

Abstract
DNA glycosylases catalyze the excision of chemically modified bases from DNA. Although most glycosylases are specific to a particular base, the 3-methyladenine (m3A) DNA glycosylases include both highly specific enzymes acting on a single modified base, and enzymes with broader specificity for alkylation-damaged DNA. Our structural understanding of these different enzymatic specificities is currently limited to crystal and NMR structures of the unliganded enzymes and complexes with abasic DNA inhibitors. Presented here are high-resolution crystal structures of the m3A DNA glycosylase from Helicobacter pylori (MagIII) in the unliganded form and bound to alkylated bases 3,9-dimethyladenine and 1,N6-ethenoadenine. These are the first structures of a nucleobase bound in the active site of a m3A glycosylase belonging to the helix–hairpin–helix superfamily. MagIII achieves its specificity for positively-charged m3A not by direct interactions with purine or methyl substituent atoms, but rather by stacking the base between two aromatic side chains in a pocket that excludes 7-methylguanine. We report base excision and DNA binding activities of MagIII active site mutants, together with a structural comparison of the HhH glycosylases.
Keywords: 3-methyladenine/base excision/DNA repair/glycosylase/helix–hairpin–helix
Introduction
In addition to the one-dimensional genetic code, DNA harbors a structural code that is read by proteins binding to specific nucleobase sequences or shapes of DNA. The stability of the genome is constantly challenged by the chemical reactivity of DNA bases, which are subject to modifications by cellular and environmental agents. Alkylation, oxidation, deamination and hydrolysis of DNA bases can cause DNA mutations or inhibit replication and thereby cause toxicity or disease (reviewed in Lindahl, 1993; Friedberg et al., 1995). It is therefore important to understand the the basis for recognition of structural damage to DNA by repair pro teins. Base excision repair (BER) is the principle mech anism by which DNA with discrete base modifications is restored to its original state. DNA glycosylases, each specific for a particular type of damage, initiate this pathway by locating and removing the modified base through cleavage of the C1′-N glycosylic bond. The importance of specificity during base excision is underscored by the fact that glycosylases must identify subtle changes in the DNA structure amidst a vast excess of unmodified DNA.
Several different architectures of DNA glycosylases have evolved that are designed to gain access to damaged bases in DNA. These enzymes distort the double helix to expose a modified nucleotide substrate to the enzyme active site by the process of base-flipping, a common strategy used by enzymes that perform chemistry on nucleotides in double-stranded DNA (reviewed in Roberts and Cheng, 1998). One framework for DNA base excision is embodied by the conserved fold of the helix–hairpin–helix (HhH) superfamily of DNA glycosylases (Nash et al., 1996). The HhH motif was first identified in the pyrimidine-specific DNA glycosylase, Escherichia coli endonuclease III (EndoIII) (Thayer et al., 1995), and it was soon discovered that the overall protein fold of EndoIII is generally conserved in other DNA glycosylases (Labahn et al., 1996; Yamagata et al., 1996) with highly divergent sequences. The HhH motif itself consists of two α-helices connected by a hairpin loop having the consensus sequence [Leu/Phe]-Pro-Gly-[Val/Ile]-Gly. This signature sequence is present in hundreds of proteins that bind to DNA in a sequence-independent manner (Doherty et al., 1996). Crystal and NMR structures of HhH superfamily glycosylases include EndoIII (Kuo et al., 1992), E.coli adenine DNA glycosylase (MutY, Guan et al., 1998), E.coli 3-methyladenine (m3A) DNA glycosylase I (TAG, Drohat et al., 2002) and II (AlkA, Labahn et al., 1996; Yamagata et al., 1996), human 8-oxoguanine (o8G) DNA glycosylase (Ogg1, Bruner et al., 2000), Methanobacterium thermoformicicum T:G mismatch DNA glycosylase (MIG, Mol et al., 2002) and the mismatch glycosylase domain of eukaryotic MBD4 (Wu et al., 2003). The crystal structures of AlkA in complex with DNA containing a 1-azaribose abasic analog (Hollis et al., 2000) and of Ogg1 bound to its DNA substrate (Bruner et al., 2000) illustrate how the HhH motif is used as a platform for base-flipping (Roberts and Cheng, 1998) to expose damaged bases in DNA.
Despite the growing number of HhH glycosylase structures, their catalytic mechanisms and features imparting their distinct substrate specificities are currently a matter of speculation. This is especially true of the alkylation-specific HhH glycosylases like E.coli AlkA and TAG. TAG removes only m3A and to a lesser extent m3G (Bjelland et al., 1993), whereas AlkA is able to excise m3A, 7-methylguanine (m7G) and other alkylated or oxidized bases from DNA (McCarthy et al., 1984; Bjelland et al., 1994; Saparbaev et al., 1995). There is no structural information showing how substrate bases bind to these HhH enzymes.
A small group of HhH glycosylases that are highly selective for alkylation-damaged DNA have recently been identified in the genomic sequences of prokaryotic organisms. The Thermatoga maritima methylpurine DNA glycosylase (MpgII) shares no significant sequence homology with other m3A glycosylases outside of the HhH motif, yet it excises both m3A and m7G (Begley et al., 1999). MagIII is a m3A DNA glycosylase from Helicobacter pylori that shares 32% identity with the sequence of MpgII. However, MagIII is unable to remove m7G or hypoxanthine from DNA (O’Rourke et al., 2000) and, in this regard, it resembles the TAG protein. MagIII presents a unique opportunity to understand enzymatic selectivity for alkylation-damaged DNA.
We present crystal structures of MagIII alone and bound to the alkylated bases 3,9-dimethyladenine (m23,9A) and 1,N6-ethenoadenine (εA), along with mutational studies of MagIII enzymatic activity and DNA binding. Specificity for alkylated bases arises from favorable π–π stacking interactions between the positively charged m3A base and aromatic side chains within a binding pocket identified in the crystal structures. The shape of the binding pocket disfavors binding of m7G. A structural comparison of the active site structures of several purine-specific DNA glycosylases provides a basis for understanding the selection of modified bases for removal from DNA.
Results and discussion
MagIII is a HhH glycosylase
The crystal structure of H.pylori MagIII was determined with experimental phases obtained by a multiwavelength anomalous diffraction (MAD) experiment (Table I, Figure 1A). A crystallographic model consisting of two MagIII molecules in the asymmetric unit has been refined against X-ray data extending to 1.64 Å resolution (Figure 1B). As predicted (Begley et al., 1999; O’Rourke et al., 2000), the fold of MagIII is representative of the HhH superfamily of DNA glycosylases (Figure 2). However, unique features within the active site can explain selectivity for m3A. MagIII comprises two α-helical domains separated by a deep cleft (Figure 1C), corresponding to the most highly conserved features of the HhH glycosylases (Figure 2). One domain of MagIII (the N/C domain) consists of an α-helix (A) from the N-terminus packed against a cluster of four C-terminal α-helices (J–M). The other domain is formed from eight remaining α-helices (B–I) including the canonical HhH motif (helices H and I), referred to here as the HhH domain.
Table I. Data collection, phasing and refinement statistics.
| Native | Hg peak | Hg remote | Hg inflection | m23,9A | εA | |
|---|---|---|---|---|---|---|
| Data collectiona | ||||||
| Beamline | CHESS A-1 | NSLS X12-C | NSLS X12-C | NSLS X12-C | Laboratory source | Laboratory source |
| Wavelength (Å) | 0.9450 | 1.000 | 0.9686 | 1.0083 | 1.542 | 1.542 |
| Resolution (Å) | 25–1.64 | 50–2.4 | 50–2.5 | 50–2.6 | 50–1.93 | 50–2.13 |
| Unique reflections | 60 643 (6066) | 38 588 (3857) | 33 760 (3402) | 29 424 (2917) | 35 778 (3327) | 28 226 (2783) |
| Completeness (%) | 99.9 (99.7) | 99.9 (100.0) | 99.9 (100.0) | 100.0 (100.0) | 95.2 (90.3) | 99.8 (99.6) |
| Redundancy | 8.1 (5.7) | 3.9 (3.5) | 3.9 (3.8) | 3.9 (3.9) | 5.2 (3.0) | 2.9 (2.9) |
| Rsym (%)b | 9.9 (33.7) | 7.4 (29.4) | 7.7 (32.3) | 7.9 (31.6) | 5.0 (29.9) | 8.4 (36.9) |
| <I>/<σ(I)> | 21.8 (4.3) | 15.2 (3.6) | 14.5 (3.4) | 14.6 (3.6) | 21.2 (2.5) | 11.5 (2.5) |
| Phasing | ||||||
| Resolution (Å) | 20–2.7 | 20–2.7 | 20–2.7 | |||
| Phasing powerc | 0.92 | 0.70 | – | |||
| Rcullis (c/a)d | 0.81/0.84 | 0.87/0.88 | – | |||
| Rcullis (anom) | 0.92 | 0.95 | 0.97 | |||
| Refinement | ||||||
| Resolution | 25–1.64 | 50–1.93 | 50–2.13 | |||
| Rcryste | 0.151 | 0.183 | 0.202 | |||
| Rfreee | 0.185 | 0.222 | 0.230 | |||
| Number of atoms | ||||||
| Protein | 3474 | 3477 | 3467 | |||
| Ligand | 0 | 24 | 24 | |||
| Solvent | 255 | 370 | 169 | |||
| Average B factor (Å2) | 22.9 | 12.5 | 13.8 | |||
| R.m.s.d. bonds (Å) | 0.021 | 0.015 | 0.013 | |||
| R.m.s.d. angles (°) | 1.602 | 1.546 | 1.508 |
aValues in parentheses refer to data in the highest resolution shell.
bRsym = ΣhklΣj |Ij – <I>| / ΣhklΣj Ij where <I> is the mean intensity of j observations of reflection hkl and its symmetry equivalents.
cPhasing power = Σhkl FH / Σhkl |FPH – FPH,calc|. Value shown is the weighted average of centric and acentric data.
dRcullis = (Σhkl ||FPH ± FP| – FH,calc|) / Σhkl |FPH – FP|. c/a, centric/acentric data.
eRcryst = Σhkl |Fobs – kFcalc| / Σhkl |Fobs|. Rfree = Rcryst for 5% of reflections that were not used in refinement (Brünger, 1992).

Fig. 1. The structure of MagIII. (A) A cross-section of the refined model and the 2.7 Å solvent-flattened experimental electron density map, contoured at 1σ. (B) Same view of the refined model against the final σA-weighted 2mFo–DFc electron density map contoured at 1σ. (C) Stereoscopic ribbon representation of MagIII with the HhH domain at the top and the N/C-terminal domain at the bottom of the figure. Helices are labeled A–M, and the HhH motif is highlighted yellow. The nucleobase binding residues are colored green and putative catalytic residues are blue. DNA intercalating (Asn42, magenta) and ‘wedge’ (Phe45, yellow) residues responsible for base flipping are also shown, as well as the carbamylated lysine residue in the N/C-terminal domain (orange).

Fig. 2. The HhH superfamily of DNA glycosylases. (A) The structure-based sequence alignment of MagIII with the HhH glycosylase structures and the MpgII sequence was prepared using the program SEQUOIA (Bruns et al., 1999). MagIII secondary structure elements are shown schematically, with the HhH motif highlighted as yellow cylinders (helices H–I of MagIII). The HhH residues that contact the DNA in the AlkA–DNA complex are boxed, and the conserved catalytic aspartic acid is shaded blue. Residues in the nucleobase binding pocket confirmed (green) or predicted (gray) to contact the target base are shaded, and the positions of the side chains that intercalate the DNA helix at the lesioned and non-lesioned strands are shaded pink and yellow, respectively. Residues that contact the orphaned DNA base opposite the modified base in AlkA and Ogg1 are colored blue, while residues shown by mutagenesis to be important for either catalysis or DNA binding are colored red. Side chains that coordinate the Fe4S4 clusters (MpgII, EndoIII, MutY, MIG) and Zn2+ ion (TAG), as well as the carbamylated lysine in MagIII are shaded orange. (B) Schematic representations of the HhH glycosylase structures are oriented as in Figure 1C. Helices are shown as red and yellow (HhH motif) cylinders, β-sheets as light blue arrows, and Fe4S4 clusters as golden CPK spheres. Side chains of functionally significant active site residues are rendered as sticks, with the conserved aspartic acid colored dark blue. (C) Solvent accessible surfaces are colored according to electrostatic potential (blue, positive; red, negative) and were created with GRASP (Nicholls et al., 1991). The substrate binding pockets at the domain interface are circled. The structures have been rotated ∼90° with respect to the views shown in (B). Larger versions of (B) and (C) are available as Supplementary data.
The HhH motif of MagIII is positioned next to the interdomain cleft and opposite an aspartic acid that is conserved in all HhH glycosylases (Figures 1C, 2A and B) (Nash et al., 1996). This location of the HhH is conserved in all seven HhH glycosylases whose structures have been determined (Figure 2B); the protein fold of MagIII superimposes nicely onto these other enzymes simply by aligning the HhH motifs. The HhH domains of these glycosylases are more structurally conserved than the N/C domains, which include additional structural elements (Figure 2B). For example, EndoIII, MutY and MIG each contain an iron–sulfur cluster (Fe4S4) that is ligated by four conserved cysteines at the C-terminus of the N/C domain. AlkA and Ogg1 don an additional β-sheet at their N-terminus that packs against the N/C domain. TAG has the basic HhH architecture, but with a different topology and a unique zinc-binding motif within its N/C domain (Kwon et al., 2003). The HhH motif of MagIII is most similar to those of the Fe4S4-containing enzymes, yet MagIII lacks a metal cluster (Figure 2B). Instead, MagIII has an unusual interaction between a carbamylated lysine (Lys205) and both Arg201 and Tyr189 that probably contributes to the stability of the N/C domain (See Supplementary figure S1, available at The EMBO Journal Online). Another notable distinction of MagIII’s N/C domain is helix M, which is considerably longer than the corresponding helix of any other HhH glycosylase (Figure 2B). Residues from helix M contribute to the substrate binding pocket and the carbamylation site of MagIII (Figure 2A). Thus, the simple HhH glycosylase fold of MagIII has several unique features that illustrate the structural variation of HhH superfamily glycosylases.
The nucleobase binding pocket of MagIII
Although the HhH glycosylases utilize a common architecture to bind DNA and expose a target base, each has a unique substrate binding pocket that is suited for a particular type of modified base. Different nucleobase modifications impart unique physical and chemical characteristics that can be selectively recognized. The substrate binding and catalytic residues of HhH glycosylases are located in the cleft between the HhH and N/C domains (Figure 2) (Schärer and Jiricny, 2001). MagIII’s DNA base binding pocket was identified within this cleft by soaking crystals with the alkylated adenine bases m23,9A (Fujii et al., 1989) and εA (Figure 3). The dialkylated base m23,9A has a formal positive charge, mimicking the electron-deficient state of a m3A DNA substrate prior to cleavage of the glycosylic bond. Although εA is a poor substrate for excision (Table II), it also binds in the pocket (Figures 3B and 4). The bound m23,9A and εA bases were readily identified by difference electron density calculated from experimental MAD phases and amplitude differences for diffraction data obtained from base-soaked and native crystals. The resulting structures of enzyme complexes with m23,9A and εA were refined against X-ray data extending to 1.9 and 2.1 Å resolution, respectively (Table I). Fo–Fc omit electron density maps calculated for both complexes show strong (>3σ) planar electron density consistent with a bound purine deep within the interdomain cleft (Figures 3A and B). No other significant difference density (>1σ) is present, indicating that these alkylated bases bind selectively to the hydrophobic pocket that is bounded by Trp24, Trp25, Pro26, Phe45 and Lys211 (Figure 3C).

Fig. 3. Structure of the MagIII active site. Alkylated bases m23,9A (A), and εA (B) are shown in the unbiased Fo–Fc annealed omit electron density contoured at 3σ. The view looks down on the N/C-terminal domain from the HhH domain, with the core of the protein at the right of the figure and the entrance to the active site at the bottom. (C) Stereo view of m23,9A bound to MagIII shows the nucleobase stacked between the aromatic side chains of Trp24 and Phe45 and bounded at its edges by Trp25, Pro26 and Lys211. Ordered waters are rendered as red spheres, and hydrogen-bonds are shown as dashed lines.
Table II. Glycosylase activities and DNA binding for wild-type and active site mutants of MagIII.
| m3A activitya |
εA·C activityb |
m7G·T activityc |
DNA bindingd |
|||||
|---|---|---|---|---|---|---|---|---|
| kobs (10–2 min–1 µM–1) | Relative activity | kcat (10–5 min–1) | Relative activity | kcat (10–5 min–1) | Relative activity | Kd (µM) | Relative affinity | |
| WT | 38.8 ± 5.3 | (1.0) | 23.2 ± 3.5 | (1.0) | 189.1 ± 38.1 | (1.0) | 0.48 ± 0.10 | (1.0) |
| D150N | 1.7 ± 0.8 | 0.04 | n.d. | ≤0.04 | n.d. | ≤0.40 | 0.54 ± 0.09 | 0.90 |
| E132Q | 34.1 ± 7.2 | 0.90 | n.d. | ≤0.04 | 0.11 ± 0.02 | 4.40 | ||
| D150N E132Q | 4.7 ± 2.6 | 0.10 | n.d. | ≤0.04 | 0.18 ± 0.01 | 2.60 | ||
| H203N | 3.0 ± 1.4 | 0.08 | n.d. | ≤0.04 | 7.03 ± 1.35 | 0.10 | ||
| H203A | 2.5 ± 0.5 | 0.07 | n.d. | ≤0.04 | 7.01 ± 3.31 | 0.10 | ||
| Y140F | 18.7 ± 3.1 | 0.50 | 12.2 ± 2.5 | 0.50 | 0.57 ± 0.10 | 0.80 | ||
| W25F | 3.1 ± 0.6 | 0.08 | n.d. | ≤0.04 | 1.26 ± 0.13 | 0.40 | ||
| W25A | 2.4 ± 0.4 | 0.06 | n.d. | ≤0.04 | n.d. | ≤0.40 | 0.80 ± 0.06 | 0.60 |
| K211A | 1.6 ± 0.7 | 0.04 | 15.7 ± 0.8 | 0.70 | 151.0 ± 8.3 | 0.80 | 3.23 ± 1.02 | 0.15 |
| F45W | 58.4 ± 1.2 | 1.50 | 41.6 ± 13.0 | 1.80 | 356.5 ± 53.6 | 1.90 | 1.66 ± 0.70 | 0.30 |
| E46K | 152.8 ± 3.1 | 3.90 | 15.0 ± 1.0 | 0.70 | 1584.1 ± 68.5 | 8.40 | 0.06 ± 0.01 | 8.40 |
| F45W E46K | 190.8 ± 27.0 | 4.90 | 32.2 ± 5.7 | 1.40 | 2312.7 ± 70.7 | 12.20 | 0.12 ± 0.00 | 4.00 |
| N42A | 104.9 ± 18.0 | 2.70 | 21.4 ± 4.8 | 0.90 | 146.7 ± 5.0 | 0.80 | 0.13 ± 0.03 | 3.70 |
| F89A | 2.9 ± 0.6 | 0.07 | 20.4 ± 1.3 | 0.90 | n.d. | ≤0.40 | 5.53 ± 0.47 | 0.10 |
aSecond-order rate constants for [3H]m3A release from N-[3H]methyl-N-nitrosourea-treated genomic DNA
bFirst-order rate constants for εA excision from a 25mer oligonucleotide duplex containing a εA·C mispair. Rates indistinguishable from a non-enzymatic control (k = 1.0 × 10–5 min–1) are labeled n.d. (none detected).
cFirst-order rate constants for m7G excision from a 25mer oligonucleotide duplex containing a m7G·T mispair. Rates indistinguishable from a non-enzymatic control (k = 71.3 × 10–5 min–1) are labeled n.d.
dDissociation constants for a 25mer oligonucleotide duplex containing a 1-azaribose AP site.
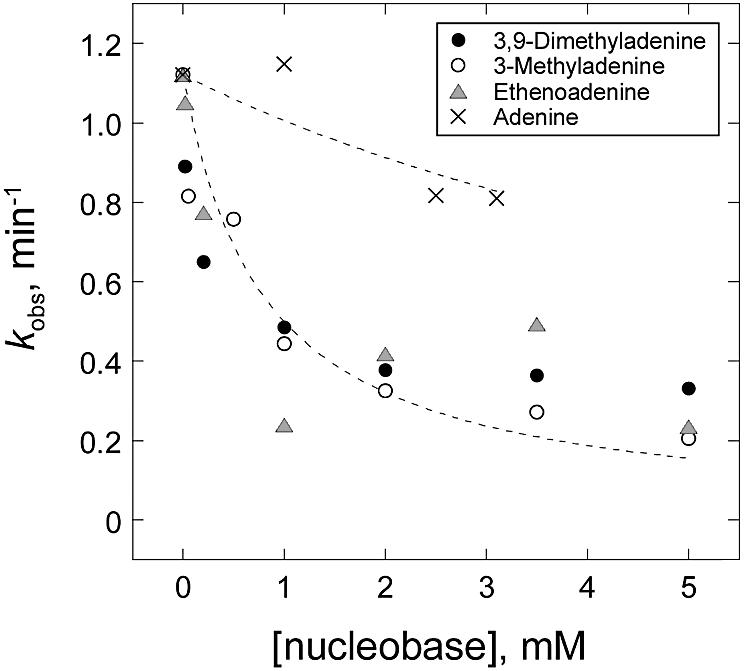
Fig. 4. Selective binding of alkylated bases to MagIII. Inhibition of MagIII excision of m3A from genomic DNA by the modified adenine bases m23,9A (closed circles), m3A (open circles), εA (shaded triangles) is shown, whereas adenine (crosses) did not inhibit MagIII. The dotted lines represent Ki = 0.8 mM (modified bases) and Ki = 8.8 mM (adenine).
The binding pocket of MagIII is perfectly shaped for modified adenine bases. The side chains of Trp24 and Phe45 pack against the faces of m23,9A and εA, whereas the edges of the bases are contacted by Trp25 and Pro26 on one side and by Lys211 on the other side. The difference electron density for m23,9A clearly reveals the positions of its exocyclic amino and methyl substituents, indicating that the base binds in one orientation (Figure 3A). The two methyl groups of m23,9A point towards the entrance to the active site where a DNA substrate would bind (Figure 3C and Supplementary figure S2). MagIII does not directly contact the aberrant methyl group of m3A, nor does it contact the exocyclic N6 of adenine. The N6 amino group is exposed to solvent through the opening between Lys211, Glu46 and Pro26 (Figure 3C). The m9 methyl of m23,9A donates a hydrogen bond to a water that participates in an extensive hydrogen bonding network linking His203, Asp150 and Glu132. The orientation of the m23,9A base is compatible with a DNA docking model that is discussed below. The shape of the electron density corresponding to εA suggests that it binds in two different orientations (Figure 3B; see Materials and methods). Although it is not well ordered, the εA base is clearly stacked between Trp24 and Phe45 with its bulky etheno group located between Lys211 and Pro26 but not directly contacting the protein (Figure 3B). The inferior quality of the electron density for this bound base suggests that the binding pocket is not well suited for εA, consistent with MagIII’s low activity towards this substrate (Table II).
The functional significance of the base binding pocket identified by our crystallographic studies was tested by measuring the potencies of alkylated and unmodified adenine bases as inhibitors of m3A glycosylase activity. The rate of m3A excision was measured using genomic DNA treated with the alkylating agent N-methyl-N-nitrosourea (Bjelland et al., 1994). This agent primarily produces m7G and m3A lesions in DNA, and MagIII selectively excises m3A but not m7G (O’Rourke et al., 2000). Figure 4 shows that under the conditions of our assay (see Materials and methods), m3A release is effectively inhibited by the alkylated bases m23,9A, m3A and εA (Ki = 0.8 ± 0.1 mM), but unaffected by adenine (Ki ≥ 8.8 mM). The selective inhibition of m3A glycosylase activity by alkylated DNA bases and not adenine agrees well with the crystallographic results for nucleobase binding.
Recognition of alkylated DNA bases
The aromatic character of MagIII’s substrate binding pocket is particularly well suited for interactions with alkylated bases. Other m3A DNA glycosylases like E.coli AlkA and human AAG feature electron-rich aromatic active sites that could stack against electron-deficient, ring-substituted purines (Labahn et al., 1996; Hollis et al., 2000; Lau et al., 2000). RNA binding proteins that specifically recognize the m7G cap of mRNAs also feature aromatic binding pockets (Hodel et al., 1997; Marcotrigiano et al., 1997; Mazza et al., 2002), suggesting this is a robust strategy for the recognition of methylated purines. Methylation or protonation of a purine enhances the π-orbital overlap between the modified base and aromatic side chains (Ishida et al., 1983, 1988b), creating a more favorable stacking interaction with the methylated base that extends beyond the surface of the methyl group itself. Molecular orbital calculations suggest that the strength of π–π stacking interactions between methylpurines and aromatic side chains decreases in the order of tryptophan, tyrosine and phenylalanine (Ishida et al., 1988a). Mutational studies of the mRNA cap binding proteins are consistent with this proposal (Hu et al., 1999; Hsu et al., 2000; Mazza et al., 2002).
An enhanced π–π interaction with m3A could account for MagIII’s enzymatic specificity. The adenine derivatives m23,9A and εA both stack between the aromatic side chains of Trp24 and Phe45 of MagIII (Figure 3). The low activity of MagIII towards the neutral base εA (Table II) could be explained by the absence of formal positive charge (Dougherty, 1996) in the π–π stacking interaction between εA and Trp24/Phe45. The base stacking residue Phe45 is a tryptophan in the closely related enzyme MpgII (Figure 2A), which excises both m3A and m7G from DNA (Begley et al., 1999). Substituting Phe45 with a larger tryptophan side chain increased MagIII’s glycosylase activity for all alkylated substrates (Table II). This enhancement is evidence that aromatic stacking plays a significant role in the recognition of alkylated bases. We also examined the effect of the Phe45Trp mutation on m7G excision. Low levels of m7G excision by wild-type MagIII can be detected for a DNA substrate containing a m7G·T mismatched base pair (Table II), whereas no detectable excision activity was seen in the context of a m7G·C substrate (not shown). However, the Phe45Trp mutant MagIII did not enhance the excision of m7G·T relative to m3A or εA (Table II), so the tryptophan at this position alone does not explain MpgII’s robust m7G excision activity.
The shape of MagIII’s substrate binding pocket suggests why m7G is a poor substrate. After superimposing m7G onto m23,9A or εA in the crystallographic models, it is evident there is no orientation of the m7G base that avoids steric clashes of either the m7 methyl group with Trp25/Pro26 or the N2 amino group with Lys211 (Figure 5). One side of the base binding pocket is bounded by the side chain of Lys211, which is fixed by its interactions with both Glu46 and Glu208 (Figure 3C). This restricts the size of the binding pocket and would appear to exclude m7G. The MpgII glycosylase is active towards m7G and m3A (Begley et al., 1999), yet it differs from MagIII in only two residues within the active site, Phe45 (Trp52 in MpgII, discussed above) and Glu46 (Lys53 in MpgII). The substitution of Glu46 with a lysine would disrupt the interaction with Lys211 and it might widen the binding pocket to accommodate m7G. In order to test this idea, we substituted Phe45 and Glu46 of MagIII with the corresponding residues from MpgII. The Phe45Trp/Glu46Lys mutant showed increased rates of excision of both m7G and m3A in comparison to wild-type MagIII, and the Glu46Lys substitution alone accounted for most of this increase in activity (Table II). These amino acid substitutions caused a slight selective advantage for m7G excision relative to other substrates.
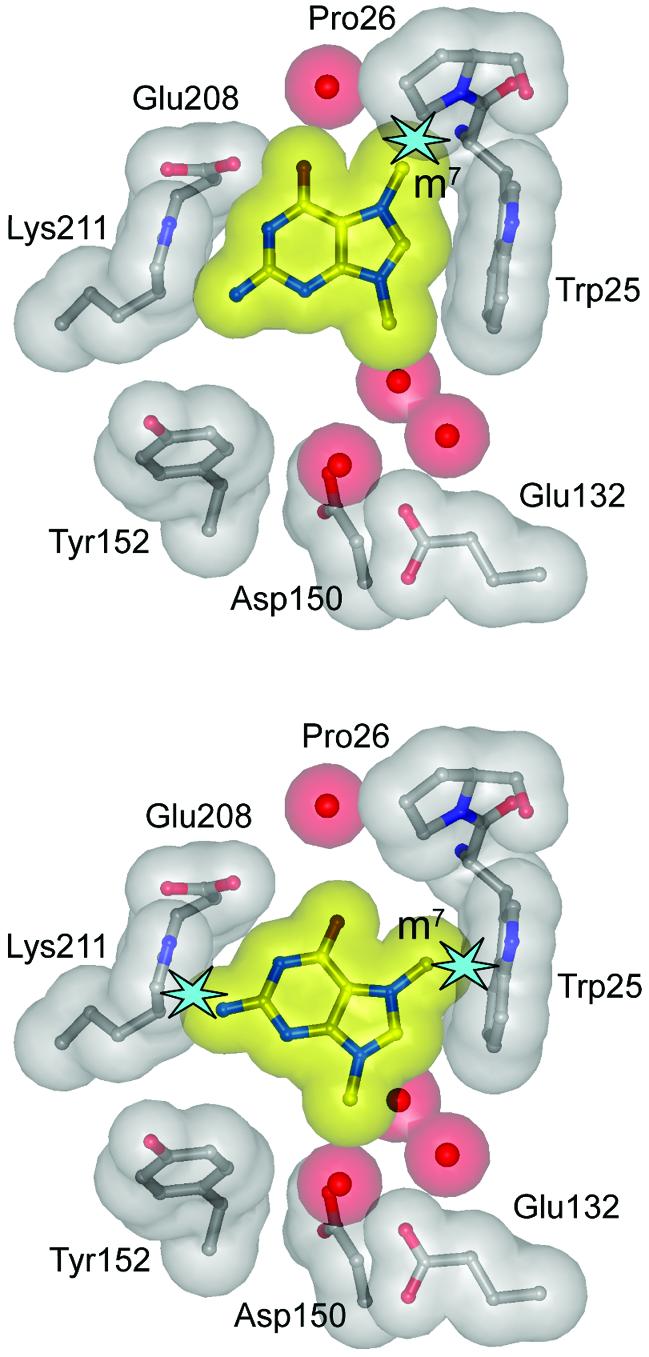
Fig. 5. Steric exclusion of m7G from the MagIII substrate binding pocket. 7,9-Dimethylguanine (m27,9G) was modeled by superimposing its purine ring onto the crystal structures of m23,9A (top) and εA (bottom) bound to MagIII. Transparent van der Waals surfaces of m27,9G (yellow), protein (gray) and waters (red) are shown. Steric clashes between atoms are highlighted as light blue stars. The views are the same as those in Figure 3A and B.
The steric exclusion of m7G from the MagIII active site was further investigated by mutating the residues that pack against the edges of the m23,9A base in the crystal structure (Figure 3). Lys211 was mutated to alanine and Trp25 was changed to either phenylalanine or alanine in order to relieve the steric constraints on base binding. The Lys211Ala mutant exhibits a 25-fold reduction in the observed rate of m3A excision, measured by necessity under subsaturating conditions (see Materials and methods). Under saturating conditions, the rate of m7G·T release was unaffected by the Lys211Ala mutation (Table II), giving the appearance that Lys211 selectively affects m3A excision. However, the Lys211Ala mutant exhibited a 10-fold reduction in DNA binding affinity (Table II). It seems likely that, under the conditions of our assay, decreased m3A excision activity resulted from the compromised DNA binding activity of Lys211Ala. Trp25 is located at the opposite side of the binding pocket, where it is important for positioning the m3A base and/or for stabilizing the fold of the pocket. The substitution of Trp25 with phenylalanine or alanine caused a loss of detectable glycosylase activity, even though the mutants are competent to bind to DNA (Table II).
Previous studies have attributed the enzymatic specificities of the HhH glycosylases to the shape and chemical characteristics of their active sites (Figure 6). The proposed binding pockets of the pyrimidine-specific glycosylases EndoIII (Thayer et al., 1995) and MIG (Mol et al., 2002) are narrower than those of purine-specific glycosylases, consistent with activity towards smaller pyrimidine bases. The narrow substrate binding pocket of Ogg1 is shaped to interact with every accessible face of its o8G substrate except at the 8-oxo carbonyl substituent (Figure 6C), and this arrangement is consistent with accommodation of the ring-opened 7,8-dihydro-8-oxoguanine (Bruner et al., 2000). In contrast, the wide active site cleft of the broadly specific AlkA glycosylase (Figure 6B) can stack against alkylated bases of many different shapes and sizes (Labahn et al., 1996; Yamagata et al., 1996). Specific hydrogen bonding interactions with substrate bases also contribute to selectivity. For example, the adenine-specific HhH glycosylase MutY forms two bidentate hydrogen bonds with the Watson-Crick and Hoogsteen faces of adenine (Figure 6D) (Guan et al., 1998). The direct read-out of o8G by Ogg1 is accomplished by a hydrogen bond between the protein and the protonated N7 nitrogen of the modified base o8G (Bruner et al., 2000). Likewise, it has been proposed that the m3A-specific TAG glycosylase (Figure 6E) gains specificity through hydrogen bonding interactions with the N1, N6 and N7 nitrogens of m3A (Drohat et al., 2002). It is therefore surprising that MagIII does not recognize adenine bases specifically through hydrogen bonding interactions, nor does it directly contact the alkylation-specific features of substrates. How, then, does MagIII bind selectively to uncharged, alkylated bases m3A and εA but not to unmodified adenine (Figure 4)? The alkyl substituents alter the shape and increase the size and hydrophobicity of adenine. It could be that the increased surface area of the modified base stacking face contributes adequate binding energy to discriminate against adenine, provided that the shape of the base fits inside the pocket. It is evident that active site features other than specific hydrogen bond donors/acceptors, (but) including the shape, aromaticity and electrostatic potential, are likely to be key determinants for the selective binding of substrates to MagIII.

Fig. 6. Characteristics of the substrate binding pockets from purine-specific HhH glycosylases. (A) MagIII-m23,9A, (B) AlkA bound to 1-azaribose AP-DNA (Hollis et al., 2000), (C) Ogg1 bound to o8G-DNA (Bruner et al., 2000), (D) MutY-adenine (Guan et al., 1998), and (E) TAG (Drohat et al., 2002). Each protein was aligned to give the same views into the substrate binding pocket from the perspective of the incoming DNA (golden sticks). At the left side of the figure are stereo views showing the specific contacts important for base recognition. Transparent van der Waals surfaces of substrate bases are colored green, and protein surfaces are colored according to electrostatic potential (blue, positive; red, negative). At the right side of the figure are the solvent accessible surfaces of the binding pockets colored according to electrostatic potential (GRASP, Nicholls et al., 1991). Substrate bases are white.
A DNA binding model
The electrostatic potential landscapes of the HhH glycosylases (Figure 2C) no doubt contribute to their interactions with DNA substrates, and the entrance to MagIII’s nucleobase binding pocket is strikingly electronegative (Figure 6A). The HhH motif contributes several acidic residues (Asp150, Glu132), which are required for efficient base excision activity (Table II). Both MagIII and the m3A-specific glycosylase TAG have an acidic patch surrounding their base binding pocket, in contrast to other HhH glycosylases (Figures 2C and 6). This charged patch might be important for the selection of positively-charged m3A nucleotides by assisting in their exposure from DNA during base-flipping. After base-flipping, the physicochemical constraints of the base binding pocket would further restrict the catalytic specificity of MagIII for m3A.
Crystal structures of AlkA (Hollis et al., 2000) and Ogg1 (Bruner et al., 2000) in complex with DNA show a common mode of DNA binding and base-flipping. Residues from the HhH motif (Figure 2A) and a nearby positively charged patch of residues (Figures 2C and 6) anchor the DNA against these enzymes and an intercalating side chain occupies the gap in the DNA left by the flipped-out nucleotide. Both enzymes bend the DNA by approximately 70° and this conformation is stabilized by the partial intercalation of a proline (AlkA) or tyrosine (Ogg1) that wedges into the non-lesioned DNA strand opposite the flipped out nucleotide.
The crystal structure of MagIII is consistent with this general model of DNA binding by HhH glycosylases. DNA from the AlkA or Ogg1 complexes superimposes well onto the fold of MagIII (Supplementary figure S2A). The DNA in this docking model has a few unfavorable clashes with MagIII that can be relieved by a small adjustment to the DNA model. Nearby residues from the HhH motif and positively charged residues flanking the active site cleft are positioned to make favorable interactions with the DNA. In the docking model, the side chain of Asn42 intercalates into the gap left by the flipped-out nucleotide, and Phe89 wedges between bases on the non-lesioned strand at the position of a sharp kink in the DNA (Supplementary figure S2B). The deoxyribose ring of the flipped-out nucleotide sits neatly inside the active site cleft next to Asp150, Glu132, Tyr152, Thr40 and several ordered water molecules (Figure 3C). The C1′ carbon of the modeled deoxyribose lies in the plane of the m23,9A base complexed to MagIII. However, the free base is located deep in the binding pocket, ∼4 Å away from C1′ of the flipped-out nucleotide. This suggests that, following cleavage of the glycosylic bond, the product base can move further into the binding pocket, possibly exiting the active site through an opening located between Pro26 and Lys211 (Figure 3C) before dissociation of the abasic DNA product.
The DNA docking model was used as a guide for mutational studies of MagIII’s DNA binding and glycosylase activities. The DNA binding affinities of wild-type and mutant MagIII enzymes for duplex DNAs containing a 1-azaribose abasic (AP) site were measured by a fluorescence anisotropy assay (Table II; Supplementary figure S3B). The positively-charged 1-azaribose (C1′→N) and pyrrolidine (O4′→N) AP-DNAs are potent inhibitors of many DNA glycosylases that were designed to mimic the positive charge of the proposed transition state for hydrolysis of the glycosidic bond (Schärer et al., 1995; Makino and Ichikawa, 1998). It is striking that MagIII binds to the 1-azaribose AP site (Kd = 0.5 µM; Table II) with >6-fold greater affinity than to the pyrrolidine AP analog (Kd > 3 µM; data not shown). Compared with other HhH glycosylases, MagIII binds weakly to the tetrahydrofuran AP site (Kd > 4 µM) and the enzymatically generated, unreduced AP site (Kd > 16 µM). This intriguing result suggests that MagIII forms stabilizing interactions with the 1-azaribose AP site that are absent from other AP–DNA complexes. Based on the DNA docking model, the candidate interactions between MagIII and the flipped-out deoxyribose are water-mediated contacts with His203, Asp150 and Glu132, as well as direct contacts to Glu132 or Thr40 (Supplementary figure S2B). Substitution of Glu132 with glutamine actually increased the binding affinity for the 1-azaribose DNA about 4-fold (Table II), probably by relieving an unfavorable interaction between Glu132 and a DNA phosphate located ∼3 Å away in the docking model. Replacing Asp150 with asparagine had no effect on DNA binding. In contrast, the substitution of His203 with either asparagine or alanine decreased DNA binding affinity by >14-fold (Table II). These amino acid changes are expected to disrupt water-mediated interactions between His203 and the deoxyribose.
Several other residues located at the DNA binding interface affect MagIII’s affinity for DNA. The loss of a DNA phosphate contact predicted by the docking model (Lys211Ala mutant) decreases affinity for DNA, whereas substituting Glu46 with a positively-charged side chain (Glu46Lys mutant) increases DNA binding affinity (Table II). Unexpectedly, the Asn42 side chain, which is predicted to intercalate into the gap left by the flipped-out nucleotide, actually seems to interfere with DNA binding. The Asn42Ala mutant binds 4-fold more tightly than wild-type MagIII to the 1-azaribose AP–DNA (Table II). This result might be indicative of an unfavorable equilibrium for flipping the substrate nucleotide from DNA bound by wild-type MagIII. This is consistent with low levels of MagIII activity towards some modified bases only in the context of a mismatch (εA·C and m7G·T, Table II; U·T, not shown). The Phe89 ‘wedge’ that is predicted to engage the non-lesioned strand and stabilize a bend in the DNA bound to MagIII contributes to DNA binding affinity. Mutating Phe89 to an alanine causes a 10-fold reduction in AP–DNA binding affinity (Table II). This is the first demonstration of the importance of this DNA-wedging residue in any HhH glycosylase.
Enzymatic catalysis of base excision
In the HhH glycosylases, a conserved aspartic acid located 8–9 residues C-terminal to the HhH motif (Figure 2A) contributes to the base excision activity of all enzymes that have been examined (Thayer et al., 1995; Labahn et al., 1996; Guan et al., 1998; Mol et al., 2002; Norman et al., 2003). This aspartate could function as a general base, activating a water or protein nucleophile for attack of the C1′ anomeric carbon (Thayer et al., 1995; Nash et al., 1997), or the aspartate might directly stabilize a carbocation intermediate formed during cleavage of the glycosylic bond (Hollis et al., 2000). The enzymatic contributions of the conserved aspartate (Asp150) and nearby glutamate (Glu132), which occupies the position of the lysine nucleophile of Ogg1 (Nash et al., 1997) and EndoIII (Thayer et al., 1995), were examined by mutational studies. Mutating Asp150 to the isosteric residue asparagine decreased the rates of m3A and εA excision by at least 25-fold (Table II; below the detection limit of our εA excision assay). This MagIII mutant retains substantially greater base excision activity than analogous aspartate mutants of other HhH glycosylases (Thayer et al., 1995; Labahn et al., 1996; Guan et al., 1998) (see Supplementary figure S3A). A relatively modest effect on base excision activity has also recently been reported for mutations of the analogous Asp268 in Ogg1 (Norman et al., 2003), suggesting that the conserved catalytic aspartate residue could play different roles during base excision by different HhH glycosylases. The Glu132Gln mutation did not measurably affect m3A excision and it reduced an already modest εA excision activity to undetectable levels (Table II). The residual glycosylase activity of the Asp150Asn mutant was unaffected by the additional mutation of Glu132 (Asp150Asn/Glu132Gln double mutant), and we conclude that Glu132 contributes little to enzymatic catalysis by MagIII. Although Asp150 has a significant role in catalysis (Table II), it is not essential for measurable glycosylase activity. This may indicate that less catalytic assistance is required for cleavage of the labile glycosylic bond of m3A (Berdal et al., 1998).
Conclusions
The crystal structures of H.pylori MagIII bound to alkylated bases provide the first structural information for understanding the selection of methyladenine lesions targeted for base excision by this and related HhH glycosylases. MagIII appears to stabilize a flipped-out m3A substrate in its active site by stacking the base between aromatic side chains of the base binding pocket, and without making hydrogen bonding interactions with the modified base. A major product of DNA methylation, m7G, is sterically excluded from the binding pocket of MagIII. The results of mutational studies support these conclusions based on the crystal structure, and they reveal the importance of, but not an absolute requirement for, the conserved aspartate (Asp150) in the base excision reaction.
Materials and methods
MagIII purification and crystallization
MagIII was expressed as a GST-fusion protein in E.coli JM109 cells at 16°C and affinity purified using Glutathione Sepharose 4B resin (Pharmacia). After cleavage of the GST-tag, MagIII was further purified by heparin affinity chromatography, followed by anion exchange on Source Q resin (Pharmacia). The protein was >99% pure at this stage, but further purification by gel filtration was necessary to obtain high-quality, diffracting crystals. MagIII was concentrated to 4 mg/ml in 20 mM Tris buffer (pH 7.5), 100 mM NaCl, 2 mM DTT and 0.1 mM EDTA.
Crystals of MagIII were grown by vapor diffusion at 4°C using a reservoir solution containing 25% PEG 4000 and 100 mM HEPES buffer (pH 7.0). Drops containing equal volumes of protein (4 mg/ml) and reservoir solution plus 2% 2-methyl-2,4-methylpentanediol (MPD) were equilibrated against reservoir which contained no MPD. The crystals grew as thin rods (∼500 × 60 × 20 µm3) in 1–2 days.
X-ray data collection, phasing and structure refinement
Prior to X-ray data collection, crystals were equilibrated at 4°C for 6 h in a harvest solution containing 28% PEG 4000, 100 mM HEPES buffer (pH 7.0) and 10% MPD and flash frozen in liquid nitrogen. Native diffraction data (Table I) were collected at Station A-1 at the Cornell High Energy Synchrotron Source (Ithaca, NY) and processed with DENZO/SCALEPACK from the HKL2000 package (Otwinowski and Minor, 1997). The crystals belong to space group C2 with unit cell dimensions a = 146.61 Å, b = 44.40 Å, c = 81.52 Å and β = 106.40°. Two MagIII protein molecules that are related by translational symmetry occupy the asymmetric unit.
Experimental X-ray phases were obtained from a multiwavelength anomalous diffraction (MAD) experiment using a single crystal that was soaked for 60 h at 4°C in harvest solution containing 0.2 mM ethyl mercury phosphate. Diffraction data (Table I) were collected at energies corresponding to the peak, inflection point and a high-energy remote setting near the LIII edge of mercury at Beamline X-12C at the National Synchrotron Light Source (Upton, NY). Two Hg atoms were evident in anomalous difference Patterson maps and they were positioned in the asymmetric unit using the program SOLVE (Terwilliger and Berendzen, 1999) (www.solve.lanl.gov). The Hg positions were refined and phases calculated to 2.7 Å using MLPHARE from the CCP4 package (Collaborative Computational Project, 1994). Solvent flattening using data from 50–2.7 Å was carried out with RESOLVE (Terwilliger, 2000), which resulted in a figure of merit of 0.60 and produced easily interpretable electron density maps. The MagIII model was built manually into the experimentally phased electron density using XtalView/Xfit (McRee, 1999).
The model was refined using all the X-ray data (25–1.64 Å) with a maximum likelihood target for experimental phases, as implemented in REFMAC 5.1 (Murshudov et al., 1997) (Table I). Improvements to the model were made by manual inspection of σA-weighted 2mFo-DFc and mFo-DFc electron density maps, and they were judged successful by a decrease in Rfree during refinement. Translation/libration/screw-rotation (TLS) refinement in REFMAC was used to model anisotropic motion of each protein domain (four in total). Individual anisotropic B-factors were derived from the refined TLS parameters and held fixed during subsequent rounds of refinement, which resulted in a 2% decrease in both R and Rfree and a noticeable improvement in the electron density maps.
The structures of m23,9A and εA bound to MagIII were determined by soaking crystals for 16 h at 4°C in harvest solution containing either 50 mM m23,9A·HCl or 2 mM εA. Diffraction data from cryocooled crystals were collected on a rotating anode Cu-Kα X-ray source. The nucleobases were identified by Fsoak–Fnat, φMAD difference Fourier maps and positioned into Fo–Fc density after refinement of the native structure against data collected on soaked crystals. Fo–Fc annealed omit electron density showed m23,9A to be well-ordered, and εA to be disordered in the plane of the base. Four possible εA orientations were modeled and subsequently refined. The unbiased Fo–Fc omit and refined 2Fo–Fc and Fo–Fc maps, as well as the behavior of Rfree after refinement, show two predominant orientations of εA in which the base is flipped about its long axis.
The model coordinates, experimental phases and structure factor amplitudes for native, m23,9A- and εA-bound MagIII structures have been deposited in the Protein Data Bank under accession numbers 1PU6, 1PU7 and 1PU8, respectively.
Mutagenesis, enzyme activity and DNA binding
Mutations were made in the MagIII expression plasmid using the Quik Change Site-Directed Mutagenesis kit (Stratagene), and they were verified by DNA sequencing. Mutant proteins were expressed and purified the same as the wild-type enzyme, but without the final gel filtration step.
DNA glycosylase activity assays for m3A excision (Supplementary figure S3A) were performed similarly to the method previously described (O’Rourke et al., 2000). The reaction mixture (50 µl) contained 3 µM enzyme and 3000 c.p.m. of N-[3H]methyl-N-nitrosourea-treated calf-thymus DNA in activity buffer (50 mM HEPES buffer (pH 7.5), 100 mM KCl, 10 mM DTT, 1 mM EDTA). Reactions were incubated at 37°C and terminated at various time points by ethanol precipitation of the DNA. The release of radioactive bases into the soluble fraction was quantitated by liquid scintillation counting. Rate constants were determined from single-exponential fits to data from three experiments. For this assay, the enzyme concentration was subsaturating with respect to substrate at the highest concentration of enzyme tested (15 µM). The failure to saturate might be caused by non-specific binding of MagIII to the vast excess of unmodified bases in the genomic DNA substrate. The observed second-order rate constants were shown to be linearly dependent on enzyme concentration up to at least 15 µM, and therefore reflect both binding and catalysis under these conditions.
The inhibition of m3A activity by modified adenine bases was measured using the same assay after pre-incubating 3 µM MagIII with m23,9A, m3A, εA or adenine at different concentrations up to the solubility limit (Figure 4). The inhibition constant (Ki) for each base was determined by fitting the data using the equation kobs = kmax/(1 + [I]/Ki), where kmax is the maximum rate in the absence of inhibitor (I).
MagIII activity towards εA and m7G was measured by alkaline cleavage of the AP–DNA product of base excision from a 25mer oligonucleotide duplex containing either εA·C or m7G·T mismatches. Oligonucleotides containing εA were chemically synthesized, while m7G was incorporated enzymatically using the appropriate primer/template and DNA polymerase I Klenow fragment (New England Biolabs) (Asaeda et al., 2000). The oligonucleotide carrying the modified base (X) in the sequence 5′-GACTACTACATGXTTCCCTACCATT was 32P-labeled at the 5′-end and annealed to excess complementary strand (5′-ATGGTAGGGAAYCATGTAGTAGTCA) containing the mismatched pyrimidine Y. In a 10 µl reaction, 2 nM radiolabeled DNA duplex was incubated with 3 µM enzyme in activity buffer. The reaction was stopped at various times by addition of 0.2 N NaOH, and heated at 70°C for 10 min. Products and remaining substrate were separated by denaturing polyacrylamide gel electrophoresis and quantitated by autoradiography. Rate constants were calculated from exponential fits to data from three experiments. Under the conditions of this assay, the enzyme concentration is saturating with respect to enzyme.
DNA binding was measured by following an increase in fluorescence anisotropy as protein was added to a fluorescently-labeled oligonucleotide (Supplementary figure S3B). Using the same 25mer DNA sequence described above, AP-containing oligonucleotide (X) was annealed in excess to the complementary strand (Y = C) containing a 6-carboxyfluorescein moiety at the 3′-end. MagIII was added over the concentration range of 0.05–5 µM to a solution containing 25 nM DNA duplex in activity buffer. Polarized fluorescence intensities using excitation and emission wavelengths of 495 and 515 nm were measured for 20 s (1/s) and averaged. Anisotropy (r) was calculated using the equation r = (Ipar–Iperp)/(Ipar+2Iperp), where Ipar and Iperp are the observed fluorescence intensities recorded through polarizers oriented parallel and perpendicular, respectively, to the direction of vertically polarized light. Dissociation constants (Kd) were derived by fitting a simple two-state binding model to data from three experiments using Kaleidagraph 3.5 (Synergy Software, PA).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
The authors thank Taisuke Itaya (Kanazawa University, Japan) for generously providing 3,9-dimethyladenine for these experiments, and the staff at beamlines X-12C and X-25 of the National Synchrotron Light Source (Upton, NY) and station A-1 at the Macromolecular Diffraction Facility of the Cornell High Energy Synchrotron Source (Ithaca, NY) for their assistance during X-ray data collection. Additional thanks to John Pascal for assistance with data collection, and to Patrick O’Brien, Tom Hollis and Eric Toth for helpful discussions. This work was funded by the National Institutes of Health (R01-GM52504 to T.E.). B.F.E. is supported by an NRSA Postdoctoral Fellowship from the National Institutes of Health. E.J.O’R. thanks the Fundación Antorchas, Buenos Aires, Argentina for financial support. T.E. is the Hsien Wu and Daisy Yen Wu Professor at Harvard Medical School.
References
- Asaeda A., Ide,H., Asagoshi,K., Matsuyama,S., Tano,K., Murakami,A., Takamori,Y. and Kubo,K. (2000) Substrate specificity of human methylpurine DNA N-glycosylase. Biochemistry, 39, 1959–1965. [DOI] [PubMed] [Google Scholar]
- Begley T.J., Haas,B.J., Noel,J., Shekhtman,A., Williams,W.A. and Cunningham,R.P. (1999) A new member of the endonuclease III family of DNA repair enzymes that removes methylated purines from DNA. Curr. Biol., 9, 653–656. [DOI] [PubMed] [Google Scholar]
- Berdal K.G., Johansen,R.F. and Seeberg,E. (1998) Release of normal bases from intact DNA by a native DNA repair enzyme. EMBO J., 17, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelland S., Bjoras,M. and Seeberg,E. (1993) Excision of 3-methylguanine from alkylated DNA by 3-methyladenine DNA glycosylase I of Escherichia coli. Nucleic Acids Res., 21, 2045–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelland S., Birkeland,N.K., Benneche,T., Volden,G. and Seeberg,E. (1994) DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli. J. Biol. Chem., 269, 30489–30495. [PubMed] [Google Scholar]
- Bruner S.D., Norman,D.P. and Verdine,G.L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature, 403, 859–866. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. (1992) Free R-Value—a Novel Statistical Quantity for Assessing the Accuracy of Crystal-Structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Bruns C.M., Hubatsch,I., Ridderstrom,M., Mannervik,B. and Tainer,J.A. (1999) Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J. Mol. Biol., 288, 427–439. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallog., D50, 760–763. [DOI] [PubMed] [Google Scholar]
- Doherty A.J., Serpell,L.C. and Ponting,C.P. (1996) The helix–hairpin–helix DNA-binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res., 24, 2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty D.A. (1996) Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr and Trp. Science, 271, 163–168. [DOI] [PubMed] [Google Scholar]
- Drohat A.C., Kwon,K., Krosky,D.J. and Stivers,J.T. (2002) 3-Methyladenine DNA glycosylase I is an unexpected helix–hairpin–helix superfamily member. Nat. Struct. Biol., 9, 659–664. [DOI] [PubMed] [Google Scholar]
- Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC. [Google Scholar]
- Fujii T., Itaya,T., Saito,T., Mohri,K., Kawanishi,M. and Nakasaka,T. (1989) Purines. XXXII. Synthesis and Ring Fission of 3,9-Dialkyladenines. Chem. Pharm. Bull., 37, 1504–1513. [Google Scholar]
- Guan Y., Manuel,R.C., Arvai,A.S., Parikh,S.S., Mol,C.D., Miller,J.H., Lloyd,S. and Tainer,J.A. (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat. Struct. Biol., 5, 1058–1064. [DOI] [PubMed] [Google Scholar]
- Hodel A.E., Gershon,P.D., Shi,X., Wang,S.M. and Quiocho,F.A. (1997) Specific protein recognition of an mRNA cap through its alkylated base. Nat. Struct. Biol., 4, 350–354. [DOI] [PubMed] [Google Scholar]
- Hollis T., Ichikawa,Y. and Ellenberger,T. (2000) DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J., 19, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.C., Hodel,M.R., Thomas,J.W., Taylor,L.J., Hagedorn,C.H. and Hodel,A.E. (2000) Structural requirements for the specific recognition of an m7G mRNA cap. Biochemistry, 39, 13730–13736. [DOI] [PubMed] [Google Scholar]
- Hu G., Gershon,P.D., Hodel,A.E. and Quiocho,F.A. (1999) mRNA cap recognition: dominant role of enhanced stacking interactions between methylated bases and protein aromatic side chains. Proc. Natl Acad. Sci. USA, 96, 7149–7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T., Shibata,M., Fujii,K. and Inoue,M. (1983) Inter- and intramolecular stacking interaction between indole and adeninium rings. Biochemistry, 22, 3571–3581. [DOI] [PubMed] [Google Scholar]
- Ishida T., Doi,M. and Inoue,M. (1988a) A selective recognition mode of a nucleic acid base by an aromatic amino acid: l-phenylalanine-7-methylguanosine 5′-monophosphate stacking interaction. Nucleic Acids Res., 16, 6175–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T., Doi,M., Ueda,H., Inoue,M. and Scheldrick,G.M. (1988b) Specific Ring Stacking Interaction on the Tryptophan 7-Methylguanine System: Comparative Crystallographic Studies of Indole-Derivatives 7-Methylguanine Base, Nucleoside and Nucleotide Complexes. J. Am. Chem. Soc., 110, 2286–2294. [Google Scholar]
- Kuo C.F., McRee,D.E., Fisher,C.L., O’Handley,S.F., Cunningham,R.P. and Tainer,J.A. (1992) Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science, 258, 434–440. [DOI] [PubMed] [Google Scholar]
- Kwon K., Cao,C. and Stivers,J.T. (2003) A novel zinc snap motif conveys structural stability to 3-methyladenine DNA glycosylase I. J. Biol. Chem., 278, 19442–19446. [DOI] [PubMed] [Google Scholar]
- Labahn J., Schärer,O.D., Long,A., Ezaz-Nikpay,K., Verdine,G.L. and Ellenberger,T.E. (1996) Structural basis for the excision repair of alkylation-damaged DNA. Cell, 86, 321–329. [DOI] [PubMed] [Google Scholar]
- Lau A.Y., Wyatt,M.D., Glassner,B.J., Samson,L.D. and Ellenberger,T. (2000) Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc. Natl Acad. Sci. USA, 97, 13573–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Makino K. and Ichikawa,Y. (1998) Synthesis of a 2-deoxy-ribose type 1-N-iminosugar. Tetrahedron Lett., 39, 8245–8248. [Google Scholar]
- Marcotrigiano J., Gingras,A.C., Sonenberg,N. and Burley,S.K. (1997) Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell, 89, 951–961. [DOI] [PubMed] [Google Scholar]
- Mazza C., Segref,A., Mattaj,I.W. and Cusack,S. (2002) Large-scale induced fit recognition of an m7GpppG cap analogue by the human nuclear cap-binding complex. EMBO J., 21, 5548–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy T.V., Karran,P. and Lindahl,T. (1984) Inducible repair of O-alkylated DNA pyrimidines in Escherichia coli. EMBO J., 3, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRee D.E. (1999) XtalView/Xfit–A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol., 125, 156–165. [DOI] [PubMed] [Google Scholar]
- Mol C.D., Arvai,A.S., Begley,T.J., Cunningham,R.P. and Tainer,J.A. (2002) Structure and activity of a thermostable thymine-DNA glycosylase: evidence for base twisting to remove mismatched normal DNA bases. J. Mol. Biol., 315, 373–384. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.J. (1997) Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallog., D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nash H.M., Bruner,S.D., Scharer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- Nash H.M., Lu,R., Lane,W.S. and Verdine,G.L. (1997) The critical active-site amine of the human 8-oxoguanine DNA glycosylase, hOgg1: direct identification, ablation and chemical reconstitution. Chem. Biol., 4, 693–702. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Norman D.P., Chung,S.J. and Verdine,G.L. (2003) Structural and biochemical exploration of a critical amino acid in human 8-oxoguanine glycosylase. Biochemistry, 42, 1564–1572. [DOI] [PubMed] [Google Scholar]
- O’Rourke E.J., Chevalier,C., Boiteux,S., Labigne,A., Ielpi,L. and Radicella,J.P. (2000) A novel 3-methyladenine DNA glycosylase from Helicobacter pylori defines a new class within the endonuclease III family of base excision repair glycosylases. J. Biol. Chem., 275, 20077–20083. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Roberts R.J. and Cheng,X. (1998) Base flipping. Annu. Rev. Biochem., 67, 181–198. [DOI] [PubMed] [Google Scholar]
- Saparbaev M., Kleibl,K. and Laval,J. (1995) Escherichia coli, Saccharomyces cerevisiae, rat and human 3-methyladenine DNA glycosylases repair 1,N6-ethenoadenine when present in DNA. Nucleic Acids Res., 23, 3750–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schärer O.D. and Jiricny,J. (2001) Recent progress in the biology, chemistry and structural biology of DNA glycosylases. BioEssays, 23, 270–281. [DOI] [PubMed] [Google Scholar]
- Schärer O.D., Ortholand,J.-Y., Ganesan,A., Ezaz-Nikpay,K. and Verdine,G.L. (1995) Specific binding of the DNA repair enzyme AlkA to a pyrrolidine-based inhibitor. J. Am. Chem. Soc., 117, 6623–6624. [Google Scholar]
- Terwilliger T.C. (2000) Maximum likelihood density modification. Acta Crystallog., D56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallog., D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer M.M., Ahern,H., Xing,D., Cunningham,R.P. and Tainer,J.A. (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J., 14, 4108–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P., Qiu,C., Sohail,A., Zhang,X., Bhagwat,A.S. and Cheng,X. (2003) Mismatch repair in methylated DNA. Structure and activity of the mismatch-specific thymine glycosylase domain of methyl-CpG-binding protein MBD4. J. Biol. Chem., 278, 5285–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y. et al. (1996) Three-dimensional structure of a DNA repair enzyme, 3-methyladenine DNA glycosylase II, from Escherichia coli. Cell, 86, 311–319. [DOI] [PubMed] [Google Scholar]