

Summary
Flap endonucleases (FENs) catalyse the exonucleolytic hydrolysis of blunt-ended duplex DNA substrates and the endonucleolytic cleavage of 5′-bifurcated nucleic acids at the junction formed between single- and double-stranded DNA. The specificity and catalytic parameters of FENs derived from T5 bacteriophage and Archaeoglobus fulgidus were studied with a range of single oligonucleotide DNA substrates. These substrates contained one or more hairpin turns and mimic duplex, 5′-overhanging duplex, pseudo-Y, nicked DNA, and flap structures. The FEN-catalysed reaction properties of nicked DNA and flap structures possessing an extrahelical 3′-nucleotide (nt) were also characterised. The phage enzyme produced multiple reaction products of differing length with all the substrates tested, except when the length of duplex DNA downstream of the reaction site was truncated. Only larger DNAs containing two duplex regions are effective substrates for the archaeal enzyme and undergo reaction at multiple sites when they lack a 3′-extrahelical nt. However, a single product corresponding to reaction 1 nt into the double stranded region occurred with A. fulgidus FEN when substrates possessed a 3′-extrahelical nt. Steady-state and pre-steady state catalytic parameters reveal that the phage enzyme is rate-limited by product release with all the substrates tested. Single-turnover maximal rates of reaction are similar with most substrates. In contrast, turnover numbers for T5FEN decrease as the size of the DNA substrate is increased. Comparison of the catalytic parameters of the A. fulgidus FEN employing flap and double-flap substrates indicate that binding interactions with the 3′-extrahelical nucleotide stabilise the ground state FEN-DNA interaction, leading to stimulation of comparative reactions at DNA concentrations below saturation with the single flap substrate. Maximal multiple turnover rates of the archaeal enzyme with flap and double flap substrates are similar. A model is proposed to account for the varying specificities of the two enzymes with regard to cleavage patterns and substrate preferences.
Keywords: DNA repair, flap endonuclease, specificity, FEN, DNA binding
Introduction
The processes of DNA replication and repair require an enzymatic activity capable of catalysing the hydrolysis of bifurcated nucleic acids. In all kingdoms of life this activity is provided by flap endonucleases (FENs) 1. FENs are divalent metal ion dependent enzymes that catalyse the hydrolysis of phosphate diesters with scission of the 3′-phosphorus oxygen bond to generate products possessing a 5′-phosphate and a 3′-hydroxyl group respectively 2. FENs catalyse both the 5′-exonucleolytic and structure specific endonucleolytic reactions of DNA substrates 3; 4. Structure specific activity occurs at the junction between single and double stranded DNA in 5′-flap containing structures (Figure 1(a)). Eight X-ray structures of FENs are available, although none contain DNA bound in the active site 5–12.
Figure 1.
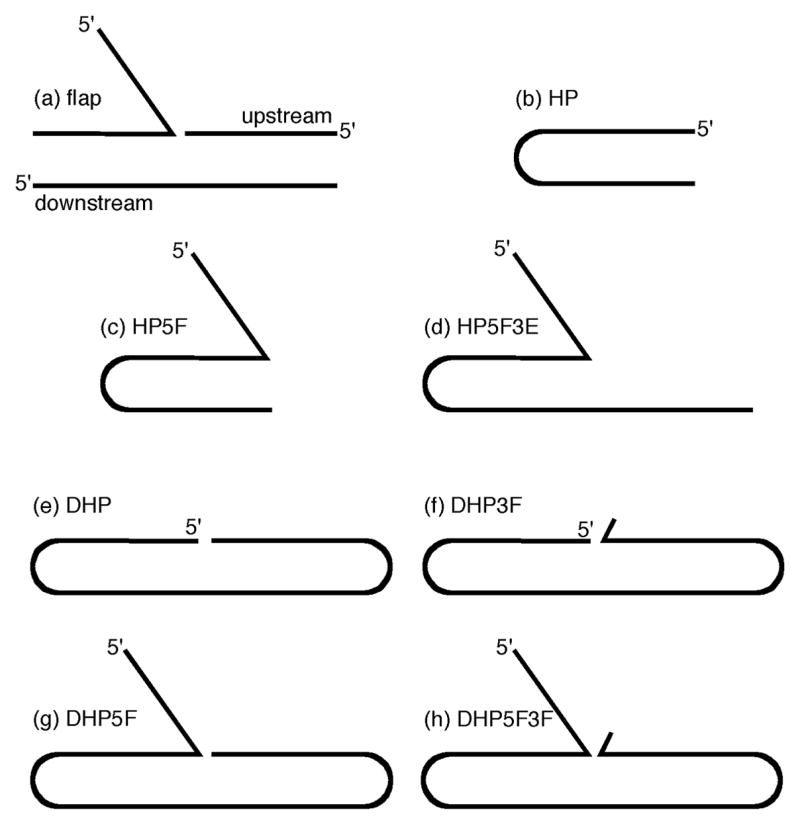
DNA substrates for flap endonucleases. (a) is a typical “flap substrate” consisting of three oligonucleotides, that form duplexes downstream and upstream of the junction between single and double stranded DNA, (b) is a single oligonucleotide (hairpin) analogue of duplex DNA (HP); three hairpin substrates are used in this study of length 18 (HP(18)), 20 (HP(20)) and 22 (HP(22)) nts, (c) is a single oligonucleotide analogue of a 5′-overhanging duplex (HP5F), (d) is a is a single oligonucleotide analogue of a 5′-overhanging duplex with a 3′-extension (pseudoY structure) (HP5F3E), (e) is a single oligonucleotide of nicked DNA (DHP), (f) is a single oligonucleotide of nicked DNA with a 3′-extrahelical nucleotide (DHP3F), (g) is a single oligonucleotide analogue of a “flap substrate” (DHP5F); two variants of this substrate were created with 7 nt (DHP5F(7)) and 2 nt (DHP5F(2)) 5′-single stranded regions, (h) is a single oligonucleotide analogue of a “flap substrate” with a 3′-extrahelical nucleotide (DHP5F3F); two variants of this substrate were created with 7 nt (DHP5F3F(7)) and 2 nt (DHP5F3F(2)) 5′-single stranded regions. The sequences used to construct the oligonucleotides used in this study are shown in table 1. The abbreviated nomenclature used for substrates as derived as follows: HP(no.) designates a blunted ended duplex with a hairpin turn, with the number of nucleotides from which it is constructed in parentheses; DHP a double hairpin with two hairpin duplexes; 5F(no.) designates a substrate with a 5′-single stranded flap with the number of single stranded nucleotides in parentheses; 3F designates a substrate with a single 3′-extrahelical nt and 3E designates a longer single stranded 3′-extension.
Studies of the FEN-DNA interaction have been greatly facilitated by the use of short oligonucleotide substrates, as these mimic the types of DNA structure that may be encountered in vivo. Similar models of the T5FEN-DNA and human FEN-DNA interaction have been generated using a combination of differing potential substrates and site directed mutagenesis 13; 14. These models define areas of the protein that interact with regions of duplex DNA upstream and downstream of the site of endonucleolytic reaction of a flap-type substrate and an area of interaction with the 5′-single-stranded overhang. Analogous conclusions have been reached about the orientation of the substrate in the DNA-Pyrococcus furiosus (Pf) FEN interaction using chemically modified flap structures containing methyl phosphonate or 2′-O-methyl nucleoside substitutions in combination with mutated proteins 15.
Broadly similar reaction specificities have been described for FENs derived from phage, bacterial, archaeal and mammalian sources on synthetic DNA flap substrates 4; 16–19. However, differences exist between the reported behaviour of FENs with smaller DNA substrates. For example T5FEN and human FEN (hFEN) have been reported to cleave duplex or hairpin DNA with a 5′-single-stranded overhang efficiently at the junction between single- and double-stranded DNA 2; 20, whereas such structures have been reported to be poor substrates for some bacterial and archaeal FENs 21. Flap substrates possessing an extrahelical 3′-nucleotide (nt) (so called “double-flap” substrates) have been reported to bring about a rate enhancement in the reactions catalysed by FENs from bacterial, archaeal, yeast and mammalian sources 20–25. Quantitative evaluation of the rate enhancement brought about by a 3′-extrahelical nucleotide revealed a 3–15 fold enhancement in Vmax for a series of bacterial and archaeal FENs compared to a flap substrate lacking the 3′-extrahelical nt 21 whereas a recent study using hFEN noted a 5-fold decrease in KM but similar turnover numbers for substrate with and without the extrahelical nucleotide 20. It is therefore unclear whether interactions between the enzyme and substrate with the 3′-extrahelical nt are used to stabilise the enzyme-substrate complex or to drive catalysis. In addition to rate acceleration, catalysis with double-flap substrates occurs with a marked enhancement in specificity for the reaction one nucleotide into the double-stranded region 20–26. The molecular basis for this specificity was revealed by an X-ray structure of Archaeoglobus fulgidus FEN (AfFEN) bound to a duplex with a 3′-extrahelical nucleotide 9. A binding pocket for the extrahelical nucleotide exists within the higher organism FENs, but the structural features that compose this binding site appear to be absent from the smaller phage enzymes (Figure 2). To date no attempt to investigate the effects of adding a 3′-extrahelical nucleotide to the substrate of the phage enzymes has been reported.
Figure 2.
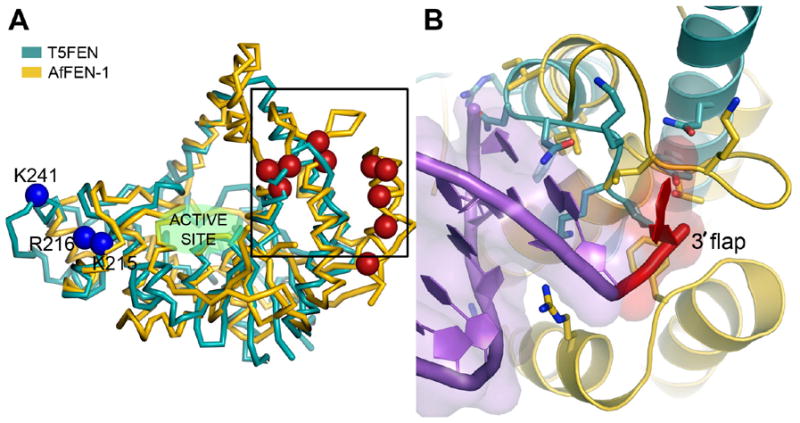
Structural comparisons of T5FEN and AfFEN proteins. (A) Structural superposition of the C-alpha atoms of T5FEN (1UT5, blue) with AfFEN (1RXW, yellow). Blue spheres indicate residues implicated in interaction of T5FEN with the downstream duplex (K241, K215, R216, left). Red spheres indicate residues of AfFEN that interact with the upstream DNA duplex (red spheres) observed crystallographically (F35, I38, L39, L47, F310, S311, R314, K317, K321, right). The boxed region highlights the key differences between T5FEN and AfFEN in the 3′ flap binding site (B). (B) T5FEN (blue) lacks the extended loop and helical regions of AfFEN (gold), which form the binding site for the 3′ flap (red) and associated upstream DNA duplex (1RXW, purple).
In an attempt to understand the factors that give rise to specificity of the FEN reaction and the relationship between catalytic parameters and effectiveness or otherwise of substrate structure we have carried out a systematic study of FENs derived from a phage and archaeal source. A series of substrates of differing sizes designed to test the effects of the presence or absence of an upstream duplex, a 5′-flap of varying length, and/or a 3′-extrahelical nucleotide but where possible identical sequence, have been employed to minimise any effects due to alteration of sequence. The specificities of the two FENs were measured with the set of substrates and steady-state catalytic parameters and maximal single-turnover rates of the reactions were evaluated under as near identical conditions as possible. The patterns of reactivity can be rationalised in terms of substrate and enzyme structure and the strength of their interactions. Within this framework, some differences in the properties of FENs from alternative sources can be rationalised in terms of structural features of the enzymes, but several general features of FENs emerge.
Results
Design of oligonucleotide substrates
A synthetic “flap substrate” typically used to assay FEN reactions is shown in Figure 1(a) and is constructed from three oligonucleotides, yielding a 5′-single stranded flap, and two regions of duplex DNA one upstream and one downstream of the sites of endonucleolytic reaction. A three-stranded flap substrate contains multiple possible FEN reaction sites, namely the single stranded 5′-terminus of the flap, the double-stranded 5′-termini of the upstream and downstream duplexes and the structure specific site at the single stranded-double stranded junction. 5′-Labelling of each of the three strands in turn allows all these activities to be observed with T5FEN (data not shown). Thus, even when monitoring the initial reactions of a single 5′-labelled flap strand, the complication that silent reactions potentially occur on other DNA strands exists.
The synthetic oligonucleotide substrates used in this study are illustrated in Figure 1 and the sequences are displayed in Table 1. These substrates have been designed to probe the cleavage specificity and catalytic parameters associated with various types of FEN activity that can potentially occur. To remove the complication of unmonitored initial reactions, substrates were constructed from single oligonucleotides possessing where appropriate, either one or two hairpin turns, and thus all contain only a single 5′-fluorescently labelled terminus. Whilst FENs display no overt sequence specificity, the possibility of alternative structures as a consequence of sequence changes could give rise to changes in reaction specificity, and therefore where possible the same sequences have been conserved throughout the substrate design. The selection of substrate oligonucleotide sequence was aided by stability and structure prediction software 27, so that no alternative folded structures were predicted to be possible. In addition, the sequence of all hairpin DNAs used in these experiments were selected to have independent predicted melting temperatures of greater than 55°C at 50 mM NaCl (0 mM MgCl2) pH 9.3, thus ensuring that under the conditions of the reaction (25 mM potassium glycinate (pH 9.3), 10 mM MgCl2, 50 mM KCl, 37 °C or 55 °C) hairpin formation was favoured. Unless otherwise indicated, a pH of 9.3 was selected for evaluation of the specificity and catalytic parameters of the FEN reactions, because both the multiple turnover number and the maximal single turnover rate of the T5 FEN catalysed reaction with a magnesium ion cofactor are maximal and pH independent at this pH value 28; 29. Substrates were synthesised using conventional methods and all contain a 5′-fluorescein (FAM) label. It has previously been demonstrated that both 5′-fluorescent and 5′-radiolabelled variants of HP5F have similar catalytic parameters and therefore the fluorophore does not interfere with the reaction 30. Oligonucleotides were purified by reversed phase HPLC (RPHPLC). Separation of the phosphorothioate diastereoisomers of the 5′-FAM labelled psHP, designated f (fast) and s (slow) in order of their elution, was also possible using RPHPLC. However, RPHPLC resolution of the intact epimers of psHP5F was not possible and so the individual phosphorothioate diastereoisomers of this oligonucleotide were assembled from smaller separable fragments by templated ligation. MALDI-TOF mass spectrometry was used to confirm the primary structure and homogeneity of all oligonucleotides.
Table 1.
The oligonucleotide sequences used to construct the substrates illustrated in Figure 1.
| Substrate | Sequence |
|---|---|
| HP(18) | FAM–d(GAACACGCTTGCGTGTTC) |
| HP(20) | FAM–d(GAACACCGCTTGCGGTGTTC) |
| HP(22) | FAM–d(GAACACACGCTTGCGTGTGTTC) |
| HP5F | FAM–d(CGCTGTCGAACACACGCTTGCGTGTGTTC) |
| HP3E | FAM–d(CGCTGTCGAACACACGCTTGCGTGTGTTCCCCAACATAGAGAGCCCC) |
| DHP | FAM–d(GAACACACGCTTGCGTGTGTTCCACAACATAGAGATATGTTGTG) |
| DHP3F | FAM–d(GAACACACGCTTGCGTGTGTTCCACAACATAGAGATATGTTGTGC) |
| DHP5F(7) | FAM–d(CGCTGTCGAACACACGCTTGCGTGTGTTCCATAACATAGAGATATGTTATG) |
| DHP5F3F(7) | FAM–d(CGCTGTCGAACACACGCTTGCGTGTGTTCCATAACATAGAGATATGTTATGC) |
| DHP5F(2) | FAM–d(TCTTACACACGCTGTCGTGTGTAACATAACATAGAGATATGTTATG) |
| DHP5F3F(2) | FAM–d(TCTTACACACGCTGTCGTGTGTAACATAACATAGAGATATGTTATGC) |
| psHP | FAM–d(GpsAACACACGCTTGCGTGTGTTC) |
| psHP5F | FAM–d(CGCTGTCGpsAACACACGCTTGCGTGTGTTC) |
| FAM-8mer | FAM–d(CGCTGTCG) |
Specificity of reactions with the DNA substrates
The reaction specificities of T5FEN catalysed reactions of the oligonucleotide substrates are summarised in Figure 3. The reactions of the various substrates were monitored utilising the 5′-FAM label. Reaction mixtures were separated and quantified by denaturing HPLC (dHPLC), equipped with a fluorescence detector. The dHPLC gradient and buffers used in this experiment were chosen to allow single nucleotide resolution of all possible products of length 1–10 nucleotides. Oligonucleotide standards of the appropriate length were used to confirm the length of 5′-fluorescent products by co-injection. In some cases the results of these studies were verified by MALDI-TOF mass spectrometry, which also allowed investigation of the nature of the unlabelled 3′-products of the reactions. The results of MALDI-TOF analysis are summarised in Materials and Methods. The data presented in Figure 3 summarise the relative amounts of reaction products observed after reaction of each substrate (100 nM) for 8 mins at an enzyme concentration of 100 pM. However, in cases where accurate determination of the relative amounts of products was possible over the entire course of the reaction, the ratios of products remained constant.
Figure 3.
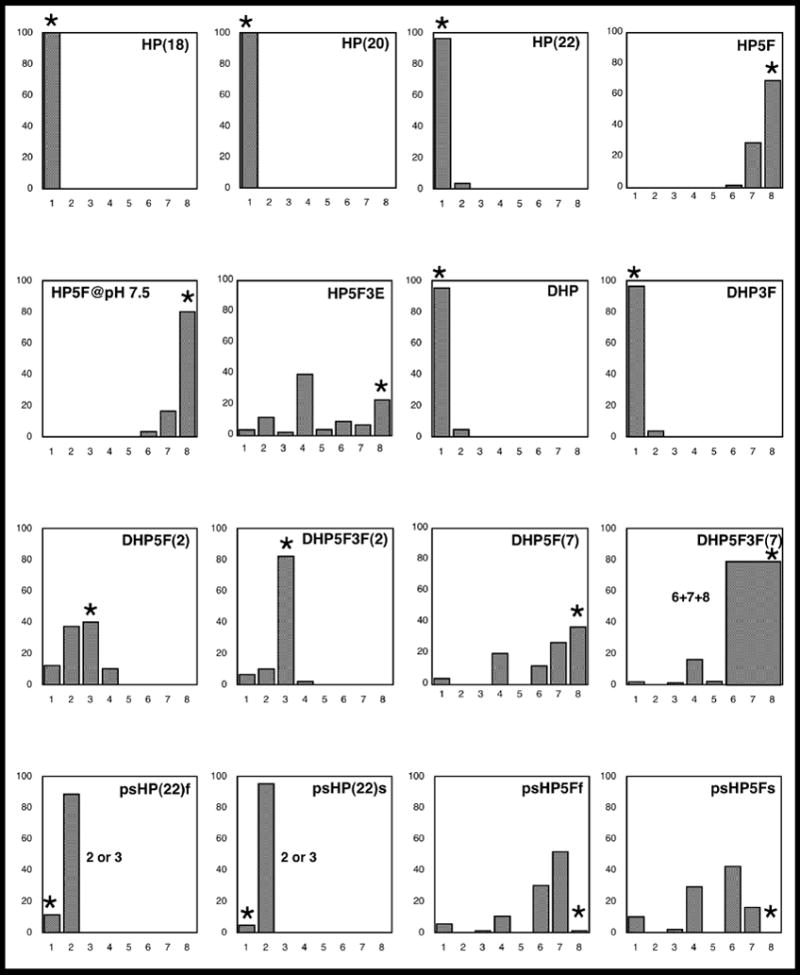
The relative amounts of differing length products produced by T5FEN catalysed hydrolysis of the substrates used in this study. Enzyme reactions (100 pM T5FEN, 100 nM substrate, 25 mM potassium glycinate (pH 9.3), 10 mM MgCl2, 50 mM KCl, 0.01 mg/ml BSA, 37°C) were allowed to progress for 8 mins. Each set of data is the average of at least three experiments. The length of product is shown on the x axis, as a percentage of total product on the y axis. An asterisk indicates the site of reaction 1 nt into the double stranded region. For DHP5F3F(7), it was impossible to quantify the individual amounts of 6, 7 and 8 mer due to peak overlap. In the case of psHPf and psHPs we do not posses the relevant phosphorothioate containing product FAM-d(GpsA) and FAM-d(GpsAA) standards and are therefore unable to assign whether the major product of these reactions is a 2 mer or 3 mer. A table of the data used to construct this figure is shown in Table 1S (Supplementary Information).
T5FEN catalysed reaction of the 5′-overhanging hairpin substrate HP5F (Figure 1(c)) has been previously reported to initially produce 8 mer and 21 mer products as a result of cleavage one nucleotide into the double stranded region 2. Although 5′-FAM-8 mer is the major 5′-reaction product (69 %), in this current study smaller amounts of fluorescent 7 mer (29 %) and 6 mer (less than 2 %) products were also observed in dHPLC traces (Figure 3). MALDI-TOF analysis of the reaction close to completion confirmed the presence of small amounts of 6, 7 mer as well as 8 mer product, but puzzlingly only detected the presence of a single phosphorylated 21 mer 3′-product (data not shown). Similar MALDI-TOF results were obtained regardless of whether HP5F was 5′-phosphorylated or was a 5′-FAM derivative, indicating that the presence of the fluorophore had not changed the reaction specificity. If the 6 and 7 mer oligomers observed by MALDI-TOF MS originated from reaction of intact HP5F it would be expected that the presence of the corresponding phosphorylated 22 mer and 23 mer products would be detected. This raised the possibility that the smaller 6 and 7 mer products could be the consequence of further reaction of the 8 mer product. However, when independently synthesised 5′-FAM-8 mer was incubated under identical conditions used for enzymatic reaction of HP5F (100 pM FEN, 100 nM FAM-8 mer, 25 mM potassium glycinate (pH 9.3), 10 mM MgCl2, 50 mM KCl, 0.01 mg/ml BSA, 37 °C) for 24 hours, there was no reaction. Small (< 2.5%) traces of 1 mer product were observed from this reaction after 72 hours. We therefore concluded that oligomers of 6 and 7 nts in length are products of reaction of intact HP5F, but that the corresponding phosphorylated 23 and 22 mer 3′-products must be substrates for T5FEN and react within the time scale of complete reaction of intact HP5F generating the phosphorylated 21 mer that is the only 3′-product detected by MS analysis. As has been previously noted for the reactions of hFEN with “flap substrates”, reducing the pH of the T5FEN catalysed reaction of HP5F enhanced the preference for reaction 1 nt into the double stranded region 26.
To study the double-stranded exonucleolytic activity of T5FEN and verify the conclusions regarding the origin of reaction products of the 5′-overhanging hairpin substrate, we also investigated the reaction of a blunt ended 22 mer hairpin, HP(22) (Figure 1(b)), of identical sequence to the hairpin component of HP5F. By analogy to the reaction of the 5′-overhanging hairpin, this proved to be an efficient substrate for T5FEN and the major product of the reaction was 5′-FAM-dG corresponding to the same major reaction site of HP5F, one nucleotide into the double stranded region. Minor traces of 5′-FAM-d(GA) were also observed during product analyses by dHPLC (less than 4 %) (Figure 3). As with reaction of HP5F, the major 3′-product of reaction is phosphorylated 21 mer, which is resistant to further enzyme catalysed reaction under the same conditions and timescale that produce reaction of HP(22). Two shorter hairpin substrates HP(20) and HP(18) produced only a single 5′-FAM-dG product (Figure 3). A single oligonucleotide nicked DNA substrate was created by addition of a second hairpin duplex to the 3′-terminus of HP to create DHP (Figure 1(e)). This did not alter the substrate specificity of the reaction and identical products were observed to those seen with HP(22), namely a major 1 mer product with trace amounts of 2 mer (Figure 3).
Extending the 3′-terminus of HP5F, produces HP5F3E (Figure 1(d)), a single oligonucleotide analogue of a pseudo-Y structure. Surprisingly the initial products of T5FEN catalysed hydrolysis of HP5F3E, were eight different 5′-FAM-oligomers ranging in length from 1–8 nts (Figure 3). The major products of this reaction were 8, 7, 6, and 4 mers, the generation of 8 mer corresponding to reaction 1 nt into the double-stranded region, whereas all other reaction sites were in the single-stranded 5′-flap region. An analogous lack of specificity has been observed with some pseudo-Y substrates and hFEN13. The single oligonucleotide analogue of a three-stranded flap structure, DHP5F (Figure 1(g)), has two hairpin turns and contains the same sequence as HP5F with the addition of an upstream duplex identical to that of DHP. The T5FEN catalysed reaction of DHP5F(7) is less specific than the reaction of the 5′-overhanging hairpin substrate HP5F producing 1, 4, 6, 7 and 8 mer. Addition of a 3′-extrahelical nt to flap structures, has been reported to enhance the specificity of reaction of flap endonucleases from several sources, producing a site specific reaction one nt into the double-stranded region 21; 23–25. However the double-flap substrate DHP5F3F(7) (Figure 1(h)), that contains a 3′ extrahelical nt, possesses further reaction sites when compared to the 5′-overhanging double-hairpin structure (DHP5F(7)) without a 3′-flap (Figure 3). Analogous results were obtained with flap and double-flap substrates with a shortened 5′-single-stranded region (DHP5F(2) and DHP5F3F(2)), except that in these cases reaction was also observed two nts into the double stranded region. These substrates produced 1, 2, 3 and 4 mer products, where the 3 mer product results from cleavage one nt into the double stranded region.
Similarly, addition of an extrahelical 3′-nucleotide to the nicked DNA substrate (DHP3F, Figure 1(f)), did not produce any change in the cleavage specificity of this reaction, resulting in the same 5′-FAM-dG major product in analogy to HP and DHP, but modest amounts of 5′-FAM-d(GA) were also observed (Figure 3).
Reactions involving both variants of DHP5F3F and DHP5F were also carried out using FEN from A. fulgidus (Af) (Figure 4). These were carried out under similar conditions to those employed for the T5 enzyme except that DTT was added to the reaction mixtures and the experiments were carried out at 55°C. The concentration of substrate and enzyme were varied from the T5 case to produce reaction on a similar timescale. In contrast to T5FEN, AfFEN produced a specific reaction with both variants of DHP5F3F hydrolysing the phosphate diester one nt into the double-stranded region. The finding that AfFEN performs a specific reaction on both DHP5F3F substrates implies that these oligomers are correctly folded in the proposed double-hairpin form, even at 55°C. Nicked DNA (DHP and DHP3F) was also cleaved by the enzyme, with a specific reaction observed with the presence of a 3′-extrahelical nt. Like T5FEN, DHP and DHP5F(2), substrates without a 3′-extrahelical nt and with either no or a shortened 5′-flap, produced a product corresponding to reaction two nts into the double stranded region in addition to reaction one nt into this region and in the single stranded flap when present. All other substrates required the presence of very elevated concentrations of enzyme and prolonged incubation to detect any evidence of reaction, which was generally non-specific. The results observed here are in line with those reported previously for a range of thermophilic FENs 21.
Figure 4.

The relative amounts (%) of differing length products produced by AfFEN catalysed hydrolysis of the substrates used in this study. Enzyme reactions (8μM substrate and 0.1μM AfFEN, 25 mM potassium glycinate (pH 9.3), 10 mM MgCl2, 50 mM KCl, 0.01 mg/ml BSA, 1 mM DTT, 55°C) were allowed to progress for 8 mins. Each set of data is the average of at least three experiments. The length of product is shown on the x axis as a percentage of total product on the y axis. An asterisk indicates the site of reaction 1 nt into the double stranded region. Smaller substrates (HP5F3E, HP5F and HP (22)) did not undergo any reaction under these conditions on this timescale. A table of the data used to construct this figure is shown in Table 2S (Supplementary Information).
Substrates where a chiral phosphorothioate linkage replaces the scissile phosphate diester could potentially be employed to monitor the stereochemical course of the FEN catalysed reaction, revealing whether the FEN reaction proceeds by direct attack of water (or hydroxide) or involves covalent catalysis. However, based on previous precedents with metalloendonucleases such as EcoRV and Vsr endonucleases 31; 32, it is likely that replacement of the scissile bond with a phosphorothioate would be both refractory to reaction and impair substrate binding. In line with these expectations, replacement of the phosphate diester that is the major site of reaction of unmodified HP(22) with a phosphorothioate linkage of either stereochemistry (psHPf and psHPs, Figure 3) results in a change in the major reaction site of T5FEN compared to the corresponding unmodified oligomer. Similarly, replacement of the phosphate diester one nt into the double stranded region in HP5F (psHP5Ff and psHP5Ff) with a phosphorothioate linkage of either stereochemistry, resulted in reactions within the single stranded region of the molecule (Figure 3).
Kinetic characterisation of the substrates
Variation in catalytic parameters of T5FEN with the length of the downstream duplex
To determine the minimal substrate requirements for the T5FEN catalysed reaction the steady state catalytic parameters of three blunt ended duplex hairpin substrates (Figure 1(b)) of varying length (HP(18), HP(20) and HP(22)) were determined and are shown in Table 2. The use of fluorescent substrates for determination of the catalytic parameters of the FEN reaction has been discussed in detail elsewhere 30. The formation of multiple products was dealt with by measuring the total amounts of all products relative to substrate at various time intervals to generate the initial rates of reaction. Analysis of the catalytic parameters of hairpin substrates of varying length, suggests the 22 mer serves as an effective minimal substrate. Decreasing the length of the duplex region by one base pair (HP(20)), has a negligible effect on catalytic efficiency. However, decreasing the length of the substrate by a further base pair (HP(18)) results in a very dramatic decrease in catalytic efficiency as a consequence of a 100-fold increase in KM.
Table 2.
The catalytic parameters of T5FEN catalysed reactions. Full experimental details are contained in the materials and methods section. Steady-state parameters were determined by measuring the initial rates of reaction of substrates at concentrations around their respective KMs. Maximal single-turnover rate constants (kc) were measured using concentrations of enzyme at least 8 X KM and at a substrate concentration of 1 nM. In the case of HP(18) the high KM precluded an evaluation of kc. Each set of data is the average of at least three experiments.
| Substrate | kcat (min−1) | KM (nM) | kcat/KM (nM−1min−1) | kc (min−1) | kc/kcat |
|---|---|---|---|---|---|
| HP(18) | 226 ± 23 | 45.6 ± 10.6 × 103 | 5.0 × 10−3 | ND | ND |
| HP(20) | 678 ± 69 | 483 ± 150 | 1.40 | 1106 ± 123 | 1.6 |
| HP(22) | 237 ± 6 | 109 ± 10 | 2.2 | 1466 ± 271 | 6.2 |
| HP5F | 79 ± 2 | 31 ± 3 | 2.5 | 329 ± 29 | 4.2 |
| HP5F3E | 154 ± 5 | 13 ± 2 | 11.6 | 840 ± 190 | 5.5 |
| DHP | 70 ± 2 | 32 ± 4 | 2.2 | 1518 ± 168 | 21.8 |
| DHP3F | 68 ± 3 | 39 ± 7 | 1.7 | 881 ± 52 | 13.1 |
| DHP5F(7) | 44 ± 2 | 7 ± 3 | 6.9 | 881 ± 121 | 20.1 |
| DHP5F3F(7) | 40 ± 1 | 7 ± 2 | 5.8 | 1195 ± 52 | 29.9 |
As it has been previously demonstrated that the T5FEN-catalysed steady-state multiple-turnover reaction of HP5F is rate-limited by product release 29; 30, maximal single turnover rates were also determined where possible using quenched flow apparatus. Maximal single turnover rates were measured using enzyme concentrations that were at least 8-fold higher than KM. The amount of enzyme was then increased until constant maximal rate was observed. The maximal single turnover rates of HP(22) and HP(20) are identical within experimental error. Small differences in the multiple turnover catalytic parameters of HP(22) and HP(20) are likely to reflect very small changes in the stability of the respective enzyme-substrate and enzyme-product complexes. Together these data suggest that a minimal hairpin substrate for T5FEN must be 20 or 22 nts in length (assumed eight or nine base pairs) and that interactions between enzyme and substrate must take place with the terminal eighth base pair of these substrates. Similar conclusions about potential interactions of PfFEN and its downstream DNA substrate have been reported 15.
Comparison of the catalytic parameters of a range of T5FEN substrates
The kinetic parameters of the T5FEN-catalysed reactions of a range of substrates of varying structure are shown in Table 2. All these substrates possess the downstream duplex HP(22) sequence. An example of the data used to construct table 2 is shown in figure 5(a). Several general trends in the steady-state catalytic parameters are evident. When compared to the minimal substrate HP(22), addition of either a 5′ single-stranded flap and/or a 3′-upstream duplex lowers the Michaelis constant. Increasing the size of the substrates also lowers the turnover number of the enzyme. A noteworthy consequence of these effects is that several substrates have similar catalytic efficiencies. Thus under conditions where blunt-ended duplexes, 5′-overhanging duplexes, nicked DNAs and “flap constructs” with a 3′-extrahelical nucleotide (HP5F, HP, DHP, DHP3F and DHP3F5F(7)) substrates are present at physiologically relevant concentrations below KM, all will react with similar efficiency.
Figure 5.

Examples of multiple turnover kinetic data used to construct tables 2 and 3. Normalised initial rates of reaction (v/[E]) were determined in triplicate at substrate concentrations around the respective KMs. Values of v/[E] were averaged and the standard error is shown as error bars. Non-linear regression fitting to the Michaelis Menten equation (equation 1 Materials and Methods) was used to generate the catalytic parameters and their associated standard errors. (a) T5 FEN catalysed hydrolysis of HP5F substrate yields catalytic parameters KM = 31 ± 3 nM, kcat = 79 ± 2 min−1. The R factor for this curve fit is 0.99. (b) (a) Af FEN catalysed hydrolysis of DHP5F3F(2) substrate yields catalytic parameters KM = 1.4 ± 0.3 μM, kcat = 104 ± 8 min−1. The R factor for this curve fit is 0.98.
The measured maximal single-turnover rates vary from 300–1500 min−1 (5–25 s−1) and are towards the higher end of values observed for other enzyme-catalysed phosphate diester hydrolysis reactions and represent a rate acceleration of approximately 1016 compared to the uncatalysed hydrolysis of phosphate diesters 33. With the exception of HP5F the maximal single-turnover rates vary only 2-fold with respect to one another and are in the region of 800–1500 min−1. This suggests that, with the exception of HP5F, T5FEN catalyses the hydrolysis of all phosphate diesters presented in the active site at equal rate regardless of substrate structure. However the maximal single-turnover rate of HP5F (329 min−1) is modestly slower than that measured for all the other substrates. This small decrease may be the consequence of subtle changes in the positioning of the scissile bond within the active site that are the result of the differing structures of the substrates.
For all the substrates tested here, maximal single turnover rates (kc) are faster than the multiple turnover number of the enzyme catalysed reaction (kcat) and product release is therefore wholly or partially rate limiting under multiple turnover conditions (Table 2). As the rates of product release are slower than the chemical step of the T5FEN-catalysed reaction, the steady state catalytic parameters kcat and KM are not true microscopic equilibrium and rate constants and therefore some caution is required in interpreting these values. In cases where kc ≫ kcat, the multiple turnover rate of the enzyme is a measure of the rate at which products are released. The ratio kc/kcat is a measure of the extent to which reaction is rate limited by product release. These values are greater for the larger substrates possessing an upstream double stranded region, which produce larger 3′-products. This reflects the greater stability of enzyme-product complexes that result from substrates that possess an upstream double stranded region.
For reactions that are rate limited by steps other than the chemical reaction under steady state conditions, KM values define the concentration of substrate required to convert half the enzyme into the form(s) preceding the rate limiting step(s) 34. Thus for the FEN catalysed reaction KM reflects the dissociation constant of ES and EP complexes and their rate of interconversion. For all substrates, with the exception of HP5F, the rate of interconversion of ES and EP (kc) is constant within the limits of experimental error. Thus KM values reflect the stabilities of ES and EP, and as might be expected, decrease with increasing size and therefore potential for interactions with the protein. Similarly, the decrease in turnover number observed with larger substrates reflects the greater stabilities of their EP complexes.
Catalytic parameters of AfFEN
The kinetic parameters of the AfFEN catalysed reactions of double hairpin variants of flap substrates are shown in Table 3. Reactions were monitored at 55 ºC to ensure that substrates remained base paired under assay conditions, although reactions of AfFEN proceed faster at more elevated temperatures 17. All other reaction conditions were identical to the phage enzyme except that 1 mM DTT was included in the buffer. AfFEN-catalysed hydrolysis of the double-flap substrate DHP5F3F(2) yields a turnover number of 104 ± 8 min−1 (Figure 5(b)). A previous determination of the catalytic parameters of AfFEN, also at 55 ºC, produced drastically lower values of maximal rates of reaction 17. However, this study did not include a reducing agent in the reaction buffer and we observed similar much lower rates of reaction when this was not present. Others have reported the stimulation of archaeal FENs when non-ionic detergents have been included in the buffer 21 and a recent publication included both detergents and DTT in the hFEN reaction mixture 20.
Table 3.
The catalytic parameters of AfFEN catalysed reactions of DHP5F(2) and DHP5F3F(2). Full experimental details are contained in the materials and methods section. Steady state parameters were determined by measuring the initial rates of reaction of substrates at concentrations around their respective KMs. In the case of DHP5F(2), the reaction did not achieve saturation at [S] = 75 μM but kcat/KM could be determined from a plot of v/[E] vs [S] when the concentration of [S] < KM (Figure 1S, supplementary information). Each set of data is the average of at least three experiments. G, the energetic penalty incurred upon removal of the 3′-extrahelical nt, was calculated as described (equation 2 Materials and Methods) 48.
| Substrate | kcat (min−1) | KM (μM) | kcat/KM (μM−1min−1) | ΔΔG (kJmol−1) |
|---|---|---|---|---|
| DHP5F(2) | >50 | >50 | 1.1 ± 0.04 | 11.5 |
| DHP5F3F(2) | 104 ± 8 | 1.4 ± 0.3 | 74.3 | - |
The value of KM observed with the corresponding single flap substrate lacking the 3′-extrahelical nucleotide (DHP5F(2)) is dramatically increased with respect to the double-flap substrate (Table 3). Whilst we were unable to determine the saturating maximal multiple turnover rate of reaction with this substrate, the normalised rate observed at 75μM DHP5F(2) (51 ± 3 min−1) is only half the kcat value of DHP5F3F(2), suggesting that a kcat value for DHP5F(2) will be close to or exceed that of the substrate possessing the 3′-extrahelical nt (Figure 1S, supplementary information).
Due to the high concentration of enzyme that would be required to achieve maximal rate under single-turnover conditions we were unable to determine whether the reactions of AfFEN are rate limited by product release, although this may not be the case at elevated temperatures. However, the large differences in catalytic efficiency observed with the flap and double-flap substrates seem likely to originate from differing stabilities of ES complex, rather than stimulation of the chemical rate of reaction by binding of the 3′-extra nucleotide. The “specificity” of the thermophilic archaeal and bacterial proteins for flap structures, over smaller substrates lacking the upstream duplex, is also likely to originate from a lack of ability to form an ES complex.
Lack of knowledge of the rate-limiting step in the AfFEN reaction, coupled with a rate-limiting product release with the phage enzyme, and the suboptimal temperature at which the AfFEN reaction had to be monitored, also complicates direct comparisons of the catalytic parameters of the phage (Table 2) and archaeal (Table 3) enzymes. However the maximal multiple turnover rate of reaction observed here in the presence of reducing agent is in line with kcat values reported for the phage enzyme, although about 10-fold less than the maximal single-turnover rates. The Michaelis constant measured for the AfFEN reaction, even with double-flap substrate DHP5F3F(2) is much greater than any measured for the phage enzymes, although those of the T5FEN are lowered to some extent by the existence of a stable EP complex. The difference in Michaelis constants for the T5 and Af enzymes with flap substrates lacking the 3′-extra nt is close to three orders of magnitude.
Discussion
An intriguing feature of the reaction of FENs is the capacity to act on a number of different substrates and catalyse both structure-specific endonucleolytic reactions of bifurcated nucleic acids structures and the 5′-exonucleolytic degradation of duplex and nicked DNA substrates. Additionally some flap endonucleases possess the ability to 5′-exonucleolytically degrade single-stranded nucleic acids under forcing conditions and the 5′-single stranded termini of bifurcated substrates under comparable conditions to those that give the endonucleolytic reactions 19; 35; 36. Moreover, any explanation of the substrate specificity of FENs must also account for the contrasting behaviour of archaeal FENs that favour the reactions of larger flap type substrates in contrast to smaller duplexes and hairpins 21. Furthermore, in the context of these multiple activities, the ability of bacterial, yeast, archaeal and mammalian FENs to produce a specific reaction with a double-flap substrate deserves consideration 21; 23–25.
To understand and rationalise the apparent multiple activities of some FENs we studied the reaction of two FENs with various substrates. Differences in observed Michealis constants but similar turnover numbers for the phage and archaeal enzymes reported here suggest the variation in the catalytic efficiency of FENs derived from differing sources with differing substrates most likely arises from their DNA binding properties. Thus the phage enzyme, in line with mammalian FENs, readily accepts blunt ended duplexes and hairpins as its minimal substrate, whereas the archaeal FENs, require much larger DNA substrates to produce a stable ES complex. It therefore seems likely that the large rate enhancements brought about by addition of an upstream duplex, previously reported to be 60,000-fold for Af and PfFENs 21, originate from stability of ES complex rather than any stimulation in the rate of phosphate diester hydrolysis as a consequence of forming additional interactions with the substrate. Similarly, the addition of a 3′-extrahelical nt, that results in a specific reaction and is reported to stimulate the activity of bacterial, archaeal, yeast and mammalian FENs does so because the FEN-DNA interaction is improved leading to a greater rate of reaction at lower concentrations of substrate, rather than an acceleration of the rate constant.
In contrast with the results obtained here where the AfFEN reaction occurred only at one site with double flap substrates (DHP5F3F(7) and DHP5F3F(2)), reactions of T5FEN occurred at multiple sites with these substrates. In the case of AfFEN crystallographic studies have revealed that interactions are formed between the protein and the phosphate diester and 3′-hydroxyl group of this extrahelical 3′ nt (Figure 2(b)) 9. This 3′-extrahelical nt binding site is a feature of the FENs of higher organisms, as similar specificities have been observed with mammalian FENs and mutation of the residues that contact the extrahelical nt relax this specificity 24. Interactions with the 3′-extrahelical nt presumably favour only a single binding mode of the substrate, by stabilising it in preference to all others. In the case of the substrate used in this study, removal of the 3′-extrahelical nt incurs an energetic penalty of 11.5 kJmol−1, consistent with the nature of the interactions observed in the X-ray structure. The regions of the Af protein that interact with the 3′-extrahelical nt are absent in the smaller bacteriophage FEN (Figure 2), explaining the failure to observe a specific reaction with double flap substrates and the phage enzyme.
Models of the interaction of FENs with their DNA substrates suggest three binding sites for structural elements of their flap DNA substrates. One region of the protein interacts with duplex DNA downstream of the scissile bond, a second region interacts with 3′-upstream DNA and a third region is responsible for contacts with the single stranded region of bifurcated structures (Figure 2(a)). In T5FEN the downstream DNA duplex is contacted by Lys 241, Lys 215 and Arg 216 (blue spheres Figure 2(a))14. Similarly located residues have been implicated in downstream substrate binding in hFEN 13. The upstream binding residues of AfFEN have been visualised crystallographically (red spheres Figure 2(a)) 9. In accord with these proposals are the finding that T5FEN R216A and K215A mutant proteins have significantly impaired catalytic efficiency compared to the wild type enzyme when HP5F is employed as a substrate, defining the downstream binding surface and confirming that HP5F occupies this site 14. The analogues of simple duplex substrates used in this study, HP, presumably also bind in this site indicating that only interaction with this portion of the protein is strictly necessary for catalysis. Whilst reaction of HP5F is formerly designated as an endonucleolytic activity whereas reaction of HP(22) is a 5′-exonucleolytic reaction, here we show that these reactions share the common feature that a preferential reaction occurs 1 nt into the double stranded region of the substrate, although both of these substrates have additional minor reaction sites. Thus the structure specific endonucleolytic activity of 5′-overhanging duplex and hairpin substrates and the exonucleolytic behaviour of FENs on duplex substrates can be explained by occupation of the same downstream duplex binding site. In contrast the nicked DNA substrates DHP and DHP3F, must occupy both the duplex binding regions, as must all the other substrates used in this study.
The 5′-single stranded DNA interaction site of FENs involves the region of the protein that contains the helical arch or clamp. Basic residues at the base of the helical arch of T5 and DNA Polymerase I FENs have been implicated in contacting the DNA substrate 25; 28; 37. The active site of FENs lies at the base of the helical arch or clamp (Figure 2(a)). Earlier models of the T5FEN-DNA interaction threaded single stranded DNA through the arch 5; 14. However the recent observation that hFEN can catalyse structure specific cleavage of bifurcated DNA structures that have an oligodeoxynucleotide hybridised to their single stranded regions and therefore no free 5′-terminus20, and that T5FEN can cleave closed circular DNA substrates 36; 38; 39, suggests that threading cannot be operational in these substrates. It would therefore appear more likely that 5′-single-stranded DNA passes in front of the helical arch, or is clamped by the helical arch folding over the DNA. Another possibility is the arch could fold backwards, or become disordered, allowing DNA to pass over. The observation of small amounts of reaction of HP(22) and DHP, two nts into the double stranded region, a reaction site not observed with any of the substrates possessing a 7 nt 5′-flap, suggests that placing DNA, even if it is base paired, in the “flap” binding site may be beneficial to interaction especially as with these substrates this must occur with the penalty of shortening the amount of duplex present in the downstream binding site. T5FEN reaction further into the duplex region also occured with HP(22) when binding and reaction is inhibited at the site one nt into the double stranded region by inclusion of a phosphorothioate linkage. Beneficial interactions in both the T5FEN and AfFEN flap-binding site may also extend further as reaction two nts into the double stranded is region is also observed with substrates with a shorter two nt 5′-overhang (DHP5F(2)).
Flap endonucleases have an absolute requirement for a divalent metal ion cofactor. The maximal single turnover rate of the T5 enzyme is proportional to the concentration of metal bound hydroxide, or its ionic equivalent, at pH values below the pKa of metal bound water within the FEN active site 29. It is also likely that metal ion(s) play the role of Lewis acid catalysis or leaving group activation in the FEN catalysed reaction, requiring the direct association of metal ion(s) to one of the non-bridging oxygens or the leaving group oxygen of the scissile bond 29. Thus phosphate diester hydrolysis must involve intimate contact between one or more metal ions and the cleaved phosphate diester. Most X-ray structures of flap endonucleases contain two divalent metal ions that are liganded by conserved active site carboxylates, located at the base of a helical arch or clamp 20–25. The separation of the metal ions differ in the various structures but generally exceeds the less than 4 Å separation demanded by nucleases that adopt a “two metal ion mechanism” 5; 7–11. Most studies support the view that metal one, closest to the helical arch or clamp, is absolutely required for reaction 25; 40–42. Thus models of the catalytically active FEN-substrate interaction require the scissile phosphate diester bond to be located adjacent to metal site one.
Multiple initial reaction sites are observed with all the DNA substrates utilised in this study with T5FEN and with all AfFEN substrates lacking the 3′-extrahelical nt. Furthermore all of these reaction products appear to originate from a reaction of the intact substrate used in these experiments as a subsequent reaction of any 5′-FAM labelled products of the FEN catalysed reaction can be ruled out as a single stranded 8 mer is not a cleaved by either FEN under the reaction conditions used. To account for the varying specificities observed, we propose that each of the DNA substrates is capable of subtly different modes of interaction with FEN within the confines of occupying the global substrate binding areas identified in previous models. The interaction modes place different phosphate diester linkages that become the scissile bond in intimate contact with metal one. The cleavage specificity of T5FEN with HP5F could be explained by a small change in position of the bound duplex region within the downstream binding site. The most favoured interaction mode of this substrate must be one that places metal one in contact with the phosphate diester one nt into the double stranded region, as the greatest amount of reaction occurs at this site. Other binding modes that place the two 5′-adjacent phosphates of the single stranded flap in intimate contact with metal one result from placing the duplex region of the substrate further away from the active site. Both of these less favoured binding modes, would still allow duplex DNA to interact with Lys 241, Lys 215 and Arg 216 in T5FEN.
Adding a further increment of potential binding energy in the form of either a single stranded 3′-extension (HP5F3E) or an upstream duplex (DHP5F and DHP5F3F) changes the specificity of the observed reactions of the substrate with T5FEN, leading to further reactions within the single stranded region. A similar lack of specificity is observed with AfFEN and the flap substrates lacking a 3′-extrahelical nt. This presumably arises because interactions with the upstream binding site stabilise additional binding modes where the downstream duplex is moved further from the active site under the reaction conditions. The flexibility of the 3′-single stranded extension in HP5F3E may allow changes in the position of the downstream DNA to be accommodated whilst still realising interactions with the upstream binding site. Despite the greater rigidity of the upstream duplex in the flap substrates compared to a 3′-single strand, we suppose it can adjust position by stabilising various interactions with the downstream DNA within its binding site, perhaps by changing the angle subtended by the two duplex regions.
Results obtained here with flap (DHP5F(7) and DHP5F3F(7)) and pseudo-Y (HP5F3E) substrates contrast with results obtained for larger two or three stranded variants of these type of substrates with T5FEN 14; 42. Enzymatic hydrolyses of these larger substrates generate initial endonucleolytic products that result from reactions around the region of the bifurcation and smaller exonucleolytic products of 3 and 5 in length, but no products of intermediate length. The length of the 5′-single stranded flaps used in these larger substrates is typically in the region of 20 nts. The difference between endonucleolytic and exonucleolytic reactions is not obvious in the substrates used in this study that have smaller single-stranded regions of 7 nt. The exonucleolytic reactions observed with substrates with longer single stranded regions may involve a looping of the 5′-single-stranded DNA, to position the favoured reaction sites located towards the 5′-terminus in the enzyme active site. A looping of the single-stranded DNA portion of bifurcated substrates has previously been suggested as a pre-requisite for threading through the helical arch 25. However, such a looping could still occur to position the exonucleolytic reaction sites over metal site one without the need to invoke threading. An alternative binding mode that positions the exonucleolytic scissile phosphate diesters close to metal site one without looping must eventually result in no interaction between the downstream double stranded DNA and the enzyme. Indeed such a looping could also be the origin of the smaller products observed in this study. Further work is required to clarify the reasons for the preferred reaction sites in substrates with larger single stranded regions.
An important finding of this work is that for all the substrates tested with T5FEN, maximal single-turnover rates are greater than those measured in the steady state, indicating that product release is the slowest step in the T5FEN reaction pathway for all substrates. A consequence of this is that simple interpretation of steady-state catalytic parameters, which assume a Michaelis Menten type mechanism are inappropriate. As a result of a rate limiting product release, the measured Michaelis constant is lower than the true dissociation constant of the ternary (protein-metal ions-DNA) complex and similarily the measured turnover number can severely underestimate the rate of the chemical reaction. Furthermore, analysis of altered enzymes and variation in reaction conditions that rely solely on the determination of steady state parameters may well produce misleading results. For example, a ten fold decrease in the rate of the chemical reaction of any of the larger substrates containing an upstream duplex, would produce a negligible change in the observed value of kcat, if the rate of product release remained the same.
At least one other FEN reaction, that of the 5′-nuclease domain of DNA polymerase I, is rate limited by product release with flap type substrates 25. It seems likely that this may be a common feature of FEN reactions, especially those that take place at 37ºC. Mammalian FENs possess binding sites for other proteins involved in DNA replication and repair, such as the Werner and Bloom syndrome helicases that stimulate the multiple turnover rates of the enzymes 43; 44. One possibility is that the binding of these proteins may stimulate release of the FEN reactions products. The FENs of archaea, yeast and mammals contain interaction sites for the sliding clamp proliferating cell nuclear antigen (PCNA), but these are not present in the phage enzymes 9; 11; 45–47. Addition of PCNA to reactions catalysed by these FENs has been demonstrated to stimulate activity. One possibility is that PCNA interaction stabilises ES complexes of archaeal proteins, although further experiments are required to clarify whether this is the case.
Materials and Methods
T5FEN and AfFEN were purified to homogeneity as described 17; 35.
Synthesis, purification and characterisation of 5′-FAM-labelled substrates
The fluorescent oligonucleotide substrates were synthesised using an ABI model 394 DNA/RNA synthesiser using 5′-fluorescein (FAM) reagent (Glen Research via Cambio) and standard reagents and conditions for other base additions. Following deprotection, the oligonucleotides were purified by RP HPLC (Alltech Alphabond C18 column; buffer A, 100 mM triethylammonium acetate (TEAAc) pH 6.5, 5% (v/v) acetonitrile; buffer B, 100 mM TEAAc pH 6.5, 65% acetonitrile) and then desalted using NAP-10 columns.
Phosphorothioate containing oligomers (5′-FAM-GsAACACACGCTTGCGTGTGTTC (psHP) and 5′-pGsAACACACGCTTGCGTGTGTTC) were synthesised using Beaucage sulfurising reagent (Glen Research via Cambio). Chemical phosphorylating reagent (Glen Research via Cambio) was used for 5′-phosphorylation of the 3′-phosphorothioate fragment of psHP5F. The fast and slow isomers of oligonucleotides containing phosphorothioate linkages were purified by RP HPLC (Waters μBondapak C18; buffer A, 100 mM triethylammonium acetate (TEAAc) pH 6.5, 5% (v/v) acetonitrile; buffer B, 100 mM TEAAc pH 6.5, 65% acetonitrile). For resolution of diastereoisomers of psHP t= 0 min 0% B, t= 30 min 100%B; retention times psHP fast 13.22min; ps HP slow 13.78min. For resolution of diastereoisomers of 5′-pGsAACACACGCTTGCGTGTGTTC t= 0 min 1% B, t= 5min 1% B. t= 35 min 40% B; retention times fast 20.36min; slow 21.60min. psHP5F substrates were then assembled by templated ligation of a 5′ fragment (5′-FAM-CGCTGCT-3′) and a 5′-phosphorylated 3′ fragment (5′-pGsAACACACGCTTGCGTGTGTTC-3′) annealed to a template oligonucleotide (5′-ACACGCAAGCGTGTGTTCGACAGCG-3′). The oligonucleotides were reacted in the ratio of 3 OD units 3′ fragment to 4 OD units template to 9 OD units 5′ fragment in a reaction (125 μL) containing T4 DNA ligase (2600 U; NE Biolabs) in 50 mM Tris HCl (pH 7.5 at 25°C), 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 25 μg/ml BSA for 72 hours at 16°C. The products were separated from the unligated oligonucleotides by 20% PAGE, followed by gel elution into deionised water. Following gel elution the oligonucleotide was desalted using a NAP-25 column.
All oligonucleotides were characterised by MALDI-TOF mass spectrometry and molecular weights were in good agreement with their calculated values.
Determination of the reaction specificity and steady-state parameters of FEN catalysed reactions
T5FEN
Reaction mixtures containing appropriate concentrations of FAM labelled oligonucleotides in 25 mM potassium glycinate pH 9.3, 50 mM KCl, 0.01 mg/mL BSA, 10 mM MgCl2 were heated to 95 °C for 1 min and cooled to 37°C to pre-fold the substrate. Enzyme stock was diluted in 125 mM potassium glycinate pH 9.3, 250 mM KCl, 0.05 mg/ml BSA and 50 mM MgCl2, and kept on ice until used. Reactions were initiated by addition of enzyme. Final concentrations of 10–1000 nM substrate (100 nM–160 μM for HP(18) and 25–3600 nM for HP(20)) were used. Enzyme concentrations ranging from and 3–200 pM were employed depending on the substrate under study. In the case of HP(18), where the KM was drastically raised, concentrations ranging from 4–100 nM enzyme were used. Aliquots were removed at eight time intervals and quenched with an equal volume of 20 mM EDTA. dHPLC (Wave® fragment analysis system with a fluorescence detector; Transgenomic, Warrington, UK) was used to separate and quantify the fluorescent product from the labelled substrate. A DNA Sep® column was used with the following buffers: buffer A, 2.5 mM tetrabutyl ammonium bromide, 1 mM EDTA, 0.1% acetonitrile; buffer B, 2.5 mM tetrabutyl ammonium bromide, 1 mM EDTA, 70% acetonitrile; gradient, t = 0 min, 5% B; t = 9 min, 50% B; t = 13.5 min, 100% B; retention times of products, fluorescent 8mer product 9.5 min, fluorescent 7mer product 9.1 min, fluorescent 6mer product 8.7 min, fluorescent 5mer product 8.0 min, fluorescent 4mer product 7.5 min, fluorescent 3mer product 6.8 min, fluorescent 2mer product 6.2 min, fluorescent 1mer product 5.6 min. Initial rates of reaction at the various substrate concentrations were determined and the kinetic parameters were calculated by non-linear regression fitting of the data to the Michaelis Menten equation.
| (equation 1) |
All curve fitting was carried out using Kaleidagraph software (Synergy Software, Reading, PA).
A comparison of the reaction of specificity of T5FEN with the varying substrates was carried out at 100 nM substrate and 100 pM enzyme.
AfFEN
Experiments with AfFEN were carried out essentially as described for the phage enzyme except that reactions were carried out at 55°C and DTT (final concentration 1 mM) was added after substrate denaturation and refolding. The specificity of the reaction was studied at concentrations of 8 μM substrate and 0.1 μM enzyme 25 mM potassium glycinate pH 9.3, 50 mM KCl, 0.01 mg/ml BSA, 10 mM MgCl2, 1 mM DTT at 55°C. Kinetic parameters were assessed using final substrate concentrations 0.05 μM – 75 μM and enzyme of 0.1 nM – 10 nM. The energetic penalty incurred by removal of the 3′ extrahelical nt ( G) was calculated using equation 2.
| (equation 2) |
Single turnover rapid quench experiments
Rapid quench experiments were carried out at 37 °C using a RQF-63 device from Hi-Tech Ltd (Salisbury, UK). An 80 μl aliquot of enzyme in reaction buffer (25 mM potassium glycinate (pH 9.3), 10 mM MgCl2, 50 mM KCl, 0.01 mg/ml BSA) was mixed with an equal volume of oligonucleotide substrate in reaction buffer. Enzyme was used at final concentrations of at least 8 X KM and then increased until a constant maximal rate of reaction was observed. Oligonucleotide substrates were used at a final concentration of 1 nM. After a controlled time delay of 9.1 milliseconds to 6.4 seconds, 80 μl of quench (1.5 M sodium hydroxide, 20 mM EDTA) was added. Solutions of enzyme, substrate and quench were held in a thermostated bath within the instrument during the reactions. The quenched reaction mixtures were recovered from the sample recovery loop and were analysed using denaturing HPLC as described previously for the steady-state analyses. The first-order rate of the reaction was determined by plotting the appearance of product against time and by non-linear regression fitting to the equation:
| (equation 3) |
where Pt is the amount of product at time t, P∞ is the amount of product at time ∞, and kc is the maximal rate of reaction under single turnover conditions.
MALDI-TOF mass spectral analysis of cleavage products
Substrate (HP5F, 100 μM) was incubated at 37 °C in a solution of 25 mM potassium glycinate (pH 9.3), 50 mM KCl, and 10 mM MgCl2. T5FEN was then added to a concentration of 80 nM to initiate the reaction. Aliquots of reaction mixture were taken at intervals from five to 60 minutes, quenched by the addition of equal volume of 20 mM EDTA. Salts were removed using Dowex beads (NH4+ form) and the sample was mixed with matrix (50 mg/ml 3-hydroxypicolinic acid 3:1 H2O:CH3CN). Samples were then submitted to MALDI-TOF mass spectrometry in positive ion mode on a Bruker Reflex III instrument to confirm their identities.
Reaction of HP5F:
HP5F calc. 9398.24 found 9401; product 21mer calc. 6477.19 found 6480; product FAM-8mer calc. 2939.07, found 2942; product FAM-7mer calc. 2609.86, found 2612; product FAM-6mer calc. 2320.68, found 2323. No peaks were observed corresponding to 22 mer or 23 mer products.
Supplementary Material
Acknowledgments
We thank Elaine Frary for expert technical assistance and Mr. Simon Thorpe for recording the mass spectra. This work was funded by BBSRC grant nos. B19316, B20079, B19466 and REI18458, a National Institutes of Health grant CA081967 (to JAT) and the Structural Cell Biology of DNA Repair Program Grant (P01 CA92584 to JAT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ryan Williams, Centre for Chemical Biology, Department of Chemistry, Krebs Institute, University of Sheffield, Sheffield, S3 7HF, UK.
Blanka Sengerová, Centre for Chemical Biology, Department of Chemistry, Krebs Institute, University of Sheffield, Sheffield, S3 7HF, UK.
Sadie Osborne, Centre for Chemical Biology, Department of Chemistry, Krebs Institute, University of Sheffield, Sheffield, S3 7HF, UK.
Karl Syson, Centre for Chemical Biology, Department of Chemistry, Krebs Institute, University of Sheffield, Sheffield, S3 7HF, UK.
Sophie Ault, Centre for Chemical Biology, Department of Chemistry, Krebs Institute, University of Sheffield, Sheffield, S3 7HF, UK.
Anna Kilgour, University of Sheffield School of Medicine and Biomedical Science, Henry Wellcome Laboratories for Medical Research, Beech Hill Rd., Sheffield, S10 2RX, UK..
Brian R Chapados, Skaggs Institute for Chemical Biology, The Scripps Research Institute, Department of Molecular Biology - MB4, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA.
John A Tainer, Skaggs Institute for Chemical Biology, The Scripps Research Institute, Department of Molecular Biology - MB4, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA.
Jon R Sayers, University of Sheffield School of Medicine and Biomedical Science, Henry Wellcome Laboratories for Medical Research, Beech Hill Rd., Sheffield, S10 2RX, UK..
Jane A Grasby, Centre for Chemical Biology, Department of Chemistry, Krebs Institute, University of Sheffield, Sheffield, S3 7HF, UK.
References
- 1.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 2.Pickering TJ, Garforth SJ, Thorpe SJ, Sayers JR, Grasby JA. A single cleavage assay for T5 5′->3′ exonuclease: determination of the catalytic parameters for wild-type and mutant proteins. Nucl Acids Res. 1999;27:730–735. doi: 10.1093/nar/27.3.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieber MR. Bioessays. Vol. 19. 1997. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair; pp. 233–240. [DOI] [PubMed] [Google Scholar]
- 4.Lyamichev V, Brow MAD, Dahlberg JE. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science. 1993;260:778–783. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 5.Ceska TA, Sayers JR, Stier G, Suck D. A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature. 1996;382:90–93. doi: 10.1038/382090a0. [DOI] [PubMed] [Google Scholar]
- 6.Kim Y, Eom SH, Wang JM, Lee DS, Suh SW, Steitz TA. Crystal structure of Thermus aquaticus DNA polymerase. Nature. 1995;376:612–616. doi: 10.1038/376612a0. [DOI] [PubMed] [Google Scholar]
- 7.Mueser TC, Nossal NG, Hyde CC. Structure of bacteriophage T4 RNase H, a 5′ to 3′ RNA-DNA and DNA-DNA exonuclease with sequence similarity to the Rad2 family of eukaryotic proteins. Cell. 1996;85:1101–1112. doi: 10.1016/s0092-8674(00)81310-0. [DOI] [PubMed] [Google Scholar]
- 8.Hosfield DJ, Mol CD, Shen BH, Tainer JA. Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: Coupling DNA and PCNA binding to FEN-1 activity. Cell. 1998;95:135–146. doi: 10.1016/s0092-8674(00)81789-4. [DOI] [PubMed] [Google Scholar]
- 9.Chapados BR, Hosfield DJ, Han S, Qiu JZ, Yelent B, Shen BH, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 10.Hwang KY, Baek K, Kim HY, Cho Y. The crystal structure of flap endonuclease I from Methanococcus jannaschii. Nat Struct Biol. 1998;5:707–713. doi: 10.1038/1406. [DOI] [PubMed] [Google Scholar]
- 11.Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, Uchida M, Ohtsuka E, Morioka H, Hakoshima T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005;24:683–693. doi: 10.1038/sj.emboj.7600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsui E, Musti KV, Abe J, Yamasaki K, Matsui I, Harata K. Molecular structure and novel DNA binding sites located in loops of flap endonuclease-1 from Pyrococcus horikoshii. J Biol Chem. 2002;277:37840–37847. doi: 10.1074/jbc.M205235200. [DOI] [PubMed] [Google Scholar]
- 13.Qiu JZ, Liu R, Chapados BR, Sherman M, Tainer JA, Shen BH. Interaction interface of human flap endonuclease-1 with its DNA substrates. J Biol Chem. 2004;279:24394–24402. doi: 10.1074/jbc.M401464200. [DOI] [PubMed] [Google Scholar]
- 14.Dervan JJ, Feng M, Patel D, Grasby JA, Artymiuk PJ, Ceska TA, Sayers JR. Interactions of mutant and wild-type flap endonucleases with oligonucleotide substrates suggest an alternative model for DNA binding. Proc Natl Acad Sci USA. 2002;99:8542–8547. doi: 10.1073/pnas.082241699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allawi HT, Kaiser MW, Onufriev AV, Ma WP, Brogaard AE, Case DA, Neri BP, Lyamichev VI. Modeling of flap endonuclease interactions with DNA substrate. J Mol Biol. 2003;328:537–554. doi: 10.1016/s0022-2836(03)00351-6. [DOI] [PubMed] [Google Scholar]
- 16.Harrington JJ, Lieber MR. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 1994;13:1235–1246. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosfield DJ, Frank G, Weng YH, Tainer JA, Shen B. Newly discovered archaebacterial flap endonucleases show a structure-specific mechanism for DNA substrate binding and catalysis resembling human flap endonuclease-1. J Biol Chem. 1998;273:27154–27161. doi: 10.1074/jbc.273.42.27154. [DOI] [PubMed] [Google Scholar]
- 18.Bhagwat M, Hobbs LJ, Nossal NG. The 5′-exonuclease activity of bacteriophage T4 RNase H is stimulated by the T4 gene 32 single-stranded DNA-binding protein, but its flap endonuclease is inhibited. J Biol Chem. 1997;272:28523–28530. doi: 10.1074/jbc.272.45.28523. [DOI] [PubMed] [Google Scholar]
- 19.Garforth SJ, Sayers JR. Structure-specific DNA binding by bacteriophage T5 5′->3′ exonuclease. Nucl Acids Res. 1997;25:3801–3807. doi: 10.1093/nar/25.19.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu R, Qiu JZ, Finger LD, Zheng L, Shen BH. The DNA-protein interaction modes of FEN-1 with gap substrates and their implication in preventing duplication mutations. Nucl Acids Res. 2006;34:1772–1784. doi: 10.1093/nar/gkl106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaiser MW, Lyamicheva N, Ma WP, Miller C, Neri B, Fors L, Lyamichev VI. A comparison of eubacterial and archaeal structure-specific 5′-exonucleases. J Biol Chem. 1999;274:21387–21394. doi: 10.1074/jbc.274.30.21387. [DOI] [PubMed] [Google Scholar]
- 22.Lyamichev VI, Brow MA, Varvel VE, Dahlberg JE. Comparison of the 5′ nuclease activities of Taq DNA polymerase and its isolated nuclease domain. Proc Natl Acad Sci USA. 1999;96:6143–6148. doi: 10.1073/pnas.96.11.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao KI, Henricksen LA, Liu Y, Bambara RA. Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J Biol Chem. 2002;277:14379–14389. doi: 10.1074/jbc.M110662200. [DOI] [PubMed] [Google Scholar]
- 24.Friedrich-Heineken E, Hubscher U. The Fen1 extrahelical 3′-flap pocket is conserved from archaea to human and regulates DNA substrate specificity. Nucl Acids Res. 2004;32:2520–2528. doi: 10.1093/nar/gkh576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Potapova O, Leschziner AE, Grindley NDF, Joyce CM. Contacts between the 5′ nuclease of DNA polymerase I and its substrate DNA. J Biol Chem. 2001;276:30167–30177. doi: 10.1074/jbc.M100985200. [DOI] [PubMed] [Google Scholar]
- 26.Qiu JZ, Bimston DN, Partikian A, Shen BH. Arginine residues 47 and 70 of human flap endonuclease-1 are involved in DNA substrate interactions and cleavage site determination. J Biol Chem. 2002;277:24659–24666. doi: 10.1074/jbc.M111941200. [DOI] [PubMed] [Google Scholar]
- 27.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucl Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pickering TJ, Garforth SJ, Sayers JR, Grasby JA. Variation in the steady state kinetic parameters of wild-type and mutant T5 5′-3′ exonuclease with pH. Protonation of Lys-83 is critical for DNA binding. J Biol Chem. 1999;274:17711–17717. doi: 10.1074/jbc.274.25.17711. [DOI] [PubMed] [Google Scholar]
- 29.Tock MR, Frary E, Sayers JR, Grasby JA. Dynamic evidence for metal ion catalysis in the reaction mediated by a flap endonuclease. EMBO J. 2003;22:995–1004. doi: 10.1093/emboj/cdg098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel DV, Tock MR, Frary E, Feng M, Pickering TJ, Grasby JA, Sayers JR. A conserved tyrosine residue aids ternary complex formation but not catalysis, in phage T5 flap endonuclease. J Mol Biol. 2002;320:1025–1035. doi: 10.1016/s0022-2836(02)00547-8. [DOI] [PubMed] [Google Scholar]
- 31.Elliott SL, Brazier J, Cosstick R, Connolly BA. Mechanism of the Escherichia coli DNA T : G-mismatch endonuclease (Vsr protein) thiophosphate-containing probed with oligodeoxynucleotides. J Mol Biol. 2005;353:692–703. doi: 10.1016/j.jmb.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 32.Thorogood H, Grasby JA, Connolly BA. Influence of the phosphate backbone on the recognition and hydrolysis of DNA by the EcoRV restriction endonuclease - A study using oligodeoxynucleotide phosphorothioates. J Biol Chem. 1996;271:8855–8862. doi: 10.1074/jbc.271.15.8855. [DOI] [PubMed] [Google Scholar]
- 33.Schroeder GK, Lad C, Wyman P, Williams NH, Wolfenden R. The time required for water attack at the phosphorus atom of simple phosphodiesters and of DNA. Proc Natl Acad Sci USA. 2006;103:4052–4055. doi: 10.1073/pnas.0510879103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutfreund F. Kinetics for the Life Sciences. Cambridge University Press; Cambridge: 1995. [Google Scholar]
- 35.Sayers JR, Eckstein F. Properties of overexpressed phage T5 D15 exonuclease - similarities with Escherichia coli DNA polymerase I 5′-3′ exonuclease. J Biol Chem. 1990;265:18311–18317. [PubMed] [Google Scholar]
- 36.Sayers JR, Eckstein F. A single-strand specific endonuclease activity copurifies with overexpressed T5 D15 exonuclease. Nucl Acids Res. 1991;19:4127–4132. doi: 10.1093/nar/19.15.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garforth S, Ceska T, Suck D, Sayers J. Mutagenesis of conserved lysine residues in T5 5′-3′ exonuclease suggests separate mechanisms of endo-and exonucleolytic cleavage. Proc Natl Acad Sci USA. 1999;96:38–43. doi: 10.1073/pnas.96.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sayers JR, Evans DJBT. Identification and eradication of a denatured DNA isolated during alkaline lysis-based plasmid purification procedures. Anal Biochem. 1996;241:186–9. doi: 10.1006/abio.1996.0397. [DOI] [PubMed] [Google Scholar]
- 39.Kiss-Toth E, Dower SK, Sayers JR. A method for enhancing the transfection efficiency of minipreps obtained from plasmid cDNA libraries. Anal Biochem. 2001;288:230–232. doi: 10.1006/abio.2000.4858. [DOI] [PubMed] [Google Scholar]
- 40.Xu Y, Derbyshire V, Ng K, Sun XC, Grindley NDF, Joyce CM. Biochemical and mutational studies of the 5′-3′ exonuclease of DNA polymerase I of Escherichia coli. J Mol Biol. 1997;268:284–302. doi: 10.1006/jmbi.1997.0967. [DOI] [PubMed] [Google Scholar]
- 41.Shen BH, Nolan JP, Sklar LA, Park MS. Functional analysis of point mutations in human flap endonuclease-1 active site. Nucl Acids Res. 1997;25:3332–3338. doi: 10.1093/nar/25.16.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng M, Patel D, Dervan JJ, Ceska T, Suck D, Haq I, Sayers JR. Roles of divalent metal ions in flap endonuclease-substrate interactions. Nat Struct Mol Biol. 2004;11:450–456. doi: 10.1038/nsmb754. [DOI] [PubMed] [Google Scholar]
- 43.Sharma S, Sommers JA, Wu L, Bohr VA, Hickson ID, Brosh RM. Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J Biol Chem. 2004;279:9847–9856. doi: 10.1074/jbc.M309898200. [DOI] [PubMed] [Google Scholar]
- 44.Brosh RM, Driscoll HC, Dianov GL, Sommers JA. Biochemical characterization of the WRN-FEN-1 functional interaction. Biochemistry. 2002;41:12204–12216. doi: 10.1021/bi026031j. [DOI] [PubMed] [Google Scholar]
- 45.Wu XT, Li J, Li XY, Hsieh CL, Burgers PMJ, Lieber MR. Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucl Acids Res. 1996;24:2036–2043. doi: 10.1093/nar/24.11.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomes XV, Burgers PMJ. Two modes of FEN1 binding to PCNA regulated by DNA. EMBO J. 2000;19:3811–3821. doi: 10.1093/emboj/19.14.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tom S, Henricksen LA, Bambara RA. Mechanism whereby proliferating cell nuclear antigen stiumlates flap endonulcease 1. J Biol Chem. 2000;275:10498–10505. doi: 10.1074/jbc.275.14.10498. [DOI] [PubMed] [Google Scholar]
- 48.Fersht AR. Enzyme Structure and Mechanism. 2. W.H. Freeman and Company; New York: 1985. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.