

Abstract
A preeminent phenotype of the infected cell protein 0 (ICP0) of herpes simplex virus 1 (HSV-1) is that it acts as a promiscuous transactivator. In most cell lines exposed to ΔICP0 mutant virus at low ratios of virus per cell infection, α genes are expressed but the transition to β and γ gene expression does not ensue, but can be enhanced by inhibitors of histone deacetylases (HDACs). Earlier studies have shown that ICP0 interacts with CoREST and displaces HDAC1 from the CoREST–REST–HDAC1/2 complex. HDAC1 and CoREST are then independently translocated to the cytoplasm. Here, we test the hypothesis that ICP0 blocks the silencing of HSV DNA by displacing HDAC1 from the CoREST–REST complex. Specifically, first, mapping studies led us to construct a truncated CoREST (CoREST146–482) that in transfected cells displaced HDAC1 from the CoREST–REST complex. Second, we constructed two viruses. In BACs encoding the entire HSV-1, we replaced the gene encoding ICP0 with AmpR to yield a ΔICP0 mutant R8501. We also replaced ICP0 with CoREST146–482 to yield recombinant R8502. The yield of R8502 mutant virus in Vero, HEp-2, and human embryonic lung cells exposed to 0.1 pfu of virus per cell was 100-, 10-, and 10-fold higher, respectively, than those of R8501 mutant virus. In Vero cells, the yield of R8502 was identical with that of wild-type virus. We conclude that CoREST146–482 functionally replaced ICP0 and that, by extension, ICP0 acts to block the silencing of viral DNA by displacing HDAC1/2 from the CoREST–REST complex.
Keywords: interactive domains, IFN signaling, protein kinases
In humans and in experimental animal systems, herpes simplex viruses 1 or 2 (HSV-1 or HSV-2) replicate at the portal of entry into the body, infect nerve endings of sensory or dorsal root ganglia, and ascend to the nucleus where, in some neurons, they establish latent infections. In the latently infected cells, all viral genes except that encoding the latency-associated RNA (LAT) are turned off (reviewed in ref. 1). These observations suggested that HSV DNA possess the properties of being activated or silenced by viral and or host factors. Indeed, a viral protein carried by the infecting virion designated α-transinducing factor or viral protein 16 (VP16) has been shown to interact with host transcriptional factors to enhance the expression of α or immediate-early genes (reviewed in ref. 2). Studies published recently have linked LAT to the maintenance of viral DNA in neuronal nuclei in a silenced form (3).
The studies described in this article center on the role of the infected cell protein 0 (ICP0). This protein, the product of the α0 gene, is made immediately after infection. It is dispensable in cells infected at a high multiplicity. With few exceptions (e.g., U2OS cell; ref. 4), in cell lines infected at low multiplicity with ΔICP0 mutants, α genes are expressed but the transition from α to β or γ genes does not ensue, and, as illustrated schematically in Fig. 1A, the virus fails to replicate (5). Related to this phenotype of ICP0 mutants is the observation that, in transfected cells, ICP0 is a promiscuous transactivator of genes introduced into cells by infection or transfection (6–8).
Fig. 1.

Schematic representation of early events in HSV-1-infected cells. (A) Cell infected with a ΔICP mutant at low pfu per cell. The schematic diagram illustrates that VP16 enters the nucleus concomitantly with the release of viral DNA from capsids at the nuclear pore. VP16 associates with cellular proteins Oct1 and HCF and transactivates α gene promoters. α proteins are made, but the expression of β and γ genes does not ensue. (B) Cell infected with wild-type virus. As reported earlier, ICP0 interacts with CoREST and displaces HDAC1 from the CoREST–REST complex. HDAC1 and CoREST are phosphorylated and translocated to the cytoplasm. β and γ genes are expressed. (C) Predicted role of a dominant-negative CoREST that dissociates HDAC1 from the CoREST–REST complex. If ICP0 enables the transition from α to β and γ gene expression by dissociating HDAC1/2 from the CoREST–REST complex, then a dominant-negative CoREST that displaces HDAC1 from the complex would substitute for ICP0 to enable enhanced replication of ΔICP0 mutant virus.
The second major function of ICP0 is associated with resistance to IFN-associated host responses to infection.
The focus of this article is on the transactivating functions of ICP0. Elsewhere (9), we reported that, first, ICP0 contains near it carboxyl-terminal domain a sequence of 79 residues homologous to a corresponding sequence near the amino-terminal domain of CoREST, a component of the histone deacetylase 1/2 (HDAC1/2)–CoREST–REST repressor complex (10–13). Second, in lysates of uninfected cells, antibody to CoREST pulled down CoREST, REST, and HDAC1. The same proteins were coprecipitated by antibody to HDAC1 from uninfected cells. In reactions with lysates from wild-type virus-infected cells, antibody to CoREST pulled down REST, but not HDAC1. Third, as illustrated schematically in Fig. 1B, viral protein kinases US3 and UL13 mediated the phosphorylation of CoREST and HDAC1 in wild-type virus-infected cells (9, 14). Fourth, in wild-type virus-infected cells, CoREST–REST complex and HDAC1 were translocated to the cytoplasm after the onset of viral DNA synthesis. In cells infected with high ratios of ΔICP0 mutant virus per cell, the entire complex HDAC1–CoREST–REST in the infected cells is also translocated to the cytoplasm (9).
HSV-1 virions carry VP16, a transactivator that, in newly infected cells interacts with Oct1 and HCF to enable the transcription of α genes (reviewed in ref. 1). To test the hypothesis that dissociation of HDAC1/2 from the HDAC1–CoREST–REST complex by ICP0 is key step in enabling the expression β and γ genes and blocking the silencing of HSV-1 DNA, we mapped the sites of the interaction between CoREST and ICP0 and between CoREST and HDAC1. On the basis of the mapping data, we identified a truncated form of CoREST that in transfected cells displaced HDAC1 from the CoREST–REST complex. We predicted, as illustrated in Fig. 1C, that insertion of the truncated CoREST into ΔICP0 mutants would at least compensate for the absence of the α0 gene.
Results
The Binding Site for CoREST in ICP0.
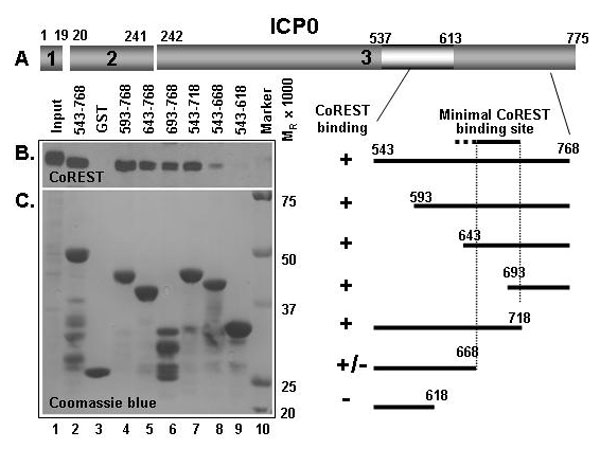
To map the binding sites of CoREST on ICP0, we constructed a set of GST fusion proteins containing various domains of ICP0. These were used to react with uninfected cell lysates. In initial experiments (data not shown), we mapped the binding domain to the carboxyl-terminal half domain of ICP0. The results of the pull-down experiments with GST-CoREST chimeras containing carboxyl-terminal domains of ICP0 are shown in supporting information (SI) Fig. 7. Briefly, as summarized in the schematic diagram shown in Fig. 2A, the binding site of CoREST was mapped to residues 668 to 718 of ICP0.
Fig. 2.

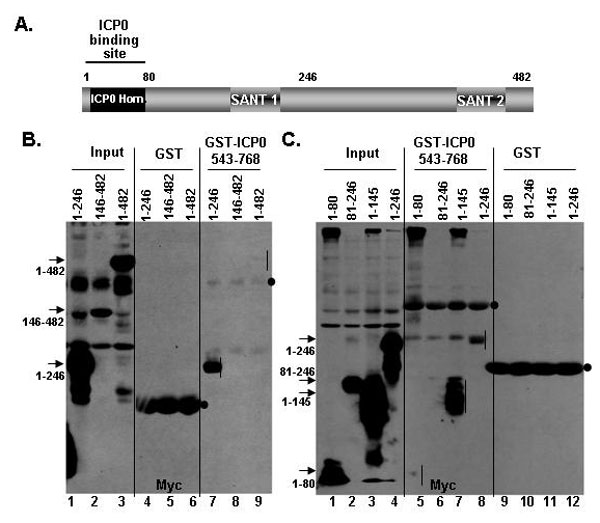
The position of key domains in CoREST and ICP0 relevant to these studies. (A) The numbers 1–3 identify the residues encoded in exons 1–3. The numbers above the bar refer to residue numbers. The sequence homologous to residue 3–79 of CoREST is at position 537–613. The binding site for CoREST determined on the basis of SI Fig. 7 is between residues 668 and 718. (B) Location of key domains in CoREST. The site of ICP0 homologous sequences (residues 3–79) are also the site of binding to ICP0 on the basis of the data shown in SI Fig. 8. The numbers below the bar indicate amino acid number. The binding site for HDAC1 is based on results shown in SI Fig. 9.
The Binding Site for ICP0 and HDAC1 on CoREST.
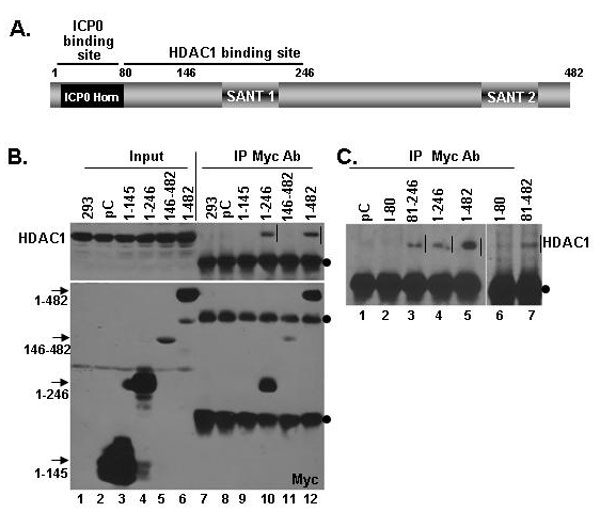
To map the binding site of ICP0 and CoREST, we reacted infected cells lysates with a set of purified GST fusion proteins containing various domains of CoREST. The results of the pull-down assays are shown in SI Fig. 8. Briefly, as summarized in Fig. 2B, ICP0 binds to residues 1–80 (i.e., to the sequences homologous to ICP0). The same CoREST constructs were used to map the binding site in CoREST for HDAC1 to residues 81–246 (SI Fig. 9) consistent with those of You et al. (11) who mapped the binding site of HDAC1 and CoREST-dependent HDAC activity to residues 75–254 of CoREST.
The Truncated CoREST Polypeptide Containing Residues 146–482 Disrupts the Endogenous CoREST–HDAC1 Interaction.
HEK 293 cells were transfected with a plasmid encoding Myc-tagged intact or truncated CoREST polypeptide. After 40 h, the cell lysates prepared were reacted with anti-HDAC1 or anti-CoREST antibody. The immune precipitates were solubilized, electrophoreticaly separated in denaturing gels, and reacted with anti-CoREST antibody (Fig. 3A) or anti-REST and anti-HDAC1 antibodies (Fig. 3B). The results show that the polypeptide CoREST146–482 disrupted the interaction between HDAC1 and CoREST inasmuch as in lysates of cells transfected with a plasmid encoding this protein antibody to HDAC1 failed to coprecipitate CoREST (Fig. 3A, lane 11) and antibody to CoREST coprecipitated REST but not HDAC1 (Fig. 3B, lane 10).
Fig. 3.

CoREST146–482 polypeptide displaces HDAC1 from the HDAC1–CoREST–REST complex. HEK 293 cells were transfected with plasmids encoding the truncated CoREST polypeptides tagged with Myc. After 40 h of incubation, the cells were harvested and lysed. The immune precipitates (IPs) obtained with polyclonal anti-HDAC1 antibody (A) or polyclonal CoREST antibody (B) were collected as described in Experimental Procedures, solubilized, electrophoretically separated in a denaturing gel, and reacted with monoclonal antibody against CoREST (A) or anti-HDAC1 or anti-REST antibodies (B).
Construction and Verification of Recombinant Viruses R8501 and R8502.
If the dissociation of HDAC1 from CoREST–REST complex by ICP0 reported earlier (9) plays a role in blocking the silencing of β and γ gene expression, then insertion of a gene encoding the truncated CoREST146–482 protein into the viral genome should overcome at least in part the inability of ΔICP0 mutant virus to replicate in cells infected at a low multiplicity of infection. To this end, by using a BAC, we first replaced both copies of the α0 gene and the inserted in place of the ampicillin resistance (AmpR) genes or the Myc-tagged CoREST146–482 genes driven by the CMV immediate-early promoter. The BAC DNA with one copy of α0 replaced with AmpR and the other copy replaced with CoREST146–482 was transfected into U2OS cells. The viruses obtained by transfection were designated R8501 and R8502, respectively (Fig. 4A).
Fig. 4.

Construction and verification of recombinant viruses. (A) Schematic representation of wild-type HSV-1, the sequence arrangements of the α0 gene encoding ICP0. The third line shows the replacement of ICP0 with AmpR. The fourth line shows the replacement of ICP0 with a Myc-tagged CoREST146–482 gene. The procedures were as described in Experimental Procedures. (B) Expression of CoREST146–482 in Vero cells infected with 10 pfu of R8501 or R8502 mutant virus per cell. As shown, lysates of R8502 reacted with antibodies to the truncated and endogenous CoREST, whereas the lysates of cells infected with the R8501 mutant reacted only with antibody to endogenous CoREST. (C) Electrophoretically separated lysates of cells harvested at times shown after infection with 10 pfu per cell were probed with antibodies to ICP0, ICP4, or ICP27.
The results shown in Fig. 4B indicate that the recombinant R8502 expressed the CoREST146–482 polypeptide identified by both anti-Myc and anti-CoREST antibody (Fig. 4B, lanes 2 and 4). Fig. 4C shows that in cells infected at a high multiplicity (10 pfu per cell), both viruses express ICP4, and ICP27, but not ICP0.
CoREST146–482 Enhances the Replication of ΔICP0 Mutant Virus in Cells Infected at a Low Ratio of pfu Per Cell.
In this series of experiments, replicate cultures of Vero, HEp-2, or human embryonic lung (HEL) cells were infected with 0.1 pfu of wild-type HSV-1(F), R8501, or R8502 virus per cell. At intervals shown, individual cultures were harvested and titered on U2OS cells. The results (Fig. 5) were as follows: In three independent experiments (Fig. 5 A–C), cells infected with R8502 yielded ≈100-fold more virus than the ΔICP0 parent virus R8501. In Vero cells, the yields of R8502 mutant approached those of wild-type virus (Fig. 5A). In HEp-2 and HEL cells, the yield of the R8502 mutant was on the average 10-fold higher than the ΔICP0 virus R8501 mutant. We conclude from these experiments that overexpression of the CoREST146–482 protein compensates for the absence of ICP0 in a cell type-dependent manner.
Fig. 5.

The replication of wild-type HSV-1 and of mutant viruses R8501 and R8502 in Vero, HEp-2, and HEL cells. Replicate cultures were exposed to 0.1 pfu per cell. The cultures were harvested at 3, 11, 24, and 48 h after infection for A; 3, 7, 24, and 48 h for B; 5, 8, 12, 24, 40, and 64 h for C; 3, 11, 24, and 48 h for D; and 3, 11, 24, and 48 h for E. The yields shown are based on titrations on USOS cells.
It is noteworthy that progeny virus continued to accumulate for at least 48–64 h and that the onset of accumulation of the progeny of R8502 mutant virus was delayed by many hours relative to that of the wild-type virus. The extended replicative cycle is not surprising inasmuch as the cells were exposed to a low ratio of pfu per cell. The delay in accumulation of infectious R8502 mutant virus may indicate that initiation of viral replication was delayed until CoREST146–482 protein reached sufficiently high levels to compete with native CoREST.
Discussion
Although ICP0 interacts with several proteins and appears to perform multiple functions, two properties stand out. First, ICP0 blocks IFN-dependent host responses designed to block viral replication (15–18). Second, it is a promiscuous transactivator of genes introduced by infection or transfection (6–8). At least some of the functions designed to block IFN-dependent responses appear to map to the RING finger domain of ICP0. Briefly, ICP0 and in particular the RING finger domain mapping in exon 2 was shown to disperse the ND10 nuclear bodies and mediate the degradation of promyelocytic leukemia (PML) protein (19). In the chain of discoveries, ICP0 RING finger was shown to acts an E3 ubiquitin ligase (20, 21). The RING finger E3 polyubiquitylates UbCH5a and UbcH6 in vitro (20, 21). Studies on dominant-negative E2 enzymes showed that dn UBcH5a but not dn UbCH6 blocked degradation of PML (22). Further studies demonstrated that IFN-α or -γ blocked viral replication in murine PML+/+ cells but not in PML−/− cells (17). By degrading PML, ICP0 in effect blocks the induction of IFN-mediated antiviral responses by the host cell. The degradation of PML by the RING finger E3 ubiquitin ligase does not exclude the possibility that ICP0 also blocks IFN signaling pathways by other domains of the protein.
The focus of this article is on the promiscuous transactivator functions of ICP0. Earlier, we reported that ICP0 interacts with CoREST and in infected cells dissociates HDAC1 from the CoREST–REST complex. The viral protein kinases US3 and both US3 and UL13 mediate the phosphorylation of HDAC1 and CoREST, respectively. At later stages of infection consistent with accumulation of products of β proteins involved in viral DNA synthesis, both HDAC1 and CoREST–REST complex are independently translocated to the cytoplasm (9).
In this article, we tested the hypothesis that the dissociation of HDACs from the CoREST–REST complex is the key event that enables the replication of ΔICP0 mutant virus in cells infected at low multiplicity. We demonstrated that the truncated CoREST146–482 polypeptide performed a function similar to that of ICP0 in that it displaced HDAC1 from the CoREST–REST complex in transformed cells. We also demonstrated that a mutant virus in which ICP0 was replaced with CoREST146–482 replicated 100-fold better than ΔICP0 mutant virus in Vero cells and at least 10-fold better in HEp-2 or HEL cells. The results indicate that a key function of ICP0 is to dissociate HDAC1/2 from CoREST–REST complex, thereby enabling the expression of viral genes in cell as depicted schematically in Fig. 1C.
Two observations made in the course of these studies are noteworthy.
Specifically, first, the replication of R8502 was significantly better in Vero cells than in HEp-2 or HEL cells (Fig. 5). One hypothesis that could explain these results is that in Vero cells the IFN pathway is unimpaired. Consistent with this hypothesis is the observation that the Δγ134.5 mutant viruses replicated better in Vero cells than in cells in which the IFN pathway is unimpaired such as HEp-2 or HEL cells (23). Whereas these mutants are highly attenuated in wild-type mice, they become virulent in mice defective in IFN pathway (24). A key conclusion of this article is that, in cells in which the IFN pathway is impaired, the truncated CoREST146–482 fully substituted for ICP0, whereas in cells with unimpaired IFN pathways CoREST146–482 only partially compensated for the absence of ICP0.
Second, ICP0 contains both the sequence homologous to the amino-terminal 79 residues of CoREST and a binding domain for these sequences (Fig. 6B). The two sequences are separated by ≈40 residues of which nearly one-half consist of serines and glycines. This is a surprising finding and suggests the possibility that ICP0 exists in two configurations. In the open configuration, the CoREST binding site interacts with the cognate site on CoREST. In the closed configuration, the binding site folds to enable the interaction with the cognate site on ICP0. The advantage of such a system is that, in the closed configuration, ICP0 could interact with host or viral proteins and perform functions that are different from those of ICP0 in the open configuration. Further studies should determine whether the hypothesis is tenable.
Fig. 6.

Schematic representations of the interactions between ICP0 and HDAC1/2–CoREST–REST complex and the predicted structure of ICP0. (A) The sequence of ICP0 showing the residues in the binding sites for CoREST (red) and the sequence homologous to residues 3–79 of CoREST (brown). (B) Hypothetical structure of ICP0 in the absence of CoREST (closed conformation) and in the presence of CoREST (open conformation).
Last, it should be pointed out that, in addition to proteins that block silencing of viral genes in the early stages of infection, HSV-1 may also encode an RNA (latency-associated transcript) that has been linked to the maintenance of viral DNA in a silent form in latently infected neurons (3).
Experimental Procedures
Cells and Viruses.
The sources and maintenance of HeLa, HEK 293, and Vero, U2OS, and human embryonic cells were reported elsewhere (9, 22). HSV-1(F) is the prototype strain used in this laboratory (25). The BAC encoding the HSV-1(F) DNA was reported elsewhere (26, 27). The wild type and mutants generated in this study were titered on U2OS cells.
Plasmid Construction for Deletion Mappings.
The primers used to PCR amplify portions of CoREST (10) are shown in SI Table 1. The PCR products were digested with StuI and PstI and ligated into plasmid pRB8501, previously designated as MTS1-Myc (22) to generate pRB8502 to pRB8509, respectively. The Myc-tagged CoREST sequences excised with EcoRI and PstI from these plasmids were ligated to EcoRI- plus PstI-double-digested pcDNA3.1(+)zeor to generate pRB8512 to pRB8519, respectively. By using MTS1α0PL (28) as template, portions of ICP0 were PCR amplified with the primers shown in SI Table 2. The PCR products were digested with EcoRI plus XhoI and ligated to EcoRI- plus XhoI-digested pGEX 4T-1 (Amersham Biosciences, Piscataway, NJ) to generate pRB8401 to pRB8407, respectively.
The GST-ICP0 polypeptides containing residues 1–19, 20–241, 245–395, and 543–768, respectively, were described in ref. 29.
Construction of ΔICP0 HSV-1 BAC.
pRB112 containing BamHI B fragment (30) was digested with BamHI and EcoRV. The 4.8-kb fragment containing ICP0 exons 2 and 3 was cloned into BamHI and blunted SpeI sites of pBluescript KS to generate pRB8520. pRB115 containing BamHI SP1 fragment (30) was digested with StuI and BamHI. The 1.4-kb fragment containing exon 1 was ligated into the BamHI- and EcoRV-double-digested pRB8520. This generates pRB8521 with a complete ICP0 gene. The 6.0-kb BamHI and HpaI fragment of pRB8521 was gel purified to serve as linear vector. pRB8517 and pRB8512 were digested with PstI and XbaI, blunt-ended, and self-ligated to eliminate the XbaI and XhoI sites between the CoREST and BGH polyadenylation site. The BglII and PvuII fragment containing the CMV promoter driven CoREST146–482 was cloned into BamHI- and HpaI-linearized pRB8521 to generate pRB8522 containing CoREST146–482 flanked by ICP0 sequence. pRB8522 was then digested with XbaI and XhoI, and the fragment containing CoREST146–482 was cloned into pKO5 plasmid (28) to generate pKO8522.
A 1.4-kb BglII and blunt-ended AlwNI fragment from pcDNA3.1 containing ampicillin resistant gene (AmpR) was cloned into the BamHI- and HpaI-linearized pRB8521. This generated pRB8523, which contains the AmpR flanked by the ICP0 sequence. A 4.5-kb fragment from XbaI- and XhoI-digested pRB8523 was cloned into pKO5 to generate pKO8523.
Escherichia coli RR1 stain harboring HSV-1 BAC was electroporated with pKO8522, and incubated at 43°C on LB plates containing 25 μg/ml zeocin (Zeo) and 20 μg/ml chloramphenicol. The colonies were diluted and plated on LB plates containing chloramphenicol (20 μg/ml) and 5% sucrose. Colonies grown on the sucrose plates were screened with PCR or by colony hybridization. The recombinant BAC8501 obtained contains one copy of ICP0 gene replaced with CoREST146–482. Competent cells of RR1 harboring BAC8501 (ICP0/CoREST146–482) were electroporated with pKO8523, the selection process was repeated, and BAC8502 (AmpR/CoREST146–482) was obtained, in which the other copy of ICP0 was replaced with AmpR gene. Plasmids DNAs isolated from E. coli were transfected into U2OS cells. The resulting viruses were plaque purified three times and analyzed to verify that in recombinant R8501 both copies of ICP0 were replaced by AmpR and R8502 in which both copies of AmpR gene were replaced by CoREST146–482.
GST Pull-Down Assay.
Plasmids pRB8401 to pRB8407 were transformed into E. coli BL21 stain. For each construct, a fresh colony was inoculated into 3 ml of LB supplemented with ampicillin (100 mg/ml) and grow at 37°C overnight. The bacterial culture was then diluted 1:100 into fresh LB plus ampicillin (100 mg/ml) and grown at 37°C until OD600 reached 0.6. Isopropyl β-d-thiogalactopyranoside (0.5 mM) was then added, and incubation was continued for 4 h. Bacterial cell pellet from 25-ml cultures was resuspended in 1.6 ml of STE buffer (10 mM Tris·HCl, pH 8, 150 mM NaCl, 1 mM EDTA) supplemented with 1× protease inhibitor mixture (Sigma, St. Louis, MO) and reacted on wet ice with 0.1 mg/ml lysozyme for 15 min. After addition of DTT (final concentration of 1 mM) and sarkosyl (final concentration of 0.5%), the bacteria were briefly sonicated to lyse the cells. The lysate was cleared by centrifugation, and the supernatant fluid was reacted with glutathione Sepharose beads (Amersham Biosciences) at 4°C for 30 min. The beads were rinsed five times each with 10 ml of PBS. Eukaryotic cell lysates prepared by brief sonication in lysis buffer (10 mM Tris, pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 1 mM DTT, 0.5% Nonidet P-40, 0.1 mM NaVO4, 10 mM NaF, 10 mM glycerol phosphate, 1× protease inhibitor mixture) and cleared of cells were reacted first with unbound glutathione Sepharose, and then overnight with GST or GST-ICP0 beads. The beads were then collected, rinsed three times with lysis buffer, and the bound proteins were eluted with 1× SDS/PAGE loading buffer.
Immunoprecipitation.
HEK 293 cells were harvested and lysed in lysis buffer and cleared of cell debris 40 h after Lipofectamine (Invitrogen, Carlsbad, CA)-dependent transfection with plasmids encoding full-length and truncated CoREST. The lysates were reacted at 4°C overnight with antibodies as stated in Results. The immunoprecipitates were harvested with protein A Sepharose CL-4B (Amersham Biosciences), rinsed three times with lysis buffer and once with lysis buffer plus 0.1% SDS, and then eluted with 1× SDS/PAGE loading buffer.
Immunoblots.
Total cell lysates, GST precipitates, or immunoprecipitates from above were electrophoretically separated on denaturing polyacrylamide gels and transferred to preequilibrated poly(vinylidene difluoride) membrane (Millipore, Bedford, MA). The membranes were blocked in TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween 20) containing 5% nonfat dry milk and reacted at 4°C overnight with primary antibody in TBST/5% dry milk, rinsed, and reacted with alkaline phosphatase- or peroxidase-conjugated secondary antibodies (Sigma). The membranes were rinsed and developed with either 5-bromo-4-chloro-3-indolyl phosphate plus nitroblue tetrazolium (Denville Scientific, Metuchen, NJ) or ECL Western blotting detection reagents (Amersham Biosciences) according to the manufacturer's instructions.
Supplementary Material
Acknowledgments
We thank Mr. Palak Desai (Rush Medical College, Chicago, IL) for the construction of several plasmids used in the mapping studies. We thank Dr. Gail Mandel (Oregon Health and Science University, Portland, OR) for the generous gifts of CoREST cDNA plasmid and anti-REST antibody. These studies were aided by National Cancer Institute Grant CA78766.
Abbreviations
- HSV
herpes simplex virus
- ICP0
infected cell protein 0
- VP16
viral protein 16
- HDAC
histone deacetylase
- AmpR
ampicillin resistance
- HEL
human embryonic lung
- PML
promyelocytic leukemia.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0707266104/DC1.
References
- 1.Roizman B, Knipe DM, Whitley RJ. In: Fields' Virology. 5th Ed. Knipe DM, Howley P, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. New York: Lippincott Williams & Wilkins; 2007. pp. 2501–2601. [Google Scholar]
- 2.Wysocka J, Herr W. Trends Biochem Sci. 2003;28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 3.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. Proc Natl Acad Sci USA. 2005;102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao F, Schaffer PA. J Virol. 1995;69:6249–6258. doi: 10.1128/jvi.69.10.6249-6258.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Silverstein S. J Virol. 1992;66:2916–2927. doi: 10.1128/jvi.66.5.2916-2927.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gelman IH, Silverstein S. Proc Natl Acad Sci USA. 1985;82:5265–5269. doi: 10.1073/pnas.82.16.5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinlan MP, Knipe DM. Mol Cell Biol. 1985;5:957–963. doi: 10.1128/mcb.5.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nabel GJ, Rice SA, Knipe DM, Baltimore D. Science. 1988;239:1299–1302. doi: 10.1126/science.2830675. [DOI] [PubMed] [Google Scholar]
- 9.Gu H, Liang Y, Mandel G, Roizman B. Proc Natl Acad Sci USA. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. Proc Natl Acad Sci USA. 1999;96:9873–9878. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.You A, Tong JK, Grozinger CM, Schreiber SL. Proc Natl Acad Sci USA. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong JA, Tapia-Ramirez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 13.Schoenherr CJ, Anderson DJ. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 14.Poon AP, Liang Y, Roizman B. J Virol. 2003;77:12671–12678. doi: 10.1128/JVI.77.23.12671-12678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melroe GT, Silva L, Schaffer PA, Knipe DM. Virology. 2007;360:305–321. doi: 10.1016/j.virol.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobol PT, Mossman KL. J Virol. 2006;80:218–225. doi: 10.1128/JVI.80.1.218-225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chee AV, Lopez P, Pandolfi PP, Roizman B. J Virol. 2003;77:7101–7105. doi: 10.1128/JVI.77.12.7101-7105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harle P, Sainz B, Jr, Carr DJ, Halford WP. Virology. 2002;293:295–304. doi: 10.1006/viro.2001.1280. [DOI] [PubMed] [Google Scholar]
- 19.Chelbi-Alix MK, de The H. Oncogene. 1999;18:935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- 20.Boutell C, Sadis S, Everett RD. J Virol. 2002;76:841–850. doi: 10.1128/JVI.76.2.841-850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagglund R, Van Sant C, Lopez P, Roizman B. Proc Natl Acad Sci USA. 2002;99:631–636. doi: 10.1073/pnas.022531599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu H, Roizman B. Proc Natl Acad Sci USA. 2003;100:8963–8968. doi: 10.1073/pnas.1533420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou J, Roizman B. Proc Natl Acad Sci USA. 1992;89:3266–3270. doi: 10.1073/pnas.89.8.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW. Proc Natl Acad Sci USA. 2000;97:6097–6101. doi: 10.1073/pnas.100415697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ejercito PM, Kieff ED, Roizman B. J Gen Virol. 1968;2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 26.Ye GJ, Roizman B. Proc Natl Acad Sci USA. 2000;97:11002–11007. doi: 10.1073/pnas.97.20.11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horsburgh BC, Hubinette MM, Tufaro F. Methods Enzymol. 1999;306:337–352. doi: 10.1016/s0076-6879(99)06022-x. [DOI] [PubMed] [Google Scholar]
- 28.Lopez P, Van Sant C, Roizman B. J Virol. 2001;75:3832–3840. doi: 10.1128/JVI.75.8.3832-3840.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang Y, Kurakin A, Roizman B. Proc Natl Acad Sci USA. 2005;102:5838–5843. doi: 10.1073/pnas.0501253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Post LE, Conley AJ, Mocarski ES, Roizman B. Proc Natl Acad Sci USA. 1980;77:4201–4205. doi: 10.1073/pnas.77.7.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}