in vivo context, in a neuronal population selectively vulnerable to PD. We will review herein the mechanisms of Akt signaling, and how they may provide future neuroprotective approaches to PD.
2. The role of JNK signaling in neuron death
2.1 Overview of the mixed lineage kinase-JNK signaling pathways
JNK (also known as stress-activated protein kinase, or SAPK) is a member of a family of kinases called the mitogen activated protein kinases (MAPKs) which also includes the p38 and the extracellular signal-regulated kinases (ERKs) (Kyriakis & Avruch, 2001; Davis, 1994). All eukaryotic cells possess multiple MAPK pathways which are activated by a wide range of stimuli including hormones and growth factors, inflammatory cytokines, and diverse environmental stresses (Kyriakis & Avruch, 2001). These pathways, in turn, have a variety of downstream effects, including the regulation of gene transcription, the cell cycle, cellular differentiation and cell death. All MAPK signaling pathways are organized in a three-tier signaling structure (Figure 1) in which the MAPKs (in the first tier) are activated by phosphorylation of Tyr and Thr residues within a conserved Thr-X-Tyr motif. This phosphorylation is catalyzed by upstream kinases (the second tier), the MAPK Kinases (MAPKKs), also known as the MAPK/extracellular signal-regulated kinases (ERK)-kinases (MEKs or MKKs) (Kyriakis & Avruch, 2001; Davis, 2000). These kinases are, in turn, regulated by Ser/Thr phosphorylation, which is catalyzed by any of several MAPK-kinase-kinase (MAPKKK) families. Regulation of MAPKKKs is achieved by membrane recruitment, oligomerization and phosphorylation. In some cellular contexts, this is mediated by GTPases of the Ras superfamily, such as Cdc42 and Rac proteins. A fundamental organizational principal of the MAPK pathways is that their complexity is managed in part by scaffold proteins which are capable of binding, sequestering, and fostering specific interactions among select components. This organization permits specific types of stimuli to produce unique and coordinated MAPK signaling responses (Kyriakis & Avruch, 2001).
Figure 1. The mitogen-activated protein kinase (MAPK) signaling pathways.
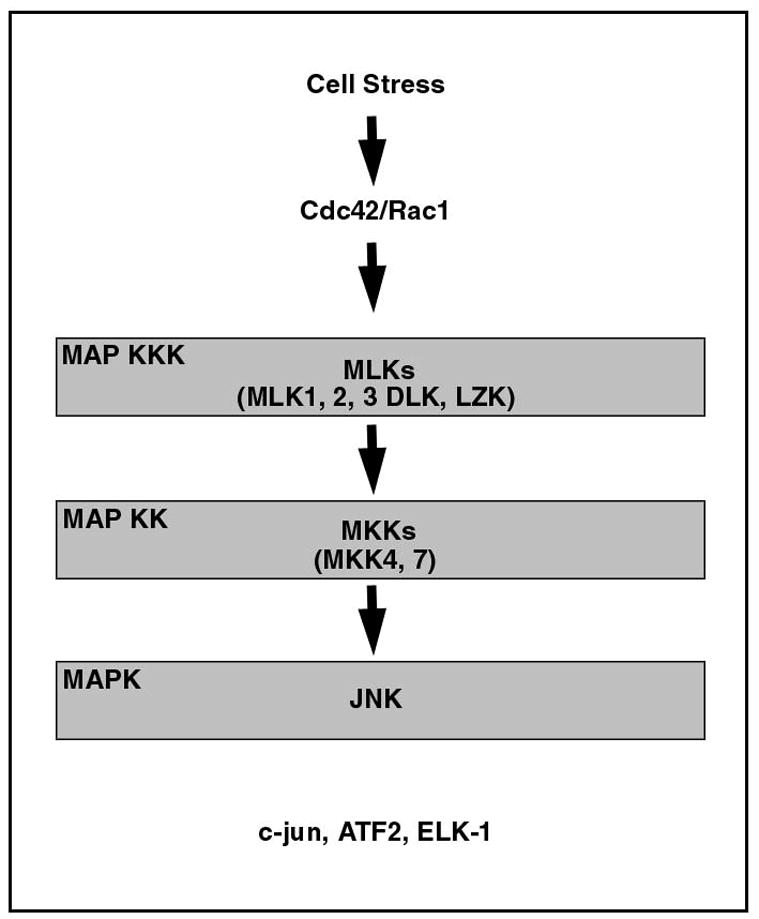
MAPK signaling pathways are organized in a three tier structure. JNK, which mediates phosphorylation and activation of c-jun, is a MAPK in the first tier. Also in this group of kinases are the p38 MAPKs and the ERKs (not shown). The MAPKs are activated by phosphorylation of Tyr and Thr residues by the MAPK kinases in the second tier. In this tier, MKK4 and MKK7, in particular, mediate activation of JNK. Upstream to the MAPKKs, in the third tier, several families of kinases have been reported to activate JNKs (see the text). Among these kinases, the MLKs have been implicated in neuron death.
JNK was first identified in 1990 as a rat hepatic kinase for microtubule-associated protein 2 (Kyriakis & Avruch, 1990). It was cloned in 1994 (Kyriakis et al., 1994; Derijard et al., 1994). There are three genes encoding JNK protein kinases. The Jnk1 and Jnk2 genes are widely expressed, whereas Jnk3 is expressed only in brain, heart and testis (Davis, 2000). Alternate splicing of the gene transcripts results in further molecular diversity. A splice site within the Jnk1 and 2 transcripts results in two splice forms; a second alternate splice for all JNK transcripts occurs at the C-terminus of the protein resulting in the 46 and 55 kDa protein isoforms. Thus 10 isoforms have been identified. JNK has numerous substrates, but it is the dominant kinase for c-jun in vivo; immunodepletion of JNKs from cell extracts removes all stress and cytokine-activated c-jun phosphorylation activity (Kyriakis & Avruch, 2001).
Upstream activation of the JNKs is mediated primarily be two MAPKKs, MKK4 (also know as SAPK/ERK kinase-1, or SEK1) and MKK7. Gene disruption of MKK4 and 7 eliminates JNK activation, indicating that they are major activators of JNK in vivo (Tournier et al., 2001). These two kinases act synergistically to phosphorylate, and activate, the JNKs. In vitro, each kinase alone activates JNKs about 5 to 10 fold, but when added together, they activate them about 100 fold (Kyriakis & Avruch, 2001). In addition, they appear to mediate JNK activation by different stimuli; MKK7 is activated primarily by cytokines (such as tumor necrosis factor (TNF) and IL-1), whereas MKK4 is primarily activated by environmental stress (Davis, 2000).
Upstream to the MAPKKs, several families of MAPKKKs have been reported to activate JNKs, including the MEK kinases (MEKKs 1–4) (MEK is an alternate name for the MKKs), apoptosis signal-regulating kinase-1 (ASK1), transforming-growth factor βactivated kinase1 (TAK1), Tumor progression locus-2 (TPL-2), and the mixed lineage kinases (MLKs) (see Kyriakis and Avruch (Kyriakis & Avruch, 2001), and Davis, (Davis, 2000) for complete reviews). The MLKs have been clearly implicated in PCD in neurons, based on studies with the MLK inhibitor CEP1347 (Maroney et al., 2001) and dominant negative genetic approaches (Xu, Maroney, Dobrzanski, Kukekov, & Greene, 2001), so we will limit this review to this family of JNK activators.
In general, protein kinases contain eleven conserved subdomains. Of these, subdomains I-VII of the MLKs resemble serine/threonine kinases, whereas subdomains VIII – XI more closely resemble tyrosine kinases. Thus, when the MLK genes were initially cloned, they were termed “mixed lineage kinases”. However, biochemical studies have shown that the MLKs serve as serine/threonine kinases. In recent years, three subfamilies of MLKs have been identified, containing seven different kinases (Gallo & Johnson, 2002). Based on an analysis of domain arrangements and sequence similarities, the three families are: the MLKs (containing MLKs 1–4); the dual-leucine-zipper-bearing kinases (DLKs) (containing DLK and leucine-zipper kinases); and the zipper sterile-α-motif kinases (ZAK) (containing ZAK alone). In rat substantia nigra (SN), mRNA for DLK is far more abundant than that for MLKs 1–3 or LZK (Ganguly et al., 2004), but in human SNpc MLK3 is the most abundant (Kholodilov, Rzhetskaya, & Burke, 2006). The domain structure for the MLK families can be exemplified by that of MLK3 and DLK (Figure 2). MLKs 1–4 have an amino-terminal Src-homology-3 (SH3) domain, which apparently serves to autoinhibit the kinase activity (Gallo & Johnson, 2002). Moving towards the C-terminal, this domain is followed by the kinase domain, containing the catalytic site for phosphorylation. There then follows a leucine zipper region, important for protein-protein interactions. It has been shown for DLK that the leucine zipper is required for self-association, phosphorylation, activation and stimulation of the JNK pathway (Gallo & Johnson, 2002). An MLK3 mutant lacking the zipper fails to autophosphorylate and activate JNK (Gallo & Johnson, 2002). In MLKs 1–4, following the leucine zipper towards the C-terminal, there is a Cdc42/Rac-interactive binding motif (CRIB), which mediates interaction with these Rho family GTPases. MLK3, for example, is able to bind to activated forms of Cdc42 and Rac; when Cdc42 and MLK 3 are co-expressed in cells, there is an increase in MLK3 activity and potentiation of JNK activation. All of the known members of the MLK family, when transfected into cells, act as MAPKKKs to activate JNK, and they do so by activation of MAPKKs such as MKK4 and MKK7 (Gallo & Johnson, 2002; Kyriakis & Avruch, 2001). While the MLKs are principally known for their ability to activate the JNKs (Kyriakis & Avruch, 2001), some have been shown to also activate p38 (Gallo & Johnson, 2002).
Figure 2. The protein domain structures of MLK3 and DLK.
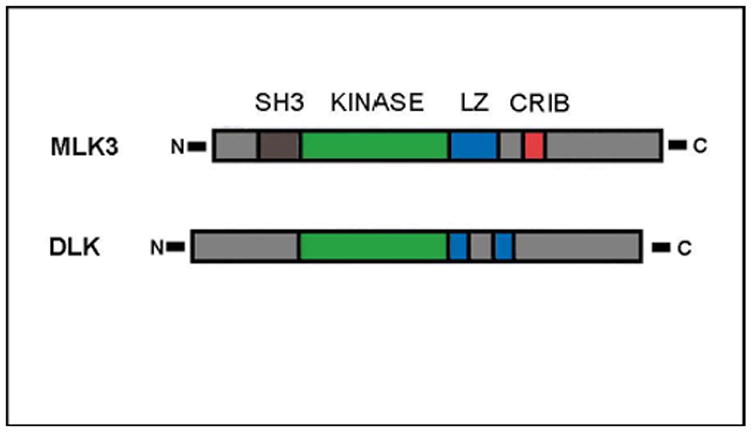
MLK3 and DLK provide representative examples of the domain structures of the MLKs. Common to all MLKs is the kinase domain (green), which mediates phosphorylation, and the leucine zipper domain (blue), which mediates protein-protein dimerization. Exemplified here by MLK3, the MLKs1–4 contain an N-terminal Src-homology-3 (SH3) domain (brown), which may be involved in autoregulatory mechanisms. MLKs1–4 also contain a Cdc42/rac-interactive binding (CRIB) motif (red) that is believed to mediate activation by Cdc42.
In the context of JNK activation by cytokines, such as members of the TNF family, the MAPKKKs are activated by the binding of the cytokine ligand to cell surface receptors of the TNF receptor superfamily (Kyriakis & Avruch, 2001; Davis, 2000). Binding induces oligomerization with signaling proteins, the TNFR-associated factor (TRAF) proteins either directly, as in the case of TNFR2, or indirectly through a TNFR-associated death domain protein (TRADD), as is the case for TNFR1. The TRAFs, in turn, can activate JNK via MAPKKKs (Kyriakis & Avruch, 2001). One of the TRAFs, TRAF2, has also been shown to mediate the activation of JNK in the setting of endoplasmic reticulum stress (Urano et al., 2000).
The ability of JNK to phosphorylate c-jun and thereby enhance its ability to transactivate other genes (Hibi, Lin, Smeal, Minden, & Karin, 1993) remains its principal recognized role (Davis, 2000). C-jun dimerizes with itself and other transcription factors (such as c-Fos and ATF) to constitute Activator Protein-1 (AP-1) transcription factors which regulate the expression of a number of stress-responsive genes. N-terminal phosphorylation of c-jun increases its stability (Musti, Treier, & Bohmann, 1997). JNK also phosphorylates and activates other AP-1 proteins, including JunB, JunD and ATF2. In addition, another transcription factor, ElK1, is a target of JNK phosphorylation; it is involved in induction of the c-fos gene. Thus, the role of JNK in mediating PCD is likely to be, at least in part, through its role in regulating gene transcription, and this role is likely to be mediated primarily through its ability to phosphorylate c-jun. This conclusion is supported by the observation that mutations of c-jun, substituting alanines for its serine 63 and 73 phosphorylation sites, lead to increased resistance to apoptosis in neurons (Behrens, Sibilia, & Wagner, 1999), as discussed further below.
However, JNK mediates its effects on apoptosis not only through its effects on gene transcription. There is increasing evidence that JNK is able to directly phosphorylate and regulate pro- and anti-apoptotic activity of members of the Bcl-2 family. A number of studies have demonstrated that JNK can phosphorylate, and diminish the anti-apoptotic activity of, both Bcl-2 (Maundrell et al., 1997; Yamamoto, Ichijo, & Korsmeyer, 1999) and Bcl-XL (Kharbanda et al., 2000). The latter study demonstrated that induction of apoptosis by irradiation is associated with translocation of JNK to mitochondria and binding to Bcl-XL. In addition, JNK is capable of phosphorylating the pro-apoptotic protein BAD at serine 128 and potentiating its pro-apoptotic effect (Donovan, Becker, Konishi, & Bonni, 2002). Similarly, JNK has been demonstrated to phosphorylate the pro-apoptotic proteins Bim and Bmf, thereby causing their release from sequestration by dynein motor complexes, with translocation to mitochondria, followed by release of mitochondrial death mediators (Lei & Davis, 2003).
A general principle in the organization of the MAPK signaling cascades is that protein interactions are orchestrated in part by scaffolding proteins which are able to bind specific protein components and foster interactions among them, thus permitting specific stimuli to produce unique signaling responses. This principle can be illustrated for JNK signaling by two such scaffolding proteins: JNK interacting protein-1 (JIP1) (Dickens et al., 1997) and POSH (plenty of SH3s) (Tapon, Nagata, Lamarche, & Hall, 1998).
JIP1 was first identified as a specific JNK interacting protein; it does not interact with the p38s or the ERKs (Dickens et al., 1997). As predicted for a scaffolding protein, it is able to bind multiple components involved in JNK activation: among the MAPKKKs, it interacts with MLK3 and DLK; among the MAPKKs, it interacts with MKK7. Coexpression of JIP1 with MLK3 or MKK7 enhances the ability of these kinases to activate JNK. JIP1 illustrates the role of scaffolding proteins to regulate the specificity of MAPK signaling, because JIP1 null animals show deficits in the activation of JNK due to excitotoxic stress or anoxia, but their neurons do not show deficits in activation due to UV radiation (Whitmarsh et al., 2001). Expression of the JNK binding domain (JBD) alone (aa 127–281) has dominant negative effects (Dickens et al., 1997), and it has been used in vivo to demonstrate the role of JNK signaling in MPTP toxicity (see below) (Xia et al., 2001).
POSH was first identified as a Rac-interacting protein by the yeast two-hybrid method, and the interaction was shown to be GTP-dependent (Tapon et al., 1998). POSH was shown to induce JNK activation and apoptosis in non-neuronal cells (Tapon et al., 1998). Xu and co-workers demonstrated that POSH expression induces JNK phosphorylation and apoptosis in neural cells (Xu, Kukekov, & Greene, 2003). Furthermore, it is capable of direct interaction with the MLKs, MKKs 4 and 7 and the JNKs, and, as would be predicted, it acts upstream to the MLKs, the MKKs and c-jun to induce cell death (Xu et al., 2003).
2.2 MAPK signaling in neuronal cell death in vitro
Over the last decade, a large body of in vitro evidence has emerged suggesting that c-jun plays an important role in the mediation of neuronal PCD. One of the principal in vitro models for the study of cell death in neurons has been the use of sympathetic neurons isolated from rat superior cervical ganglia (SCG). Following the withdrawal of nerve growth factor (NGF), these neurons undergo apoptosis (Edwards & Tolkovsky, 1994; Martin et al., 1988; Deckwerth & Johnson, 1993). Early studies demonstrated upregulation of c-jun protein (Ham et al., 1995) and mRNA (Estus et al., 1994) in this model. Microinjection of a dominant-negative form of c-jun protected SCG neurons from NGF withdrawal-induced cell death, whereas overexpression of wild-type c-jun protein resulted in significant induction of apoptosis even in the presence of NGF (Ham et al., 1995). Similarly, microinjection of neutralizing antibodies for c-jun protein significantly reduced neuronal death following NGF withdrawal (Estus et al., 1994). Identical treatment using neutralizing antibodies directed against other members of the AP-1 family, such as JunB, and JunD, failed to demonstrate a protective effect. These studies clearly illustrated that c-jun plays a significant role in the death of sympathetic neurons following trophic factor withdrawal. Similar results have also been reported in cerebellar granule cells following survival signal withdrawal (Watson et al., 1998), and in differentiated PC-12 cells following NGF withdrawal (Mesner, Winters, & Green, 1992).
Activation of the c-jun pathway upon induction of PCD has also been identified in tissue culture in cells that more closely resemble the dopaminergic phenotype. Holtz and O’Malley have investigated MN9D cells, which display many properties of living dopaminergic neurons including the synthesis and storage of dopamine (Choi et al., 1991), and primary mesencephalic cultures (Holtz & O'Malley, 2003). They identified upregulation of both c-jun mRNA and c-jun phosphorylation in MN9D cells, and increased phosphorylation in primary cultures, following treatment with the catecholamine-specific neurotoxin 6-hydroxydopamine (6OHDA).
As the principal kinase for c-jun, JNK has also been implicated in PCD in tissue culture models. Withdrawal of NGF from PC12 cells leads to sustained activation of JNK (Xia, Dickens, Raingeaud, Davis, & Greenberg, 1995). In SCG cells, withdrawal of NGF induces increased JNK activity, serine 63 phosphorylation of c-jun, and transcriptional activation of the c-jun promoter (Eilers, Whitfield, Babij, Rubin, & Ham, 1998). Prevention of c-jun phosphorylation protects PC-12 cells from apoptosis following NGF withdrawal (Le Niculescu et al., 1999).
Upstream regulators of JNK activity, such as members of the MLK family have also been shown to modulate c-jun activation and apoptosis. Overexpression of MLKs induced apoptotic cell death in PC12 cells (Xu et al., 2001) and SCG neurons (Mota, Reeder, Chernoff, & Bazenet, 2001). Expression of dominant negative MLKs blocked NGF withdrawal-induced apoptosis (Xu et al., 2001; Mota et al., 2001).
The possibility of targeting the MLKs in particular for neuroprotective therapeutics has been supported by studies with the MLK inhibitor CEP1347. CEP1347 is a derivative of the indolocarbazole alkaloid K-252a, a natural product isolated from Nocardiopsis bacteria, which had previously been shown to promote cell survival in vitro (Knusel & Hefti, 1992). CEP1347 has been shown to be neuroprotective in vitro using an array of cellular insults (reviewed in (Saporito, Hudkins, & Maroney, 2002)). Maroney and colleagues (Maroney et al., 1998) demonstrated that, following trophic factor withdrawal, rat motoneuron cultures treated with CEP1347 displayed enhanced survival. Survival in this study was shown to correlate with the inhibition of JNK1 activity. These findings were confirmed and extended in a subsequent study demonstrating that pre-treatment with CEP1347 prevented NGF-withdrawal-, UV-irradiation-, and oxidative stress-induced death in neuronally differentiated PC-12 cells and rat sympathetic neurons (Maroney et al., 1999). The fact that these cellular insults induce death via three distinct pathways suggested that the mechanism of CEP1347 involved a shared molecular component, most likely, the activation of the JNK pathway (Saporito et al., 2002).
The role of the JNK cascade in CEP1347-mediated neuroprotection was further elucidated by findings demonstrating that CEP1347 inhibited both JNK activation and cell death induced by members of the MLK family in vitro (Maroney et al., 2001). These findings suggested that the protective effect of CEP1347 was a result of the inhibition of the MLKs. Mathiasen et al expanded these findings by demonstrating that treatment with CEP1347 or a dominant-negative MLK3 adenoviral construct inhibited MPP+-induced cell death as well as JNK signaling in neuronally differentiated human neuroblastoma SH-SY5Y cells (Mathiasen et al., 2004).
2.3 MAPK signaling in neuronal cell death in vivo
Initial studies of c-jun expression in the central nervous system of living animals in models of injury were difficult to interpret in relation to cell death, because early studies in peripheral systems had shown that expression could be upregulated by regenerative processes (Jenkins & Hunt, 1991). Such was also the case in some contexts of central injury; in a fimbria-fornix axotomy model, for example, in which death of medial basal forebrain neurons does not occur, there is a sustained increase in c-jun mRNA and protein expression (Haas, Deller, Naumann, & Frotscher, 1996). Therefore, in the earliest studies of c-jun expression at the regional level in injury models accompanied by neuron death, it was difficult in the diverse neuronal populations to specifically attribute cell death to c-jun expression. Nevertheless, early studies in a variety of ischemia models noted close associations in time and regional location between c-jun mRNA or protein expression and neuron death (Dragunow et al., 1993; Gubits, Burke, Casey McIntosh, Bandele, & Munell, 1993; Wessel, Joh, & Volpe, 1991). One particular study, by Dragunow and co-workers, noted expression of c-jun in neurons undergoing a delayed neuronal death as opposed to early necrotic death in a neonatal hypoxia-ischemia model, and suggested that the former may be a form of PCD (Dragunow et al., 1994).
The earliest studies specifically within the substantia nigra (SN) in models of death induced by 6OHDA (Jenkins, O'Shea, Thomas, & Hunt, 1993) and by axotomy (Leah, Herdegen, Murashov, Dragunow, & Bravo, 1993) noted substantial and sustained increases in c-jun expression but these changes were interpreted to be related to a role in regeneration. In the 6OHDA model, however, the maximal expression of c-jun, at 4–8 days post-lesion (Jenkins et al., 1993) is when other investigators subsequently showed that cell death is maximal (Sauer & Oertel, 1994).
With increased awareness of apoptosis as a distinct morphology of PCD (Kerr, Gobe, Winterford, & Harmon, 1995), and the ability to detect it by nuclear staining, it became clear that c-jun expression could be correlated at the cellular level with this form of cell death in living animals. This was true in the context of naturally occurring cell death in the peripheral (Messina, Jaworowski, & Bell, 1996) and central (Ferrer, Olive, Ribera, & Planas, 1996) nervous systems, and in models of induced naturally occurring cell death (Ferrer, Olive, Blanco, Cinos, & Planas, 1996). Similarly, in the SN, close correlations could be made between c-jun expression and markers of apoptosis. Herdegen et al (Herdegen et al., 1998) demonstrated in the adult axotomy model a close regional and temporal association between prolonged c-jun expression and TUNEL labeling for apoptosis (Gavrieli, Sherman, & Ben-Sasson, 1992). Oo et al (Oo, Henchcliffe, James, & Burke, 1999) demonstrated in a postnatal model of apoptosis in the SNpc, induced by early target deprivation, that c-jun and JNK expression could be correlated at a cellular level with apoptotic morphology. Thus, these morphologic studies of apoptotic cell death suggested a clear correlation with c-jun expression.
The first principal evidence for a functional role for JNK/c-jun signaling in cell death in living animals derived from studies in JNK null animals. Yang and co-investigators reasoned that since the JNK3 isoform is selectively expressed in the nervous system, it may play a role in neuronal death. They showed that JNK3 null mice are indeed resistant to kainic acid-induced seizures and associated hippocampal neuron apoptosis (Yang et al., 1997). These animals also demonstrated diminished levels of c-jun phosphorylation and AP-1 transcriptional activity. While this study demonstrated a clear role for this JNK isoform in mediating cell death, it remained an open question whether c-jun itself was the relevant substrate for this effect. To address the precise role of c-jun, Behrens and colleagues created mice by homologous recombination in which the endogenous c-jun gene was replaced by an altered gene in which the serines at positions 63 and 73 were replaced by alanines, which cannot be phosphorylated (Behrens et al., 1999). Mice homozygous for this mutant, non-phosphorylatable form of c-jun were also resistant to seizures and hippocampal neuron apoptosis induced by kainate. Thus, the phosphorylation of c-jun by JNK appears to be responsible for apoptosis in this model.
A functional role for c-jun in mediating death specifically within dopamine neurons has been supported by studies using viral vector gene transfer approaches. Crocker et al (Crocker et al., 2001) have demonstrated in an axotomy model that adenovirus-mediated expression of a c-jun dominant-negative construct not only prevents the loss of dopamine neurons in the SN, but also the loss of dopaminergic fibers in the striatum. A functional role for JNK/c-jun signaling in dopamine neuron death is also supported by the demonstration that gene transfer of the JNK binding domain of JIP-1 (which inhibits JNK activation) protects dopamine neurons from chronic MPTP toxicity (Xia et al., 2001). Again, this approach not only prevented the loss of SN dopamine neurons, but also their striatal terminals, as assessed by catecholamine levels.
In view of this evidence that phosphorylation of c-jun plays a role in the mediation of cell death in dopamine neurons, it would be anticipated that JNK isoforms would also play a role. Hunot and co-investigators have shown in a model of acute MPTP toxicity that both JNK2 and JNK3 homozygous null animals are resistant; each genotype shows only about a 50% reduction of SN dopaminergic neurons, much less than controls (Hunot et al., 2004). JNK1 null animals were not protected. Compound mutant JNK2 and 3 homozygous nulls were even more protected showing only a 15% loss of neurons. Thus both JNK2 and JNK3 play a role in cell death in this model. The compound null mutation also protected dopaminergic fibers in the striatum. These investigators postulated that increased transcriptional activity mediated by JNK phosphorylation of c-jun may mediate cell death, and they found, by microarray analysis, that the immune mediator cyclooxgenase-2 is upregulated. JNK was shown to be necessary for this upregulation, as it was abolished in the compound JNK mutants. Thus, JNK may ultimately act, at least in part, in the acute MPTP model by upregulation of cycloogenase-2, which has been implicated as a death mediator in this model (Teismann et al., 2003).
These results cannot, however, be construed as direct evidence of a role for JNK as a cell-autonomous mediator of apoptotic death within dopamine neurons. The principal reason is that apoptosis does not occur in the acute MPTP model (Jackson-Lewis, Jakowec, Burke, & Przedborski, 1995), whereas it does in the chronic MPTP model (Tatton & Kish, 1997). Another important difference between the two models is that a major inflammatory component occurs in the acute model, whereas it is much less in the chronic model (Furuya et al., 2004). Therefore, while it is clear that JNK plays an important role in dopamine neuron death in the presence of inflammation in the acute model, it remains to be determined if it is necessary for cell-autonomous apoptotic death within dopamine neurons.
These studies, based on either gene transfer or transgenesis in mice, indicating a functional role for MAPK signaling in the mediation of neuron death in living animals, have received much support from pharmacologic studies using the specific MLK inhibitors CEP1347, described earlier, and its analogue CEP11004 (Murakata et al., 2002). As summarized above, there is much evidence that CEP1347 can abrogate PCD in a variety of tissue culture models utilizing many different types of cellular insult. Efficacy of CEP1347 to forestall PCD has also been observed in diverse living animal models. Glicksman et al demonstrated that application of CEP1347 prevented naturally occurring cell death in spinal motor neurons in both embryonic chicks and postnatal rats (Glicksman et al., 1998). This compound has also been demonstrated to forestall pathologic cell death in injury models. In an excitotoxic injury model, induced by intracerebral injection of ibotenate, CEP1347 protected basal forebrain cholinergic neurons (Saporito et al., 1998). In a model of apoptosis induced in auditory hair cells by noise trauma, CEP1347 diminished the loss of cells, and protected hearing (Pirvola et al., 2000).
These MLK inhibitors have also been shown to be protective in animal models of parkinsonism. In a single dose model of MPTP toxicity, Saporito and co-investigators demonstrated that CEP1347 attenuated the loss of dopaminergic terminal markers and cell bodies in SN (Saporito, Brown, Miller, & Carswell, 1999). In the MPTP single dose model, there is increased phosphorylation of JNK and the upstream kinase MKK4, and these increases are attenuated by CEP1347 (Saporito, Thomas, & Scott, 2000). A similar ability to inhibit the phosphorylation of MKK4 and prevent the loss of dopaminergic terminals in the single dose MPTP model was also demonstrated for CEP11004 (Murakata et al., 2002). In the acute MPTP model, Teismann et al (Teismann et al., 2003) demonstrated that CEP11004 inhibited phosphorylation of c-jun, diminished the loss of TH-positive neurons, and prevented increases in cyclooxygenase-2. Since these models did not directly examine the occurrence of cell death, and since, as discussed above, apoptosis does not occur in the acute MPTP model, it remained to be determined whether MLK inhibition could directly forestall apoptotic death within SN dopamine neurons. In a model characterized by the exclusive induction of apoptosis in these neurons by intrastriatal injection of 6OHDA in postnatal rats, CEP11004 diminished the number of dopaminergic apoptotic profiles (Ganguly et al., 2004). This death is mediated, at least in part, by the intrinsic mitochondrial pathway, because it is associated with an induction of the activated cleaved form of caspase 9. CEP11004 acts upstream to this point, because it diminishes the number of caspase-9-positive profiles in proportion to overall protection from cell death (Ganguly et al., 2004). A notable result of this study was an almost complete protection of striatal TH-positive fibers.
The ability of CEP1347 to protect SN dopamine neurons from MPTP has also been demonstrated in primates (Saporito et al., 2002). Cynomologous monkeys were administered MPTP on a weekly basis, and those treated with CEP1347 showed less parkinsonism than vehicle-treated animals. In addition, they showed a preserved number of SN dopamine neurons. Overall, these studies demonstrate a clear neuroprotective effect of these MLK inhibitors in a variety of living animal models of parkinsonism.
2.4 MAPK signaling in human postmortem brain in neurodegenerative disease
To date, evidence for the involvement of c-jun in human neurodegenerative disease has been limited. Nevertheless, a number of studies have implicated this pathway in the pathophysiology of human neurodegenerative disease, particularly AD. Anderson et al first reported increased intensity of c-jun immunostaining in the hippocampus and entorhinal cortex of AD brains, in comparison to controls (Anderson, Cummings, & Cotman, 1994). The c-jun immunoreactivity was co-localized with staining for paired helical filaments. These investigators subsequently also observed a relationship between c-jun immunostaining and TUNEL labeling for DNA strand breaks (Anderson, Su, & Cotman, 1996). MacGibbon et al (MacGibbon et al., 1997) also found some evidence for increased immunostaining for c-jun in AD postmortem hippocampus. Using a quantitative approach, Marcus et al also identified an increased number of c-jun-positive profiles in AD hippocampus, as compared to age-matched controls (Marcus et al., 1998). Such differences were not observed in a control region, the cerebellum.
Evidence of a role for MAPK signaling in PD brain has been mixed. Recently, Hunot and colleagues (Hunot et al., 2004) have reported evidence of c-jun activation in postmortem tissue from idiopathic Parkinson’s disease patients. A quantitative analysis of c-jun-positive, pigmented neurons in SNpc revealed that a greater proportion showed a nuclear localization among the PD patients. Ferrer and colleagues identified phosphorylated JNK immunoreactivity rarely in the cytoplasm of some neurons, in the vicinity of Lewy bodies, in the brain stem of patients with PD or dementia with Lewy bodies (Ferrer et al., 2001). However, no association was observed between immunostaining and either DNA breaks or activated caspase-3. In an analysis of 4 PD brains, Jellinger did not observe any difference in the expression of c-jun as compared to controls (Jellinger, 2000).
2.5 MAPK signaling: Lessons from a clinical trial in PD
The pre-clinical data indicating that CEP1347 is neuroprotective in a variety of PD models, particularly the MPTP primate model, in conjunction with proven safety and tolerability in PD patients (Parkinson Study Group, 2004), provided a sound basis for a Phase II/III trial of its ability to provide neuroprotection. This trial, named PRECEPT, was carried out with 806 early PD patients not yet requiring dopaminergic therapy (presented as a Late Breaking Abstract, I. Shoulson for the Parkinson Study Group, Annual Meeting of the American Academy of Neurology, 2006). It was a double-blind, prospective comparison of placebo to three doses of CEP1347: 10, 25 and 50 mg bid, utilizing an end point of disability requiring levodopa therapy. The study was terminated early after an average of 21.4 months of follow-up when a planned interim analysis demonstrated that it would be futile to continue treatment. At that time, 57% of patients on placebo had reached endpoint, while 65%, 59% and 64% of those randomized to 10, 25, and 50mg bid, respectively, had reached endpoint. These disappointing negative results call for a critical re-assessment of the hypotheses and pre-clinical experimental data which provided the scientific rationale for the trial. Based on the trial’s intent, what appears at first glance to be the most straight-forward, and the most disappointing, set of conclusions would be that in spite of effective inhibition of the MLKs in diseased dopamine neurons, PD patients continued to progress. If we propose that progression of the disease is directly related to dopamine neuron loss, then we can conclude that the MLKs do not play a role in dopamine neuron death in PD, the pre-clinical data notwithstanding.
However, this set of conclusions cannot be drawn from the negative results of the trial, based on considerations related to its implementation and our current state of knowledge of CEP1347 and PD. First, it is not known whether any of the doses of CEP1347 actually achieved inhibition of MLKs in the SN or in any brain region. The doses selected were based approximately on the effective doses used in the MPTP animal model studies (Waldmeier et al., 2006). However, in the primate pre-clinical studies, it is not known if MLK inhibition was achieved in brain. Even if we accept, without evidence, that MLK inhibition was achieved, it is unknown whether the drug or any of its metabolites have other effects which may impact a possible response to MLK inhibition when administered to human patients.
If we ignore the above concerns, and assume that in the trial CEP1347 provided in brain an effective and selective inhibition of the MLKs, which did not prevent progression of parkinsonism, we still would not be able to conclude that it failed to abrogate dopamine neuron death, for the reason that we do not know the precise relationship between ongoing dopamine neuron loss and the progression of parkinson signs. This relationship is unlikely to be a simple one. Parkinson signs do not appear until about 50% of SN dopamine neurons are lost (Marsden, 1990). It is likely that clinical signs do not appear with early lower levels of neuron loss because there are compensatory changes in the nigro-striatal system at the striatal dopaminergic terminal level, as described in experimental models of parkinsonism (Zigmond, Abercrombie, Berger, Grace, & Stricker, 1990). These neurochemical changes include increased synthesis and release of dopamine, diminished re-uptake, and the development of postsynaptic receptor supersensitivity. In addition to these neurochemical changes to compensate for dopamine neuron loss, there are likely to be anatomical responses, such as regeneration of damaged axons. These compensatory changes are likely to provide a “buffer” between neuron loss and the eventual appearance of clinical signs. Therefore, the appearance of worsening parkinson signs with increasing disease duration may, within a limited timeframe, not be due to neuronal loss, but instead due to failure of these compensatory process, or cellular dysfunction resulting in diminished phenotype (Hirsch, Graybiel, & Agid, 1988), or axonal degeneration.
If, in spite of these considerations, if we accept for the sake of discussion that progression of clinical signs may be directly related to the loss of SN neurons, it would remain difficult within the timeframe of the trial to detect protection. Based on the work of Fearnley and Lees (Fearnley & Lees, 1991), about 45% total SN pigmented neurons are lost within the first decade with an exponential time course; interpolation of their data suggests that about a 15% loss would take place within the 21 month timeframe of the PRECEPT trial early in the course of the disease. If MLK inhibition blocks 50% of neuron death, it would be a substantial effect. But within this timeframe, this magnitude of effect would only reduce a 15% loss to a 7.5 % loss. Thus, a substantial protective effect would have a minimal impact on the numbers of neurons lost, and may not be detectable by assessment of clinical signs.
It is important for this discussion to realize that the preclinical studies of the MLK and JNK signaling cascade principally demonstrated neuroprotection at the cell body level. In these studies, abrogation of these cascades blocked PCD and the resulting cell soma destruction. While some studies demonstrated axonal protection, it is now known that the pathways of axonal destruction are likely to be different from those of PCD (Raff et al., 2002), and the precise role, if any, of MLK-JNK signaling in axonal degeneration is unknown.
Therefore, although the outcome of the PRECEPT trial is a disappointment, it should not lead us to the premature conclusions that the JNK cascade, or PCD altogether, are irrelevant to neuron death in PD. Nor can it be taken as definitive evidence that MLK inhibition is a failed strategy. Some of the limitations on interpretation of the PRECEPT trial related to our lack of understanding of the neural basis of progression of PD, particularly our minimal understanding of the relationship between neuron loss and progression, apply, of course, not only to the PRECEPT trial, but also to other trials of neuroprotective approaches.
2.6 Mutations in leucine-rich repeat kinase (LRRK2) cause an autosomal dominant form of PD
Mutations in the gene LRRK2 (the protein product is also known as dardarin) (Paisan-Ruiz et al., 2004; Zimprich et al., 2004)are an important cause of familial and some sporadic cases of PD. Mutations in LRRK2 cause an autosomal dominant form of PD, and they have attracted great interest because, unlike mutations in α-synuclein, which also cause autosomal dominant PD, they are common, particularly in select ethnic groups (Lesage et al., 2006; Ozelius et al., 2006). The LRRK2 gene encodes a 2527 amino acid protein which contains several domains, beginning from the N-terminal: the leucine-rich repeat region, a Roc (Ras of complex proteins) domain belonging to the Ras/GTPase superfamily, a COR (C-terminal of Roc) domain, a non-receptor tyrosine kinase-like domain, and a WD40 domain. The kinase domain most closely resembles kinases of the MLK family, and LRRK2 has been demonstrated to have MLK-like activity (West et al., 2005) (reviewed in (Mata, Wedemeyer, Farrer, Taylor, & Gallo, 2006)).
West and co-investigators have demonstrated, by autophosphorylation assay, that both the G2019S and R1441C mutations associated with PD increase LRRK2 kinase activity (West et al., 2005). This observation is of great interest, given the known role of the MLKs in mediating PCD, as discussed above. Smith and colleagues have shown that mutant forms of LRRK2 are toxic to cells, both human SH-SY5Y cells and mouse primary cortical neurons, and induce death by apoptosis (Smith et al., 2005). Greggio and colleagues likewise observed an increase in the kinase activity of the G2019S mutant, but not in the R1441C or Y1699C mutations (Greggio et al., 2006). They observed two phenotypes in transiently transfected cells: the formation of cellular inclusions, and the induction of cell death. Using wildtype and mutant kinase-dead constructs, they demonstrated that both of these phenotypes depend on kinase activity. Smith and co-investigators have likewise demonstrated that cellular toxicity depends on kinase activity (Smith et al., 2006). In addition, they showed that the kinase domain is regulated by GTP interaction with the GTPase Roc domain. Gloeckner and colleagues demonstrated a modest increase in kinase activity of the I2020T mutant (Gloeckner et al., 2006). They further demonstrated that, like other MLKs, LRRK2 molecules are capable of dimerization and autophosphorylation. Investigation of the LRRK2 homologue LRRK1 confirms that it is an active kinase, and that it is also regulated by GTP binding within the Roc domain (Korr et al., 2006). However, when the PD-causing mutations of LRRK2 are incorporated into the sequence of LRRK1, they result in decreased kinase activity. Thus, the precise role of the LRRK2 kinase activity in pathogenesis of PD will require further investigation, but it clearly has the potential to become a therapeutic target.
3. The Akt signaling pathway and neuron survival
3.1 Overview of Akt signaling
Akt is a serine/threonine kinase with diverse roles related to the regulation of cell growth, proliferation, migration, glucose metabolism, transcription, protein synthesis, angiogenesis and cell survival (Brazil & Hemmings, 2001). For the purpose of this review, we will focus only on its role as a mediator of cell survival by inhibition of apoptosis; for more general treatments, the reader is referred to excellent reviews (Brazil, Yang, & Hemmings, 2004; Vivanco & Sawyers, 2002; Brazil & Hemmings, 2001). The discovery of Akt began in 1977 with the isolation of transforming murine leukemia virus, termed AKT8, by Staal and co-workers (Staal, Hartley, & Rowe, 1977). Subsequently, in 1991 the oncogene encoded by the virus, v-akt, was cloned (Bellacosa, Testa, Staal, & Tsichlis, 1991). The same gene was cloned independently that year by two other groups and referred to as “related to A and C kinases” (RAC) and protein kinase B (PKB) (Jones, Jakubowicz, Pitossi, Maurer, & Hemmings, 1991; Coffer & Woodgett, 1991). There are now known to be three isoforms of Akt, encoded by three separate genes in mammalian cells: Akt1/PKBα, Akt2/PKBβ, Akt3/PKBγ (Figure 3). All three isoforms are widely expressed, but Akt1 is predominant in most tissues (Kandel & Hay, 1999). The Akts are in the family of the AGC kinases (PKA, PKG, AND PKC related kinases), which are characterized by a central kinase domain and a C-terminal hydrophobic motif (see Figure 3).
Figure 3. The domain structure of the AGC kinases.
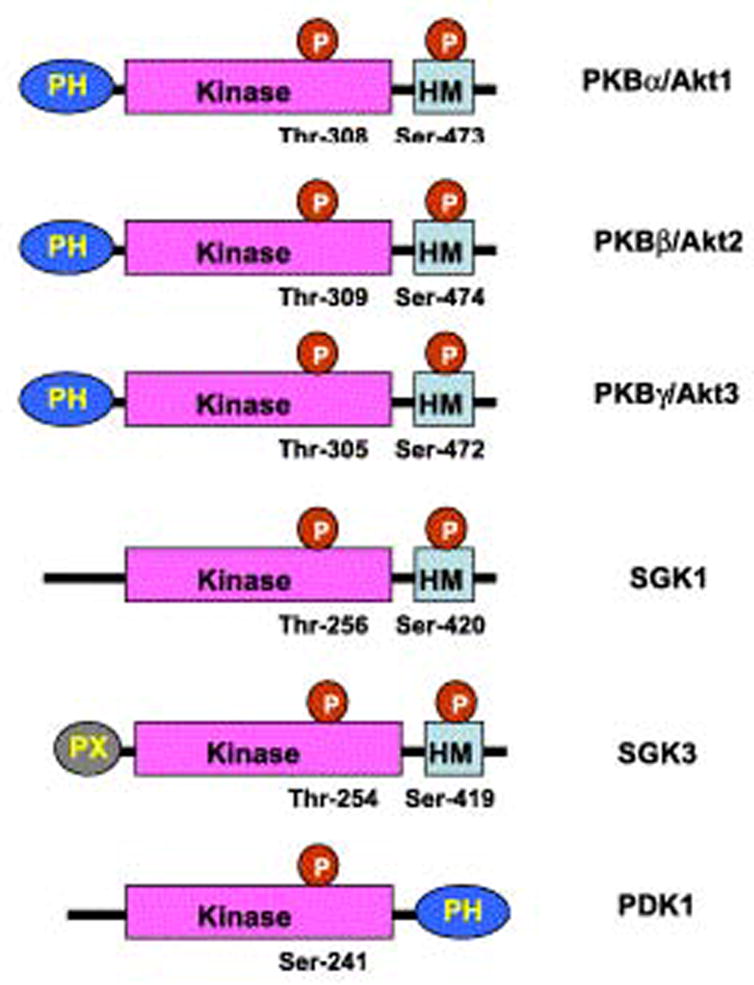
The protein isoforms of Akt, and other AGC kinases, have a central kinase domain, and a C-terminal hydrophobic domain. Abbreviations: PH: pleckstrin homology domain, HM: hydrophobic motif, SGK1: serum and glucocorticoid induced protein kinase, PDK1: 3-phosphoinsitide-dependent kinase 1. Adapted from (Hanada et al., 2004), with permission.
Activation of Akt occurs following the binding of a protein growth factor to its receptor on the surface of the cell (See Figure 4). Ligand binding induces autophosphorylation of tyrosine residues in the cytoplasmic portion of the receptor, resulting in the recruitment and activation of phosphatidylinositol 3-kinase (PI3K). PI3K phosphorylates phosphatidylinositol 4,5 biphosphate (PtdIns(4,5)P2) to phosphatidylonositol 3,4,5 triphosphate (PtdIns (3,4,5)P3), which mediates localization of Akt to the inner surface of the cell membrane by interaction with its pleckstrin homology (PH) domain. PtdIns(3,4,5)P3 can be de-phosphorylated by PTEN (phosphatase and tensin homologue deleted on chromosome ten), which thus serves as a negative regulator of Akt activation. Once localized to the inner surface of the cell membrane, Akt is activated by phosphorylation at two critical residues: Thr 308 in the kinase domain and Ser473 in the hydrophobic motif. The Thr308 kinase is 3-phosphoinsitide-dependent kinase 1 (PDK1); like Akt it is localized to the inner surface of the cell membrane by an interaction between PtdIns(3,4,5)P3 and its PH domain (Hanada, Feng, & Hemmings, 2004). Like Akt, PDK1 is in the family of AGC kinases (Hanada et al., 2004) (Figure 3). The kinase for the Ser473 residue in the hydrophobic motif had been elusive and the subject of debate for many years, but recently Sarbassov and colleagues have provided compelling evidence that it is a complex consisting of mammalian target of rapamycin (mTOR), G-protein β-subunit-like protein (GβL) and rictor (Sarbassov, Guertin, Ali, & Sabatini, 2005; Bayascas & Alessi, 2005).
Figure 4. Akt signaling pathways.
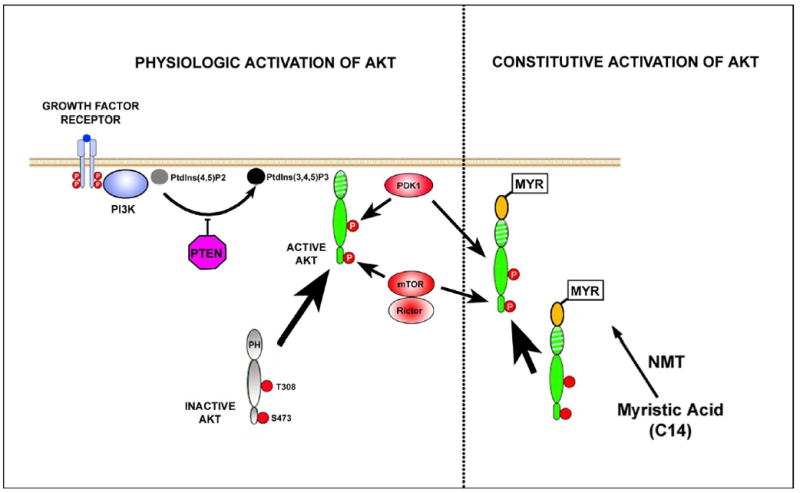
The physiologic pathways for the activation of Akt, as described in the text, are shown on the left side of the Figure. On the right side are shown the pathways for constitutive activation of Akt following N-terminal incorporation of a myristoylation signal (yellow oval). The myristoylation signal is targeted by N-myristoyltransferase (NMT) for the post-translational transfer of myristate onto the N-terminal glycine of the modified protein. Myristic acid is a C14 saturated fatty acid which targets the protein to the inner surface of the cell membrane, where it is phosphorylated, even in the absence of PI3K activation. This myristoylated form of Akt was used in our in vivo studies on SN dopamine neurons, described in the text (Ries et al., 2006).
The first evidence that PI3K/AKT signaling plays a role in supporting the survival of neurons was obtained in studies of NGF-treated PC12 cells (Yao & Cooper, 1995). Yao and Cooper demonstrated that the ability of NGF to block apoptosis in these cells was abrogated by treatment with two specific inhibitors of PI3K. In addition they showed that the ability of platelet-derived growth factor (PDGF) to prevent apoptosis was blocked in cells expressing a mutant PDGF receptor which failed to activate PI3K (Yao & Cooper, 1995). Subsequently, other investigators confirmed that PI3K signaling could prevent cell death in a variety of other tissue culture models utilizing cerebellar, sympathetic (Crowder & Freeman, 1998), sensory, cortical and motor neurons (reviewed in (Kaplan & Miller, 2000)). A role for Akt in mediating neuronal survival was first demonstrated by Dudek and colleagues (Dudek et al., 1997) in a primary postnatal cerebellar granule cell culture model, in which apoptosis is induced by either low potassium or growth factor withdrawal (D'Mello, Galli, Ciotti, & Calissano, 1993). Dudek et al demonstrated that transfection of these neurons with either of two dominant negative forms of Akt blocked the ability of insulin to promote survival, which they showed was mediated by activation of PI3K (Dudek et al., 1997). They further demonstrated that survival was enhanced by transfection with wild-type Akt. Since these initial observations, a large number of studies have demonstrated that Akt protects from apoptosis due to a wide variety of death-inducing stimuli, including the withdrawal of growth factors, UV irradiation, matrix detachment, cell cycle disturbance, DNA damage, and treatment of cells with anti-Fas antibody (reviewed in (Datta, Brunet, & Greenberg, 1999). Conversely, Luo and colleagues have shown that in a variety of tissue culture models, deactivation of Akt accompanies cell death induced by many different agents (Luo et al., 2003).
3.2 Inhibition of apoptosis by Akt in vitro
Akt has been demonstrated to inhibit apoptosis by many mechanisms affecting diverse apoptotic pathways at multiple levels, from upstream signaling pathways which regulate transcriptional activity, to downstream targets, such as caspase-9 (Figure 5). It is beyond the scope of the present review to consider all of these mechanisms in detail. For a more thorough overview, the reader is referred to several comprehensive reviews (Datta et al., 1999; Brunet, Datta, & Greenberg, 2001; Downward, 2004). For the purposes of this review, we will illustrate some of the mechanisms by which Akt acts to inhibit pro-apoptotic transcriptional activation by c-jun and the forkhead family of transcription factors. In addition, we will briefly outline the mechanism by which Akt activates eIF-4E in a recently defined, novel role as an inhibitor of apoptosis by regulating mitochondrial cytochrome c release.
Figure 5. Anti-apoptotic mechanisms of Akt.
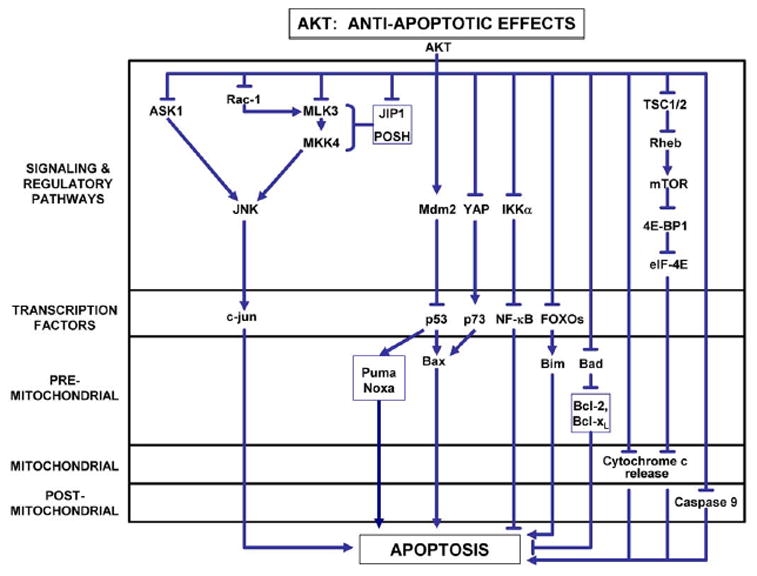
Akt has been described to inhibit apoptosis at multiple levels: signaling pathways, transcription factors, and, in the intrinsic pathway of PCD, at pre-mitochondrial, mitochondrial, and post-mitochondrial levels. Detailed mechanisms for some of these pathways are presented in the text. For more complete reviews, see (Datta et al., 1999; Brunet et al., 2001; Downward, 2004).
Akt negatively regulates the phosphorylation and activation of c-jun by a number of mechanisms (Figure 5). As previously discussed in relation to MLK-JNK signaling, in some cellular contexts, MAPKKKs are activated by the small GTP binding proteins Rac1 or Cdc42 (see Figure 1). Both of these signaling proteins have been shown to participate in NGF-withdrawal-induced apoptosis in sympathetic neurons (Bazenet, Mota, & Rubin, 1998) and in PC12 cells (Xu et al., 2001). The relevance of Cdc42 signaling to the death of dopamine neurons in vivo is supported by the demonstration in an axotomy model that transduction of these neurons with a dominant negative form protects them from neurodegeneration (Crocker et al., 2006). Akt has been shown to phosphorylate Rac1 at serine 71, and thereby reduce its ability to bind GTP, as required for activation (Kwon, Kwon, Chun, Kim, & Kang, 2000).
Downstream of Rac1 and Cdc42, Akt also negatively regulates MLK3. Barthwal and colleagues have demonstrated that Akt interacts directly with MLK3, via a C-terminal domain (Barthwal et al., 2003). Within this domain, Akt phosphorylates serine 674, resulting in diminished JNK activation by MLK3, and decreased cell death. In the model examined, the ability of insulin to attenuate MLK3, MKK7 and JNK activation was dependent on PI3K. Thus, in this system, the pro-survival effects of insulin appear to be mediated by PIK3/AKT signaling, and dependent on Akt phosphorylation of MLK3. Further downstream of the MLKs, Akt also interacts with and phosphorylates MKK4 at serine 78 (Park et al., 2002). Phosphorylation at this residue inhibits MKK4-mediated apoptosis.
MLK3 is not the only MAPKKK target of Akt. Apoptosis signaling kinase (ASK1) is a MAPKKK which activates both JNK and p38 (Ichijo et al., 1997). Kim and co-workers have demonstrated that Akt binds to ASK1, phosphorylates it at serine 83, and thereby reduces its kinase activity (Kim, Khursigara, Sun, Franke, & Chao, 2001). This modification of ASK1 results in reduced activation of JNK, and a reduction of apoptosis in cell lines.
The scaffold proteins JIP1 and POSH, described earlier, are also Akt targets. Akt1 binds to JIP1 in primary neurons and inhibits its ability to potentiate JNK activation (Kim et al., 2002). Similarly, Figueroa and colleagues have shown that Akt2 binds to POSH and negatively regulates its ability to activate JNK (Figueroa, Tarras, Taylor, & Vojtek, 2003). This inhibition appears to be mediated by phosphorylation of MLK3, resulting in its dissociation from the POSH signaling complex.
Whereas Akt inhibition of c-jun activation is achieved by phosphorylation of these diverse upstream activators, its inhibition of the forkhead box O (FoxO) family of transcription factors is achieved by direct interaction with, and phosphorylation of, the proteins (Figure 5). The FoxO family of transcription factors plays a role in diverse cellular functions, including initiation of apoptosis by the induction of the pro-apoptotic genes FasL (Brunet et al., 1999) and Bim (Dijkers, Medema, Lammers, Koenderman, & Coffer, 2000), (reviewed in van der Heide et al (van der Heide, Ramakers, & Smidt, 2006)). To date, four members of the FoxO family have been identified in mammalian cells: FoxO1, FoxO3, FoxO4, and FoxO6. All but FoxO6 are expressed in brain. The FoxOs all contain three Akt phosphorylation motifs. Brunet and colleagues demonstrated that Akt phosphorylates FoxO3 (also known as FKHRL1) at T32, S253 and S315 in cells, which enables an interaction with protein 14-3-3 (Brunet et al., 1999). This interaction sequesters FoxO3 in the cytoplasm, preventing it from migrating to the nucleus and activating pro-apoptotic gene transcription. The critical role of phosphorylation of these residues in FoxO3 to abrogate its pro-apoptotic effects was confirmed by creating a triple mutant form in which non-phosphorylatable alanines were substituted. The triple mutant form of FoxO3 translocated to the nucleus and induced apoptosis even in the presence of the survival factor IGF1 (Brunet et al., 1999).
Akt also inhibits apoptosis at the post-transcriptional level by phosphorylation of pro-apoptotic proteins, including pre-mitochondrial mediators, such as Bad (Datta et al., 1997) and post-mitochondrial mediators such as caspase-9 (Zhou, Li, Meinkoth, & Pittman, 2000). More recently, an anti-apoptotic effect of Akt has been demonstrated to be mediated through activation of mTor (Wendel et al., 2004; McCormick, 2004). Wendel and colleagues have demonstrated in a murine lymphoma model that the anti-apoptotic effects of Akt can be blocked by rapamycin, an inhibitor of mTor. The anti-apoptotic effect of Akt could be restored by eIF-4E, and this effect was not blocked by rapamycin, indicating that eIF-4E is downstream. While eIF-4E is best known for its role as an elongation initiation factor for the translation of protein from mRNA, it has more recently been shown to play a role in blocking apoptosis as well. Li and co-workers have demonstrated that eIF-4E is capable of blocking apoptosis induced by c-myc (Li et al., 2003). They show that eIF-4E increases both the abundance and translation of the mRNA of the anti-apoptotic Bcl-XL, and thereby decreases mitochondrial cytochrome c release (Li et al., 2003).
Activation of eIF-4E by mTor is mediated by phosphorylation and inhibition of 4E-BP1, a negative regulator of eIF-4E (reviewed in (Schmelzle & Hall, 2000; Manning & Cantley, 2003). Activation of mTor by Akt is achieved indirectly by Akt phosphorylation and inhibition of tuberin (tuberous sclerosis complex (TSC)2). Tuberin/TSC2 functions in a complex with hamartin/TSC1 as a GTPase-activating protein (GAP) to inhibit a Ras-related small GTPase Rheb (Manning & Cantley, 2003). Rheb is positive regulator of Tor signaling. Thus, Akt ultimately activates mTor by blocking negative regulation of Rheb (Manning & Cantley, 2003).
3.3 Effects of Akt in vivo
In spite of the extensive information available about the mechanisms of the anti-apoptotic effects of Akt derived from studies in tissue culture, there have been remarkably few studies of its effects on neurons in vivo. In one promising study, Namikawa and colleagues demonstrated in a postnatal axotomy model that transduction of motor neurons with a constitutively active form of Akt improved their survival and accelerated the regeneration of their axons (Namikawa et al., 2000).
We have recently demonstrated that a constitutively active, myristoylated form of Akt (Myr-Akt) has potent anti-apoptotic effects on dopamine neurons of the substantia nigra in vivo. Transduction of these neurons by use of an adeno-associated virus (AAV) vector with Myr-Akt inhibited apoptosis by about 80% in an intrastriatal 6-hydroxydopamine (6OHDA) model of parkinsonism (Ries et al., 2006). This inhibition of dopamine neuron death resulted in an almost complete preservation of these neurons; in the presence of AAV Myr-Akt there was only a 13% (not significant) loss, whereas in AAV-GFP controls, there was an 80% loss, typical of this model. AAV-Myr-Akt was able to protect dopamine neurons in this model not only when it was administered before 6OHDA, but also when it was administered 3 weeks after. In this delayed-delivery paradigm, only 14% of dopamine neurons survived in AAV GFP controls, whereas 50% survived in the AAV Myr-Akt group.
Many anti-apoptotic approaches have been demonstrated to successfully protect neuronal cell bodies, but unfortunately they often do not preserve axons (Eberhardt et al., 2000; Silva et al., 2005). Myr-Akt, however, not only preserved neuronal cells bodies, but also dopaminergic striatal projections (Ries et al., 2006). Following 6OHDA lesion, striatal dopaminergic innervation was reduced by 74% in control mice treated with AAV GFP, whereas in mice treated with AAV Myr-GFP, it was reduced by only 26%. Even when AAV Myr-Akt was injected into the SN 3 weeks after 6OHDA, it was still highly protective of striatal dopaminergic fibers. In AAV GFP-treated control mice, fiber-density was reduced by 90%, whereas in AAV-Myr-Akt-treated mice, it was reduced only 50%. This morphologic preservation of striatal dopaminergic innervation was associated with preserved striatal dopaminergic biochemical indices and behavioral recovery (Ries et al., 2006).
In addition to these striking neuroprotective effects in the context of a neurotoxin lesion, Myr-Akt also had remarkable trophic effects on unlesioned dopamine neurons in normal adult and aged mice. Neurotrophic effects included an increase in the size of individual dopamine neurons, the regional volume of the SNpc, and an increase in the expression of the phenotypic protein tyrosine hydroxylase, associated with an increased abundance of dopamine. In addition, there was an induction of sprouting into the striatum, with an associated increase in striatal dopamine turnover and an augmented response to amphetamine (Ries et al., 2006). Neurotrophic effects on dopamine neurons of the SNpc in aged mice are illustrated Figure 6.
Figure 6. Effect of AAV Myr-Akt on SN dopaminergic neurons in a 22 month old mouse.
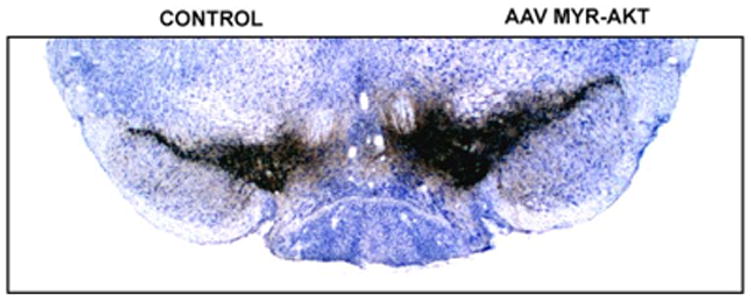
Aged mice were injected into the substantia nigra with either AAV GFP or AAV Myr-Akt and sacrificed for morphologic analysis 7 weeks later. Myr-Akt induced a 31% increase in the total volume of the SN as compared to the contralateral non-injected SN. There was also an increase in the size of individual TH-positive neurons. Myr-Akt also induced a vigorous sprouting response in aged mice, with a 37% increase in striatal TH optical density values on the AAV Myr-Akt injected side in comparison to contralateral control and both striata of AAV GFP injected mice (not shown).
4.0 Conclusions
As reviewed herein, there is an abundance of evidence derived from both in vitro and in vivo studies to suggest that MAPK signaling pathways mediate PCD in neurons, including dopamine neurons, and that inhibition of them can abrogate neuron death. In spite of this compelling pre-clinical evidence, and a well-powered Phase II/III clinical trial, an MLK inhibitor failed to provide neuroprotection in PD. As disappointing as this result is, it should not lead as to the conclusion that this is a failed approach, for the simple reason that we do not know if kinase inhibition was achieved in brain. Nevertheless, the trial does suggest important lessons, and it does encourage a more critical analysis of our goals in providing neuroprotection. We know that CEP1347 was very effective in preventing progression of parkinson signs in MPTP-treated primates. The negative results of the trial may therefore suggest that this neurotoxin model does not provide a reliable prediction of neuroprotective efficacy.
The aforementioned pre-clinical data in almost all instances investigated the ability of MAPK inhibition to prevent neuron cell body death as the primary outcome measure. In the clinical trial, in which the primary outcome measure was the progression of parkinsonian signs, it was assumed that prevention of neuron death, as demonstrated in the pre-clinical studies, would translate into a slowing of disease progression in the clinical context. But this assumption is unlikely to be true; we know that extensive neuron loss occurs prior to the appearance of any parkinson signs. While at some level, there is likely to ultimately be a relationship between dopamine neuron loss and clinical progression, the relationship is unlikely to be simple linear one, and it may change over the course of the disease. It is, for example, possible that after the disease is first diagnosed, over the course of the first year or two (the period which was the subject of the PRECEPT trial), the major determinant of progression is axonal degeneration or regenerative failure, and not neuron loss.
Another possible lesson of the PRECEPT trial is that it may be insufficient to attempt to inhibit only one pathway of PCD. There is such diversity and redundancy in these pathways that the only approaches likely to be effective will simultaneously target many of them. An example of such an approach is the use of the survival signaling kinase Akt, which shows initial promise in rodent studies.
Whether or not medical or gene therapy approaches to inhibition of MAPK signaling ever prove useful as therapies for neuroprotection in PD, the discovery of disease-causing mutations in the MLK-like kinase LRRK2 clearly indicates the importance of these related pathways in regulating the viability of adult dopamine neurons. Thus, these pathways are exceedingly likely to yield important future therapeutic targets.
Acknowledgments
The author is supported by NIH grants NS26836, NS38370, The Department of Defense grant DAMD17-03-1-0492, The Parkinson's Disease Foundation, The Lowenstein Foundation, and the Michael J Fox Foundation.
Abbreviations
- 6OHDA
6-hydroxydopamine
- AAV
adeno-associated virus
- AD
Alzheimer’s disease
- AGC kinases
PKA, PKG, AND PKC related kinases
- AP-1
Activator protein-1
- ASK1
apoptosis signaling kinase-1
- DLK
dual-leucine-zipper-bearing kinases
- JBD
JNK binding domain
- JIP1
JNK-interacting protein-1
- JNK
c-jun N-terminal kinase
- LRRK2
leucine-rich repeat kinase
- MAPK
mitogen activated protein kinase
- MLK
mixed lineage kinase
- mTOR
mammalian target of rapamycin
- NGF
nerve growth factor
- PCD
programmed cell death
- PD
Parkinson’s disease
- PDGF
platelet-derived growth factor
- PDK1
3-phosphoinsitide-dependent kinase 1
- PH
pleckstrin homology domain
- PI3K
phosphatidylinositol 3-kinase
- PKB
protein kinase
- B POSH
plenty of SH3s
- PTEN
phosphatase and tensin homologue deleted on chromosome ten
- SCG
superior cervical ganglion SN, substantia nigra
- TH
tyrosine hydroxylase
- TNF
tumor necrosis factor
- TSC1/2
tuberous sclerosis complex 1 and 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson AJ, Cummings BJ, Cotman CW. Increased immunoreactivity for Jun- and Fos-related proteins in Alzheimer's disease: association with pathology. Exp Neurol. 1994;125:286–295. doi: 10.1006/exnr.1994.1031. [DOI] [PubMed] [Google Scholar]
- Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. Journal of Neuroscience. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthwal MK, Sathyanarayana P, Kundu CN, Rana B, Pradeep A, Sharma C, et al. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J Biol Chem. 2003;278:3897–3902. doi: 10.1074/jbc.M211598200. [DOI] [PubMed] [Google Scholar]
- Bayascas JR, Alessi DR. Regulation of Akt/PKB Ser473 phosphorylation. Mol Cell. 2005;18:143–145. doi: 10.1016/j.molcel.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Bazenet CE, Mota MA, Rubin LL. The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc Natl Acad Sci USA. 1998;95:3984–3989. doi: 10.1073/pnas.95.7.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Influence of strict, intermediate, and broad diagnostic criteria on the age- and sex-specific incidence of Parkinson's disease. Movement Disorders. 2000;15:819–825. doi: 10.1002/1531-8257(200009)15:5<819::aid-mds1009>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Choi HK, Won LA, Kontur PJ, Hammond DN, Fox AP, Wainer BH, et al. Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 1991;552:67–76. doi: 10.1016/0006-8993(91)90661-e. [DOI] [PubMed] [Google Scholar]
- Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991;201:475–481. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Hayley SP, Smith PD, Mount MP, Lamba WR, Callaghan SM, et al. Regulation of axotomy-induced dopaminergic neuron death and c-Jun phosphorylation by targeted inhibition of cdc42 or mixed lineage kinase. Journal of Neurochemistry. 2006;96:489–499. doi: 10.1111/j.1471-4159.2005.03568.x. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Lamba WR, Smith PD, Callaghan SM, Slack RS, Anisman H, et al. c-Jun mediates axotomy-induced dopamine neuron death in vivo. Proc Natl Acad Sci USA. 2001;98:13385–13390. doi: 10.1073/pnas.231177098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. Journal of Neuroscience. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Davis RJ. MAPKs: new JNK expands the group. TIBS. 1994;19:470–473. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of Nerve Growth Factor. Journal of Cell Biology. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, et al. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–40949. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Beilharz E, Sirimanne E, Lawlor P, Williams C, Bravo R, et al. Immediate-early gene protein expression in neurons undergoing delayed death, but not necrosis, following hypoxic-ischaemic injury to the young rat brain. Molecular Brain Research. 1994;25:19–33. doi: 10.1016/0169-328x(94)90274-7. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Young D, Hughes P, MacGibbon G, Lawlor P, Singleton K, et al. Is c-Jun involved in nerve cell death following status epilepticus and hypoxic-ischaemic brain injury? Molecular Brain Research. 1993;18:347–352. doi: 10.1016/0169-328x(93)90101-t. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Eberhardt O, Coelln RV, Kugler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E, et al. Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Journal of Neuroscience. 2000;20:9126–9134. doi: 10.1523/JNEUROSCI.20-24-09126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SN, Tolkovsky AM. Characterization of apoptosis in cultured rat sympathetic neurons after Nerve Growth Factor withdrawal. Journal of Cell Biology. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. Journal of Neuroscience. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RE, Yuan J, Horvitz HR. Mechanisms and functions of cell death. Annual Review of Cell Biology. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM. Altered gene expression in neurons during programmed cell death identification of c-jun as necessary for neuronal apoptosis. Journal of Cell Biology. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Puig B, Barrachina M, Gomez C, et al. Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson's disease and Dementia with Lewy bodies. J Neural Transm. 2001;108:1383–1396. doi: 10.1007/s007020100015. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Olive M, Blanco R, Cinos C, Planas AM. Selective c-Jun overexpression is associated with ionizing radiation-induced apoptosis in the developing cerebellum of the rat. Molecular Brain Research. 1996;38:91–100. doi: 10.1016/0169-328x(95)00334-o. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Olive M, Ribera J, Planas AM. Naturally occurring (programmed) and radiation-induced apoptosis are associated with selective c-jun expression in the developing rat brain. European Journal of Neuroscience. 1996;8:1286–1298. doi: 10.1111/j.1460-9568.1996.tb01297.x. [DOI] [PubMed] [Google Scholar]
- Figueroa C, Tarras S, Taylor J, Vojtek AB. Akt2 negatively regulates assembly of the POSH-MLK-JNK signaling complex. J Biol Chem. 2003;278:47922–47927. doi: 10.1074/jbc.M307357200. [DOI] [PubMed] [Google Scholar]
- Furuya T, Hayakawa H, Yamada M, Yoshimi K, Hisahara S, Miura M, et al. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Journal of Neuroscience. 2004;24:1865–1872. doi: 10.1523/JNEUROSCI.3309-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Oo TF, Rzhetskaya M, Pratt R, Yarygina O, Momoi T, et al. CEP11004, a novel inhibitor of the mixed lineage kinases, suppresses apoptotic death in dopamine neurons of the substantia nigra induced by 6-hydroxydopamine. J Neurochem. 2004;88:469–480. doi: 10.1046/j.1471-4159.2003.02176.x. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. Journal of Cell Biology. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glicksman MA, Chiu AY, Dionne CA, Harty M, Kaneko M, Murakata C, et al. CEP-1347/KT7515 prevents motor neuronal programmed cell death and injury-induced dedifferentiation in vivo. J Neurobiol. 1998;35:361–370. [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, et al. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Gubits RM, Burke RE, Casey McIntosh G, Bandele A, Munell F. Immediate early gene induction after neonatal hypoxia-ischemia. Brain Res Mol Brain Res. 1993;18:228–238. doi: 10.1016/0169-328x(93)90194-t. [DOI] [PubMed] [Google Scholar]
- Haas CA, Deller T, Naumann T, Frotscher M. Selective expression of the immediate early gene c-jun in axotomized rat medial septal neurons is not related to neuronal degeneration. Journal of Neuroscience. 1996;16:1894–1903. doi: 10.1523/JNEUROSCI.16-05-01894.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, et al. A c-jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochim Biophys Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, et al. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N- terminal kinases after neuronal injury. Journal of Neuroscience. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Graybiel AM, Agid YA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2004;101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6,-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Jain S, Wood NW, Healy DG. Molecular genetic pathways in Parkinson's disease: a review. Clin Sci (Lond) 2005;109:355–364. doi: 10.1042/CS20050106. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Cell death mechanisms in Parkinson's disease. J Neural Transm. 2000;107:1–29. doi: 10.1007/s007020050001. [DOI] [PubMed] [Google Scholar]
- Jenkins R, Hunt SP. Long-term increase in the levels of c-jun mRNA and jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neurosci Lett. 1991;129:107–110. doi: 10.1016/0304-3940(91)90731-8. [DOI] [PubMed] [Google Scholar]
- Jenkins R, O'Shea R, Thomas KL, Hunt SP. c-jun expression in substantia nigra neurons following striatal 6-hydroxydopamine lesions in the rat. Neuroscience. 1993;53:447–455. doi: 10.1016/0306-4522(93)90208-w. [DOI] [PubMed] [Google Scholar]
- Johnson EM, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Annual Review of Neuroscience. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci USA. 1991;88:4171–4175. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kerr JFR, Gobe GC, Winterford CM, Harmon BV. Anatomical methods in cell death. In: Schwartz LM, Osborne BA, editors. Methods in Cell Biology: Cell Death. New York: Academic Press; 1995. pp. 1–27. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- Kholodilov N, Rzhetskaya M, Burke RE. Analysis of the relative abundance of the mixed lineage kinases (MLK) in human substantia nigra (SN) using real-time PCR. Abstracts Society for Neuroscience 2006 [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, et al. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35:697–709. doi: 10.1016/s0896-6273(02)00821-8. [DOI] [PubMed] [Google Scholar]
- Knusel B, Hefti F. K-252 compounds: modulators of neurotrophin signal transduction. Journal of Neurochemistry. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- Korr D, Toschi L, Donner P, Pohlenz HD, Kreft B, Weiss B. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–920. doi: 10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Kwon T, Kwon DY, Chun J, Kim JH, Kang SS. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem. 2000;275:423–428. doi: 10.1074/jbc.275.1.423. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J Biol Chem. 1990;265:17355–17363. [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson's disease. First of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Le Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leah JD, Herdegen T, Murashov A, Dragunow M, Bravo R. Expression of immediate early gene proteins following axotomy and inhibition of axonal transport in the rat central nervous system. Neuroscience. 1993;57:53–66. doi: 10.1016/0306-4522(93)90111-r. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, et al. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- Li S, Takasu T, Perlman DM, Peterson MS, Burrichter D, Avdulov S, et al. Translation factor eIF4E rescues cells from Myc-dependent apoptosis by inhibiting cytochrome c release. J Biol Chem. 2003;278:3015–3022. doi: 10.1074/jbc.M208821200. [DOI] [PubMed] [Google Scholar]
- Luo HR, Hattori H, Hossain MA, Hester L, Huang Y, Lee-Kwon W, et al. Akt as a mediator of cell death. Proc Natl Acad Sci USA. 2003;100:11712–11717. doi: 10.1073/pnas.1634990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGibbon GA, Lawlor PA, Walton M, Sirimanne E, Faull RL, Synek B, et al. Expression of Fos, Jun, and Krox family proteins in Alzheimer's disease. Exp Neurol. 1997;147:316–332. doi: 10.1006/exnr.1997.6600. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28:573–576. doi: 10.1016/j.tibs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Marcus DL, Strafaci JA, Miller DC, Masia S, Thomas CG, Rosman J, et al. Quantitative neuronal c-fos and c-jun expression in Alzheimer's disease. Neurobiol Aging. 1998;19:393–400. doi: 10.1016/s0197-4580(98)00077-3. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Bozyczko-Coyne D, O'Kane TM, Neff NT, Tolkovsky AM, et al. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. Journal of Neurochemistry. 1999;73:1901–1912. [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, et al. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight EJ, Murphy CA, et al. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. Journal of Neuroscience. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD. Parkinson's disease. Lancet. 1990;335:948–952. doi: 10.1016/0140-6736(90)91006-v. [DOI] [PubMed] [Google Scholar]
- Martin DP, Schmidt RE, DiStefano P, Lowry O, Carter J, Johnson E. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. Journal of Cell Biology. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends in Neurosciences. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Mathiasen JR, McKenna BA, Saporito MS, Ghadge GD, Roos RP, Holskin BP, et al. Inhibition of mixed lineage kinase 3 attenuates MPP(+)-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2004;1003:86–97. doi: 10.1016/j.brainres.2003.11.073. [DOI] [PubMed] [Google Scholar]
- Maundrell K, Antonsson B, Magnenat E, Camps M, Muda M, Chabert C, et al. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem. 1997;272:25238–25242. doi: 10.1074/jbc.272.40.25238. [DOI] [PubMed] [Google Scholar]
- Mayeux R. Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- McCormick F. Cancer: survival pathways meet their end. Nature. 2004;428:267–269. doi: 10.1038/428267a. [DOI] [PubMed] [Google Scholar]
- Mesner PW, Winters TR, Green SH. Nerve growth factor withdrawal-induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. Journal of Cell Biology. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina A, Jaworowski A, Bell C. Detection of jun but not fos protein during developmental cell death in sympathetic neurons. J Comp Neurol. 1996;372:544–550. doi: 10.1002/(SICI)1096-9861(19960902)372:4<544::AID-CNE4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Mota M, Reeder M, Chernoff J, Bazenet CE. Evidence for a role of mixed lineage kinases in neuronal apoptosis. Journal of Neuroscience. 2001;21:4949–4957. doi: 10.1523/JNEUROSCI.21-14-04949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakata C, Kaneko M, Gessner G, Angeles TS, Ator MA, O'Kane TM, et al. Mixed lineage kinase activity of indolocarbazole analogues. Bioorg Med Chem Lett. 2002;12:147–150. doi: 10.1016/s0960-894x(01)00690-4. [DOI] [PubMed] [Google Scholar]
- Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, et al. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. Journal of Neuroscience. 2000;20:2875–2886. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo TF, Henchcliffe C, James D, Burke RE. Expression of c-fos, c-jun, and c-jun N-terminal kinase (JNK) in a developmental model of induced apoptotic death in neurons of the substantia nigra. Journal of Neurochemistry. 1999;72:557–564. doi: 10.1046/j.1471-4159.1999.0720557.x. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annual Review of Neuroscience. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, et al. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der BM, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Park HS, Kim MS, Huh SH, Park J, Chung J, Kang SS, et al. Akt (protein kinase B) negatively regulates SEK1 by means of protein phosphorylation. J Biol Chem. 2002;277:2573–2578. doi: 10.1074/jbc.M110299200. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. The safety and tolerability of a mixed lineage kinase inhibitor (CEP-1347) in PD. Neurology. 2004;62:330–332. doi: 10.1212/01.wnl.0000103882.56507.20. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, et al. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci 2000. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland RJ, During MJ, et al. Oncoprotein Akt/PKB: Trophic effects in murine models of Parkinson's Disease. Proc Natl Acad Sci USA. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J Pharmacol Exp Ther. 1999;288:421–427. [PubMed] [Google Scholar]
- Saporito MS, Brown ER, Carswell S, DiCamillo AM, Miller MS, Murakata C, et al. Preservation of cholinergic activity and prevention of neuron death by CEP-1347/KT-7515 following excitotoxic injury of the nucleus basalis magnocellularis. Neuroscience. 1998;86:461–472. doi: 10.1016/s0306-4522(98)00059-1. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Hudkins RL, Maroney AC. Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem. 2002;40:23–62. doi: 10.1016/s0079-6468(08)70081-x. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Thomas BA, Scott RW. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. Journal of Neurochemistry. 2000;75:1200–1208. doi: 10.1046/j.1471-4159.2000.0751200.x. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6 hydroxydopamine a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. Journal of Neurochemistry. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, et al. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci USA. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal SP, Hartley JW, Rowe WP. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc Natl Acad Sci USA. 1977;74:3065–3067. doi: 10.1073/pnas.74.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Nagata K, Lamarche N, Hall A. A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-kappaB signalling pathways. EMBO J. 1998;17:1395–1404. doi: 10.1093/emboj/17.5.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labeling and acridine orange. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, et al. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci USA. 2003;100:5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15:1419–1426. doi: 10.1101/gad.888501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- van der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol. 2006;79:205–221. doi: 10.1016/j.pneurobio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL. Recent clinical failures in Parkinson's disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem Pharmacol. 2006 doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. Journal of Neuroscience. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- Wessel TC, Joh TH, Volpe BT. In situ hybridization analysis of c-fos and c-jun expression in the rat brain following transient forebrain ischemia. Brain Res. 1991;567:231–240. doi: 10.1016/0006-8993(91)90800-b. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, et al. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001;15:2421–2432. doi: 10.1101/gad.922801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:10433–10438. doi: 10.1073/pnas.181182298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xu Z, Kukekov NV, Greene LA. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 2003;22:252–261. doi: 10.1093/emboj/cdg021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483–494. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends in Neurosciences. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]