

Abstract
To evaluate the number and time of the migration(s) that colonized the New World we analyzed all available sequences of the first hypervariable segment of the human mitochondrial DNA control region, including 544 Native Americans. Sequence and population trees showed that the Amerind, Na-Dene, and Eskimo are significantly closer among themselves than anyone is to Asian populations, with the exception of the Siberian Chukchi, that in some analyses are closer to Na-Dene and Eskimo. Nucleotide diversity analyses based on haplogroup A sequences suggest that Native Americans and Chukchi originated from a single migration to Beringia, probably from east Central Asia, that occurred ≈30,000 or ≈43,000 years ago, depending on which substitution rate is used, with 95% confidence intervals between ≈22,000 and ≈55,000 years ago. These results support a model for the peopling of the Americas in which Beringia played a central role, where the population that originated the Native Americans settled and expanded. Some time after the colonization of Beringia they crossed the Alberta ice-free corridor and peopled the rest of the American continent. The collapse of this ice-free corridor during a few thousand years ≈14,000–20,000 years ago isolated the people south of the ice-sheets, who gave rise to the Amerind, from those still in Beringia; the latter originated the Na-Dene, Eskimo, and probably the Siberian Chukchi.
Keywords: Native Americans, Amerindian origins, human evolution, population genetics
Although the peopling of the New World has been the object of a great number of archeologic, linguistic, and genetic studies, the few agreements achieved to date are their origin by migration from Asia during the last glaciation [at least 12,000 years before present (ybp)], through the Bering Strait (1, 2), but even the latter point is not yet entirely settled (3, 4). The number and age of the migration(s) into the Americas are major questions under extended discussion (5–10). An important related issue is the origin of the biological and cultural diversity now found in aboriginal Americans, if it is indigenous or if it predates the occupation of the continent. Questions of origin notwithstanding, most of the recent analyses distinguish three groups of aboriginal Americans, usually equating them to three language phyla: Amerind, Na-Dene, and Eskimo-Aleut (11).
A recent model for the peopling of the Americas is the so-called “three-wave” hypothesis (5), which stated that the continent was settled by three distinct migrations from Siberia, which corresponded exactly to the above three language groups. According to this model, the first migration gave rise to the so-called Paleo-Indians, represented today by Amerind speakers, and two subsequent migrations originated the Na-Dene and Eskimo, but the order and age of the latter two were not consensual among the proponents of such hypothesis (10). This model raised considerable debate, with evidences for (12, 13) and against it (7–10, 14). A different scenario has been proposed by Szathmary (15, 16), who found a very close relationship between Na-Dene and Eskimo and suggested (10, 17, 18) that their differentiation from the Amerind occurred in Beringia. Their divergence was prompted by the blocking of the Alberta ice-free corridor during a few thousand years that isolated the people south of the glaciers (who gave rise to the Amerind) from those still in Beringia (who gave rise to the Na-Dene and Eskimo). This model requires an early occupation of Beringia, at least a few thousand years before the coalescence of the glaciers, which occurred around 18,000 ybp (18).
Despite having been addressed in several mitochondrial DNA (mtDNA) studies, no clear picture of the New World colonization problem has yet emerged. For example, a different three (or four)-migration model has been suggested (6, 19), based mainly on results from mtDNA restriction site polymorphism (RFLP) data. This hypothesis proposed that the Amerind originated from two migrations, the first carrying the so-called Native American mtDNA haplogroups A, C, and D (13), and the second mtDNA sequences from haplogroup B only. A third migration would have given rise to the Na-Dene speakers, which are postulated to be more recent because they have been found to retain much lower haplogroup A diversity than the Amerind. Nothing was said about the origin of the Eskimo-Aleuts. Alternatively, based on mtDNA control region (CR) sequences, it was proposed (20) that each of the four Native American mtDNA haplogroups derived from a different migration from Asia. Another set of mtDNA CR sequences showed that populations from Beringia (Na-Dene, Eskimo, and the Siberian Chukchi) are very closely related and may have had a recent common origin (14). Recent reanalyzes of all relevant mtDNA RFLP information (8), and new mtDNA CR sequence data from Mongolians (9), indicated that, contrary to what had been previously proposed (12, 13), these results would suggest a single migration scenario. These data and the distribution of the human T-lymphotropic virus, type II infection in Asia and the New World (7) indicated that the people now in east Central Asia (e.g., Mongolia) are more closely related to Native Americans than those who now inhabit northeast Siberia. In this paper we address the peopling of the Americas problem by means of a complete reanalysis of all available Native American and Asian mtDNA CR sequences, using several phylogenetic and population genetic methods.
MATERIALS AND METHODS
Populations and Sequences Studied.
We first constructed global phylogenetic trees using all available sequences of the first hypervariable segment of the human mtDNA CR (HVS-I, about 360 bp; ref. 21), consisting of more than 1000 different sequences described from about 2500 individuals. From these global trees, the set of sequences that clustered in the Amerindian haplogroup A (13) was selected for further investigation (since it represents the highest number of sequences found in Eskimo, Na-Dene, and Chukchi populations). The sequences were easily aligned by hand, and the positions that were considered insertions in relation to the reference sequence (22) were not used. This criterion was also applied to the region from positions 16184 to 16193, shown to be prone to somatic mutations in the number of C’s (23). For most analyses all sites with deletions or unknown nucleotides in any sequence were not used, although the results were not different using the pairwise-deletion approach (24).
In general, two sets of population analyses were performed, one using all available sequences for each population and another using haplogroup A sequences only. All available populations of Native Americans and relevant Asians (Siberians, Chukchi, Mongolians, and East Asians) were used, as well as a European and two Africans as outgroup populations. Data on two South American indigenous populations (20) were not used because they were not sequenced for the first 100 bases of the HVS-I. Below are the populations used, for each major geographic region and population group (n = sample size), and the reference for the sequence data: Amerind: South America (n = 171): Xavante (n = 25) (25), Zoró (n = 30) (25), Gavião (n = 27) (25), Wai-Wai (n = 26) (unpublished data), Suruí (n = 24) (unpublished data), Mapuche (Argentina) (n = 39) (26); Central America (n = 136): Huetar (n = 27) (27), Ngöbé (n = 46) (28), Kuna (n = 63) (29); North America (n = 145): Nuu-Chah-Nulth (n = 63) (30), Yakima (n = 42) (14), BellaCoola (n = 40) (31); Na-Dene (n = 70): Haida (n = 41) (31), Athapaskan (n = 21) (14), several populations (n = 8) (12); Eskimo (n = 22): Inupiaq Eskimo (n = 5) (14), West Greenland Eskimo (n = 17) (14); Siberian Chukchi (n = 7) (14); Siberia (n = 33): Altai (n = 17) (14), several populations (n = 16) (19); Mongolia (n = 103) (9); East Asia (n = 99) (12, 32, 33); Africa (n = 42): Mbuty Pygmy (n = 30) (32, 33), Yoruba (n = 12) (33); Europe (n = 77) (34).
Phylogenetic Analysis.
For all tree constructions, we used several DNA sequence distances (24), and the neighbor-joining (NJ) method (35), but all distances gave essentially the same results, so only those with the p-distance (proportion of differences) are presented. The global phylogenetic trees of all sequences were constructed using the njboot program (provided by N. Takezaki, National Institute of Genetics, Tokyo). The NJ trees of the haplogroup A sequences and the confidence probability values for their branches were estimated using the mega program (24). The population trees were constructed using the dA and dXY population distances (36) and the sendbs program (provided by N. Takezaki). The reliability of the branches in the population trees was estimated using the bootstrap approach (37), resampling 100 times the sites. Minimum spanning trees (MSTs) of all haplogroup A sequences used in the NJ trees were constructed by the minspnet program (provided by L. Excoffier, University of Geneva), that also estimate all alternative links. All distances tested gave the same tree.
Diversity and Divergence Estimates.
The age of the populations and the time of divergence between them were estimated by using two methods. The first was the mismatch distribution approach (38), which calculates the mean time of expansion within and between (intermatch) populations through the distribution of the pairwise differences. The time estimates were calculated using Roger’s method of moments (39), and the 95% confidence interval (CI) (±2 × TMSE) for these times was calculated by using the Redd et al. (32) approach and the following formula:
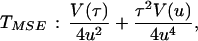 |
1 |
where TMSE is the mean square error of the mean expansion or separation time; u is the total mutation rate over all sites (given below); τ (tau) is the time in units of mutation, calculated from the sample as described (39); and V(u) and V(τ) are their sampling variances. V(u) was obtained using the methods and data given in ref. 40, as detailed below, and V(τ) was calculated by a method we developed (unpublished data) using the jackknifing approach (37). Briefly, we first compute τ using all individuals (n) of the data set. Then we eliminate the first one, compute τ again, and denominate it τ1. We repeat the procedure for each individual and denominate the new values τ2, τ3 … τn. The variance of τ is computed by
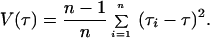 |
2 |
The second approach for age estimation uses the average nucleotide diversity within (π) and between (dXY) populations (36). The standard error (SE) of these estimates was calculated with a bootstrap method by 100 replicates over sites using the sendbs program. As the distances were very small and all distances gave very similar results, only the results with the p-distance are presented. The 95% CI for the π and dXY values was calculated using the usual ± 2 × SE, and the 95% CI for the time of origin and divergence of the populations were estimated using an approach similar to that described in Eq. 1.
For the time estimates two substitution rates for the HVS-I were used. One was the recently estimated (40) slow rate of 10.3% per million years, whose SE of 1.35% was calculated here using their approach (40) and data provided by N.Takahata. The usual fast rate of 15% per million years (ref. 30; M. Stoneking, personal communication) was also used, with an associated SE of 1.97%, calculated from the SE of the slow rate, based on the proportion between the rates.
RESULTS
Haplogroup A Polymorphisms.
The HVS-I sequences of the Amerindian haplogroup A (13) generally shared the following polymorphisms (numbered in relation to the reference sequence; ref. 22): 16111(T), 16223(T), 16290(T), 16319(A), and 16362(C). Nucleotides 16111(T), 16290(T), and 16319(A) are characteristic of haplogroup A only, 16362(C) is shared with haplogroup D, and 16223(T) is shared with haplogroups C and D. Despite the about 2000 non-Native Americans that have been sequenced for the HVS-I to date, only two (two Siberian Chukchi; ref. 14) presented the above set of five markers, while the joint occurrence of four markers was found in six (non-Chukchi) Asians: two Mongolians, two east Asians, one Siberian, and one Tahitian. Among the 281 Native Americans harboring haplogroup A sequences, ≈81% have five and ≈17% four markers. The figures for each linguistic group and Chukchi are as follows: Amerind (n = 189), ≈77% have five and ≈23% four markers; Na-Dene (n = 68), ≈90% have five and ≈7% four markers; Eskimo (n = 24), ≈88% have five and ≈4% four markers; Chukchi (n = 6), 50% have five and ≈33% four markers.
Sequence Phylogenies.
The phylogenetic trees were constructed with all Native American and Asian haplogroup A sequences, and one Amerind sequence representative of each of the other three haplogroups. In these trees (Fig. 1) almost all Na-Dene and Eskimo sequences clustered with Amerind sequences, not with Asian or other sequences. Interestingly, most Na-Dene (mainly Athapaskan), four Eskimo and two Chukchi sequences, clustered together in a group that shared a T in position 16192. The main exceptions are four Haida sequences, which together with an Eskimo and two Amerind, clustered with other Amerind. Also, it is clear from the tree that the Na-Dene and Eskimo present high sequence diversity, similar to the Amerind. Finally, of the six Siberian Chukchi sequences, only one (nonhaplogroup A) clustered with Asian sequences, the other five clustered with or are identical to Native American sequences. Similarly to the NJ trees, in the MSTs (not shown) almost all Amerind, Na-Dene, Eskimo, and Chukchi sequences radiate from ancestral sequences not shared with Asians, while no Asian sequence is derived from these ancestral sequences.
Figure 1.

NJ tree based on the p-distance for haplogroup A sequences from Native Americans and Asian populations, plus one representative of the other three haplogroups. Shared sequences have multiple labels. The number on the branches are confidence probability values (24).
Population Trees.
In addition to the individual sequences approach above, we have calculated several DNA distance matrices comparing populations or groups of populations (see below) and with them constructed NJ population trees. These population trees are not to be interpreted in a strict “phylogenetic” context, but as a useful way to summarize the relationship presented in the distance matrices. In all these analyses we considered the Chukchi as a separate group due to the peculiar position of its sequences in the phylogeny (Fig. 1). The NJ population tree considering all sequences (Fig. 2a) show the Na-Dene, Eskimo, and Chukchi, whose sequences are mainly from haplogroup A, clustered in a tight group with a high confidence value, and this group clustered with the Amerind also with high confidence. A similar result can be seen in the NJ tree constructed with haplogroup A sequences only (Fig. 2b), except that the Chukchi are now placed outside the Native American cluster. But these four groups still cluster together with a high confidence value. We also constructed trees considering each Native American (plus the Chukchi) population separately (data not shown), with very similar results; the Athapaskan clustered with the Eskimo and Chukchi and these clustered with most of the other Native American populations.
Figure 2.

NJ trees for the populations based on the dA distance using (a) all sequences and (b) haplogroup A sequences only. The numbers on the branches are bootstrap values based on 100 replications.
Diversity Analyses.
Diversity values within populations (such as π) have been used for inferences about their age (13), although it has been shown that under a neutral model and constant population size, this parameter measures historical population size, not age (41). However, it has been demonstrated (42) that if a population suffered a prolonged, severe bottleneck, followed by size expansion, genetic diversity would measure population age, specifically the postbottleneck time. In such case, the shape of the distribution of the number of pairwise differences in a sample (also called the mismatch distribution) has been used to estimate some population parameters, especially the occurrence and time since a population size expansion (42–44). This approach has also been applied to a monophyletic group of sequences, in contrast to a defined population (32). Interestingly, it has been found that high mutation rate heterogeneity (α parameter of the gamma distribution < 0.1) mimics the effects of population expansion and may disguise a stationary population as an expanding one (45). Nonetheless, a significantly negative Tajima’s D statistics (46) is a clear signature of a large population expansion.
It is clear that, unless the size of the Native American founding population was unreasonably high, the peopling of the continent should have been characterized by a dramatic population expansion. Assuming the maximum number of Native Americans at any time was 10 million, a conservative estimate (47), a 1000-fold size expansion would imply a colonizer population of up to 10,000 individuals. A dramatic postcolonization Native American expansion is corroborated here by the smooth unimodal shape of their haplogroup A mtDNA mismatch distribution (Fig. 3), the branching patterns of the sequence tree (Fig. 1), and the MSTs (not presented), which show most Native American sequences radiating from one node. Furthermore, as Native American haplogroup A sequences present a significantly negative Tajima’s D value (equal to −2.28, n = 85, P < 0.01), an α parameter of 0.47 (48), and the indication of at least a 100-fold expansion, the demographic parameters estimated here would be only slightly affected by the mutation rate heterogeneity problem (see table 1 in ref. 45).
Figure 3.

Mismatch and intermatch distributions of haplogroup A sequences from Native American populations and a merged Asian group. Fi indicates the relative frequency of pairs of sequences that differ by i nucleotide sites.
Table 1 shows the nucleotide diversity (π) and divergence (dxy) for several groups of populations and their associated age, based on both substitution rates and, for the first time (to our knowledge) in this kind of analysis, a 95% CI for these estimates. These values were calculated using haplogroup A sequences only. If the sequences from the other three Amerindian haplogroups were also used the initial divergence among these haplogroups, which occurred in Asia thousands of years before the colonization of the Americas (refs. 8 and 30; S.L.B., M. Stoneking, C.E.A. Coimbna, Jr., R.V. Sandas, and F.M.S., unpublished results), they would inflate the estimate. Interestingly, the population ages estimated by the π values and by the mismatch distribution approach (data not shown) were identical or very similar. This further indicates that the peopling of Americas is compatible with a large population expansion, as both age estimations should coincide unless the initial diversity was high, which would indicate a large founding population. Amerind, Na-Dene, and Eskimo haplogroup A sequences show very similar mean diversity values, around 0.0085 (95% CI = 0.0066–0.01), and mean expansion ages around 28,000 ybp (95% CI ≈ 18,000–37,000, 15% rate) and 41,000 ybp (95% CI ≈ 27,000–54,000, 10.3% rate). The Chukchi diversity and ages were lower than the three Native American groups, but these differences were not significant. When we merged as one group the Amerind, Na-Dene, Eskimo, and Chukchi sequences, this merged population diversity (0.0088) and mean ages (≈29,000 and ≈42,000 ybp) were very similar to the values calculated for each population separately and to the Native Americans as a whole (Table 1). The divergence calculated for all pairs of Native American populations were similar among themselves and also to the within-population values (Table 1), and although Na-Dene and Eskimo were always closer to each other than to the Amerind, this was not significant. The similar mismatch and intermatch distributions among Native American populations (Fig. 3) are indicative that their expansion occurred before or coincidentally with their separation (43).
Table 1.
Nucleotide diversity and divergence, and age estimates based on haplogroup A sequences only
| Population | n* | Ns† | Nucleotide diversity, 95% CI (%) | Mean age, 95% CI (years)
|
|
|---|---|---|---|---|---|
| 10.3% rate | 15% rate | ||||
| Within | π | Expansion | |||
| Amerind | 44 | 32 | 0.85 (0.77–0.92) | 41,067 (29,725–52,408) | 28,199 (20,397–36,001) |
| Na-Dene | 24 | 19 | 0.83 (0.66–1.00) | 40,238 (26,892–53,585) | 27,630 (18,454–36,806) |
| Eskimo | 17 | 10 | 0.85 (0.73–0.98) | 41,505 (29,145–53,864) | 28,500 (20,000–37,000) |
| Native Americans | 85 | 57 | 0.89 (0.81–0.97) | 43,339 (31,348–55,329) | 29,759 (21,511–38,008) |
| Chukchi | 5 | 5 | 0.64 (0.41–0.87) | 31,045 (17,233–44,858) | 21,318 (11,827–30,809) |
| Asia | 10 | 10 | 1.38 (1.05–1.70) | 66,795 (43,216–90,373) | 45,866 (29,657–62,074) |
| Between | dXY | Divergence | |||
| Amerind vs. Na-Dene | 68 | 51 | 0.91 (0.80–1.03) | 44,393 (31,513–57,272) | 30,483 (21,639–39,327) |
| Amerind vs. Eskimo | 61 | 42 | 0.96 (0.88–1.03) | 46,466 (33,728–59,204) | 31,907 (23,160–40,654) |
| Amerind vs. Chukchi | 49 | 37 | 0.78 (0.67–0.88) | 37,652 (26,632–48,672) | 25,854 (18,287–33,422) |
| Amerind vs. Asia | 54 | 42 | 1.42 (1.23–1.61) | 68,826 (48,605–89,047) | 47,260 (33,375–61,146) |
| Na-Dene vs. Eskimo | 41 | 29 | 0.88 (0.77–1.00) | 42,853 (30,274–55,432) | 29,426 (20,788–38,064) |
| Na-Dene vs. Chukchi | 29 | 24 | 0.77 (0.64–0.90) | 37,278 (25,637–48,919) | 25,598 (17,604–33,591) |
| Na-Dene vs. Asia | 34 | 29 | 1.47 (1.27–1.67) | 71,263 (50,270–92,257) | 48,934 (34,519–63,349) |
| Eskimo vs. Chukchi | 22 | 15 | 0.77 (0.65–0.89) | 37,354 (25,996–48,712) | 25,650 (17,851–33,449) |
| Eskimo vs. Asia | 27 | 20 | 1.43 (1.25–1.61) | 69,313 (49,151–89,474) | 47,595 (33,751–61,439) |
| Chukchi vs. Asia | 15 | 15 | 1.08 (0.86–1.30) | 52,213 (34,851–69,574) | 35,853 (23,931–47,774) |
| Native Americans vs. Asia | 95 | 71 | 1.43 (1.25–1.62) | 69,611 (49,326–89,897) | 47,800 (33,871–61,729) |
*Number of individuals.
Number of different sequences.
Table 1 also shows the divergence values and ages between the Native American groups and a combined Asian population (Central Siberia plus Mongolia plus East Asia). The divergence values between any pair of Native American groups was always significantly lower than the divergence between them and the combined Asia group; the divergence times between the whole Native American and the combined Asian group were about 48,000 (15% rate) and 70,000 (10.3% rate) ybp. In the comparisons dealing with the individual Asian populations this same pattern was found, although only the divergence values, not the ages estimates, were always significantly lower (data not shown). Again, the Chukchi were the exception, being in general as similar to Native American populations as to Siberians. These results held even when we used all sequences (not only haplogroup A), as can be seen in the population tree of Fig. 2a. In this case, the Na-Dene, Eskimo, and also the Chukchi were all very closely related among themselves and more distantly related to the Amerind. The intermatch between the combined east Central Asian population and the Native Americans (Fig. 3) is intermediate between each mismatch distribution, and this is consistent with the Native Americans having grown like an Asian subpopulation (43).
DISCUSSION
Previous studies have found that the Na-Dene and Eskimo have mtDNA mainly from haplogroup A, while the Amerind have the four New World haplogroups (6, 8). Haplogroup A mtDNAs are absent or occur in a low frequency in Asian populations, from zero in most Siberian populations to 5% in Mongolia and Siberian Evenk to 11% in Tibetans (8, 9, 19). If the presence of non-A sequences in Na-Dene and Eskimo groups is only due to admixture with Amerinds or may also have existed since the origin of these two populations is a matter of dispute (6, 8). As we cannot distinguish between these two hypotheses, most of our results were based on analyses of haplogroup A sequences only, while some were based on all available sequences. If the second hypothesis proves to be correct the results and conclusions presented here, suggesting a single and early colonization event, will turn out to be even stronger (8, 9). The low frequency of haplogroup A sequences in Asians has led several authors to point out how unlikely it would be that two or three different migrations from Asia would carry these low frequency sequences to America (7–9) resulting, in the case of the Na-Dene and Eskimo, in almost fixation. This argument received further strength here as we showed that most (81%) of the haplogroup A sequences in Native Americans present five characteristic polymorphisms that, as a set, were absent or found in a very low frequency (only three Chukchi) in Asia. Furthermore, in the sequence tree (Fig. 1) and the MSTs, the majority of the Native American and some Chukchi sequences clustered together while the Asian sequences were found outside this Native American group. If the Na-Dene and Eskimo (together or independently) came from Asia more recently than the Amerind, we would expect to find some Asian haplogroup A sequences located in the tree between the majority Na-Dene plus Eskimo cluster and the majority Amerind cluster. However, only one Chukchi sequence is in such a position.
Torroni et al. (13) found that the Na-Dene have a much lower haplogroup A diversity than the Amerind and presented this as an evidence for an independent migration of the Na-Dene to the Americas. However, as can be seen in Table 1, Na-Dene haplogroup A sequences do not have a significantly lower diversity than the Amerind or Eskimo. Differently from them, we did not consider the within-population frequency of each sequence, only their between-population frequency. This is because the within-population frequency is affected mainly by recent demographic history, and its use to estimate the diversity of a much larger group during most of its history would artificially lower the result, especially if the sample includes just a few local populations, as is the case here. But this issue do not fully explain the large difference between the two results. While Torroni et al. (13) found that haplogroup A diversity in Amerinds was 3.4 times higher than in the Na-Dene, we found, even using as they have done the within- and between-population frequencies, a value only 1.4 times higher. The discordance between the results may have been caused by the different populations and regions of the mtDNA genome investigated.
The divergence values (Table 1) and the population trees (Fig. 2) show that generally the Amerind, Na-Dene, and Eskimo are significantly closer among themselves than any one is to Siberian (non-Chukchi), Mongolian or East Asian populations. This closer relationship among the three Native American groups is suggestive of a single migration model, and it is not parsimonious to consider that an ancestral Asian population disappeared leaving no traces. Similarly to other investigations (14–16), we also found the Na-Dene and Eskimo to be more closely related to each other than any one is to other populations (Fig. 2, Table 1). While these results were not statistically significant in the comparisons involving haplogroup A sequences only (Table 1), they were significant when all four Amerind haplogroups were used (not shown).
The relationship of the Siberian Chukchi to the Native American groups was ambiguous, because in some analyses they were located outside the Native American cluster (Fig. 2a), and in others they were more closely related to the Na-Dene and Eskimo than to the Amerind (Fig. 2b). Their close affinity with Eskimo and Na-Dene is further corroborated by classical genetics studies (5, 17, 49) and by language (17), suggesting they all may have a common origin. It should be noted that the Chukchi sample used here, although it was not specified (14), seems to be from the Coast Chukchi, since like them (T. G. Schurr, E.B. Starikovskaya, R.I. Sukernik, and D.C. Wallace, personal communication), most of their mtDNA sequences were from haplogroup A. Although the differences were not significant and the sample size is small, Chukchi diversity and expansion times (Table 1) are smaller than those of the Amerind and, especially, than the Na-Dene and Eskimo, and this is consistent with their having a more recent origin (14, 18).
Our estimates for the mean time of expansion of Native American ancestral population, based on haplogroup A diversity, are ≈30,000 or ≈43,000 ybp (Table 1), depending if we use the 15% or 10.3% substitution rate, respectively. Although the 10.3% rate seems to be the best one (40), its estimated dates may be slightly inflated because this ancestral population likely started to differentiate during the journey from somewhere in east Central Asia to Beringia (7, 9). Therefore, the actual date for their origin probably lies between the above fast and slow rates estimates, that have 95% CIs between about 22,000 and 55,0000 ybp, respectively (Table 1).
The “three-wave” (5) and most of the other colonization models regards Beringia just as a “corridor” or a “bridge” for a migration to America, centering the origin of the different migration(s) somewhere in Asia. Our results support a different model that, on the contrary, puts Beringia in a central role, where the population that originated the Native Americans settled and diversified before the further colonization of the rest of the American continent. It could be called an “out of Beringia” model, in contrast to the traditional “out of Asia” models, and is based on some of Szathmary’s works (10, 18), especially the key hypotheses that the glaciers’ coalescence was the isolation barrier that promoted the differentiation of the Amerind from the Na-Dene plus Eskimo, and that the latter diverged on Beringia. At least part of Beringia was a vast arctic steppe, consisting of the exposed continental shelves of the Chukchi and Bering seas and part of Siberia and Alaska, most of the time between 60,000 and 11,000 ybp (1, 50) and could have sustained human settlements (1, 18). It is also important to note that, in the context of the present model, questions such as in which continent, Asia or America, Native Americans started to diverge, become inapplicable, since the primary colonization and differentiation should have occurred in a single, merged land mass.
The picture that we suggest is that some time after the colonization of Beringia the population expanded and crossed the Alberta ice-free corridor that connected Beringia to the south of North America or, alternatively, followed a coastal route (51). The collapse of the Laurentide and Cordilleran ice sheets around 14,000 (18) or 20,000 ybp (51) blocked the Alberta corridor and isolated Beringia from the rest of the American continent during some time [6000 (10) or 2000 (52) years]. If, as our data imply, both Beringia and the region south of the glaciers were already colonized by this time, the two populations became isolated from each other during this time. This isolation may explain the close affinity among the Beringian groups (Na-Dene, Eskimo, and perhaps the Chukchi) on one side (10), and the more distant relationship of this group to the Amerind inhabiting the rest of the Americas on the other. This model would also explain why Beringian groups have a lower mtDNA diversity (primarily haplogroup A sequences) (7, 14) than the Amerind. While the people that colonized the south of the continent had an enormous area and a more favorable climate to expand, the people that lived north of the glaciers should have suffered with the deterioration of the climate and the reduction of their habitable area, that was never very large. The above conditions should have caused the inevitable loss of mtDNA sequence diversity (53), while the expanding southern populations maintained, as a whole, most of the original diversity. With the temperature rise after about 14,000 ybp, the habitable area in the American side of Beringia increased accordingly, probably followed by population growth and a much smaller rate of mtDNA lineage extinction (53). The differentiation of the Beringians in Na-Dene, Eskimo, and Chukchi may have occurred through several mechanisms, like the ice-free refugia model (52), and/or differential adaptation to the coastal and inland life stiles (15), the Chukchi probably representing a remnant population that stayed in the west side of Beringia, northeastern Siberia, after the final rise of the Bering Strait (17).
Acknowledgments
We thank E. Eizirik for critical reading the manuscript; N. Takezaki and L. Excoffier for supplying computer programs; N. Takahata for help in computing the SE of the HVS-I substitution rate; and M. Stoneking and A. Redd for providing their mtDNA data files. Financial support was provided by Financiadora de Estudos e Projetos and Conselho Nacional de Desenvolvimento Cientifico e Tecnologics.
ABBREVIATIONS
- ybp
years before present
- mtDNA
mitochondrial DNA
- CR
control region
- HVS-I
first hypervariable segment
- NJ
neighbor-joining
- CI
confidence interval
- MST
minimum spanning tree
References
- 1.Hoffecker J F, Powers W R, Goebel T. Science. 1993;259:532–536. doi: 10.1126/science.259.5091.46. [DOI] [PubMed] [Google Scholar]
- 2.Cavalli-Sforza L L, Piazza A, Menozzi P. History and Geography of Human Genes. Princeton: Princeton Univ. Press; 1994. [Google Scholar]
- 3.Bonatto S L, Redd A J, Salzano F M, Stoneking M. Am J Hum Genet. 1996;59:253–256. [PMC free article] [PubMed] [Google Scholar]
- 4.Cann R L, Lum J K. Am J Hum Genet. 1996;59:256–258. [Google Scholar]
- 5.Greenberg J H, Turner C G, Zegura S L. Curr Anthropol. 1986;27:477–498. [Google Scholar]
- 6.Wallace D C. Am J Hum Genet. 1995;57:201–223. [PMC free article] [PubMed] [Google Scholar]
- 7.Neel J V, Biggar R J, Sukernik R I. Proc Natl Acad Sci USA. 1994;91:10737–10741. doi: 10.1073/pnas.91.22.10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merriwether D A, Rothhammer F, Ferrell R E. Am J Phys Anthropol. 1995;98:411–430. doi: 10.1002/ajpa.1330980404. [DOI] [PubMed] [Google Scholar]
- 9.Kolman C J, Sambuughin N, Bermingham E. Genetics. 1995;142:1321–1334. doi: 10.1093/genetics/142.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szathmary E J E. In: Prehistoric Mongoloid Dispersals. Akazawa T, Szathmary E J E, editors. Oxford: Oxford Univ. Press; 1996. pp. 149–164. [Google Scholar]
- 11.Greenberg J H. Language in the Americas. Stanford: Stanford Univ. Press; 1987. [Google Scholar]
- 12.Torroni A, Schurr T G, Cabell M F, Brown M D, Neel J V, Larsen M, Smith D G, Vullo C M, Wallace D C. Am J Hum Genet. 1993;53:563–590. [PMC free article] [PubMed] [Google Scholar]
- 13.Torroni A, Schurr T G, Yang C-C, Szathmary E J E, Williams R C, Schanfield M S, Troup G A, Knowler W C, Lawrence D N, Weiss K M, Wallace D C. Genetics. 1992;130:153–162. doi: 10.1093/genetics/130.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shields G F, Schmiechen A M, Frazier B L, Redd A, Voevoda M I, Reed J K, Ward R H. Am J Hum Genet. 1993;50:758–765. [PMC free article] [PubMed] [Google Scholar]
- 15.Szathmary E J E, Ossenberg N S. Curr Anthropol. 1978;19:673–701. [Google Scholar]
- 16.Szathmary E J E. In: The First Americans: Origins, Affinities, and Adaptations. Laughlin W S, Harper A B, editors. New York: Gustav Fischer; 1979. pp. 185–210. [Google Scholar]
- 17.Szathmary E J E. Yearb Phys Anthropol. 1981;24:37–73. [Google Scholar]
- 18.Szathmary E J E. Evol Anthropol. 1993;1:202–220. [Google Scholar]
- 19.Torroni A, Sukernik R I, Schurr T G, Starikovskaya Y B, Cabell M F, Crawford M H, Comuzzie A G, Wallace D C. Am J Hum Genet. 1993;53:591–608. [PMC free article] [PubMed] [Google Scholar]
- 20.Horai S, Kondo R, Nakagawa-Hattori Y, Hayashi S, Sonoda S, Tajima K. Mol Biol Evol. 1993;10:23–47. doi: 10.1093/oxfordjournals.molbev.a039987. [DOI] [PubMed] [Google Scholar]
- 21.Vigilant L, Pennington R, Harpending H, Kocher T D, Wilson A C. Proc Natl Acad Sci USA. 1989;86:9350–9354. doi: 10.1073/pnas.86.23.9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson S, Bankier A T, Barrell B G, de Bruijn M H L, Coulson A R, Drouin J, Eperon I C, Nierlich D P, Roe B A, Sanger F, Schreier P H, Smith A J H, Staden R, Young I G. Nature (London) 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 23.Bendall K E, Sykes B C. Am J Hum Genet. 1995;57:248–256. [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar, S., Tamura, K. & Nei, M. (1993) mega (Pennsylvania State Univ., University Park), Version 1.01.
- 25.Ward R H, Salzano F M, Bonatto S L, Hutz M H, Coimbra C E A, Jr, Santos R V. Am J Hum Biol. 1996;8:317–323. doi: 10.1002/(SICI)1520-6300(1996)8:3<317::AID-AJHB2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 26.Ginther C, Corach D, Penacino G A, Rey J A, Carnese F R, Hutz M H, Anderson A, Just J, Salzano F M, King M C. In: DNA Fingerprinting: State of the Science. Penna S D J, Chakraborty R, Epplen J T, Jeffreys A J, editors. Basel: Birkhäuser; 1993. pp. 211–219. [DOI] [PubMed] [Google Scholar]
- 27.Santos M, Ward R H, Barrantes R. Hum Biol. 1994;66:963–977. [PubMed] [Google Scholar]
- 28.Kolman C J, Bermingham E, Cooke R, Ward R H, Arias T D, Guionneau-Sinclair F. Genetics. 1995;140:275–283. doi: 10.1093/genetics/140.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batista O, Kolman C J, Bermingham E. Hum Mol Genet. 1995;4:921–929. doi: 10.1093/hmg/4.5.921. [DOI] [PubMed] [Google Scholar]
- 30.Ward R H, Frazier B L, Dew-Jager K, Pääbo S. Proc Natl Acad Sci USA. 1991;88:8720–8724. doi: 10.1073/pnas.88.19.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward R H, Redd A, Valencia D, Frazier B, Pääbo S. Proc Natl Acad Sci USA. 1993;90:10663–10667. doi: 10.1073/pnas.90.22.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Redd A J, Takezaki N, Sherry S T, McGarvery S T, Sofro A S M, Stoneking M. Mol Biol Evol. 1995;12:604–615. doi: 10.1093/oxfordjournals.molbev.a040240. [DOI] [PubMed] [Google Scholar]
- 33.Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson A C. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- 34.DiRienzo A, Wilson A C. Proc Natl Acad Sci USA. 1991;88:1597–1601. [Google Scholar]
- 35.Saitou N, Nei M. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 36.Nei M, Jin L. Mol Biol Evol. 1989;6:290–300. doi: 10.1093/oxfordjournals.molbev.a040547. [DOI] [PubMed] [Google Scholar]
- 37.Efron B. The Jackknife, the Bootstrap, and Other Resampling Plans. Philadelphia: Soc. of Industrial and Applied Mathematics; 1982. [Google Scholar]
- 38.Rogers A, Harpending H. Mol Biol Evol. 1992;9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 39.Rogers A. Evolution. 1995;49:608–615. doi: 10.1111/j.1558-5646.1995.tb02297.x. [DOI] [PubMed] [Google Scholar]
- 40.Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N. Proc Natl Acad Sci USA. 1995;92:532–536. doi: 10.1073/pnas.92.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tajima F. Genetics. 1993;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogers A, Jorde L. Hum Biol. 1995;67:1–36. [PubMed] [Google Scholar]
- 43.Harpending H, Sherry S T, Rogers A, Stoneking M. Curr Anthropol. 1993;34:483–496. [Google Scholar]
- 44.Sherry S T, Rogers A, Harpending H, Soodyall H, Jenkins T, Stoneking M. Hum Biol. 1994;66:761–775. [PubMed] [Google Scholar]
- 45.Aris-Brosou D, Excoffier L. Mol Biol Evol. 1996;13:494–504. doi: 10.1093/oxfordjournals.molbev.a025610. [DOI] [PubMed] [Google Scholar]
- 46.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salzano F M, Callegari-Jacques S M. South American Indians: A Case Study in Evolution. Oxford: Clarendon; 1988. [Google Scholar]
- 48.Wakeley J. J Mol Evol. 1993;37:613–623. doi: 10.1007/BF00182747. [DOI] [PubMed] [Google Scholar]
- 49.Spuhler J N. In: The First Americans: Origins, Affinities, and Adaptations. Laughlin W S, Harper A B, editors. New York: Gustav Fischer; 1979. pp. 135–184. [Google Scholar]
- 50.Hopkins D M. In: The First Americans: Origins, Affinities, and Adaptations. Laughlin W S, Harper A B, editors. New York: Gustav Fischer; 1979. pp. 275–292. [Google Scholar]
- 51.Rogers R A, Rogers L A, Martin L D. Hum Biol. 1992;64:281–302. [PubMed] [Google Scholar]
- 52.Meltzer D J. Evol Anthropol. 1993;1:157–169. [Google Scholar]
- 53.Avise J C, Neigel J E, Arnold J J. J Mol Evol. 1984;20:99–105. doi: 10.1007/BF02257369. [DOI] [PubMed] [Google Scholar]