

Abstract
Base excision repair (BER) proteins act upon a significantly broad spectrum of DNA lesions that result from endogenous and exogenous sources. Multiple sub-pathways of BER (short-path or long-patch) and newly designated DNA repair pathways (e.g., SSBR and NIR) that utilize BER proteins complicate any comprehensive understanding of BER and its role in genome maintenance, chemotherapeutic response, neurodegeneration, cancer or aging. Herein, we propose a unified model of BER, comprised of three functional processes: Lesion Recognition/Strand Scission, Gap Tailoring and DNA Synthesis/Ligation, each represented by one or more multiprotein complexes and coordinated via the XRCC1/DNA Ligase III and PARP1 scaffold proteins. BER therefore may be represented by a series of repair complexes that assemble at the site of the DNA lesion and mediates repair in a coordinated fashion involving protein-protein interactions that dictate subsequent steps or sub-pathway choice. Complex formation is influenced by post-translational protein modifications that arise from the cellular state or the DNA damage response, providing an increase in specificity and efficiency to the BER pathway. In this review, we have summarized the reported BER protein-protein interactions and protein post-translational modifications and discuss the impact on DNA repair capacity and complex formation.
1. Introduction of a unifying BER model
We propose a unifying model for base excision repair (BER) that describes the processing of a multitude of DNA lesions conducted by BER proteins. This model includes repair of classical oxidation and alkylation base modifications and encompasses the repair of single-strand breaks (processed through a single-strand break sub-pathway), oxidative lesions processed via alternate sub-pathways such as NIR and repair initiated by the NEIL-family of bi-functional DNA glycosylases. As will be described, BER functions via a series of transient repair complexes that assemble at the site of the DNA lesion [1]. As the lesion is processed, additional proteins are recruited and exchanged to advance the repair process [2]. BER protein complex formation is further influenced by post-translational protein modifications that provide an increase in specificity and efficiency to the BER pathway.
BER is the predominant DNA damage repair pathway for the processing of small base lesions, derived from oxidation and alkylation damage. An estimated rate of 104 damaging events/mammalian cell/day underscores the importance of the BER pathway [3-5]. BER is normally defined as DNA repair initiated by a lesion-specific DNA glycosylase (mono- or bi-functional) and completed by either of two sub-pathways: short-patch BER; a mechanism whereby only 1 nucleotide is replaced or long-patch BER; a mechanism whereby 2-13 nucleotides are replaced. The majority of repair is currently thought to occur via the short-patch pathway initiated by either a mono-functional or bi-functional glycosylase. The paradigm for the short-patch BER pathway initiated by a mono-functional glycosylase involves base lesion removal and then AP site hydrolysis by AP endonuclease (APE1) [6], catalyzing the incision of the damaged strand, leaving a 3’OH and a 5’deoxyribose-phosphate moiety (5’dRP) at the margins. DNA polymerase ß (pol ß) hydrolyzes the 5’dRP moiety and fills the single nucleotide gap, preparing the strand for ligation by either DNA Ligase I (LigI) or a complex of DNA Ligase IIIα (LigIIIα) and XRCC1 (Fig. 1).
Fig. 1. Classic BER pathway.
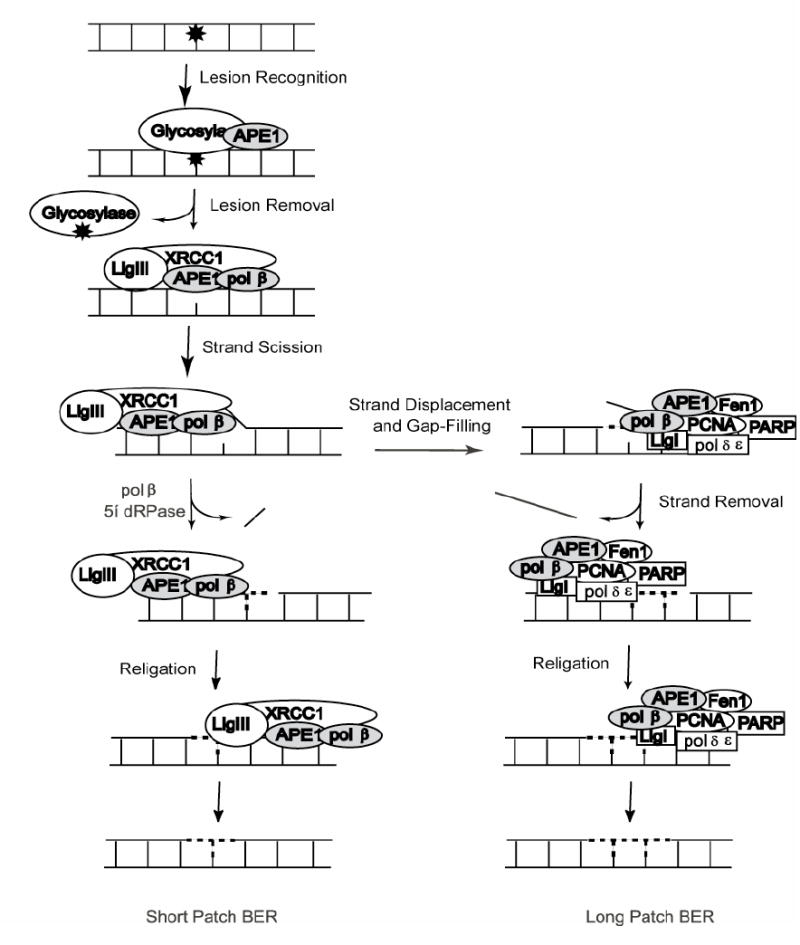
Short-patch pathway (left) depicts sub-pathway initiation by glycosylase activity followed by strand scission by APE1. Gap tailoring (5’dRP lyase) and nucleotide incorporation are accomplished by pol ß. The resulting nick is ligated by a complex of XRCC1 and LigIIIα to complete the pathway. Long-patch pathway (right) depicts a sub-pathway of BER responsible for repair under conditions of 5’lesions refractory to pol ß cleavage. In this case, BER complex formation shifts, nucleotide incorporation is conducted either by pol ß or is transferred to pol-δ or pol-•. The refractory 5’ moiety is removed as part of a flap of DNA by Fen1 and re-ligation is completed by LigI.
However, oxidative base lesions are primarily removed by bi-functional DNA glycosylases that also encode an additional 3’AP lyase activity. Upon recognition of a base lesion by a bi-functional DNA glycosylase, the lesion (e.g., 8-oxodG) is excised from the DNA strand in a mechanism similar to mono-functional DNA glycosylases as depicted above. However, the DNA backbone can then be incised 3’ to the damage site, leaving a 3’ α,ß-unsaturated aldehyde (after ß-elimination) and a 5’phosphate at the termini. A 3’ phosphodiesterase activity, supplied by APE1 [7], cleaves this terminus in preparation for polymerase extension by pol ß and ligation by LigI or XRCC1/LigIIIα.
Long-patch BER is initiated in a fashion similar to short-patch BER to produce a nicked DNA intermediate. Repair completion requires a 3’OH moiety for proper nucleotidyl transfer and chain elongation. In cases where the 5’lesion is refractory to pol ß lyase activity [8], polymerase δ, ε or ß, coupled with proliferating cell nuclear antigen (PCNA) and a variety of other proteins including the structure specific flap endonuclease (Fen1), poly(ADP-ribose)polymerase 1 (PARP1) and LigI synthesizes DNA to fill the gap, resulting in a displaced DNA flap of 2-13 bases in length [8-10]. DNA synthesis and strand displacement by pol ß is stimulated by the combined presence of Fen1 and PARP1 [11, 12]. The Werner Syndrome Protein helicase (WRN) is also observed to stimulate strand displacement activities of pol ß. Fen1 then catalyzes the removal of the ensuing DNA flap, leaving a nick that has been transferred 2-13 nucleotides downstream of the original damage site. Finally, the intact DNA strand is restored by LigI (Fig. 1).
Although this longstanding BER paradigm has been useful in delineating the protein players and DNA intermediates of the pathway, increasingly it has become clear that the pathway handles a broader spectrum of lesions than this paradigm can accurately depict. For example, the single-strand break repair (SSBR) pathway utilizes many of the same proteins as BER, namely APE1, pol ß and LigIIIα, along with the scaffold proteins PARP1 and XRCC1 [13]. The principal difference between SSBR and BER is the pathway initiation step (Table I). In each case repair may occur at the single nucleotide level (i.e., short-patch repair) or as a longer patch of repair. BER as outlined above is initiated via DNA glycosylase activity that results in the removal of a damaged base. SSBR, on the other hand, is defined specifically for the repair of single-strand breaks in DNA arising from irradiation, incomplete topoisomerase action or ROS byproducts of metabolism [14]. PARP1 recognizes the single-strand break, signaling recruitment of repair proteins to the damaged site [15]. One of the first proteins recruited is XRCC1, a protein closely associated with BER pathway coordination. PARP1 interacts with XRCC1 to form the protein scaffold upon which the repair complex is built [16]. Furthermore, PARP1 has been linked more directly to BER as it interacts both physically and functionally with pol ß [12, 17] and LigIIIα [18], placing it as a member of the short-patch BER pathway (Table II). In addition, PARP1 coordinates with long-patch BER proteins to facilitate the repair of longer stretches of DNA [12, 19].
Table I.
Possible entry mechanisms of a unified base excision repair model
| Substrate Recognized | Enzyme/Event Creating the Break | DNA Ends Flanking the Gap | Gap Tailoring Protein Required | Current Sub-Pathway |
|---|---|---|---|---|
| Alkylation Damage
(e.g. 3Me-adenine) |
Monofunctional Glycosylase | 3’-OH
5’-dRP |
pol ß | BER |
| Spontaneous Base Loss
(e.g. AP Site) |
APE1 | 3’-OH
5’-dRP |
pol ß | BER |
| Oxidative Damage
(e.g. 8-oxo-guanine) |
Bifunctional Glycosylase | 3’-α,ß unsaturated aldehyde
5’- PO4 |
APE1 | BER |
| Oxidative Damage
(e.g. 5-OH-cytosine) |
APE1 | 3’-OH
5’-OH cytosine residue |
FEN1 | NIR or LP BER |
| Oxidative Damage
(e.g. 5-OH-uracil) |
NEIL1/NEIL2 | 3’-PO4
5’- PO4 |
PNKP | NEIL-directed BER |
| Supercoiled DNA | Topoisomerase I malfunction | 3’-tyrosyl residue
5’-PO4 |
Tdp1 | SSBR |
| DNA | Radiation | 5’phosphate, 3’phosphate, 3’phosphoglycolate (and others)[138] | APE1
FEN1 PNKP |
SSBR |
Table II.
Protein-protein interactions among base excision repair proteins
| BER Proteins | Interacting Proteins | Citation |
|---|---|---|
| UNG1 | NDa | - |
| UNG2 | PCNA, RPA, Vpr | [139-141] |
| SMUG | Vpr | [141] |
| TDG | APE1, p300, XPC/HR23B | [26, 27, 142] |
| MBD4 | ND | |
| Aag/MPG | XRCC1, HR23A, MBD1 | [28, 143, 144] |
| OGG1 | XRCC1, CSB | [34, 145] |
| OGG2 | ND | - |
| MYH | APE1, PCNA, RPA | [32, 146] |
| MTH | ND | - |
| NTH1 | XPG | [147, 148] |
| NEIL1 | pol ß, LigIIIα | [23] |
| NEIL2 | pol ß, LigIIIα, PNKP, XRCC1 | [23] |
| NEIL3 | ND | - |
| APE1 | TDG, MYH, pol ß, LigI, Fen1, PCNA, p53, GzmA | [26, 38, 40, 42, 43, 142, 149-151] |
| APE2 | ND | - |
| pol ß | APE1, LigI, Fen1, PCNA, PARP1, PARP2, PNKP, NEIL1, NEIL2, Aprataxin, p53, Trf2, p300, XRCC1, PRMT1, PRMT6 | [12, 17, 23, 63, 66, 75, 88, 113, 119, 128, 129, 149, 150, 152-157] |
| LigI | APE1, pol ß, PCNA | [43, 152, 158, 159] |
| LigIIIα | XRCC1, PARP1, PARP2, PNKP, NEIL1, NEIL2, Aprataxin | [15, 18, 63, 66, 75, 113] |
| Fen1 | APE1, pol ß, PCNA, PARP1, p300, WRN, BLM | [12, 42, 43, 123, 160-164] |
| PCNA | APE1, pol ß, PARP1, XRCC1, p300, CSB, WRN | [19, 42, 139, 140, 146, 155, 158-161, 165-170] |
| XRCC1 | MPG, OGG1, APE1, pol ß, LigIIIα, PARP1, PARP2, PNKP, Tdp1, Aprataxin | [15, 23, 28, 34, 40, 60, 61, 63, 66, 72-76, 113] |
| PNKP | pol ß, LigIIIα, XRCC1, NEIL1, NEIL2 | [63] |
| PARP1 | pol ß, PCNA, XRCC1, p300, CSB, WRN, Ku, LigIIIα | [12, 16-19, 74, 87, 113, 157, 171-173] |
| PARP2 | pol ß, LigIIIα, PARP1, XRCC1 | [113] |
| Aprataxin | pol ß, LigIIIα, PARP1, XRCC1 | [72-76] |
| Tdp1 | XRCC1 | [61] |
ND – No interacting proteins identified to date.
Over the past several years, two additional sub-pathways of BER or alternate repair mechanisms have been described that utilize unique combinations of BER proteins to repair base damage. For example, APE1 has been found to initiate DNA glycosylase-independent repair of oxidized cytosines [20-22]. This alternate repair pathway has been defined as Nucleotide Incision Repair (NIR), yet can also be considered a minor sub-pathway of BER with a unique APE1-mediated repair initiation event. More recently, Mitra and colleagues have defined an APE1-independent BER sub-pathway for repair of oxidized bases initiated by the DNA glycosylase/AP lyases NEIL1 and NEIL2 [23, 24]. Although each of the BER sub-pathways described above are unique, each pathway is similar in several general biochemical steps, as depicted in Table I and Fig. 2. This unified BER model acknowledges the mechanistic similarities among these varied BER sub-pathways yet broadens the pathway entry points to reflect both the variety of DNA damage able to enter the pathway as well as the plethora of proteins necessary to process the resulting repair intermediates (Table I). In the proposed model (Fig. 2) the five canonical BER steps have been reorganized into three distinct phases: Lesion Recognition/Strand Scission; DNA Gap Tailoring; and DNA Synthesis/Ligation, each coordinated via the XRCC1/DNA Ligase IIIα and PARP1 scaffold protein complexes and associated protein-protein interactions (Table II and Fig. 2).
Fig. 2. A unified model of BER.
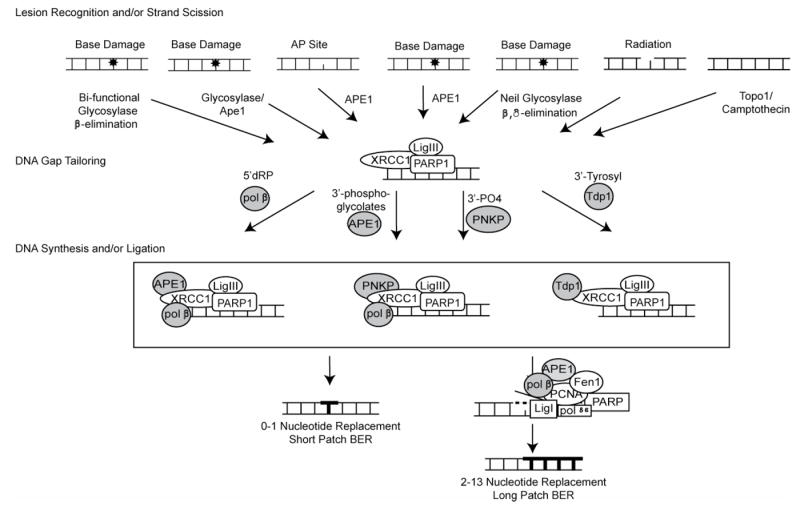
This model depicts protein complexes for repair of oxidation and alkylation base modifications and encompasses the repair of single-strand breaks, oxidative lesions processed via alternate sub-pathways such as NIR and repair initiated by the NEIL-family of bi-functional DNA glycosylases.
2. Initiation of BER by lesion recognition / strand scission
The common denominator in all of these BER sub-pathways is a single-strand break. We propose to combine all of these sub-pathways into one unifying pathway by broadening the entry points and focusing on the protein complexes formed as a function of the chemistry of the resulting repair intermediate. Thus, we propose that this unified BER pathway encompasses numerous entry points contingent on the type of damage encountered. Entry points include bi-functional glycosylase activity and 3’AP lyase activity with ß-elimination; mono-functional glycosylase activity and APE1 endonuclease incision; spontaneous base loss followed by APE1 endonuclease incision; direct APE1 endonuclease activity on oxidative damage in a glycosylase independent manner; NEIL-associated glycosylase activity followed by 3’ AP lyase activity via ß,δ-elimination; mutagen induced oxidative (e.g. irradiation) damage generating single-strand DNA breaks with a variety of possible ends; and finally incomplete topoisomerase function that freezes the covalently-bound protein onto the DNA. Each of these entry points is represented in Table I which delineates the specific DNA ends and in Fig. 2 under lesion recognition / strand scission, from left to right respectively.
2.1 Mono-functional and Bi-functional DNA glycosylases and AP-site repair
As mediators of the initial step of BER, glycosylases are critical to the recognition and removal of DNA base lesions. Their mechanism of action has been extensively studied on a number of different levels. Kinetic data, with purified protein, suggests that glycosylases may be inhibited by their own AP site products (for review, see [25]). Evaluation of protein partnerships, such as with APE1, demonstrate a stimulation of overall catalytic efficiency through glycosylase displacement resulting in an alleviation of product inhibition. Importantly, regulation of glycosylase activity via protein-protein interactions has been shown to facilitate improved lesion recognition and to ensure that initiated repair may be completed via the formation of lesion-specific repair complexes. For example, thymine DNA glycosylase (TDG) is a mono-functional DNA glycosylase that recognizes G:T and G:U mismatches, among other types of damage. TDG interacts physically with APE1 [26], the subsequent enzyme in the BER pathway. This interaction enhances the ability of TDG to excise damaged bases. Together TDG and APE1 produce the 3’hydroxyl (3’OH) and 5’deoxyribose phosphate (5’dRP). TDG activity may also be enhanced through other protein partnerships. For example, functional interactions have been detected between TDG and xeroderma pigmentosum group C protein (XPC-HR23B), where XPC-HR23B was shown to promote dissociation from AP sites, stimulating TDG turn-over (Table II) [27]. N-methylpurine DNA glycosylase (MPG) is the general DNA glycosylase for most types of DNA alkylation damage. Recently MPG was reported to interact physically with the human scaffolding protein XRCC1, stimulating activity of MPG and supporting the hypothesis that single-strand alkylation damage is processed via XRCC1 scaffold protein coordination [28].
Bi-functional glycosylases add an additional level of complexity due to their 3’ AP lyase activity, yielding a replication-blocking 3’ ß-eliminated unsaturated aldehyde and a 5’phosphate at the margins of the repair gap. Until recently, glycosylase action and catalysis to yield the ß-elimination product were thought to occur concomitantly. However, studies with bi-functional glycosylases such as human endonuclease III (NTH1) and mammalian 8-oxoguanine-DNA N-glycosylase homolog (OGG1), suggest a dissociation of the two activities. Studies of hNTH1 (activity and binding affinity to thymine glycol (Tg) containing DNA) revealed that the 3’AP lyase activity of the enzyme is dependent on the binding affinity of the enzyme to its product AP site [29]. Indeed, APE1-assisted dissociation of NTH1 was so effective in abrogating the 3’lyase activity that upon activation of APE1, only 5’dRP (APE1 product) was detectable, not the products of ß-elimination. Therefore, under physiological conditions (i.e. abundant APE1), NTH1 may act as a mono-functional glycosylase on Tg:A damaged DNA, shuttling damage towards cleavage by APE1, which has a greater capacity to cleave 5’ to the AP site than 3’ to the ß-elimination product [30]. Similarly, OGG1and MYH showed an enhancement of glycosylase activity and suppression of the glycosylase 3’ lyase function upon addition of APE1, suggesting that circumventing the 3’lyase activity is commonly observed for bi-functional glycosylases in vivo [31-33]. This reduction of the 3’lyase activity of bi-functional glycosylases is further supported by the interaction of OGG1 with XRCC1 [34]. OGG1 interacts physically and functionally with XRCC1 to stimulate base hydrolysis without inhibiting the subsequent APE1 strand incision step, resulting in an overall increase in processing of toxic intermediates through the pathway and highlighting the orchestrating abilities of XRCC1 [34]. Nevertheless, should the 3’unsaturated aldehyde present as a repair blocking lesion, APE1 3’phosphodiesterase activity has the ability to remove it to restore active repair [30].
AP sites are detrimental cellular DNA lesions that give rise to an increase in genetic mutations and other genetic rearrangements [35]. AP sites are generated through spontaneous base loss in addition to glycosylase action [5]. The sheer quantity of this lesion suggests that a tightly controlled system is necessary for accurate repair. APE1 recognizes these AP sites and incises the DNA 5’ to the lesion, leaving the 3’OH and 5’phosphate residues, as described above. Mice with a targeted homozygous null mutation in the APE1 (Ref-1) gene die during early embryonic development [36]. Detailed analysis of APE1 null embryos (pre-implantation APE1 null embryos) following ionizing radiation indicate a role for APE1 in the repair of ionizing-induced DNA damage [37]. No other APE1-deficient mammalian cellular models are available, making in vivo studies in mammalian systems challenging. The recent demonstration that siRNA may be used to down-regulate APE1 does however show promise for future studies to define the role of APE1 in mammalian cells in vivo [38]. However, complete depletion may be impossible as it is shown to be essential for cellular survival [39]. Although AP-site specific 5’endonucleolytic activity is the major function of APE1, there are a number of minor functions associated with APE1 as well, including 3’phosphodiesterase, 3’phosphoglycolase, 3’phosphatase and 3’-5’exonuclease capabilities. Many of these alternate functions participate in the gap-tailoring of 3’ blocked termini within a repair patch. Physical and functional partnerships with APE1 include a number of DNA repair proteins such as LigI, Fen1, PCNA, pol ß and most importantly, XRCC1 (see Table II). APE1 physically interacts with XRCC1, enhancing endonucleolytic incision rates and coordinating AP site repair initiation with subsequent gap-tailoring enzymes [40].
2.2 Direct APE1-mediated nucleotide incision as an initiator of BER
APE1 directly incises the damaged DNA duplex upstream of an oxidatively damaged base, leaving a 3’OH and a 5’phosphate-containing damaged nucleotide at the margins. In other words, for specific types of oxidative damage, APE1 has the ability to cleave the damaged DNA strand in a glycosylase-independent manner. This novel function of APE1 was the driving force behind the development of the alternative BER pathway called nucleotide incision repair (NIR) [41]. Although kinetic data suggest that 5-hydroxy-2’-deoxycytidine (5OH-C) residues are inefficiently repaired by DNA glycosylases NTH1 and NEIL1 coupled with either APE1 3’phosphodiesterase or 3’phosphotase activity, Daviet et. al. determined that NIR activity was predominant towards 5OH-C in cell-free extracts, presumably due to the exceptionally high concentration of APE1 in the cellular milieu [20]. The resulting 5’modified nucleotide would be a substrate for Fen1, the flap endonuclease, associated with long-patch BER. APE1 partners physically with Fen1, resulting in a marginally increased activity of Fen1 5’ flap removal [42]. Moreover, APE1 stimulated the endonucleolytic activity of Fen1 on DNA flaps of increasing length, suggesting that the APE1 influence on Fen1 catalytic capacity increases as more strand displacement occurs [43], and that repair of these lesions is more likely to be completed through long-patch BER. APE1 also stimulated the resealing capability of LigI [43] and has been demonstrated to interact physically with PCNA [42]. All of these proteins are essential to long-patch repair, implying a coordination of the long-patch pathway by APE1 and that 5OH-C damage may proceed through a long-patch BER repair mechanism as depicted in Fig. 2.
2.3 NEIL2 DNA glycosylase initiated repair
In the past several years, a distinct subdivision of bi-functional glycosylases has been reported [44, 45]. The human bi-functional glycosylases NEIL1 and NEIL2 process oxidized base damage in a fashion similar to OGG1 and NTH1, however NEIL1/2 further processes the 3’-unsaturated aldehyde through a ß,δ-elimination mechanism to yield a 3’phosphate and 5’phosphate at the margins (Table I) [46]. While APE1 can efficiently process the ß-elimination product of OGG1 and NTH1, its DNA 3’-phosphatase activity needed for the processing of the ß,δ-elimination product produced by NEIL1/2 is extremely weak [24]. Therefore NEIL-mediated repair is independent of APE1 function [24]. Instead, NEIL-mediated repair is dependent on polynucleotide kinase phosphatase (PNKP) for removal of the 3’ phosphate [24]. PNKP is a 3’DNA phosphatase and 5’DNA kinase that is traditionally associated with the SSBR pathway [47]. Finally, both NEIL1 and NEIL2 interact directly with pol ß, Lig IIIα and XRCC1 and indirectly with PNKP through formation of a larger complex with these same BER proteins [23]. NEIL-mediated repair clearly falls within the expanded BER pathway as represented in Fig. 2. NEIL glycosylases hydrolyze the oxidized lesion, leaving a 3’phosphate that directs complex formation of NEIL, pol ß, XRCC1, LigIIIα and PNKP for the subsequent gap-tailoring step.
2.4 Radiation-induced DNA damage
Single Strand Break Repair (SSBR) describes the repair of single-strand breaks generated by endogenous oxidative metabolism such as reactive oxygen species (ROS) and environmental agents such as ionizing radiation in the form of X-rays or gamma-rays. Key steps within the pathway include PARP1 recognition of and adherence to single-strand breaks. The PARP1:DNA complex then recruits the scaffold protein XRCC1. The remainder of the pathway mimics BER both in proteins involved and sub-pathway options available, i.e., short-patch and long-patch. The hallmark agent for entry into the SSBR pathway is ionizing radiation, inducing genomic DNA mutations and chromosomal aberrations. Persistent genomic instability is thought to be a critical step toward the transformation of cells to a cancerous state [48]. Ionizing radiation generates a bolus of oxygen radical in the water surrounding the DNA molecule, resulting in a clustering effect of the damage [49]. Interestingly, these radiation induced breaks are reported to exist primarily as 5’-phosphate and equal amounts of either 3’-phosphate or 3’-phosphoglycolates at the margins of the gap [50-52]. Therefore subsequent gap-tailoring would require enzymatic activity by either PNKP or APE1.
2.5 Strand-breaks generated by Topoisomerase poisons
A new entry point into BER is emerging. In this case, a trapped DNA:topoisomerase 1 (Top1) complex comprises the lesion. Top1 is essential for the relaxation of DNA supercoiling ahead of the replication machinery [53, 54]. If Top1 is inhibited either chemically (for example through the action of the chemotherapeutic drug camptothecin) or as a result of clustered ROS damage, the protein remains bound to the DNA creating a toxic single-strand break in actively replicating cells [55]. Tyrosyl DNA phosphodiesterase (Tdp1) catalyzes the hydrolysis of Top1 from the 3’ terminus of DNA, resulting in a gap with a 3’phosphate and a 5’hydroxyl group at the margins [56], although there are alternative mechanisms of repair for this lesion involving Mre11 or Mus81 [57]. After lesion removal by Tdp1, PNKP can then transfer the phosphate group from the 3’ to the 5’ terminus, thus preparing the DNA ends for repair [58]. If any gap-filling activity is required, pol ß is considered a likely candidate [54, 59-62]. Interestingly, both PNKP and pol ß are known to interact with XRCC1 and it has recently been reported that Tdp1, critical for the repair of these Top1-mediated lesions, co-immunoprecipitated with XRCC1 [61]. Further, XRCC1 complementation enhanced Tdp1 activity in EM9 cell lines (deficient in XRCC1 activity) [61]. The PNKP-mediated transfer of a phosphate group was also stimulated by XRCC1 association [63].
Each of the seven entry points described above depicts a unique type of DNA damage that results in either the enzymatic or environmentally-induced hydrolysis of the sugar-phosphate backbone in DNA. The ensuing ends dictate further processing by gap-tailoring enzymes and thus determine protein complex formation. Furthermore, XRCC1 orchestrates these protein partnerships by acting as a scaffold upon which the necessary protein complexes can form, and in the process often stimulating repair.
3. Lesion-driven complex formation
3.1 Scaffold proteins
XRCC1 was initially cloned in 1990 and found to affect cellular sensitivity to ionizing radiation [64]. Soon thereafter, a stable complex of XRCC1 and LigIIIα was discovered [65] and by the late 1990s XRCC1 was established as a BER scaffold protein [66] in partnership with pol ß and PARP1 [16]. The list of cellular components that interact with XRCC1 has been expanded to include nicked DNA [67, 68] and AP site containing DNA, both intermediates in the BER pathway [69]. In the past several years, XRCC1 coordination was further expanded to include pathway initiation enzymes (e.g.; MPG, OGG1 and NEIL2) as well as APE1, firmly establishing XRCC1 in the orchestration of the entire BER pathway [23, 28, 34, 40]. Thus, XRCC1 interacts with most, if not all components of the BER short-patch pathway.
Likewise, XRCC1 impacts repair of single-strand breaks via the ternary complex of XRCC1, LigIIIα and PARP1 [15, 18]. PARP1 is believed to be involved in the sensing of DNA nicks (BER intermediates) [70], acting in conjunction with XRCC1 as a scaffold for the recruitment of gap-tailoring enzymes. Furthermore, in vivo real time imaging of irradiated or oxidized cells showed the necessary coordination or recruitment of pol ß by XRCC1, supporting the unified pathway model proposed here [71]. PNKP also forms a functional complex with XRCC1, pol ß, and LigIIIα [63], establishing PNKP as the kinase/phosphatase responsible for DNA gap-tailoring in response to ROS and irradiation. Likewise, Tdp1 was immunoprecipitated in a complex with XRCC1 that exhibited both Tdp1 and PNKP activity, suggesting a functional interaction between these three proteins [61]. Therefore, all the gap-tailoring enzymes proposed herein interact physically with XRCC1, including APE1, pol ß, PNKP and Tdp1 (Table II). Finally, aprataxin, a protein that confers cellular resistance to methyl methanesulfonate and DNA ssDNA breaks, forms a similar complex including XRCC1, pol ß, and LigIIIα [72-76]. This places aprataxin as a possible gap-tailoring enzyme and will be discussed in the next section. The nicked DNA is broadly categorized into two groups: those requiring 5’ processing such as 5’dRP and those requiring 3’ processing such as 3’PO4 and 3’unsaturated aldehydes. BER protein complex formation is then dictated by the type of DNA ends present in the repair gap.
3.2 Gap tailoring
There are currently four enzymes known to modify DNA ends in the context of BER. Pol ß is the classical BER protein for removal of the 5’dRP moiety resulting from APE1 cleavage. Interestingly, both DNA polymerase lambda (pol λ) and DNA polymerase iota (pol ι) encode a 5’dRP lyase function [77, 78]. Each has been shown capable of removing the 5’dRP lesion subsequent to APE1 strand cleavage [77, 78] but a deficiency in pol λ or pol ι does not render cells alkylation sensitive [79], confirming that pol ß is the predominant activity in mouse embryonic fibroblasts (MEFs) [79]. However, it was demonstrated that pol λ deficient MEFs are sensitive to hydrogen peroxide [80], suggesting that the participation of either pol λ or pol ι in BER may be lesion specific. Complementation studies will be required to determine the precise role, if any, of these alternate BER polymerases in cell survival following genotoxic stress.
The reaction products of bi-functional glycosylases, 3’-α,ß unsaturated aldehydes, can be processed by APE1 [81, 82]. There is evidence that the coordinated activities of bi-functional glycosylases, APE1 and XRCC1 suppress 3’lyase activity of the glycosylase through a product inhibition mechanism, implying that oxidized base damage is shuttled through BER in a fashion similar to alkylated base damage [83]. In other words, the 3’phosphoglycolate products of 3’lyase activity may not be generated in significant quantities by BER initiation. However, these types of lesions may be generated by radiation damage and could be processed by APE1 to generate a clean 3’OH end.
Alternatively, the processing of strand-breaks induced by oxidative damage, which includes radiation damage, can be accomplished by PNKP [58]. PNKP converts 3’PO4 groups to the 3’OH groups necessary for DNA synthesis [58]. PNKP has also been implicated in the processing of DNA ends resulting from NEIL glycosylase activity. As described above, NEIL glycosylase substrates undergo ß,δ-elimination to yield phosphate groups on both the 3’ and 5’ ends. PNKP efficiently removes this replication block generating a 3’OH moiety [46]. Furthermore, a stable complex of NEIL2, PNKP, XRCC1, LigIIIα, and pol ß was isolated as a ‘repairosome’ from human cells [23].
Strand-breaks generated by Top1 poisons are unique in that they are the result of a covalent attachment between the DNA and Top1. As such, their removal appears to require a unique enzyme, Tdp1, which functions within the same XRCC1 scaffold framework as depicted in Fig 2. Tdp1 removes the 3’tyrosyl moiety leaving the 3’OH necessary for DNA synthesis [56] and accomplishes this activity through a physical association with XRCC1 that included a functional dependence on PNKP [61].
The NIR pathway is initiated through APE1 recognition of oxidized base damage followed by direct cleavage 5’ to the lesion, resulting in a 3’OH and a 5’ modified base. The end would be refractory to pol ß 5’dRP lyase activity and therefore is unlikely to be repaired through the XRCC1-mediated short-patch pathway. Instead, removal of this 5’blocking lesion is most likely the result of strand displacement and Fen1-mediated hydrolysis and would be shuttled into the long-patch complex depicted in Fig 2, bottom right complex.
In 2001, the gene responsible for ataxia-ocular apraxia 1 (AOA1), a neurological disorder similar to ataxia-telangiectasia, was identified. The protein was named aprataxin (APTX) and is found to share a distant homology with PNKP [84]. Interestingly, APTX also interacts in vitro and in vivo with XRCC1 [72-76] and co-immunoprecipitated with PARP1 [73, 74]. Moreover, APTX was identified in a complex comprised of XRCC1 and LigIIIα [75], and ataxia-oculomotor apraxia type 1 patient cells exhibit a marked increase in genome instability as a function of increasing camptothecin dose, clearly supporting APTX inclusion as a gap-tailoring protein in the unified BER model proposed herein. Finally, in vitro investigations determined that APTX can remove 5’ blocking groups to generate a clean 5’phophate end [85].
Werner Syndrome protein (WRN) may also be included as a potential gap tailoring protein that functions on a 3’lesion. To date there is no evidence that WRN interacts with XRCC1 although it does form a complex with PARP1 [86, 87]. WRN is classified as a RecQ family DNA helicase but has the unique feature of containing a 3’-5’ exonuclease function. WRN was reported to interact both physically and functionally with pol ß [88]. Recently, the exonucleolytic capacity of WRN has been assessed as a function of the pol ß interaction. Specifically, WRN exonuclease activity can act in conjunction with pol ß, a protein with no intrinsic 3’-5’ proofreading activity [89]. WRN also partners with PARP1, the DNA single-strand nick sensing protein [87].
Each of these proteins has the ability to tailor the ends of DNA in preparation for subsequent synthesis and ligation. Furthermore, each of these proteins is reported to interact with XRCC1/LigIIIα [34, 60, 61, 90], (and in some cases PARP1) supporting the idea that XRCC1/LigIIIα, together with PARP1, acts as a scaffold to coordinate a broad range of substrates and proteins through the BER pathway.
3.3 Repair DNA synthesis and DNA ligation
In this unified BER model pol ß is shown as the primary polymerase responsible for nucleotidyl transfer. However, DNA pols δ, ε, γ, ι and λ have all been suggested to participate in BER as determined by analysis of cell extracts or purified proteins. Pol ß however remains the primary BER DNA polymerase and the complexes formed are represented in the boxed section of Fig. 2. The complexes are representative of the protein associations possible depending on DNA end processing requirements and incorporate gap-tailoring protein, scaffold proteins and pol ß for the nucleotidyl transfer activity. In this unifying model, the DNA Synthesis/Ligation steps were combined to include substrates requiring ligation without necessarily requiring DNA synthesis, as depicted in the structure on the right [61]. Specifically, Tdp1-mediated removal of a 3’tyrosyl residue from the DNA would leave an intact nucleotide complete with a 3’OH group ready for ligation. Should nucleotide replacement prove necessary, pol ß is the most likely polymerase candidate.
Apoptosis may be initiated in the event that 3’ blocking lesions prove resilient to removal by these gap-tailoring proteins. A persistent 5’ blocking lesion however can be removed via the long-patch BER pathway. Long patch BER has long been established as a complex of proteins associated to repair larger nucleotide gaps. Fen1, the flap endonuclease responsible for removal of the 5’blocking lesion, functions in conjunction with PARP1, PCNA, APE1, and pol δ, pol •, or pol ß. Oxidized bases recognized by the NIR pathway, namely the direct cleavage of DNA strands containing damage by APE1, may be shuttled directly through the long-patch pathway as there is no known association between XRCC1 and Fen1.
4. BER protein post-translational modifications
Each sub-pathway of BER relies on the formation of protein complexes that assemble at the site of the DNA lesion and facilitate repair in a coordinated fashion [83]. Table II outlines the known protein-protein interactions of the proteins involved in BER. This coordination of protein complex formation that is driven by the lesion or repair intermediate was originally compared to the passing of a baton, where the repair product of each enzyme in the BER pathway is “handed” to the sequential enzyme in the pathway [2]. These protein-protein partnerships can regulate pathway efficacy, dictating subsequent steps or sub-pathway choice. This mechanism or process of complex formation appears to provide an increase in specificity and efficiency to the BER pathway, thereby facilitating the maintenance of genome integrity by preventing the accumulation of highly toxic repair intermediates [83]. Decreased concentrations of just one protein within this pathway could significantly alter the balance of complex formation, resulting in reduced repair capacity and an increased exposure to toxic BER intermediates, such as has been observed in pol ß deficiency in mouse cells [79, 91-93], in animal models [94] and in human cells [62].
The regulation of BER is further refined through post-translational modifications (PTM). PTMs can take the form of phosphorylation by protein kinases such as ATM, ATR and DNA-PK, acetylation by proteins like p300 and CBP, sumoylation, mono- or poly-ribosylation, mono- and poly-ubiquitylation or methylation, among others [95]. PTMs can alter the binding characteristics, turnover rates, sub-cellular localization and/or overall efficacy of the target protein. Therefore, if modified correctly, the function of the protein is streamlined to meet the needs of the individual cellular environment. However, if modification is slow or non-existent, overall pathway efficacy can be greatly reduced, leading to greater longevity of toxic BER intermediates, impacting mutational load, genomic stability and cellular senescence. In this section, we describe a few examples of BER protein PTMs to emphasize the significance of PTMs on BER capacity and have summarized known BER protein PTMs in Table III.
Table III.
Post translational modifications of BER proteins
| Function | Protein | Phosphorylation | Acetylation | Sumoylation | Ubiquitylation | Methylation |
|---|---|---|---|---|---|---|
| Lesion Recognition and Strand Scission | TDG | C--[26] | A--[99-101, 174] | |||
| UNG2 | A--[175]; C--[176, 177] | B--[176] | ||||
| MPG | A--[178] | |||||
| MYH | A--[98] | |||||
| NEIL2 | B--[102] | |||||
| OGG1 | B--[96]*; C--[96, 179] | |||||
| APE1 | B--[107]**; C--[103-107, 126] | C--[109, 110] | ||||
|
| ||||||
| BER scaffold | XRCC1 | A--[75, 115, 116] | C--[117] | |||
| Parp1 | A--[180] | A--[181] | C--[117] | [182] | ||
|
| ||||||
| Gap Tailoring | pol ß | A--[127]; [126] | B--[119] | |||
| Fen1 | B--[121] | B--[122]; C--[123] | ||||
| WRN | B--[183-187] | A--[125] | C--[188]## and [189] | |||
|
| ||||||
| DNA Synthesis and Ligation | pol ß | A--[127] | A--[128]# | |||
| LigI | C--[130] | |||||
| LigIIIα | C--[131] | |||||
| PCNA | A--[190] | A--[156] | C--[191, 192] | A--[193]; C--[194] | ||
A - Denotes Increased Catalytic Efficiency
B - Denotes Decreased Catalytic Efficiency
C - No Effect Shown
Specific for Cdk4 phosphorylation only
Specific for CKII phosphorylation only
Stimulated DNA binding and processivity; single nucleotide insertion and 5’dRP lyase activity were not affected
Mouse homolog identified as substrate for modification
4.1 Modification of proteins that initiate BER via lesion recognition and strand scission
The most common PTM of BER proteins is phosphorylation. As delineated in Table III, most of the critical proteins involved in the BER pathway are post-translationally phosphorylated. This could be the result of cellular signaling cascades that are designed to activate checkpoint pathways and thus prevent the cell from proceeding with cell division while simultaneously enhancing DNA repair. Recently, post-translational phosphorylations of OGG1, the predominant glycosylase for oxidative damage, have been reported [96]. Two independent kinases were shown to phosphorylate OGG1 in vitro [96]. C-Abl-mediated phosphorylation of hOGG1 had no effect on activity. However, Cdk4 phosphorylation affected the catalytic efficiency, resulting in a 2.5 fold increase in 8-oxodG incision activity [96], suggesting that the modulation of OGG1 activity is dependent on PTM location [96].
Of the remaining DNA glycosylases only MYH and UNG2 are phosphorylated. In each case, this modification has been observed to increase catalytic activity and hence may regulate BER initiation [97, 98]. On the other hand, TDG, MPG and NEIL2 are modified and regulated by acetylation (Table III). Whereas acetylation does not appear to regulate TDG, SUMO modification acts as a regulatory switch by altering its subcellular localization [99-101]. NEIL2 is acetylated by p300 [102]. In this case, acetylation of the Lys49 residue on NEIL2 abrogates both the DNA excision and the AP lyase activities of the enzyme, effectively inactivating BER initiation through modification of this glycosylase. Meanwhile acetylation of Lys153 had no effect on either activity. Although studies identifying PTMs are in their infancy, it appears that the type and location of PTMs are essential in determining the overall ability to affect DNA glycosylase-mediated lesion removal. This in turn refines the efficiency of BER pathway initiation and provides tighter regulation of the BER pathway.
A similar story has unfolded for the APE1 protein, in which all reports agree that APE1 is phosphorylated by a variety of protein kinases [103-107], but conflict as to the effect of this modification. Early studies showed that phosphorylation by the serine/threonine casein kinase I (CKI) and protein kinase C had no effect while modification by CKII abolished repair capacity completely [107]. Others reported that phosphorylation by CKII did not alter the repair capacity of APE1 but did stimulate its redox capabilities [103]. Independent of its role in BER, APE1 was purified from HeLa cell extracts as a protein capable of activating the transcription factor AP-1 via reduction of an oxidized cysteine residue [108]. The multiple roles of APE1 obfuscate the specific effects that post-translational modifications have on the protein, rendering the ramifications of specific modifications elusive. Nevertheless, it seems clear that the PTM-mediated regulation of APE1 is dependent on the type and site (amino acid residue) of the modification. Additionally, APE1 is acetylated by the transcriptional co-activator p300; although this modification has yet to be linked to modulation of the repair capacity of APE1 and appears to influence its redox potential exclusively [109, 110].
4.2 BER scaffold proteins and post-translational modifications
The BER mechanism can be viewed as a multitude of protein complexes unique to the initiating lesion (Table I), with XRCC1 (in concert with its binding partner LigIIIα) acting as a scaffold to orchestrate the movement of DNA repair intermediates from repair initiation and strand scission (DNA glycosylase and APE1) to gap tailoring (APE1 and/or pol ß) and facilitating repair completion (LigIIIα) through a series of protein interactions [34, 111, 112]. For example, an interaction between XRCC1 and PNKP, a protein necessary for the initial processing of DNA single-strand break termini into ends that are amenable to re-ligation, was shown to be involved in BER following oxidative stress [58, 63]. Interestingly, XRCC1 stimulated both the kinase and phosphatase activity of PNKP at damaged termini, resulting in an overall acceleration of single-strand break repair [63], supporting our contention that SSBR may be considered a sub-pathway of BER. Further, PARP1 interacts exclusively with the BRCT-I region of XRCC1 and preferentially binds when it is in an active poly(ADP-ribosyl)ated state. Indeed, XRCC1 has increased affinity for modified PARP1 [113]. Furthermore, PARP1 has been shown to actively recruit XRCC1 at sites of oxidative stress [114], suggesting that PARP1 senses damage and recruits XRCC1 to act as the scaffold upon which a repair complex can be constructed.
As with most of the BER proteins, XRCC1 and PARP1 present with several different post-translational modifications that ultimately impact BER function, either directly, by altering enzymatic activity or indirectly, by altering protein-protein interactions (Table III). XRCC1 is phosphorylated on at least four amino acid residues (S518, T519, T523 and S371) [75, 115, 116]. CK2-mediated phosphorylation of amino acid residues S518, T519 and T523 facilitates the binding of XRCC1 to aprataxin and increased protein stability of XRCC1 due to the aprataxin complex formation [75]. Phosphorylation of S371 by DNA-PK appears to be related to a role for XRCC1 in double-strand break repair [115]. This modification has not yet been evaluated with regard to its role in BER. XRCC1 was also recently identified as a substrate for SUMO-modification but has not been characterized further [117]. PARP1 is most abundantly auto-modified by ADP-ribosylation and further modified by phosphorylation, SUMO and acetylation (Table III). The reader is referred to an excellent and thorough review of PARP-family members for more detail [118].
4.3 Post-translational modifications of BER gap tailoring proteins
DNA pol ß interacts with the transcriptional co-activator p300, suggesting that this protein may regulate BER via a post-translational mechanism [119]. p300 was found to acetylate pol ß at an amino acid critical for the 5’dRP lyase function: lysine 72. Acetylation at this site blocks formation of the Schiff-base intermediate and results in abrogation of the 5’dRP lyase activity of pol ß but leaves its gap-filling and DNA binding functions unaffected [119]. Whether acetylated pol ß interacts with other BER proteins in the same manner as unacetylated pol ß remains to be seen. Nevertheless, p300 may have a novel regulatory role by modulating the critical pol ß 5’dRP lyase activity that is required for cellular survival following stress [93]. Given the dire consequences of abrogation of the pol ß 5’dRP lyase activity (cellular sensitivity to alkylating agents) [79, 92, 93], it seems likely that a de-acetylase, designed to reactivate pol ß 5’dRP lyase function, is present in vivo but to date has not been identified. It is tempting to speculate that the reason for 5’dRP lyase abolition is to force repair through long-patch BER, however, the cellular conditions necessary and the advantage gained from a preference for long-patch repair remains to be determined.
As described above, the long-patch sub-pathway utilizes the gap-tailoring properties of Fen1 to remove the displaced DNA flap. Fen1 functionally and physically interacts with pol ß to promote strand displacement and flap hydrolysis [11, 120]. The functionality of Fen1 is further modified by phosphorylation [121] and acetylation [122, 123], each modification providing a different level of regulation. Whereas phosphorylation prevents Fen1-mediated stimulation of PCNA, acetylation by p300 appears to reduce the ability of Fen1 to bind to DNA and to function as a nuclease, with no effect on its PCNA interaction (Table III). WRN also stimulates strand displacement activities of pol ß [88, 89] and is included herein as a gap tailoring protein since it removes 3’lesions via its 3’⇒5’ exonuclease activity [124] and acts as a participant in promoting long-patch BER. Similar to Fen1, phosphorylation of WRN decreases its catalytic activity (Table III). However, p300-mediated acetylation appears to augment the ability of WRN to translocate to nucleoplasmic foci, suggesting that modification regulates its function by altering cellular localization [125]. Finally, WRN is also SUMO-modified, via a p53-independent interaction with p14 ARF and Ubc9, although it has not yet been established how or if SUMO-modification may alter the BER-related functions of WRN (See Table III and references within). None of the other proteins we have listed as gap-tailoring BER enzymes have been reported to undergo post-translational modification.
4.4 BER DNA synthesis and ligation proteins
Evidence for a phosphorylated form of pol ß has been reported. In both instances, rat pol ß was phosphorylated and exhibited higher BER activity than the un-phosphorylated form [126, 127]. The site of phosphorylation is not known but it was determined that the modification is a serine residue [126]. It is not clear as to the exact nature of the catalytic effect of the phosphate-modification or if phosphorylation impacts protein-protein interactions. Recently, pol ß was found to form a complex with and is modified by protein arginine methyltransferase 6 (PRMT6), resulting in methylation of R83 and R152. Methylation of these residues stimulated processive polymerase activity. Single-nucleotide insertions and 5’dRP lyase activity remained static, however, implying that methylation of pol ß preferentially shuttles repair through long-patch BER [128]. More recently, pol ß was reported to be methylated (in vitro and in vivo) by PRMT1 but methylation was at amino acid residue 137 [129]. In this case, modification of arginine 137 did not alter the DNA polymerase activity or 5’ dRP lyase activity but the interaction with PCNA was diminished.
Both BER DNA Ligases (I and IIIα) undergo post-translational modification in the form of phosphorylation (Table III). The DNA-PK-mediated phosphorylation of LigI activates the enzyme and is observed both in vitro and in vivo [130]. The observed in vivo phosphorylation of LigI corresponds with the generation of double-stranded DNA breaks and may be specific for the role of LigI in non-homologous end joining. Any role for post-translational modifications of LigI in BER has not been identified. On the other hand, LigIIIα is phosphorylated on Ser123 by Cdk2 in a cell-cycle specific fashion and is de-phosphorylated in an ATM-dependent mechanism linked to the onset of oxidative stress [131]. PCNA is also listed as a BER DNA synthesis and ligation related protein and is modified in many ways, including phosphorylation, acetylation, SUMO and ubiquitin, as listed in Table III. There are many detailed reviews that characterize the varied post-translational modifications of PCNA and the impact of these modifications on PCNA-protein interactions [132-134].
5. Conclusion
We describe a BER model that incorporates the many varied sub-pathways of BER, including short-patch, long-patch, the repair of single-strand breaks, NIR and repair initiated by the NEIL-family of DNA glycosylases. As with most DNA repair processes, BER appears to function via a series of repair complexes that assemble at the site of the DNA lesion [1, 135]. BER protein complex formation is further influenced by post-translational protein modifications that arise from the cellular state or the DNA damage response, providing an increase in specificity and efficiency to the BER pathway. There are many post-translational modifications of BER and/or genome stability proteins (Table III), and more PTMs are reported in the literature regularly. This information, however, is just the beginning of our understanding of how PTMs influence overall biological processes. Much work needs to be completed before a clear picture emerges from the impact PTMs have on catalytic efficacy and biological function with regard to BER. Post-translational modifications play key roles in modulating protein function [136]. Such post-translational modifications must be identified by direct protein characterization techniques. In addition, a single protein can play many different roles within a cell; therefore it is important to characterize its relationship with other proteins to gain a true understanding of its function(s) at any particular time.
Mouse whole-animal and cellular knockout models have been instrumental in understanding the cellular and biological role of many BER proteins beyond the biochemical characterization of substrate identity and specificity. In particular, mouse embryonic fibroblasts (MEFs) have been a workhorse reagent towards understanding the cellular function of BER proteins with regard to cell survival, genome integrity, mutagenesis and genotoxicity and for the validation of and discovery of protein-protein complexes. Clear differences of cellular response between mouse and human cells, as well as the varied responses from cells of different organ types or various states of differentiation necessitate continued evaluation of BER proteins isolated from human cells (tumor cells, stem cells and terminally-differentiated cells). Future studies will require detailed analysis of how these BER complexes and PTMs impact repair of DNA damage from endogenous sources to maintain cellular genome integrity. In addition, a valuable next step is an appreciation of the impact of these complexes and PTM-mediated regulation towards repair of DNA damage from exogenous sources to evaluate the cellular response to environmental and chemotherapeutic genotoxins.
Many of the proteins that participate in BER, as determined by in vitro analysis of purified proteins and cell extracts or in vivo analysis (whole cell and animal studies) have been identified [6]. The multiple and varied complexes that form to direct repair of a host of DNA lesions (Table II) are still being discovered. Further, the interplay of the in situ repair complexes, the mechanisms of regulation (transcriptional, translational, epigenetic, post-translational or mediated by micro- or small RNAs) and the impact of repair regulation and cellular function is an exciting area of current and future studies. We are only beginning to appreciate the biological impact of mutations that effect BER protein function, protein complex formation, PTM or all of these. The discovery that MYH variants predispose to somatic mutations and colorectal tumors underscores the significance of the BER pathway in genome maintenance [137]. Detailed analyses are forthcoming, for example, to determine if inheritable mutations in other BER proteins impact human health (e.g., cancer predisposition, neurological function) and whether these mutations affect BER capacity through disruption of complex formation or regulation due to defects in post-translational modification.
Acknowledgments
This research was supported by a Research Scholar grant (RSG-05-246-01-GMC) from the American Cancer Society, grants from the Susan G. Komen Breast Cancer Foundation (Grant # BCTR0403276), NIH (1 R01 AG24364-01; P20 CA103730), the UPMC Health System Competitive Medical Research Fund, the University of Pittsburgh Cancer Institute and a Cancer Research award from the Elsa U Pardee Foundation to RWS and by the RI-INBRE Grant # P20 RR016457 from NCRR/NIH to KHA. We would like to thank Dr. B. Wittschieben (Department of Pharmacology and UPCI, University of Pittsburgh) for critically evaluating this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair (Amst) 2007 doi: 10.1016/j.dnarep.2006.10.004. In Press. [DOI] [PubMed] [Google Scholar]
- 2.Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nature Structural Biology. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Research. 1998;58:222–225. [PubMed] [Google Scholar]
- 4.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 6.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 7.Wallace SS. Enzymatic processing of radiation-induced free radical damage in DNA. Radiation Research. 1998;150:S60–S79. [PubMed] [Google Scholar]
- 8.Gary R, Kim K, Cornelius HL, Park MS, Matsumoto Y. Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. Journal of Biological Chemistry. 1999;274:4354–4363. doi: 10.1074/jbc.274.7.4354. [DOI] [PubMed] [Google Scholar]
- 9.Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry. 1998;37:3575–3580. doi: 10.1021/bi972999h. [DOI] [PubMed] [Google Scholar]
- 10.Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hubscher U, Dogliotti E. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene. 1998;17:835–843. doi: 10.1038/sj.onc.1202001. [DOI] [PubMed] [Google Scholar]
- 11.Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase ß mediates an excision step in mammalian long patch base excision repair. Journal of Biological Chemistry. 2000;275:4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- 12.Prasad R, Lavrik OI, Kim SJ, Kedar P, Yang XP, Vande Berg BJ, Wilson SH. DNA polymerase ß-mediated long patch base excision repair: Poly(ADP-ribose)polymerase-1 stimulates strand displacement DNA synthesis. Journal of Biological Chemistry. 2001;276:32411–32414. doi: 10.1074/jbc.C100292200. [DOI] [PubMed] [Google Scholar]
- 13.Fan J, Wilson DM., 3rd Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radic Biol Med. 2005;38:1121–1138. doi: 10.1016/j.freeradbiomed.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2. Washington, D.C: ASM Press; 2006. [Google Scholar]
- 15.Caldecott KW, Aoufouchi S, Johnson P, Shall S. XRCC1 polypeptide interacts with DNA polymerase ß and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Research. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Molecular and Cellular Biology. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 18.Leppard JB, Dong Z, Mackey ZB, Tomkinson AE. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Molecular and Cellular Biology. 2003;23:5919–5927. doi: 10.1128/MCB.23.16.5919-5927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frouin I, Maga G, Denegri M, Riva F, Savio M, Spadari S, Prosperi E, Scovassi AI. Human Proliferating Cell Nuclear Antigen, Poly(ADP-ribose) Polymerase-1, and p21waf1/cip1: A DYNAMIC EXCHANGE OF PARTNERS. Journal of Biological Chemistry. 2003;278:39265–39268. doi: 10.1074/jbc.C300098200. [DOI] [PubMed] [Google Scholar]
- 20.Daviet S, Couve-Privat S, Gros L, Shinozuka K, Ide H, Saparbaev M, Ishchenko AA. Major oxidative products of cytosine are substrates for the nucleotide incision repair pathway. DNA Repair (Amst) 2007;6:8–18. doi: 10.1016/j.dnarep.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Gros L, Ishchenko AA, Ide H, Elder RH, Saparbaev MK. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 2004;32:73–81. doi: 10.1093/nar/gkh165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishchenko AA, Deprez E, Maksimenko A, Brochon JC, Tauc P, Saparbaev MK. Uncoupling of the base excision and nucleotide incision repair pathways reveals their respective biological roles. Proc Natl Acad Sci U S A. 2006;103:2564–2569. doi: 10.1073/pnas.0508582103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das A, Wiederhold L, Leppard JB, Kedar P, Prasad R, Wang H, Boldogh I, Karimi-Busheri F, Weinfeld M, Tomkinson AE, Wilson SH, Mitra S, Hazra TK. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair (Amst) 2006;5:1439–1448. doi: 10.1016/j.dnarep.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Almeida KH, Sobol RW. Increased Specificity and Efficiency of Base Excision Repair through Complex Formation. In: Siede W, Doetsch PW, Kow YW, editors. DNA Damage Recognition. Marcel Dekker Inc.; New York: 2005. pp. 33–64. [Google Scholar]
- 26.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Molecular Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu Y, Iwai S, Hanaoka F, Sugasawa K. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO Journal. 2003;22:164–173. doi: 10.1093/emboj/cdg016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campalans A, Marsin S, Nakabeppu Y, O’Connor TR, Boiteux S, Radicella JP. XRCC1 interactions with multiple DNA glycosylases: a model for its recruitment to base excision repair. DNA Repair (Amst) 2005;4:826–835. doi: 10.1016/j.dnarep.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 29.Marenstein DR, Chan MK, Altamirano A, Basu AK, Boorstein RJ, Cunningham RP, Teebor GW. Substrate specificity of human endonuclease III (hNTH1): Effect of human APE1 on hNTH1 activity. Journal of Biological Chemistry. 2003;278:9005–9012. doi: 10.1074/jbc.M212168200. [DOI] [PubMed] [Google Scholar]
- 30.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annual Review of Biochemistry. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 31.Vidal AE, Hickson ID, Boiteux S, Radicella JP. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: bypass of the AP lyase activity step. Nucleic Acids Research. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, Clendenin WM, Wong D, Demple B, Slupska MM, Chiang JH, Miller JH. Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Research. 2001;29:743–752. doi: 10.1093/nar/29.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Research. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marsin S, Vidal AE, Sossou M, Menissier-De Murcia J, Le Page F, Boiteux S, De Murcia G, Radicella JP. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 2003;278:44068–44074. doi: 10.1074/jbc.M306160200. [DOI] [PubMed] [Google Scholar]
- 35.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annual Review of Genetics. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 36.Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proceedings of the National Academy of Science. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, Meneses J, Pedersen RA, Chen DJ. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutation Research. 1998;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- 38.Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, Pommier Y, Lieberman J. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nature Immunology. 2003;4:145–153. doi: 10.1038/ni885. [DOI] [PubMed] [Google Scholar]
- 39.Izumi T, Brown DB, Naidu CV, Bhakat KK, Macinnes MA, Saito H, Chen DJ, Mitra S. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci U S A. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO Journal. 2001;20:6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature. 2002;415:183–187. doi: 10.1038/415183a. [DOI] [PubMed] [Google Scholar]
- 42.Dianova VA, Bohr II, Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40:12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 43.Ranalli TA, Tom S, Bambara RA. AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. Journal of Biological Chemistry. 2002;277:41715–41724. doi: 10.1074/jbc.M207207200. [DOI] [PubMed] [Google Scholar]
- 44.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 45.Hazra TK, Izumi T, Maidt L, Floyd RA, Mitra S. The presence of two distinct 8-oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucleic Acids Research. 1998;26:5116–5122. doi: 10.1093/nar/26.22.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proceedings of the National Academy of Science. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caldecott KW. Polynucleotide kinase: a versatile molecule makes a clean break. Structure. 2002;10:1151–1152. doi: 10.1016/s0969-2126(02)00833-x. [DOI] [PubMed] [Google Scholar]
- 48.Sieber OM, Heinimann K, Tomlinson IP. Genomic instability--the engine of tumorigenesis? Nat Rev Cancer. 2003;3:701–708. doi: 10.1038/nrc1170. [DOI] [PubMed] [Google Scholar]
- 49.Gros L, Saparbaev MK, Laval J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene. 2002;21:8905–8925. doi: 10.1038/sj.onc.1206005. [DOI] [PubMed] [Google Scholar]
- 50.Henner WD, Rodriguez LO, Hecht SM, Haseltine WA. gamma Ray induced deoxyribonucleic acid strand breaks. 3’ Glycolate termini. J Biol Chem. 1983;258:711–713. [PubMed] [Google Scholar]
- 51.Henner WD, Grunberg SM, Haseltine WA. Sites and structure of gamma radiation-induced DNA strand breaks. J Biol Chem. 1982;257:11750–11754. [PubMed] [Google Scholar]
- 52.Henner WD, Grunberg SM, Haseltine WA. Enzyme action at 3’ termini of ionizing radiation-induced DNA strand breaks. J Biol Chem. 1983;258:15198–15205. [PubMed] [Google Scholar]
- 53.Bjornsti MA, Osheroff N. Introduction to DNA topoisomerases. Methods in Molecular Biology. 1999;94:1–8. doi: 10.1385/1-59259-259-7:1. [DOI] [PubMed] [Google Scholar]
- 54.Pommier Y, Redon C, Rao VA, Seiler JA, Sordet O, Takemura H, Antony S, Meng L, Liao Z, Kohlhagen G, Zhang H, Kohn KW. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutation Research. 2003;532:173–203. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 55.Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP. Mechanism of action of camptothecin. Annals of the New York Acadamy of Science. 2000;922:1–10. doi: 10.1111/j.1749-6632.2000.tb07020.x. [DOI] [PubMed] [Google Scholar]
- 56.Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proceedings of the National Academy of Science. 2001;98:12009–12014. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu C, Pouliot JJ, Nash HA. Repair of topoisomerase I covalent complexes in the absence of the tyrosyl-DNA phosphodiesterase Tdp1. Proceedings of the National Academy of Science. 2002;99:14970–14975. doi: 10.1073/pnas.182557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jilani A, Ramotar D, Slack C, Ong C, Yang XM, Scherer SW, Lasko DD. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3’-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J Biol Chem. 1999;274:24176–24186. doi: 10.1074/jbc.274.34.24176. [DOI] [PubMed] [Google Scholar]
- 59.R. Trivedi, R.W. Sobol. Unpublished, (2004)
- 60.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair. 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 61.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 62.R. Trivedi, R.W. Sobol. Unpublished, (2007)
- 63.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 64.Thompson LH, Brookman KW, Jones NJ, Allen SA, Carrano AV. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cellular Biology. 1990;10:6160–6171. doi: 10.1128/mcb.10.12.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Molecular and Cellular Biology. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase ß and the XRCC1 protein. EMBO Journal. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 67.Marintchev A, Mullen MA, Maciejewski MW, Pan B, Gryk MR, Mullen GP. Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat Struct Biol. 1999;6:884–893. doi: 10.1038/12347. [DOI] [PubMed] [Google Scholar]
- 68.Rice PA. Holding damaged DNA together. Nat Struct Biol. 1999;6:805–806. doi: 10.1038/12257. [DOI] [PubMed] [Google Scholar]
- 69.Nazarkina ZK, Khodyreva SN, Marsin S, Lavrik OI, Radicella JP. XRCC1 interactions with base excision repair DNA intermediates. DNA Repair (Amst) 2007;6:254–264. doi: 10.1016/j.dnarep.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 70.Ziegler M, Oei SL. A cellular survival switch: poly(ADP-ribosyl)ation stimulates DNA repair and silences transcription. Bioessays. 2001;23:543–548. doi: 10.1002/bies.1074. [DOI] [PubMed] [Google Scholar]
- 71.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proceedings of the National Academy of Science. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, Bieganowski P, Brenner C, Moreira MC, Taylor AM, Caldecott KW. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst) 2004;3:1493–1502. doi: 10.1016/j.dnarep.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 73.Date H, Igarashi S, Sano Y, Takahashi T, Takahashi T, Takano H, Tsuji S, Nishizawa M, Onodera O. The FHA domain of aprataxin interacts with the C-terminal region of XRCC1. Biochem Biophys Res Commun. 2004;325:1279–1285. doi: 10.1016/j.bbrc.2004.10.162. [DOI] [PubMed] [Google Scholar]
- 74.Gueven N, Becherel OJ, Kijas AW, Chen P, Howe O, Rudolph JH, Gatti R, Date H, Onodera O, Taucher-Scholz G, Lavin MF. Aprataxin, a novel protein that protects against genotoxic stress. Hum Mol Genet. 2004;13:1081–1093. doi: 10.1093/hmg/ddh122. [DOI] [PubMed] [Google Scholar]
- 75.Luo H, Chan DW, Yang T, Rodriguez M, Chen BP, Leng M, Mu JJ, Chen D, Songyang Z, Wang Y, Qin J. A new XRCC1-containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol Cell Biol. 2004;24:8356–8365. doi: 10.1128/MCB.24.19.8356-8365.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sano Y, Date H, Igarashi S, Onodera O, Oyake M, Takahashi T, Hayashi S, Morimatsu M, Takahashi H, Makifuchi T, Fukuhara N, Tsuji S. Aprataxin, the causative protein for EAOH is a nuclear protein with a potential role as a DNA repair protein. Ann Neurol. 2004;55:241–249. doi: 10.1002/ana.10808. [DOI] [PubMed] [Google Scholar]
- 77.Bebenek K, Tissier A, Frank EG, McDonald JP, Prasad R, Wilson SH, Woodgate R, Kunkel TA. 5’-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science. 2001;291:2156–2159. doi: 10.1126/science.1058386. [DOI] [PubMed] [Google Scholar]
- 78.Garcia-Diaz M, Bebenek K, Kunkel TA, Blanco L. Identification of an intrinsic 5’-deoxyribose-5-phosphate lyase activity in human DNA polymerase lambda: a possible role in base excision repair. J Biol Chem. 2001;276:34659–34663. doi: 10.1074/jbc.M106336200. [DOI] [PubMed] [Google Scholar]
- 79.Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. The Role of Base Excision Repair in the Sensitivity and Resistance to Temozolomide Mediated Cell Death. Cancer Res. 2005;65:6394–6400. doi: 10.1158/0008-5472.CAN-05-0715. [DOI] [PubMed] [Google Scholar]
- 80.Braithwaite EK, Kedar PS, Lan L, Polosina YY, Asagoshi K, Poltoratsky VP, Horton JK, Miller H, Teebor GW, Yasui A, Wilson SH. DNA polymerase lambda protects mouse fibroblasts against oxidative DNA damage and is recruited to sites of DNA damage/repair. J Biol Chem. 2005;280:31641–31647. doi: 10.1074/jbc.C500256200. [DOI] [PubMed] [Google Scholar]
- 81.Wilson DM., 3rd Properties of and substrate determinants for the exonuclease activity of human apurinic endonuclease Ape1. Journal of Molecular Biology. 2003;330:1027–1037. doi: 10.1016/s0022-2836(03)00712-5. [DOI] [PubMed] [Google Scholar]
- 82.Wong D, DeMott MS, Demple B. Modulation of the 3’->5’-Exonuclease Activity of Human Apurinic Endonuclease (Ape1) by Its 5’-incised Abasic DNA Product. Journal of Biological Chemistry. 2003;278:36242–36249. doi: 10.1074/jbc.M306065200. [DOI] [PubMed] [Google Scholar]
- 83.Almeida KH, Sobol RW. Increased Specificity and Efficiency of Base Excision Repair through Complex Formation. In: Siede W, Doetsch PW, Kow YW, editors. DNA Damage Recognition. Marcel Dekker Inc.; New York: 2005. [Google Scholar]
- 84.Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29:189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- 85.Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- 86.Adelfalk C, Kontou M, Hirsch-Kauffmann M, Schweiger M. Physical and functional interaction of the Werner syndrome protein with poly-ADP ribosyl transferase. FEBS Lett. 2003;554:55–58. doi: 10.1016/s0014-5793(03)01088-3. [DOI] [PubMed] [Google Scholar]
- 87.von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, Cheng WH, Bohr VA. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Molecular and Cellular Biology. 2003;23:8601–8613. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harrigan JA, Opresko PL, von Kobbe C, Kedar PS, Prasad R, Wilson SH, Bohr VA. The Werner syndrome protein stimulates DNA polymerase ß strand displacement synthesis via its helicase activity. J Biol Chem. 2003;278:22686–22695. doi: 10.1074/jbc.M213103200. [DOI] [PubMed] [Google Scholar]
- 89.Harrigan JA, Wilson DM, 3rd, Prasad R, Opresko PL, Beck G, May A, Wilson SH, Bohr VA. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006;34:745–754. doi: 10.1093/nar/gkj475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marintchev A, Robertson A, Dimitriadis EK, Prasad R, Wilson SH, Mullen GP. Domain specific interaction in the XRCC1-DNA polymerase ß complex. Nucleic Acids Research. 2000;28:2049–2059. doi: 10.1093/nar/28.10.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sobol RW. DNA polymerase ß null mouse embryonic fibroblasts harbor a homozygous null mutation in DNA polymerase iota. DNA Repair (Amst) 2007;6:3–7. doi: 10.1016/j.dnarep.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-ß in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 93.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein ß-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 94.Cabelof DC, Guo Z, Raffoul JJ, Sobol RW, Wilson SH, Richardson A, Heydari AR. Base excision repair deficiency caused by polymerase ß haploinsufficiency: accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003;63:5799–5807. [PubMed] [Google Scholar]
- 95.Krueger KE, Srivastava S. Posttranslational protein modifications: current implications for cancer detection, prevention, and therapeutics. Mol Cell Proteomics. 2006;5:1799–1810. doi: 10.1074/mcp.R600009-MCP200. [DOI] [PubMed] [Google Scholar]
- 96.Hu J, Imam SZ, Hashiguchi K, de Souza-Pinto NC, Bohr VA. Phosphorylation of human oxoguanine DNA glycosylase (alpha-OGG1) modulates its function. Nucleic Acids Res. 2005;33:3271–3282. doi: 10.1093/nar/gki636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell. 2004;15:621–634. doi: 10.1016/j.molcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 98.Parker AR, O’Meally RN, Sahin F, Su GH, Racke FK, Nelson WG, DeWeese TL, Eshleman JR. Defective human MutY phosphorylation exists in colorectal cancer cell lines with wild-type MutY alleles. J Biol Chem. 2003;278:47937–47945. doi: 10.1074/jbc.M306598200. [DOI] [PubMed] [Google Scholar]
- 99.Mohan RD, Rao A, Gagliardi J, Tini M. SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol Cell Biol. 2007;27:229–243. doi: 10.1128/MCB.00323-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Steinacher R, Schar P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr Biol. 2005;15:616–623. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 101.Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO Journal. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–3039. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fritz G, Kaina B. Phosphorylation of the DNA repair protein APE/REF-1 by CKII affects redox regulation of AP-1. Oncogene. 1999;18:1033–1040. doi: 10.1038/sj.onc.1202394. [DOI] [PubMed] [Google Scholar]
- 104.Hsieh MM, Hegde V, Kelley MR, Deutsch WA. Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic Acids Res. 2001;29:3116–3122. doi: 10.1093/nar/29.14.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McKenzie JA, Strauss PR. A quantitative method for measuring protein phosphorylation. Anal Biochem. 2003;313:9–16. doi: 10.1016/s0003-2697(02)00464-5. [DOI] [PubMed] [Google Scholar]
- 106.Orii A, Masutani H, Nikaido T, Zhai YL, Kato K, Kariya M, Konishi I, Yodoi J, Fujii S. Altered post-translational modification of redox factor 1 protein in human uterine smooth muscle tumors. J Clin Endocrinol Metab. 2002;87:3754–3759. doi: 10.1210/jcem.87.8.8734. [DOI] [PubMed] [Google Scholar]
- 107.Yacoub A, Kelley MR, Deutsch WA. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res. 1997;57:5457–5459. [PubMed] [Google Scholar]
- 108.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. Embo J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bhakat KK, Yang SH, Mitra S. Acetylation of human AP-endonuclease 1, a critical enzyme in DNA repair and transcription regulation. Methods Enzymol. 2003;371:292–300. doi: 10.1016/S0076-6879(03)71022-2. [DOI] [PubMed] [Google Scholar]
- 110.Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. Embo J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Caldecott KW. Protein-protein interactions during mammalian DNA single-strand break repair. Biochemical Society Transactions. 2003;31:247–251. doi: 10.1042/bst0310247. [DOI] [PubMed] [Google Scholar]
- 112.Beernink PT, Hwang M, Ramirez M, Murphy MB, Doyle SA, Thelen MP. Specificity of protein interactions mediated by BRCT domains of the XRCC1 DNA repair protein. J Biol Chem. 2005;280:30206–30213. doi: 10.1074/jbc.M502155200. [DOI] [PubMed] [Google Scholar]
- 113.Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. Journal of Biological Chemistry. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 114.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Research. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levy N, Martz A, Bresson A, Spenlehauer C, de Murcia G, Menissier-de Murcia J. XRCC1 is phosphorylated by DNA-dependent protein kinase in response to DNA damage. Nucleic Acids Res. 2006;34:32–41. doi: 10.1093/nar/gkj409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, Sarno S, Meggio F, Pinna LA, Caldecott KW. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 117.Gocke CB, Yu H, Kang J. Systematic identification and analysis of mammalian small ubiquitin-like modifier substrates. J Biol Chem. 2005;280:5004–5012. doi: 10.1074/jbc.M411718200. [DOI] [PubMed] [Google Scholar]
- 118.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hasan S, El-Andaloussi N, Hardeland U, Hassa PO, Burki C, Imhof R, Schar P, Hottiger MO. Acetylation regulates the DNA end-trimming activity of DNA polymerase ß. Molecular Cell. 2002;10:1213–1222. doi: 10.1016/s1097-2765(02)00745-1. [DOI] [PubMed] [Google Scholar]
- 120.Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- 121.Henneke G, Koundrioukoff S, Hubscher U. Phosphorylation of human Fen1 by cyclin-dependent kinase modulates its role in replication fork regulation. Oncogene. 2003;22:4301–4313. doi: 10.1038/sj.onc.1206606. [DOI] [PubMed] [Google Scholar]
- 122.Friedrich-Heineken E, Henneke G, Ferrari E, Hubscher U. The acetylatable lysines of human Fen1 are important for endo- and exonuclease activities. J Mol Biol. 2003;328:73–84. doi: 10.1016/s0022-2836(03)00270-5. [DOI] [PubMed] [Google Scholar]
- 123.Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, Hubscher U, Hottiger MO. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Molecular Cell. 2001;7:1221–1231. doi: 10.1016/s1097-2765(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 124.Huang S, Beresten S, Li B, Oshima J, Ellis NA, Campisi J. Characterization of the human and mouse WRN 3’-->5’ exonuclease. Nucleic Acids Res. 2000;28:2396–2405. doi: 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blander G, Zalle N, Daniely Y, Taplick J, Gray MD, Oren M. DNA damage-induced translocation of the Werner helicase is regulated by acetylation. J Biol Chem. 2002;277:50934–50940. doi: 10.1074/jbc.M210479200. [DOI] [PubMed] [Google Scholar]
- 126.Luo Y, Ji X, Ling F, Li W, Zhang F, Cao G, Chen J. Impaired DNA repair via the base-excision repair pathway after focal ischemic brain injury: a protein phosphorylation-dependent mechanism reversed by hypothermic neuroprotection. Front Biosci. 2007;12:1852–1862. doi: 10.2741/2193. [DOI] [PubMed] [Google Scholar]
- 127.Kotake M, Nakai A, Nagasaka A, Itoh M, Hidaka H, Yoshida S. Hormonal regulation of DNA polymerase beta activity and expression in rat adrenal glands and testes. Mol Cell Endocrinol. 2002;192:127–132. doi: 10.1016/s0303-7207(02)00080-1. [DOI] [PubMed] [Google Scholar]
- 128.El-Andaloussi N, Valovka T, Toueille M, Steinacher R, Focke F, Gehrig P, Covic M, Hassa PO, Schar P, Hubscher U, Hottiger MO. Arginine methylation regulates DNA polymerase ß. Molecular Cell. 2006;22:51–62. doi: 10.1016/j.molcel.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 129.El-Andaloussi N, Valovka T, Toueille M, Hassa PO, Gehrig P, Covic M, Hubscher U, Hottiger MO. Methylation of DNA polymerase beta by protein arginine methyltransferase 1 regulates its binding to proliferating cell nuclear antigen. Faseb J. 2007;21:26–34. doi: 10.1096/fj.06-6194com. [DOI] [PubMed] [Google Scholar]
- 130.Bhat KR, Benton BJ, Ray R. DNA ligase I is an in vivo substrate of DNA-dependent protein kinase and is activated by phosphorylation in response to DNA double-strand breaks. Biochemistry. 2006;45:6522–6528. doi: 10.1021/bi051504i. [DOI] [PubMed] [Google Scholar]
- 131.Dong Z, Tomkinson AE. ATM mediates oxidative stress-induced dephosphorylation of DNA ligase IIIalpha. Nucleic Acids Res. 2006;34:5721–5279. doi: 10.1093/nar/gkl705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Watts FZ. Sumoylation of PCNA: Wrestling with recombination at stalled replication forks. DNA Repair (Amst) 2006;5:399–403. doi: 10.1016/j.dnarep.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 133.Lehmann AR. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005;579:873–876. doi: 10.1016/j.febslet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 134.Lehmann AR. Translesion synthesis in mammalian cells. Exp Cell Res. 2006;312:2673–2676. doi: 10.1016/j.yexcr.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 135.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase Beta. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 136.Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 137.Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 138.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. Washington, D.C: ASM Press; 2006. [Google Scholar]
- 139.Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE. Post-replicative base excision repair in replication foci. EMBO Journal. 1999;18:3834–3844. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Muller-Weeks SJ, Caradonna S. Specific association of cyclin-like uracil-DNA glycosylase with the proliferating cell nuclear antigen. Experimental Cell Research. 1996;226:346–355. doi: 10.1006/excr.1996.0235. [DOI] [PubMed] [Google Scholar]
- 141.Schrofelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. 2005;79:10978–10987. doi: 10.1128/JVI.79.17.10978-10987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. Journal of Biological Chemistry. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 143.Miao F, Bouziane M, Dammann R, Masutani C, Hanaoka F, Pfeifer G, O’Connor TR. 3-Methyladenine-DNA glycosylase (MPG protein) interacts with human RAD23 proteins. Journal of Biological Chemistry. 2000;275:28433–28438. doi: 10.1074/jbc.M001064200. [DOI] [PubMed] [Google Scholar]
- 144.Watanabe S, Ichimura T, Fujita N, Tsuruzoe S, Ohki I, Shirakawa M, Kawasuji M, Nakao M. Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase link transcriptional repression and DNA repair in chromatin. Proceedings of the National Academy of Science. 2003;100:12859–12864. doi: 10.1073/pnas.2131819100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tuo J, Chen C, Zeng X, Christiansen M, Bohr VA. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair. 2002;1:913–927. doi: 10.1016/s1568-7864(02)00116-7. [DOI] [PubMed] [Google Scholar]
- 146.Parker A, Gu Y, Mahoney W, Lee SH, Singh KK, Lu AL. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. Journal of Biological Chemistry. 2001;276:5547–5555. doi: 10.1074/jbc.M008463200. [DOI] [PubMed] [Google Scholar]
- 147.Bessho T. Nucleotide excision repair 3’ endonuclease XPG stimulates the activity of base excision repairenzyme thymine glycol DNA glycosylase. Nucleic Acids Research. 1999;27:979–983. doi: 10.1093/nar/27.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Klungland A, Hoss M, Gunz D, Constantinou A, Clarkson SG, Doetsch PW, Bolton PH, Wood RD, Lindahl T. Base excision repair of oxidative DNA damage activated by XPG protein. Molecular Cell. 1999;3:33–42. doi: 10.1016/s1097-2765(00)80172-0. [DOI] [PubMed] [Google Scholar]
- 149.Bennett RAO, Wilson DM, 3rd, Wong D, Demple B. Interaction of human apurinic endonuclease and DNA polymerase ß in the base excision repair pathway. Proceedings of the National Academy of Science. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Masuda Y, Bennett RA, Demple B. Rapid dissociation of human apurinic endonuclease (Ape1) from incised DNA induced by magnesium. Journal of Biological Chemistry. 1998;273:30360–30365. doi: 10.1074/jbc.273.46.30360. [DOI] [PubMed] [Google Scholar]
- 151.Gaiddon C, Moorthy NC, Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO Journal. 1999;18:5609–5621. doi: 10.1093/emboj/18.20.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkinson AE, Wilson SH. Specific interaction of DNA polymerase ß and DNA ligase I in a multiprotein base excision repair complex from bovine testis. Journal of Biological Chemistry. 1996;271:16000–16007. doi: 10.1074/jbc.271.27.16000. [DOI] [PubMed] [Google Scholar]
- 153.Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. EMBO Journal. 2001;20:914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Muftuoglu M, Wong HK, Imam SZ, Wilson DM, 3rd, Bohr VA, Opresko PL. Telomere repeat binding factor 2 interacts with base excision repair proteins and stimulates DNA synthesis by DNA polymerase beta. Cancer Res. 2006;66:113–124. doi: 10.1158/0008-5472.CAN-05-2742. [DOI] [PubMed] [Google Scholar]
- 155.Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase ß and proliferating cell nuclear antigen. Journal of Biological Chemistry. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- 156.Naryzhny SN, Lee H. The post-translational modifications of proliferating cell nuclear antigen (PCNA): acetylation, not phosphorylation, plays an important role in the regulation of its function. Journal of Biological Chemistry. 2004 doi: 10.1074/jbc.M312850200. In Press. [DOI] [PubMed] [Google Scholar]
- 157.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate: Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. Journal of Biological Chemistry. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 158.Levin DS, McKenna AE, Motycka TA, Matsumoto Y, Tomkinson AE. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Current Biology. 2000;10:919–922. doi: 10.1016/s0960-9822(00)00619-9. [DOI] [PubMed] [Google Scholar]
- 159.Tom S, Henricksen LA, Park MS, Bambara RA. DNA ligase I and proliferating cell nuclear antigen form a functional complex. Journal of Biological Chemistry. 2001;276:24817–24825. doi: 10.1074/jbc.M101673200. [DOI] [PubMed] [Google Scholar]
- 160.Tom S, Henricksen LA, Bambara RA. Mechanism whereby proliferating cell nuclear antigen stimulates flap endonuclease 1. Journal of Biological Chemistry. 2000;275:10498–10505. doi: 10.1074/jbc.275.14.10498. [DOI] [PubMed] [Google Scholar]
- 161.Wu X, Li J, Li X, Hsieh CL, Burgers PM, Lieber MR. Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Research. 1996;24:2036–2043. doi: 10.1093/nar/24.11.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Brosh RM, Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO Journal. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sharma S, Sommers JA, Wu L, Bohr VA, Hickson ID, Brosh RM., Jr Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J Biol Chem. 2004;279:9847–9856. doi: 10.1074/jbc.M309898200. [DOI] [PubMed] [Google Scholar]
- 164.Sharma S, Otterlei M, Sommers JA, Driscoll HC, Dianov GL, Kao HI, Bambara RA, Brosh RM., Jr WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol Biol Cell. 2004;15:734–750. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gary R, Ludwig DL, Cornelius HL, MacInnes MA, Park MS. The DNA repair endonuclease XPG binds to proliferating cell nuclear antigen (PCNA) and shares sequence elements with the PCNA-binding regions of FEN-1 and cyclin-dependent kinase inhibitor p21. Journal of Biological Chemistry. 1997;272:24522–24529. doi: 10.1074/jbc.272.39.24522. [DOI] [PubMed] [Google Scholar]
- 166.Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature. 2001;410:387–391. doi: 10.1038/35066610. [DOI] [PubMed] [Google Scholar]
- 167.Hong R, Chakravarti D. The human proliferating Cell nuclear antigen regulates transcriptional coactivator p300 activity and promotes transcriptional repression. Journal of Biological Chemistry. 2003;278:44505–44513. doi: 10.1074/jbc.M303138200. [DOI] [PubMed] [Google Scholar]
- 168.Vairapandi M, Liebermann DA, Hoffman B, Duker NJ. Human DNA-demethylating activity: a glycosylase associated with RNA and PCNA. Journal of Cellular Biochemistry. 2000;79:249–260. doi: 10.1002/1097-4644(20001101)79:2<249::aid-jcb80>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 169.Boldogh I, Milligan D, Lee MS, Bassett H, Lloyd RS, McCullough AK. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Research. 2001;29:2802–2809. doi: 10.1093/nar/29.13.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochimica et Biophysica Acta. 2000;1471:M43–56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 171.Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. Journal of Biological Chemistry. 2003;278:45145–45153. doi: 10.1074/jbc.M307957200. [DOI] [PubMed] [Google Scholar]
- 172.Flohr C, Burkle A, Radicella JP, Epe B. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Research. 2003;31:5332–5337. doi: 10.1093/nar/gkg715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Galande S, Kohwi-Shigematsu T. Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J Biol Chem. 1999;274:20521–20528. doi: 10.1074/jbc.274.29.20521. [DOI] [PubMed] [Google Scholar]
- 174.Mohan RD, Rao A, Gagliardi J, Tini M. SUMO-1 dependent allosteric regulation of Thymine DNA Glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol Cell Biol. 2006 doi: 10.1128/MCB.00323-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell. 2004;15:621–634. doi: 10.1016/j.molcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 176.Fischer JA, Muller-Weeks S, Caradonna S. Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during the S phase of the cell cycle. DNA Repair (Amst) 2004;3:505–513. doi: 10.1016/j.dnarep.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 177.Caradonna S, Muller-Weeks S. The nature of enzymes involved in uracil-DNA repair: isoform characteristics of proteins responsible for nuclear and mitochondrial genomic integrity. Curr Protein Pept Sci. 2001;2:335–347. doi: 10.2174/1389203013381044. [DOI] [PubMed] [Google Scholar]
- 178.Likhite VS, Cass EI, Anderson SD, Yates JR, Nardulli AM. Interaction of estrogen receptor alpha with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J Biol Chem. 2004;279:16875–16882. doi: 10.1074/jbc.M313155200. [DOI] [PubMed] [Google Scholar]
- 179.Dantzer F, Luna L, Bjoras M, Seeberg E. Human OGG1 undergoes serine phosphorylation and associates with the nuclear matrix and mitotic chromatin in vivo. Nucleic Acids Res. 2002;30:2349–2357. doi: 10.1093/nar/30.11.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kauppinen TM, Chan WY, Suh SW, Wiggins AK, Huang EJ, Swanson RA. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci U S A. 2006;103:7136–7141. doi: 10.1073/pnas.0508606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, Gersbach M, Imhof R, Hottiger MO. Acetylation of PARP-1 by p300/CBP regulates coactivation of NF-kappa B-dependent transcription. J Biol Chem. 2005 doi: 10.1074/jbc.M507553200. [DOI] [PubMed] [Google Scholar]
- 182.Masdehors P, Glaisner S, Maciorowski Z, Magdelenat H, Delic J. Ubiquitin-dependent protein processing controls radiation-induced apoptosis through the N-end rule pathway. Exp Cell Res. 2000;257:48–57. doi: 10.1006/excr.2000.4870. [DOI] [PubMed] [Google Scholar]
- 183.Cheng WH, von Kobbe C, Opresko PL, Fields KM, Ren J, Kufe D, Bohr VA. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol Cell Biol. 2003;23:6385–6395. doi: 10.1128/MCB.23.18.6385-6395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Karmakar P, Bohr VA. Cellular dynamics and modulation of WRN protein is DNA damage specific. Mech Ageing Dev. 2005;126:1146–1158. doi: 10.1016/j.mad.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 185.Karmakar P, Piotrowski J, Brosh RM, Jr, Sommers JA, Miller SP, Cheng WH, Snowden CM, Ramsden DA, Bohr VA. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J Biol Chem. 2002;277:18291–18302. doi: 10.1074/jbc.M111523200. [DOI] [PubMed] [Google Scholar]
- 186.Pichierri P, Rosselli F, Franchitto A. Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene. 2003;22:1491–1500. doi: 10.1038/sj.onc.1206169. [DOI] [PubMed] [Google Scholar]
- 187.Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, Campisi J, Chen DJ. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem. 2001;276:38242–38248. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- 188.Kawabe Y, Seki M, Seki T, Wang WS, Imamura O, Furuichi Y, Saitoh H, Enomoto T. Covalent modification of the Werner’s syndrome gene product with the ubiquitin-related protein, SUMO-1. J Biol Chem. 2000;275:20963–20966. doi: 10.1074/jbc.C000273200. [DOI] [PubMed] [Google Scholar]
- 189.Woods YL, Xirodimas DP, Prescott AR, Sparks A, Lane DP, Saville MK. p14 Arf promotes small ubiquitin-like modifier conjugation of Werners helicase. J Biol Chem. 2004;279:50157–50166. doi: 10.1074/jbc.M405414200. [DOI] [PubMed] [Google Scholar]
- 190.Prosperi E, Scovassi AI, Stivala LA, Bianchi L. Proliferating cell nuclear antigen bound to DNA synthesis sites: phosphorylation and association with cyclin D1 and cyclin A. Exp Cell Res. 1994;215:257–262. doi: 10.1006/excr.1994.1341. [DOI] [PubMed] [Google Scholar]
- 191.Haracska L, Torres-Ramos CA, Johnson RE, Prakash S, Prakash L. Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:4267–4274. doi: 10.1128/MCB.24.10.4267-4274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 193.Solomon DA, Cardoso MC, Knudsen ES. Dynamic targeting of the replication machinery to sites of DNA damage. J Cell Biol. 2004;166:455–463. doi: 10.1083/jcb.200312048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]