

Abstract
In mammals, nonsense-mediated mRNA decay (NMD) is a quality-control mechanism that degrades mRNA harboring a premature termination codon to prevent the synthesis of truncated proteins. To gain insight into the NMD mechanism, we identified NMD inhibitor 1 (NMDI 1) as a small molecule inhibitor of the NMD pathway. We characterized the mode of action of this compound and demonstrated that it acts upstream of hUPF1. NMDI 1 induced the loss of interactions between hSMG5 and hUPF1 and the stabilization of hyperphosphorylated isoforms of hUPF1. Incubation of cells with NMDI 1 allowed us to demonstrate that NMD factors and mRNAs subject to NMD transit through processing bodies (P-bodies), as is the case in yeast. The results suggest a model in which mRNA and NMD factors are sequentially recruited to P-bodies.
Introduction
Nonsense-mediated mRNA decay (NMD) is a quality-control process found in all eukaryotic organisms studied to date (Maquat, 2004a; Conti and Izaurralde, 2005). One role of this process is to degrade mRNA harboring a premature termination codon (PTC) to prevent the synthesis of truncated proteins that could be nonfunctional or whose function may be deleterious to cells. The NMD pathway has been shown to be involved in the regulation of gene expression in yeast, Drosophila, and mammals (Sureau et al., 2001; He et al., 2003; Mendell et al., 2004; Wollerton et al., 2004; Rehwinkel et al., 2005).
In mammalian cells, NMD takes place after pre-mRNA splicing and in most cases is mediated by a protein complex deposited 20–24 nucleotides upstream of exon–exon junctions (Maquat, 2004b; Conti and Izaurralde, 2005; Lejeune and Maquat, 2005). This protein complex called the exon junction complex (EJC) is thought to recruit the evolutionarily conserved UPF proteins that play an essential but still not fully characterized role in NMD. During what is referred to as the “pioneer round of translation” (Ishigaki et al., 2001), PTCs are recognized and the targeted mRNAs are degraded by both 5′–3′ decay involving decapping and 5′–3′ exoribonucleases such as hXRN1 and hXRN2/hRAT1, and by 3′–5′ decay involving deadenylation and the exosome (Chen and Shyu, 2003; Lejeune et al., 2003; Couttet and Grange, 2004).
NMD implicates the participation of hUPF proteins such as hUPF1, hUPF2, hUPF3 (also named hUPF3a), and hUPF3X (also called hUPF3b). The function of these hUPF proteins is still unclear. However, it has been proposed that they are sequentially recruited by the EJC: hUPF3/3X first, followed by hUPF2, and finally hUPF1 in mammalian cells (Lykke-Andersen et al., 2001; Kim et al., 2005). Interestingly, the function of hUPF2 has been demonstrated to be dispensable in some NMD cases, suggesting the existence of different pathways to elicit NMD (Gehring et al., 2005).
UPF1 is a phosphoprotein that undergoes phosphorylation/dephosphorylation cycles during NMD (Page et al., 1999; Pal et al., 2001; Ohnishi et al., 2003). UPF1 has been shown to interact with release factors in yeast (Czaplinski et al., 1998) and mammals (Kashima et al., 2006), and could link the EJC and translation termination complex. A direct interaction between hUPF1 and the cap-binding protein CBP80 has also recently been demonstrated in mammalian cells (Hosoda et al., 2005), indicating that hUPF1 establishes a complex interaction network either before or during the pioneer round of translation. Phosphorylation of hUPF1 has been shown to be performed by hSMG1, a phosphoinositide 3-kinase–related kinase (Page et al., 1999; Pal et al., 2001; Yamashita et al., 2001), and requires the presence of hUPF2 and hUPF3 (Kashima et al., 2006). In contrast, dephosphorylation of hUPF1 requires the presence of a multiprotein complex composed of hSMG5, hSMG6, hSMG7, and protein phosphatase 2A (Chiu et al., 2003; Ohnishi et al., 2003). For the most part, hSMG5 and hSMG7 proteins are distributed evenly throughout the cytoplasm, but a fraction is also present in processing bodies (P-bodies; Unterholzner and Izaurralde, 2004). hSMG6 is a cytoplasmic protein that concentrates in cytoplasmic foci distinct from P-bodies and whose nature is still unclear (Unterholzner and Izaurralde, 2004).
P-bodies have been described in lower and higher eukaryotic cells (Ingelfinger et al., 2002; van Dijk et al., 2002; Sheth and Parker, 2003; Cougot et al., 2004). In mammals, these cytoplasmic structures contain many factors involved in mRNA decay, including components of the decapping machinery such as decapping protein 1a (DCP1a; Ingelfinger et al., 2002), DCP2 (Ingelfinger et al., 2002; van Dijk et al., 2002), GE1 (also called Hedls; Fenger-Gron et al., 2005; Yu et al., 2005), p54/RCK (Cougot et al., 2004), the deadenylase CCR4 (Cougot et al., 2004), XRN1 (Bashkirov et al., 1997), the LSM1-7 complex involved in different aspects of RNA processing (Cougot et al., 2004; Ingelfinger et al., 2002), and the hUPF1, hSMG5, and hSMG7 components of the NMD machinery (Unterholzner and Izaurralde, 2004; Fukuhara et al., 2005). The function of P-bodies is still unclear but they may serve as a storage compartment for both untranslated RNAs and proteins involved in RNA decay (Brengues et al., 2005; Pillai et al., 2005; Teixeira et al., 2005; Franks and Lykke-Andersen, 2007), and/or as a site for RNA decay (Cougot et al., 2004; Sheth and Parker, 2006).
In a recent work, we showed that hydrophobic tetracyclic indole derivatives block the function of specific splicing factors (Soret et al., 2005). In light of these findings, we decided to look further at this collection to determine if certain of these compounds also inhibit NMD. The underlying idea was that such small molecule inhibitors could represent powerful tools to decipher the NMD process. In this paper, we report the identification of one such molecule, nonsense-mediated mRNA decay inhibitor 1 (NMDI 1), that inhibits nucleus- associated as well as cytoplasmic NMD. The inhibitory mechanism appears to be caused by the loss of the interaction between hSMG5 and hUPF1, thereby leading to the stabilization of the hyperphosphorylated forms of hUPF1 and to its concentration in P-bodies. Interestingly, NMDI 1–mediated inhibition revealed that other NMD factors and PTC-containing mRNA can traffic through P-bodies as is the case in yeast (Sheth and Parker, 2006).
Results
Identification of a novel NMD inhibitor
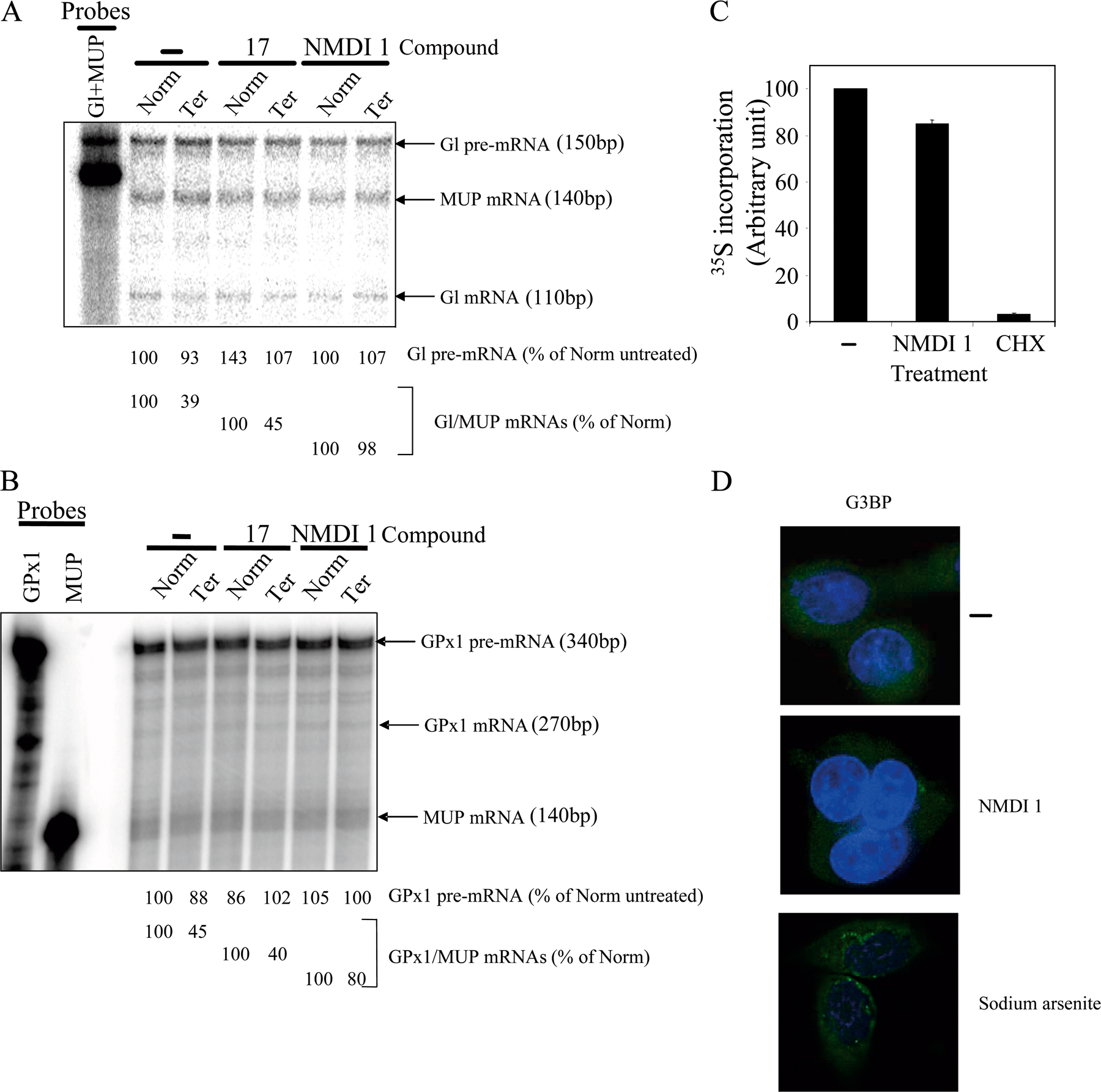
In a previous paper we identified a series of polycyclic indole derivatives that block the function of specific splicing factors (Soret et al., 2005). Because certain molecules from this family inhibit the function of proteins involved in mRNA maturation, we decided to assess their capacity to inhibit NMD. HeLa cells were transfected by two test plasmids coding for β globin (Gl) and glutathione peroxidase 1 (GPx1) mRNA, either with (Ter) or without (Norm) a PTC. Gl mRNA was subject to nucleus- associated NMD in nonerythroid cells (Thermann et al., 1998; Zhang et al., 1998) whereas GPx1 mRNA was subject to cytoplasmic NMD (Moriarty et al., 1998). Additionally, a reference plasmid coding for the mouse major urinary protein (MUP) mRNA was also introduced into the cells (Ishigaki et al., 2001). 24 h after transfection, cells were incubated for 20 h with 5 μM of indole compound (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200611086/DC1) or DMSO(−) as a control. Then, total RNAs were purified and analyzed by RT-PCR (Fig. 1) to measure NMD efficiency. Among the 25 indole derivatives tested, only compound 70 (NMDI 1) stabilized the Gl Ter mRNA level about threefold, indicating that this molecule is an inhibitor of nucleus-associated NMD (Fig. 1 A and not depicted). Interestingly, NMDI 1 also stabilized the level of GPx1 Ter mRNA by approximately twofold (Fig. 1 B and not depicted). To confirm these results, we measured the NMD inhibition by RNase protection assay (RPA), as it represents a more reliable approach for RNA quantification. The results are presented in Fig. S1 (A and B) and confirm the two- to threefold NMD inhibition by NMDI 1. Altogether, these data allowed us to conclude that NMDI 1 is an inhibitor of nucleus-associated as well as cytoplasmic NMD. Notably, the inhibition level obtained with NMDI 1 is similar to that observed with other NMD inhibitors such as cycloheximide (CHX) or to the down-regulation of hDCP2 or hPARN (Ishigaki et al., 2001; Lejeune et al., 2003). To show a more direct correlation between NMDI 1 and NMD inhibition, we measured NMD efficiency on PTC-containing Gl or GPx1 mRNA in cells that were treated with an increasing amount of NMDI 1 (Fig. 1 C). Interestingly, we observed a progressive NMD inhibition from 0 to 5 μM NMDI 1 for both Gl and GPx1 constructs. At >5 μM, we were unable to get a substantially stronger inhibition, suggesting that NMD cannot be 100% eliminated in our experimental conditions or that the 20–30% of mRNA that escaped from NMD inhibition represents the fraction of mRNAs already engaged in the NMD process at the time of NMDI 1 treatment. In all subsequent experiments, we used 5 μM NMDI 1 as our working concentration. Notably, NMDI 1 does not exhibit any cellular toxicity, as measured by trypan blue staining, even at concentrations as high as 125 μM (unpublished data).
Figure 1.

Identification of an NMD inhibitor. (A) RT-PCR analysis of globin and MUP mRNA. 106 HeLa cells were transfected with a plasmid that codes for MUP mRNA and with test and reference plasmids that code for globin mRNA wild type (Norm) or harbor a PTC (Ter). After transfection, cells were incubated with DMSO(−) or 5 μM of a chemical compound for 20 h. Purified RNA was reverse-transcribed to serve as a substrate for specific amplification by PCR. The three leftmost lanes correspond to serial twofold dilutions of PCR template to ensure that the amplification conditions are quantitative. (B) RT-PCR analysis of GPx1 mRNA wild type (Norm) or cells harboring a PTC (Ter). The experiment was performed as described in A. The measure of the level of Gl or GPx1 mRNA was normalized with the level of MUP mRNA. The level of each Gl or GPx1 Ter was normalized with the level of the corresponding Gl or GPx1 Norm and is reported as a percentage of Norm (number below each lane). (C) Dose-response effect of NMDI 1 on Gl or GPx1 Ter. Hela cells were transfected with pmCMV-Gl Ter or pmCMV-GPx1 Ter and with phCMV-MUP. After 24 h, cells were incubated with an increasing amount of NMDI 1. NMD was measured by quantitative radiolabeled RT-PCR and confirmed by RPA. (D) Measure of Fluc activity. Cells were transfected with pRLuc and pFluc expression vectors, and then treated with DMSO(−), NMDI 1, or CHX. Fluc activity was measured by a luminometer and normalized according to the expression level of Fluc and Rluc mRNA. (E) NMDI 1 does not inhibit miRNA-induced mRNA decay. HeLa cells were transfected with either pRL-3XBulgeMut (RLm) or pRL-Perf (RLp; Pillai et al., 2005) and incubated for 24 h with DMSO(−) or NMDI 1. Histogram represents the ratio of Rluc/Fluc. Results were normalized to RLm, which was set at 10 arbitrary units. All results are representative of at least three independent experiments. Error bar denotes SD.
At this stage, some controls were performed to investigate the specificity of inhibition mediated by NMDI 1. First, NMDI 1 failed to have any effect on splicing of several pre-mRNA reporter transcripts (Soret et al., 2005) and did not affect the level of pre-mRNA (Fig. S1, A and B). Second, unlike CHX, which inhibits translation, NMDI 1 does not alter the expression of transfected firefly luciferase (Fluc; Fig. 1 D), suggesting that NMDI 1 is not a general translation inhibitor. To further demonstrate the absence of any effects of NMDI 1 on translation, we performed metabolic labeling of proteins with [35S]methionine in HeLa cells and showed that treatment with NMDI 1 had no effect on 35S incorporation (Fig. S1 C). Third, to assay the integrity of the microRNA (miRNA) decay pathway in the presence of NMDI 1, we used a Renilla luciferase (Rluc) construct that is subject to degradation by let- 7 miRNA (pRL-Perf) or immune to miRNA decay pathway (pRL-3XBugleMut; Pillai et al., 2005). Our results indicate that NMDI 1 does not increase Renilla activity, which is under the control of let-7 miRNA, confirming that targeted mRNA degradation by miRNA is not altered by NMDI 1 (Fig. 1 E). Finally, we also tested whether NMDI 1 could induce the formation of the stress granules that provides a sensitive assay for proper mRNA metabolism. Indeed, these structures are aggregates of messenger RNPs that form when cells are subjected to several stresses, including mild translational inhibition. Unlike sodium arsenite treatment that is commonly used to induce stress granule formation (Kedersha et al., 2005), NMDI 1 treatment did not change the localization of G3BP protein, a well-characterized marker of stress granules (Fig S1 D; Tourriere et al., 2003). Collectively, these results indicate that NMDI 1 is a new and specific NMD inhibitor.
NMDI 1 abrogates NMD upstream of hUPF1 functions
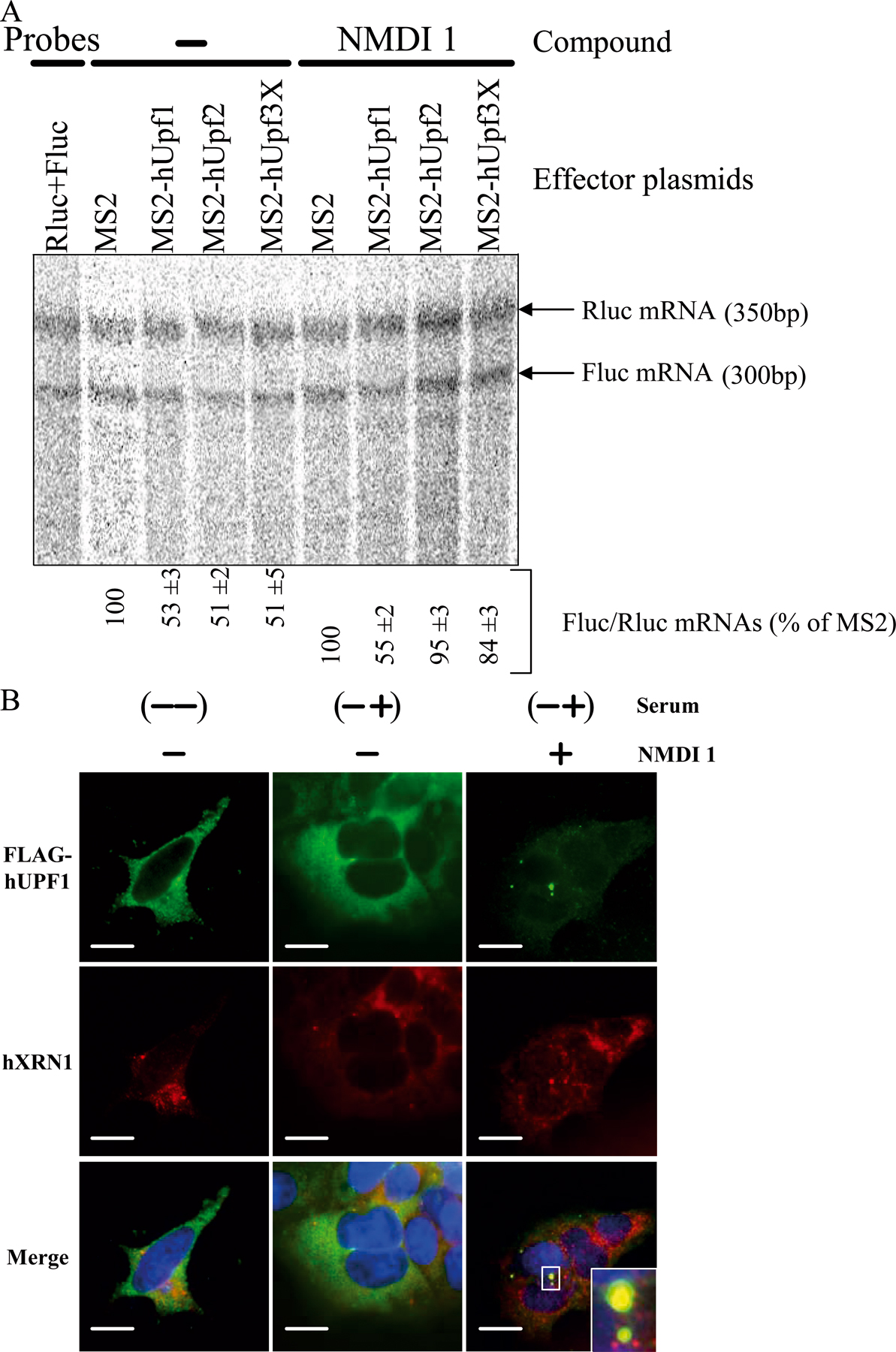
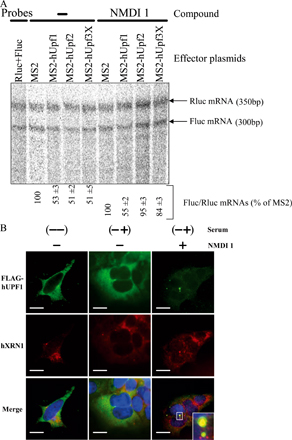
To gain insight into the mode of inhibition of NMDI 1, we analyzed its effects on a tethering system that mimics the sequential recruitment of NMD factors on mRNA (Lykke-Andersen et al., 2000; Kim et al., 2005). Cells were transfected with two types of constructs. The first codes for a Fluc mRNA containing eight binding sites for the MS2 protein in its 3′ untranslated region and the second codes for either the MS2 protein or one of the following fusions: MS2-hUPF1, MS2-hUPF2, or MS2-hUPF3X. Additionally, we transfected HeLa cells with a construct coding for the Rluc mRNA to normalize the amount of analyzed RNA. Cells were then incubated for 20 h with NMDI 1 or DMSO(−) as a negative control, and Rluc as well as Fluc mRNA levels were measured by RT-PCR as described previously (Hosoda et al., 2005). The expression of each MS2 fusion was controlled by Western blot to verify that the observed effects were not caused by a variation in protein expression (Fig. 2 A). In each case, the compound did not affect expression of the MS2 fusion, which was itself never higher than the level of the endogenous protein. As expected, the control experiment performed in the presence of DMSO revealed that the level of Fluc mRNA was lower in cells expressing one of the MS2-hUPF fusion proteins compared with cells expressing only MS2 (Fig. 2 B). Remarkably, NMDI 1 counteracted the degradation induced by MS2-hUPF2 or MS2-hUPF3X but had no effect against MS2-hUPF1 (Fig. 2 B). Notably, the inhibition levels obtained with NMDI 1 were very similar to those observed when NMD was inhibited through down-regulation of hCBP80 (Hosoda et al., 2005). To obtain a more accurate measure of the NMD inhibition, Rluc and Fluc mRNA levels were also measured by RPA. The results are presented in Fig. S2 A (available at http://www.jcb.org/cgi/content/full/jcb.200611086/DC1) and reproduce the quantification of mRNA levels by RT-PCR (Fig. 2 B). Altogether, these results indicate that NMDI 1 inhibits NMD downstream of hUPF3X or hUPF2 recruitment and upstream of hUPF1 functions.
Figure 2.

NMDI 1 inhibits NMD before the functions of hUPF1. (A) HeLa cells were transiently transfected with plasmids that encode the Rluc mRNA, the Fluc mRNA containing MS2 binding sites in its 3′ untranslated region, and the mRNA coding for MS2 protein either alone or fused with one of the hUPF proteins. 24 h after transfection, HeLa cells were incubated either with DMSO(−) or NMDI 1 for 20 h. The expression level of each MS2 fusion protein was determined by Western blot. 10 μg of protein extract was loaded on 10% SDS–polyacrylamide gel and transferred to a nitrocellulose membrane before incubation with antibodies against each of the hUPF proteins. The position of endogenous and exogenous proteins is indicated on the right. (B) The level of Fluc mRNA was normalized with the level of the corresponding Rluc mRNA and is reported as a percentage of normalized Fluc mRNA when only MS2 protein was expressed. The three leftmost lanes correspond to serial twofold dilutions of PCR template to show that the amplification conditions are quantitative.
NMDI 1 does not prevent the interactions between hUPF1 and hUPF3X
In the light of the results described in the previous paragraph, we hypothesized that NMDI 1 could prevent the recruitment of hUPF1 to the EJC via its interactions with the other hUPF proteins. To test this, we immunoprecipitated hUPF1 from HeLa cell extracts under conditions that preserve the integrity of messenger RNPs (Lejeune and Maquat, 2004). NMDI 1 or DMSO(−) was added to the cell culture 20 h before immunoprecipitation (IP). Because hUPF2 was shown to be dispensable in some NMD cases (Gehring et al., 2005), we focused our analysis on the presence of hUPF3X protein in each IP (Fig. 3 A). As a control for IP specificity, we did not detect tubulin protein in any of the hUPF1 IPs and no proteins were present in the IP performed with normal rabbit serum. The results show that hUPF3X was present in hUPF1 IP even when cells were incubated with our NMD inhibitor. Thus, these data demonstrate that the interaction between hUPF1 and hUPF3X is not abolished by NMDI 1 and suggest that NMDI 1 would not prevent the recruitment of hUPF1 to the EJC.
Figure 3.

NMDI 1 modifies the cellular distribution of hUPF proteins and stabilizes hyperphosphorylated isoforms of FLAG-hUPF1. (A) Endogenous hUPF1 protein was immunopurified using rabbit anti-hUPF1 antibodies from HeLa cell extracts that were incubated with DMSO(−) or 5 μM NMDI 1(+). In parallel, an immunopurification control was performed using normal rabbit serum to verify the specificity of the immunopurification protocol. The three leftmost lanes correspond to serial twofold dilutions of a whole HeLa cell extract. The asterisk marks an uncharacterized band that presumably represents a degradation product. (B) 2D gel analysis of the FLAG-hUPF1 phosphorylation level. 106 293T cells were transfected with 1 μg pCI-neo-FLAG-hUPF1 plasmid. 12 h later, the serum was removed from the culture medium for 24 h, and then either DMSO or 5 μM NMDI 1 was added to the culture medium for 3 h before adding back 10% serum (sample (−+) except in sample (−−)). Proteins were extracted and loaded on a 2D gel analysis system (see Materials and methods). (C) Immunofluorescence assay. HeLa cells were transfected with pCI-neo-FLAG-hUpf1 and either pYFP-hSmg7, pGFP-Ge1, or pCFP-hDcp1a. 24 h after transfection, cells were incubated with DMSO(−) or 5 μM NMDI 1 for 20 h. The blue staining visible in the merge of the two leftmost set of images corresponds to nuclei staining by Hoechst stain. (D) Immunofluorescence assay. HeLa cells were transfected with pYFP-hSmg5, pYFP-hSmg6, or pYFP-hSmg7 and with pCFP-hDcp1a expression vectors. 24 h after transfection, cells were cultured in the presence of DMSO(−) or 5 μM NMDI 1 for 20 h. The white squares are magnifications of cell areas. Bars, 10 μm.
NMDI 1 stabilizes hyperphosphorylated forms of hUPF1
Because hUPF1 requires a cycle of phosphorylation and dephosphorylation during NMD (Ohnishi et al., 2003), we next investigated the possibility that NMDI 1 may affect hUPF1 function by interfering with its phosphorylation level. We thus measured the level of hUPF1 phosphorylation in cells incubated with NMDI 1 or, as a control, with DMSO(−) by 2D gel analysis. Because hUPF1 phosphorylation is influenced by serum (Pal et al., 2001), we used 293T cells that, unlike HeLa cells, can be synchronized by serum deprivation. Cells were transfected with the expression vector pCI-neo-FLAG-hUpf1 (Sun et al., 1998) and synchronized for 24 h by serum deprivation 12 h after transfection. Finally DMSO or 5 μM NMDI 1 was added for 3 h before adding back serum for 1 h. Our results show that when serum was not added back to the cell culture, the FLAG-hUPF1 protein electrofocalized in one spot corresponding to the unphosphorylated protein (Fig. 3 B; Pal et al., 2001). After serum addition, we observed a mild phosphorylation of FLAG-hUPF1 protein in the presence of DMSO and the stabilization of hyperphosphorylated isoforms of FLAG-hUPF1 when cells were incubated with NMDI 1 (Fig. 3 B). We concluded that NMDI 1 stabilized hyperphosphorylated isoforms of hUPF1.
As it has been proposed that hUPF1 would localize to P-bodies when hyperphosphorylated (Unterholzner and Izaurralde, 2004; Fukuhara et al., 2005), we analyzed the cellular localization of FLAG-hUPF1 in HeLa cells in the absence or presence of NMDI 1 (Fig. 3 C). With the exception of coexpression experiments with hSMG7, which induces the recruitment of hUPF1 to P-bodies (Fig. 3 C; Unterholzner and Izaurralde, 2004), exogenous hUPF1 was equally distributed through the cytoplasm when cells were incubated with DMSO(−) as previously reported for untreated cells (Fig. 3 C; Lykke-Andersen et al., 2000; Mendell et al., 2002). When cells were treated with NMDI 1, we observed cytoplasmic concentrations of FLAG-hUPF1 in some structures that colocalize with the three commonly used markers of P-bodies: GFP-GE1, YFP-hSMG7, or CFP-hDCP1a (Fig. 3 C). We also verified that FLAG-hUPF1 accumulated in P-bodies in the presence of NMDI 1 in 293T cells under the same experimental conditions used to study the phosphorylation level of FLAG-hUPF1 (Fig. S2 B). We used hXRN1 protein as a P-body marker to avoid any additional transfected DNA. After addition of serum, we observed some FLAG-hUPF1 cytoplasmic concentrations that colocalize with hXRN1 only when cells were treated with NMDI 1 but not in its absence. To definitively demonstrate that hyperphosphorylated isoform of hUPF1 accumulates in P-bodies, HeLa cells were treated for 20 h with either DMSO(−) or NMDI 1, and the cellular localization of endogenous phosphorylated hUPF1 was determined using a specific antibody raised against a phosphoepitope of this protein (Ohnishi et al., 2003). The results presented in Fig. S3 (available at http://www.jcb.org/cgi/content/full/jcb.200611086/DC1) indicate that in the presence of NMDI 1, phosphorylated hUPF1 isoforms colocalize with CFP-hDCP1a foci. We conclude that NMDI 1 induces the accumulation of hyperphosphorylated hUPF1 isoforms in P-bodies. This may occur either via stimulation of phosphorylation or by blocking dephosphorylation.
To distinguish between these two possibilities, we subsequently investigated the association of hUPF1 with other NMD partners in HeLa cells treated or untreated with NMDI 1 (Fig. 3 A). We first analyzed the interaction of hUPF1 with its dephosphorylation complex. Immunoprecipitation of hUPF1 allowed recovery of hSMG5, hSMG6, and hSMG7 from DMSO-treated cells. However, after treatment of HeLa cells with NMDI 1 only hSMG6 and hSMG7 but not hSMG5 were still associated with hUPF1 (Fig. 3 A). Thus, we conclude that NMDI 1 destabilizes the interaction between hUPF1 and hSMG5. The fact that NMDI 1 does not alter the association of hUPF1 with hSMG1 and hUPF3X strongly suggests that NMDI 1 does not influence the interactions between hUPF1 and its phosphorylation complex (Fig. 3 A). Altogether, our results indicate that the hyperphosphorylation of hUPF1 is most likely caused by a failure in dephosphorylation because of the loss of interaction between hUPF1 and hSMG5 rather than an activation of phosphorylation. This conclusion is consistent with findings showing that hSMG5 is essential for hUPF1 dephosphorylation (Ohnishi et al., 2003).
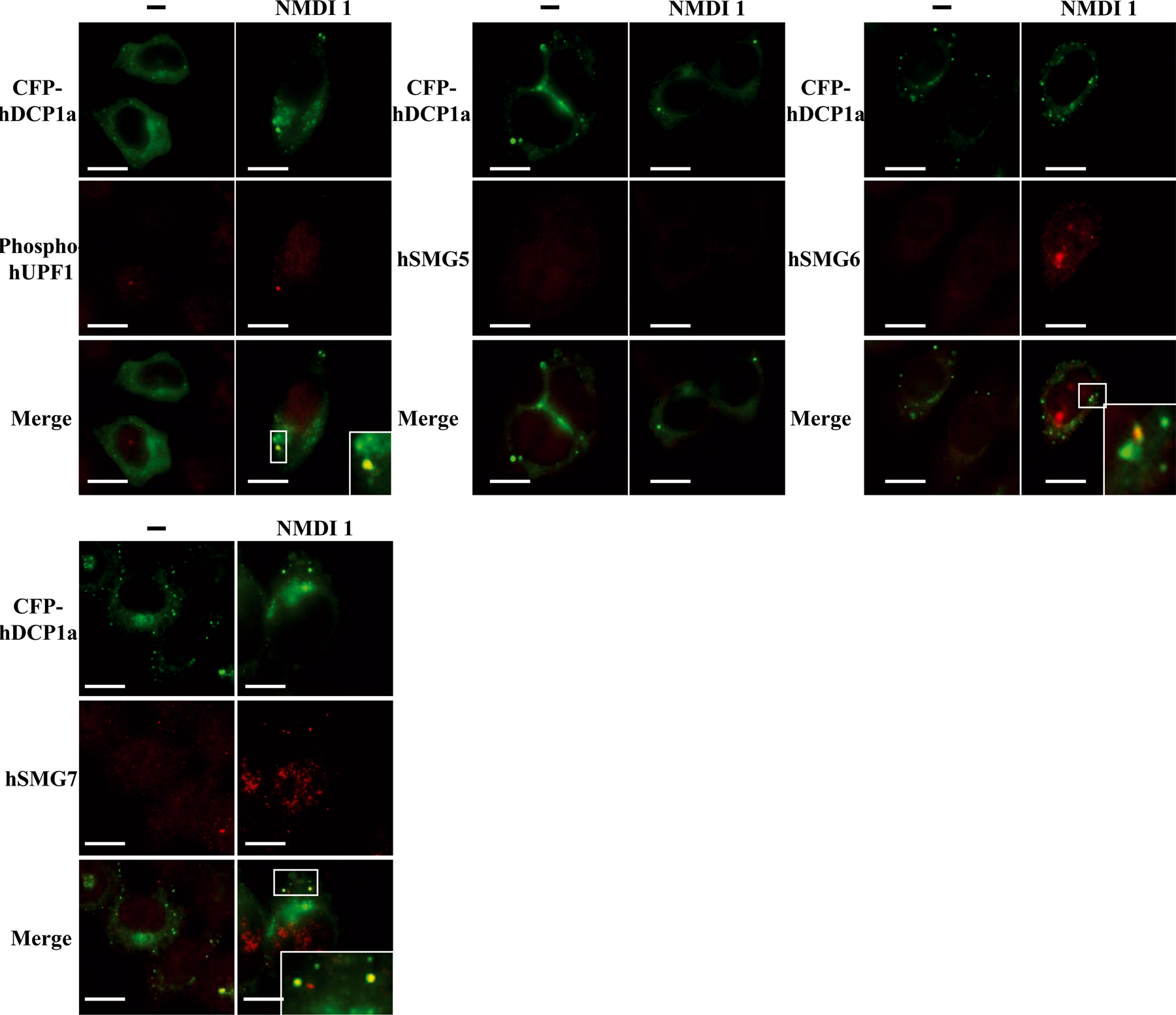
hSMG5 is excluded from P-bodies in the presence of NMDI 1
Because NMDI 1 induces the localization of hUPF1 in P-bodies, hUPF1 hyperphosphorylation, and the destabilization of interactions between hUPF1 and hSMG5, we assessed the cellular localization of the hUPF1 dephosphorylation complex during NMDI 1 treatment. hSMG5 and hSMG7 have been shown to localize mainly in the cytoplasm and particularly in P-bodies as shown by colocalization experiments with the endogenous LSM4 for hSMG7 and with hSMG7 for hSMG5 (Unterholzner and Izaurralde, 2004). hSMG6 similarly localizes mainly in the cytoplasm and also in some cytoplasmic foci that do not contain endogenous LSM4 (Unterholzner and Izaurralde, 2004). We transfected HeLa cells with expression vectors encoding YFP-hSMG5, YFP-hSMG6, or YFP-hSMG7 (Unterholzner and Izaurralde, 2004) and CFP-hDCP1a as a P-body marker. After 24 h, we added DMSO(−) or 5 μM NMDI 1 to the cells. As previously shown, in the absence of the inhibitor, YFP-hSMG5, YFP-hSMG6, and YFP-hSMG7 were concentrated in cytoplasmic foci (Unterholzner and Izaurralde, 2004; Fukuhara et al., 2005), which, for a substantial fraction of them (33, 72, and 100%, respectively), colocalized with CFP-hDCP1a (Fig. 3 D). The fact that we observed hSMG6 in P-bodies unlike what was previously observed (Unterholzner and Izaurralde, 2004) was likely caused by the different markers used for detection of P-bodies and may reflect heterogeneity of P-bodies in their protein composition (see Discussion). In the presence of NMDI 1, the cytoplasmic foci containing YFP-hSMG6 or YFP-hSMG7 perfectly colocalized with CFP-hDCP1a P-bodies. Interestingly, hSMG5 was no longer observed in cytoplasmic foci and became evenly distributed in the cytoplasm in cells treated with NMDI 1 (Fig. 3 D).
Endogenous hSMG5, hSMG6, or hSMG7 cannot be detected in cytoplasmic foci because of their weak expression (Unterholzner and Izaurralde, 2004). Because NMDI 1 inhibits NMD and induces the accumulation of hUPF1 in P-bodies as shown with exogenous as well as endogenous hUPF1 (Figs. 3 C, S2 B, and S3), we tested whether the three hSMG proteins would also accumulate in cytoplasmic foci of treated cells. The results shown in Fig. S3 indicated that these three proteins were not detected in cytoplasmic foci in DMSO-treated cells. However, when cells were incubated with NMDI 1, both hSMG6 and hSMG7 colocalized with CFP-hDCP1a in P-bodies. In agreement with the transfection experiment (Fig. 3 D) under the same conditions, hSMG5 was not detected in cytoplasmic foci. Altogether, our results indicate that NMDI 1 modifies the cellular localization of hSMG5 by excluding it from P-bodies. This is consistent with the failure of hUPF1 antibodies to immunoprecipitate hSMG5 from NMDI 1–treated cells (Fig. 3 A).
hUPF3 and hUPF3X localize in P-bodies when NMD is blocked by NMDI 1
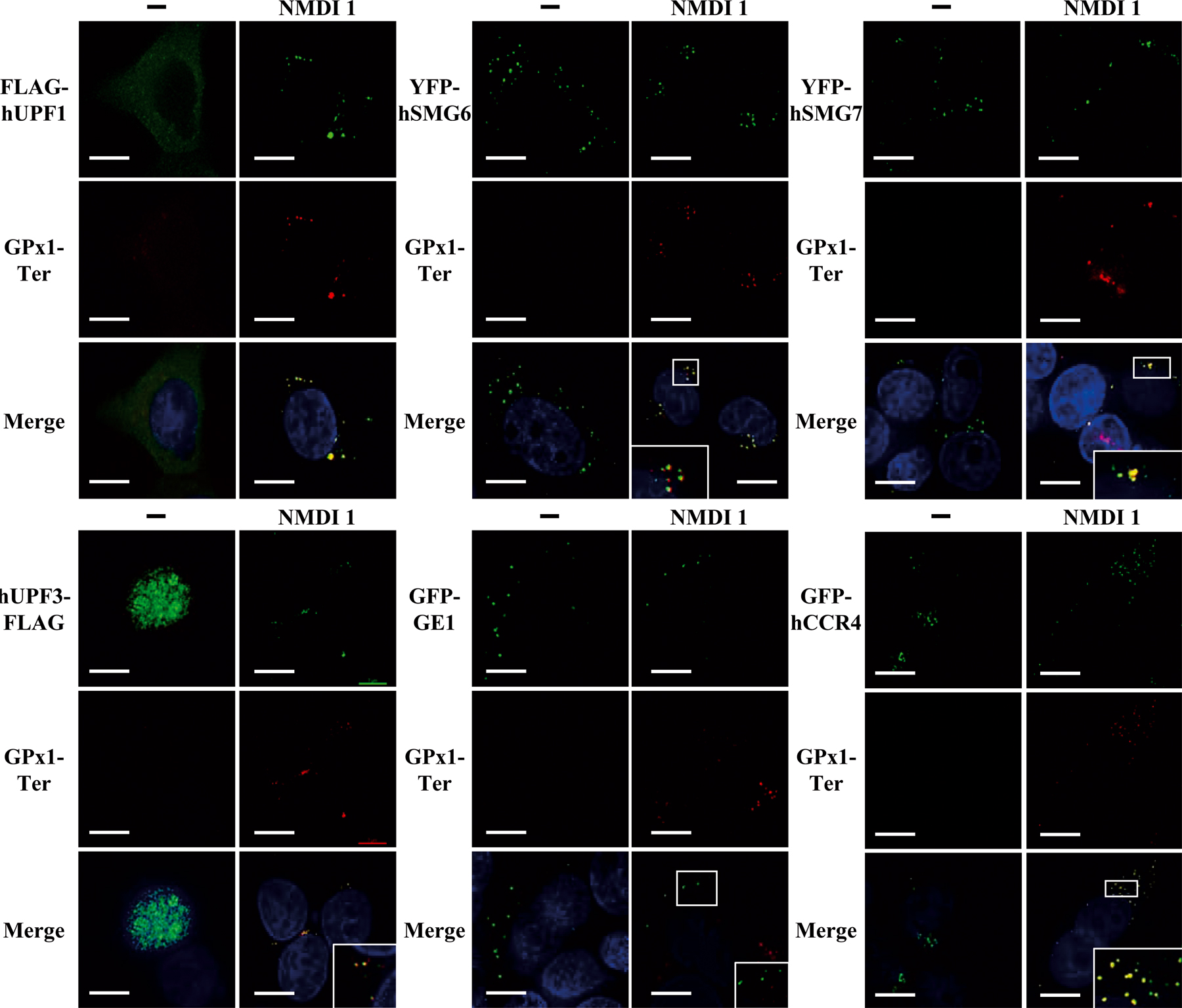
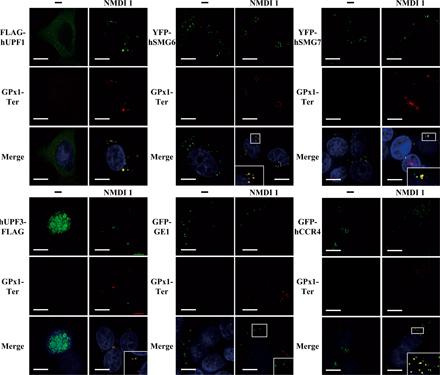
Because some NMD factors such as hUPF1, hSMG5, hSMG6, or hSMG7 localize to P-bodies (Unterholzner and Izaurralde, 2004, this paper) we envisaged that other NMD factors may pass through P-bodies in a transient manner. As NMDI 1 is able to block NMD at a step where hUPF1 is confined to P-bodies, we investigated the cellular localization of hUPF3 and hUPF3X in both treated and untreated cells. These two proteins have been previously shown to be primarily nuclear proteins in untreated cells (Serin et al., 2001). We transfected HeLa cells with expression vectors that code for hUPF3-FLAG or hUPF3X-FLAG together with one of the P-body markers YFP-hSMG6, YFP-hSMG7, GFP-GE1, or CFP-hDCP1a. The cells were treated with DMSO or NMDI 1 before we performed indirect immunofluorescence experiments (Fig. 4 and not depicted for CFP-hDCP1a). As for untreated cells (Lykke-Andersen et al., 2000; Serin et al., 2001), hUPF3 or hUPF3X localized primarily in the nucleus when cells were incubated with DMSO(−) (Fig. 4). However, when cells were grown in the presence of NMDI 1, we observed a cytoplasmic localization of hUPF3 as well as hUPF3X, with some accumulation in foci. To further characterize these foci, we analyzed their colocalization with cotransfected P-body markers (Fig. 4 and not depicted). hUPF3-FLAG as well as hUPF3X-FLAG proteins colocalized with YFP-hSMG6 (68 and 57%, respectively) or YFP-hSMG7 (50 and 51%, respectively) in cells treated with NMDI 1. Surprisingly, unlike with FLAG-hUPF1, GFP-GE1 did not colocalize with hUPF3-FLAG or hUPF3X-FLAG. We conclude that hUPF3/3X proteins can translocate to the cytoplasm, which is consistent with their shuttling properties (Lykke- Andersen et al., 2000; Serin et al., 2001), and can reach a subset of P-bodies.
Figure 4.

hUPF3 localizes to P-bodies when NMD is inhibited. HeLa cells were transfected with a pcDNA3-hUpf3-FLAG or pcDNA3-hUpf3X-FLAG plasmid and either pGFP-Ge1, pYFP-hSmg6, or pYFP-hSmg7. 24 h after transfection, cells were incubated either with DMSO(−) or with 5 μM NMDI 1 for 20 h. In the merge, nuclei are visualized by Hoechst staining in blue. The white squares are magnifications of cell areas. Bars, 10 μm.
PTC-containing mRNAs accumulate in P-bodies in the presence of NMDI 1
Because NMD factors accumulate in P-bodies when NMD is inhibited, we were interested in determining whether NMD substrates also accumulate in P-bodies. In yeast, it has recently been shown that PTC-containing mRNAs accumulate in P-bodies when NMD is blocked (Sheth and Parker, 2006). We speculated that our NMD inhibitor would allow us to reach the same conclusion in mammalian cells. HeLa cells were transfected with pmCMV-Gl Ter or pmCMV-GPx1 Ter and the localization of the resulting mRNAs was analyzed with several P-body markers: GFP-GE1, YFP-hSMG6, YFP-hSMG7, CFP-hDCP1a, GFP-hCCR4, FLAG-hUPF1, hUPF3-FLAG, or hUPF3X-FLAG (Figs. 5 and S4, available at http://www.jcb.org/cgi/content/full/jcb.200611086/DC1). As expected, in the absence of the inhibitor we were unable to detect PTC-containing mRNAs, most likely because of their rapid decay by NMD. However, in the presence of NMDI 1, PTC-containing mRNAs were stabilized and detected mainly in cytoplasmic aggregates. Interestingly, our results indicated a substantial colocalization between PTC-containing Gl or GPx1 mRNA and each tagged version of the tested hUPF proteins CFP-hDCP1a, YFP-hSMG6, or YFP-hSMG7 (Figs. 5 A and S4). Although the colocalization between Gl Ter or GPx1 Ter mRNAs and hUPF3/3X was total (Fig. 5 and not depicted for hUPF3X), it was only partial with other P-body components such as FLAG-hUPF1 (63 and 74%, respectively), YFP-hSMG6 (76 and 81%, respectively), or YFP-hSMG7 (71 and 94%, respectively), and infrequently with GFP-hCCR4 (11 and 33%, respectively; Figs. 5 A and S4). Notably, we often observed that RNA foci and P-bodies did not overlap perfectly. Additionally, we were unable to observe a colocalization between PTC-containing mRNAs and GFP-GE1 (Figs. 5 A and S4). Altogether, our results show that PTC-containing mRNAs were present in and adjacent to P-bodies when NMD was inhibited by NMDI 1. In addition, these data indicate heterogeneity in the composition of P-bodies because some markers colocalize with PTC-containing mRNAs while others do not.
Figure 5.

PTC-containing mRNAs accumulate in P-Bodies under conditions of NMD inhibition. (A) HeLa cells were transfected with pmCMV-Gl Ter and an expression vector encoding a P-body component: pCI-neo-FLAG-hUpf1, pYFP-hSmg6, pYFP-hSmg7, pcDNA3-hUpf3-FLAG, pGFP-Ge1, or pGFP-hCcr4. 24 h after transfection, DMSO(−) or 5 μM NMDI 1 was added to the culture medium for 20 h. (B) U2OS cells were transfected with a pGl-Ter MS2 plasmid and a pCFP-hDcp1a expression vector. These cells were used for their tendency to highly express transfected genes, which is crucial for this approach. Cells were treated as described in A. Nuclei are shown in the merge by Hoechst staining in blue. The white squares are magnifications of cell areas. Bars, 10 μm.
The accumulation of PTC-containing mRNAs in P-bodies when NMD is blocked in mammalian cells was confirmed by a more resolutive approach in U2OS cells. In this setting, we tagged the mRNA with a 24× MS2 binding site repeat because this approach can efficiently detect single mRNA molecules by in situ hybridization (Fusco et al., 2003; Fig. 5 B). In control cells, PTC-containing mRNAs were mostly detected in the nucleus, and the cytoplasmic molecules that were detected did not accumulate in P-bodies labeled with CFP-hDCP1a. When NMD was inhibited with NMDI 1, higher levels of Gl-Ter MS2 mRNAs were detected in the cytoplasm, and these molecules accumulated in structures that colocalized with P-bodies. As previously observed, mRNAs did not perfectly colocalize with CFP-hDCP1a, but were instead adjacent and formed a ring at the periphery of P-bodies, similar to what was found with miRNA targets (Pillai et al., 2005). These data confirmed that mRNAs subjected to NMD accumulate in P-bodies when their degradation is inhibited, and this conclusion seems not be cell type specific.
We also analyzed the localization of Gl or GPx1 Norm mRNAs in HeLa and U2OS cells treated with NMDI 1 (Fig. 6). Our results showed no specific accumulation of these mRNAs in cytoplasmic bodies typified by YFP-hSMG6, GFP-GE1, or CFP-hDCP1a in the presence of NMDI 1. In contrast, we did not see any wild-type mRNAs in P-bodies either because Norm mRNAs do not go to P-bodies or because their degradation pathway is not affected by either DMSO or NMDI 1. These results support the idea that NMDI 1 is an NMD inhibitor rather than a general RNA decay inhibitor.
Figure 6.

Wild-type mRNAs do not accumulate in P-bodies when NMD is inhibited by NMDI 1. (A) HeLa cells were transfected with pmCMV-Gl Norm and an expression vector encoding a P-body component: pYFP-hSmg6 or pGFP-Ge1. Cells were submitted to the same treatment as in Fig. 5 A. (B) Same as in Fig. 5 B except that cells were transfected with a pGl Norm MS2 plasmid rather than pGl-Ter MS2. The white squares are magnifications of cell areas. Bars, 10 μm.
Only some steps of the NMD process occur in P-bodies
To determine whether the accumulation of NMD factors in P-bodies can be triggered when any step of the NMD process is inhibited, we decided to interfere with the NMD process in three different ways. The first one relies on the down-regulation of hUPF2 using siRNA (Kim et al., 2005; Fig. 7 A). According to the current model of NMD in mammalian cells (Maquat, 2004b), depletion of hUPF2 will block NMD at an earlier step than the one induced by NMDI 1. Interestingly, hUPF2 down-regulation does not induce the accumulation of FLAG-hUPF1, hUPF3-FLAG, or hUPF3X-FLAG P-bodies. Additionally, whereas hUPF2 depletion induces a stabilization of Gl-Ter mRNA because of the inhibition of NMD, Gl-Ter mRNA was homogenously distributed in the cytoplasm with no accumulation in P-bodies (Fig. 7 B). Thus, inhibition of NMD by hUPF2 depletion does not trigger accumulation of NMD factors and substrates in P-bodies.
Figure 7.

Down-regulation of hUPF2 or hXRN1 does not lead to the accumulation of NMD factors in P-bodies. (A and C) The efficiency of the hUPF2 (A) or hXRN1 (C) down-regulation was evaluated by Western blot from 10 μg of total protein. The three leftmost lanes represent 2× dilutions of total extract from HeLa cells that were not transfected. (B) HeLa cells were transfected with pCFP-hDcp1a and either pCI-neo-FLAG-hUPF1, pcDNA3-hUPF3-FLAG, pcDNA3-hUPF3X-FLAG, or pmCMV-Gl Ter in the presence of siRNA luciferase (Luc) or siRNA hUPF2 (Upf2). (D) Same as B except siRNA hXRN1 replaced siRNA hUPF2, and pcDNA3-hUPF3X-FLAG was not tested. The white squares are magnifications of cell areas. Bars, 10 μm.
The second one is based on the down-regulation by siRNA of a protein involved in a late step of the NMD process such as hXRN1 (Cougot et al., 2004; Fig. 7 C). As for the down-regulation of hUPF2, the cellular localization of NMD factors including FLAG-hUPF1 or hUPF3-FLAG was not modified by the cellular lack of the hXRN1 protein (Fig. 7 D), whereas Gl-Ter RNA was detected in P-bodies. This result may indicate that the recycling of NMD factors had already occurred before the function of hXRN1. We conclude that the presence of NMD factors in P-bodies depends on the NMD step, i.e., only some steps in the NMD process occur in P-bodies.
According to our results, hUPF1 dephosphorylation is one of the NMD steps that occur in P-bodies. In the presence of NMDI 1, NMD is inhibited because hUPF1 is stalled in a hyperphosphorylated form caused by the release of hSMG5 from P-bodies. To further confirm this model, we aimed to mimic the effect of NMDI 1 by analyzing the cellular localization of NMD factors in the presence of the hUPF1 mutant protein (HA-hUPF1dNT) that has been shown to prevent its interaction with hSMG5 because it lacks an N- terminal, or in the presence of hSMG5 mutant proteins (HA-hSMG5dCT and HA-hSMG5DA) that cannot dephosphorylate hUPF1 because of either the lack of a C-terminal or the substitution of aspartic acid 860 by an alanine (Ohnishi et al., 2003). As shown in Fig. 8 A, when HeLa cells express HA-hUPF1dNT protein, hUPF3-FLAG, hUPF3X-FLAG proteins, Gl Ter, and GPx1 Ter mRNA localize to P-bodies as shown by the colocalization with CFP-hDCP1a. This result is similar to what we observed when cells were treated with NMDI 1. Interestingly, YFP-hSMG5 was not detected in P-bodies, suggesting that by destabilizing the interaction between hSMG5 and hUPF1, hSMG5 is excluded from P-bodies. Additionally, expression of either HA-SMG5dCT or HA-SMG5DA induced accumulation of FLAG-hUPF1, hUPF3-FLAG, hUPF3X-FLAG proteins, Gl Ter, and GPx1 Ter mRNA into P-bodies (Fig. 8, B and C, respectively). Thus, by specifically blocking the dephosphorylation of hUPF1, NMD factors and substrates concentrate into P-bodies.
Figure 8.

NMDI 1 effect on the localization of NMD factors and substrates can be reproduced by using hUPF1 or hSMG5 mutants that prevent the phosphorylation of hUPF1. HeLa cells were transfected with pCFP-hDcp1a and pHA-hUpf1dNT (A), pHA-hSmg5dCT (B) or pHA-hSmg5DA (C), and either pCI-neo-FLAG-hUpf1, pcDNA3-hUpf3-FLAG, pcDNA3-hUpf3X-FLAG, pmCMV-Gl-Ter, pmCMV-GPx1-Ter, pYFP-hSmg5 (A), or pCI-neo-FLAG-hUpf1 (B and C). The white squares are magnifications of cell areas. Bars, 10 μm.
Discussion
In this study, we have characterized an indole derivative, NMDI 1, as an NMD inhibitor. This molecule has allowed us to study specific steps of NMD. The power of this approach lies in the ability to freeze NMD at a step when hUPF1 and hUPF3/3X are detected in P-bodies (Figs. 3 and 4). Using biochemical and cellular biology approaches, we have determined the precise event blocked by NMDI 1 and established that this inhibitor prevents the interactions between hUPF1 and hSMG5, resulting in the subsequent exclusion of hSMG5 from P-bodies and the stabilization of hyperphosphorylated isoforms of hUPF1. Unlike other approaches, such as transfection-mediated down-regulation of NMD factors, where only a fraction of cells are subject to the inhibition of NMD, small chemical molecules have the ability to diffuse across the cell membrane and affect most cells in culture. Therefore, such inhibitors should enable NMD to be inhibited in more physiologically complex environments such as tissue or multicellular organisms to study NMD mechanism in vivo and to evaluate their potential therapeutic capacities (Kuzmiak and Maquat, 2006).
As in yeast (Sheth and Parker, 2006), PTC-containing mRNAs, hUPF1, and hUPF3/3X proteins are found in P-bodies of mammalian cells when NMD is prevented (Figs. 4, 5, and 8). Undoubtedly, yeast and human P-bodies share some similarities in their protein compositions and functions but clear differences can also be seen. Unlike in yeast, in mammalian cells PTC- containing mRNAs accumulate in P-bodies, or more precisely at the periphery of P-bodies, suggesting that P-bodies can be formed by several compartments. This observation is consistent with a recent paper (Pillai et al., 2005) showing that RNA can be localized at the periphery of P-bodies where it might be stored before being degraded or released from the P-bodies. Another difference between yeast and mammalian P-bodies is that a down-regulation of hUPF2 in mammals does not lead to the accumulation of hUPF1 into P-bodies (Fig. 7) as it does in yeast (Sheth and Parker, 2006). Our results suggest that hUPF2 is involved in the NMD process before the transit of NMD factors and substrates through P-bodies. These differences likely reflect a divergence in the process of NMD in yeast and in mammalian cells. Surprisingly, when we blocked NMD at a late step when RNAs are going to be degraded, (i.e., by down-regulating hXRN1) we did not detect NMD factors in P-bodies but we did observe PTC-containing mRNAs in P-bodies (Fig. 7). This confirms that NMD involves trafficking to P-bodies and suggests that by inhibiting the RNA degradation step, we did not prevent the recycling of NMD factors from P-bodies.
Interestingly, we did not observe any differences in the cellular distribution and in the protein composition between P-bodies containing Gl Ter mRNA and those containing GPx1 Ter mRNA, even though these two mRNAs are subject to nucleus-associated or cytoplasmic NMD, respectively. However, we cannot exclude the possibility that NMDI 1 would freeze a series of dynamic events that occurs during NMD and that this would induce a drift of nucleus-associated P-bodies to the cytoplasm. It is also possible that P-bodies with nucleus-associated NMD substrates and P-bodies with cytoplasmic NMD substrates have different biochemical or physical properties that would lead to the cosedimentation of one with the nuclear fraction and of the other with the cytoplasmic fraction. Further investigations will be necessary to clarify this point.
NMDI 1 allowed us to show that P-bodies display a large degree of variability in their NMD factor composition. For example, whereas hSMG6 colocalizes with CFP-hDCP1a or FLAG-hUPF1 when NMD is inhibited (Fig. 3 and not depicted), it only shows a partial overlap with CFP-hDCP1a in DMSO-treated cells (Fig. 3) and no colocalization with LSM4 (Unterholzner and Izaurralde, 2004). As another example, we detected hUPF3/3X-FLAG proteins in P-bodies positive for YFP-hSMG6 or YFP-hSMG7 but not in P-bodies containing GFP-GE1 (Fig. 4). Similarly, PTC-containing mRNA is found in all P-bodies holding hUPF3/3X-FLAG proteins and in most P-bodies containing FLAG-hUPF1, YFP-hSMG6, or YFP-hSMG7, but rarely or never in P-bodies stained with GFP-hCCR4 or GFP-GE1 (Figs. 5 and S4). Because the factors that do not colocalize upon NMDI 1 treatment, such as hUPF3/3X (or PTC- containing mRNAs) and GE1, still form foci that colocalize with other P-body markers (such as hSMG7), these data suggest that P-bodies can exist in several “flavors” or forms that would differ in protein composition (at least in mammalian cells). These variations in P-body composition could reflect different functional states. In this view, hUPF3/3X, hSMG6, hSMG7, hUPF1, and hDCP1a would be involved at early steps of RNA processing in P-bodies or, as has been recently proposed, would nucleate the formation of P-bodies on the PTC-containing mRNA (Franks and Lykke-Andersen, 2007). hCCR4 would then join the structure, followed by GE1 that would induce degradation of the PTC-containing mRNA and recycling of hUPF3/3X. Then other NMD factors are recycled for a new turn of NMD (Fig. 9). This evolution in P-body composition could arise by fusion of different subcategories of P-bodies, by the shuttling of individual components, or by a combination of these processes. An attractive approach to answer these questions would be to characterize NMD inhibitors that target other steps than the dephosphorylation of hUPF1.
Figure 9.

A model of P-body composition dynamics. (1) At the early stage, phosphorylated hUPF1, hUPF3/3X, hSMG5, hSMG6, hSMG7, and PTC-containing mRNAs transit to P-bodies. (2) hCCR4 accumulates later, followed by GE1, which induces the degradation of PTC-containing mRNAs, and (3) the recycling of hUPF3/3X. Finally, other NMD factors will be recycled, in particular hUPF1.
Materials and methods
All results presented in this article are representative from at least three independent experiments.
Chemical molecules library
All the polycyclic indole compounds studied in this paper issue from the Institut Curie–Centre National de la Recherche Scientifique compound library. Each compound was suspended at 20 mg/ml in DMSO and then prepared at a 5-μM working dilution in 10% DMSO (vol/vol). The synthesis and the purification of these compounds has been described previously (Rivalle et al., 1981).
Constructs
Gl Norm and Ter fused to MS2 binding sites constructs (Figs. 5 B and 6 B) were obtained by PCR amplification from Gl wild-type and NS39 constructs (provided by M. Hentze and N. Gehring, European Molecular Biology Laboratory, Heidelberg, Germany; Thermann et al., 1998) using a sense primer (5′-GCAACCTCAAGCTTACACCATGGTGCACCTGAC-3′) and an antisense primer (5′-AGAAAGCAGATCTGCTTAGTGATACTTGTG-3′). The amplified fragment was cloned in HindIII–BglII of a modified pRSVbgal plasmid containing 24× MS2 sites (Fusco et al., 2003).
NMD measurements by RT-PCR
HeLa cells were cultured in 60-mm dishes in DME (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum at 37°C and 5% CO2. 106 cells were cotransfected with 3 μg of test plasmid pmCMV-Gl (Norm or 39Ter; Sun et al., 1998) or 3 μg of test plasmid pmCMV-GPx1 (Norm or 46Ter; Moriarty et al., 1998) and 1 μg of reference plasmid phCMV-MUP (all provided by L. Maquat, University of Rochester, Rochester, NY; Belgrader and Maquat, 1994) using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. 24 h after transfection, cells were treated for 20 h with 5 μM of chemical molecules or 0.01% DMSO (vol/vol) as a control. Total RNA was purified using the TRI reagent (Sigma- Aldrich), and Gl, GPx1, and MUP mRNA were reverse transcribed before amplification by PCR in the presence of 32P-radiolabeled dCTP. The PCR conditions and analysis method have been previously described (Ishigaki et al., 2001). PCR products were quantified with an imaging system (Typhoon 9200; GE Healthcare).
Luciferase activity for translation efficiency assay
HeLa cells were transfected with 2 μg pFluc and 1 μg pRluc. 24 h after transfection, cells were incubated with 0.01% DMSO (vol/vol), 5 μM NMDI 1 for 20 h, or 100 μg/ml CHX for 4 h before being harvested. Luciferase activity was quantified on 2 × 105 cell equivalent on a luminometer (MicroLumat LB 96P; Berthold Technologies) using the Dual-Glo luciferase assay kit (Promega) according to the manufacturer's instructions. The luciferase activity was normalized according to the level of Fluc and Rluc mRNA level measured by RPA.
Luciferase activity for miRNA decay pathway integrity
Cells were grown in six-well plates and transfected with Lipofectamine Plus reagent using 50 ng RLperfect RNA reporter (Pillai et al., 2005), 200 ng pGl3 plasmid coding for Fluc, and 4 μg pTzU6 plasmid (Good et al., 1997). 24 h after transfection, NMDI 1 was added to corresponding wells. 48 h after transfection, luciferase activity was measured with a Dual-Glo luciferase assay kit according to the manufacturer's instructions.
Measure of FLAG-hUPF1 phosphorylation level by 2D gel analysis
106 293T cells were transfected with 1 μg pCI-neo-FLAG-hUpf1 (provided by L. Maquat; Sun et al., 1998) using Lipofectamine Plus reagent according to the manufacturer's instructions. After 12 h, serum was removed from the culture medium for 24 h before adding 5 μM NMDI 1 or 0.01% DMSO (vol/vol) as a control for 3 h at 37°C and 5% CO2. 10% serum was added back for 1 h at 37°C and 5% CO2. Total proteins were purified in a lysis buffer containing 8 M urea, 2% CHAPS, and 40 mM Tris base. The first dimension was performed according to the protocol guide from GE Healthcare for 2D electrophoresis with immobilized pH gradient. 18-cm Immobiline DryStrip, pH 3–10 (GE Healthcare), was used to separate proteins according to their isoelectric point. The second dimension was done by loading the first dimension on a 10% SDS-PAGE. Finally, proteins were transferred to a nitrocellulose membrane before incubation with 1 μg of mouse anti–α-FLAG antibody (Sigma-Aldrich) in TBS containing 0.05% Tween overnight at 4°C followed by incubation with a peroxidase-conjugated goat anti–mouse antibody (Pierce Chemical Co.). Proteins were then detected using SuperSignal West Femto maximum sensitivity substrate (Pierce Chemical Co.).
Tethering assay
This experiment was performed as described previously (Hosoda et al., 2005).
Immunofluorescence, FISH assays, and image analysis
HeLa cells were cultured on 12-mm glass coverslips in 10% DME (vol/vol) FBS. Cells (105) were transiently transfected with 500 ng of plasmids pGFP-Ge1 (provided by D. Bloch, Harvard Medical School, Charleston, MA; Yu et al., 2005), pYFP-hSmg5, pYFP-hSmg6, pYFP-hSmg7 (all provided by E. Izaurralde, Max Planck Institute for Developmental Biology, Tuebingen, Germany; Unterholzner and Izaurralde, 2004), pGFP-hCcr4, pCFP-hDcp1a (both provided by B. Séraphin, Centre National de la Recherche Scientifique, Montpellier, France; Cougot et al., 2004), pCI-neo-FLAG-hUpf1 (Sun et al., 1998), pcDNA3-hUpf3-FLAG, pcDNA3-hUpf3X-FLAG (both provided by L. Maquat; Lykke-Andersen et al., 2000), pmCMV-Gl (Norm or 39Ter; Sun et al., 1998), pmCMV-GPx1 (Norm or 46Ter; Moriarty et al., 1998), pHA-hUpf1dNT, pHA-hSmg5dCT, or pHA-hSmg5DA (all three provided by S. Ohno and A. Yamashita, Yokohama City University School of Medicine, Yokohama, Japan; Ohnishi et al., 2003). 24 h after transfection, cells were treated with 5 μM NMDI 1 or 0.01% DMSO (vol/vol) as a control. 20 h later cells were fixed using formalin solution (Sigma-Aldrich) for 10 min at room temperature and permeabilized in 70% ethanol overnight at 4°C. For immunofluorescence assays, fixed cells were incubated with a mouse anti-FLAG antibody (Sigma-Aldrich) for 2 h at room temperature, washed three times with PBS, and incubated with Cy3- or FITC-conjugated goat anti–mouse antibody (Jackson ImmunoResearch Laboratories) for 1 h at room temperature. Finally, cells were washed three times with PBS and incubated with 2 ng/μl of Hoechst stain (Sigma-Aldrich) for 2 min at room temperature. For FISH experiments, fixed cells were incubated in a prehybridization buffer (125 μg/ml tRNA, 500 μg/ml herring DNA, 1 mg/ml BSA, 0.1 g/ml dextran sulfate, 50% formamide, and 2× sodium saline citrate [SSC] buffer) at 37°C for 1 h in a tissue culture incubator. Fixed cells were incubated overnight in a tissue culture incubator with a hybridization buffer (prehybridization buffer with Cy3-labeled probes), washed three times in 2× SSC at 37°C and three times in 1× SSC at room temperature, and incubated with 2 ng/μl of Hoechst stain (Sigma-Aldrich) for 2 min at room temperature. A Cy3 5′ and 3′ end–labeled probe (5′-CGATCTGCGTTCTACGGTGGT-3′) was used to detect Gl or GPx1 mRNA Ter. For Figs. 5 B and 6 B, the MS2 probe sequence was 5′-AT*GTCGACCTGCAGACAT*GGGTGATCCTCAT*GTTTTCTAGGCAATT*A-3′ (the asterisks indicate an internal Cy3 modification).
Fixed cells were observed in VECTASHIELD mounting medium (Vector Laboratories) with a microscope (DMRA; Leica) with an oil objective (PL APO 63×, NA 1.32; Leica) and A4 (for Hoechst stain), GFP, and Y3 (for Cy3) filter sets (Leica). Images were captured at 0.3-μm intervals along the z axis using a piezzo stepper (E662 LVPTZ amplifier; Servo Products) and a cooled charge-coupled device camera (MicroMAX, 1,300 × 1,030 pixels, RS; Princeton Instruments Inc.) driven by MetaMorph v6.2 (Universal Imaging Corp.). Pixel sizes were 106 × 106 nm and voxel sizes were 106 × 106 × 100 nm. For deconvolution and image reconstruction, xyz image stacks of fixed cells were processed using deconvolution software (Huygens 2.3; Scientific Volume Imaging) using a maximum likelihood estimation algorithm. 3D restored stacks were processed with software (Imaris 4; Bitplane) for volume rendering and quantification.
Down-regulation of hUPF2 or hXRN1
105 HeLa cells were transfected with 100 nM siRNA hUpf2 (Eurogentec; Kim et al., 2005) using a JetPEI reagent (PolyPlus Transfection) for 48 h before being harvested. The down-regulation efficiency was then analyzed by Western blot or by immunofluorescence.
Down-regulation of hXRN1 (provided by J. Lykke-Andersen, University of Colorado, Boulder, CO) has been previously described (Cougot et al., 2004).
Immunopurification and Western blot analysis
hUPF1 immunopurification and Western blot analysis were performed according to the protocol described in Lejeune and Maquat (2004) using rabbit anti–α-hUPF1 antibody (provided by S. Ohno and A. Yamashita; Ohnishi et al., 2003). Western blot analyses were performed using 1:250 rabbit anti–α-hUPF1, α-hSMG5, α-hSMG6, or α-hSMG7 antibodies (provided by S. Ohno and A. Yamashita; Ohnishi et al., 2003); 1:1,000 rabbit α-hUPF3/3X (Ishigaki et al., 2001); or 1:1,000 mouse anti–α-tubulin (Sigma-Aldrich). For Fig. 2, Western blot analysis was done with 1:1,000 rabbit anti–α-hUPF1, 1:1,000 rabbit anti–α-hUPF2, and 1:1,000 rabbit anti–α-hUPF3/3X (Ishigaki et al., 2001). Proteins were detected using SuperSignal West Pico chemiluminescent substrate or SuperSignal West Femto maximum sensitivity substrate (Pierce Chemical Co.).
Online supplemental material
Fig. S1 shows that pre-mRNA splicing and mRNA translation are not affected by NMDI 1. Fig. S2 confirms the results of Fig. 2 by RPA approach and presents the evidence of the accumulation of FLAG-hUPF1 in P-bodies of 293T cells in the presence of NMDI 1 and serum. Fig. S3 shows that endogenous phosphorylated hUpf1, hSmg6, and hSmg7 are detected in P-bodies in the presence of NMDI 1. Fig. S4 demonstrates that GPx1- Ter mRNA is present in P-bodies when NMD is blocked by NMDI 1. Finally, Table S1 shows the structures of compounds that have been used in this study. Online supplemental material is available at http://www.jcb.org/content/full/jcb.200611086/DC1.
Supplementary Material
Acknowledgments
The authors sincerely thank Dr. Lynne Maquat for the pmCMV-Gl (Norm and Ter), pmCMV-GPx1 (Norm and Ter), phCMV-MUP, pCI-neo-FLAG-hUpf1, pcDNA3-hUpf3-FLAG, and pcDNA3-hUpf3X-FLAG plasmids and anti-hUPF1, anti-hUPF2, and anti-hUPF3/3X antibodies; Dr. Elisa Izaurralde for providing the pYFP-hSmg5, pYFP-hSmg6, and pYFP-hSmg7 plasmids; and Dr. Jens Lykke-Andersen for providing plasmids for tethering experiments and the anti-hXRN1 antibody. We want also to thank Dr. Donald Bloch for providing the pGFP-Ge1 plasmid; Dr. Matthias Hentze and Dr. Niels Gehring for the Gl wild-type and NS39 constructs; and Dr. Shigeo Ohno and Dr. Akia Yamashita for providing us with anti-hSMG5, anti-hSMG6, anti-hSMG7, the anti-phosphorylated isoform of hUPF1 and anti-hUPF1 antibodies, and the pHA-hUpf1dNT, pHA-Smg5DA, and pHA-Smg5dCT vectors. We further thank Dr. Bertrand Séraphin for the pCFP-hDcp1a and pGFP-hCcr4 plasmids. Finally, we would like to thank Dr. Johann Soret, Dr. Oliver Mühlemann, and Dr. Naomi Taylor for critical reading of the manuscript and the Montpellier RIO Imaging facility for help in microscopy analysis.
S. Durand was supported by a graduate fellowship from the Ministère Délégué à la Recherche et aux Technologies. F. Lejeune was supported for nine months by the Fondation pour la recherche médicale. This work was supported by a grant from the Association Française contre les Myopathies to F. Lejeune and a grant from Agence Nationale pour la Recherche (ANR 05-BLAN-0261-01) to J. Tazi.
Abbreviations used in this paper: CHX, cycloheximide; DCP, decapping protein; EJC, exon junction complex; Fluc, firefly luciferase; Gl, β globin; GPx1, glutathione peroxidase 1; IP, immunoprecipitation; miRNA, microRNA; MUP, major urinary protein; NMD, nonsense-mediated mRNA decay; NMDI 1, NMD inhibitor 1; P-bodies, processing bodies; PTC, premature termination codon; Rluc, Renilla luciferase; RPA, RNase protection assay; SSC, sodium saline citrate.
References
- Bashkirov, V.I., H. Scherthan, J.A. Solinger, J.M. Buerstedde, and W.D. Heyer. 1997. A mouse cytoplasmic exoribonuclease (mXRN1p) with preference for G4 tetraplex substrates. J. Cell Biol. 136:761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgrader, P., and L.E. Maquat. 1994. Nonsense but not missense mutations can decrease the abundance of nuclear mRNA for the mouse major urinary protein, while both types of mutations can facilitate exon skipping. Mol. Cell. Biol. 14:6326–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brengues, M., D. Teixeira, and R. Parker. 2005. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science. 310:486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.Y., and A.B. Shyu. 2003. Rapid deadenylation triggered by a nonsense codon precedes decay of the RNA body in a mammalian cytoplasmic nonsense-mediated decay pathway. Mol. Cell. Biol. 23:4805–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S.Y., G. Serin, O. Ohara, and L.E. Maquat. 2003. Characterization of human Smg5/7a: a protein with similarities to Caenorhabditis elegans SMG5 and SMG7 that functions in the dephosphorylation of Upf1. RNA. 9:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti, E., and E. Izaurralde. 2005. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 17:316–325. [DOI] [PubMed] [Google Scholar]
- Cougot, N., S. Babajko, and B. Seraphin. 2004. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couttet, P., and T. Grange. 2004. Premature termination codons enhance mRNA decapping in human cells. Nucleic Acids Res. 32:488–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplinski, K., M.J. Ruiz-Echevarria, S.V. Paushkin, X. Han, Y. Weng, H.A. Perlick, H.C. Dietz, M.D. Ter-Avanesyan, and S.W. Peltz. 1998. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 12:1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenger-Gron, M., C. Fillman, B. Norrild, and J. Lykke-Andersen. 2005. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol. Cell. 20:905–915. [DOI] [PubMed] [Google Scholar]
- Franks, T., and J. Lykke-Andersen. 2007. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 21:719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara, N., J. Ebert, L. Unterholzner, D. Lindner, E. Izaurralde, and E. Conti. 2005. SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol. Cell. 17:537–547. [DOI] [PubMed] [Google Scholar]
- Fusco, D., N. Accornero, B. Lavoie, S.M. Shenoy, J.M. Blanchard, R.H. Singer, and E. Bertrand. 2003. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr. Biol. 13:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring, N.H., J.B. Kunz, G. Neu-Yilik, S. Breit, M.H. Viegas, M.W. Hentze, and A.E. Kulozik. 2005. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol. Cell. 20:65–75. [DOI] [PubMed] [Google Scholar]
- Good, P.D., A.J. Krikos, S.X. Li, E. Bertrand, N.S. Lee, L. Giver, A. Ellington, J.A. Zaia, J.J. Rossi, and D.R. Engelke. 1997. Expression of small, therapeutic RNAs in human cell nuclei. Gene Ther. 4:45–54. [DOI] [PubMed] [Google Scholar]
- He, F., X. Li, P. Spatrick, R. Casillo, S. Dong, and A. Jacobson. 2003. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol. Cell. 12:1439–1452. [DOI] [PubMed] [Google Scholar]
- Hosoda, N., Y.K. Kim, F. Lejeune, and L.E. Maquat. 2005. CBP80 promotes interaction of Upf1 with Upf2 during nonsense-mediated mRNA decay in mammalian cells. Nat. Struct. Mol. Biol. 12:893–901. [DOI] [PubMed] [Google Scholar]
- Ingelfinger, D., D.J. Arndt-Jovin, R. Luhrmann, and T. Achsel. 2002. The human LSm1-7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA. 8:1489–1501. [PMC free article] [PubMed] [Google Scholar]
- Ishigaki, Y., X. Li, G. Serin, and L.E. Maquat. 2001. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 106:607–617. [DOI] [PubMed] [Google Scholar]
- Kashima, I., A. Yamashita, N. Izumi, N. Kataoka, R. Morishita, S. Hoshino, M. Ohno, G. Dreyfuss, and S. Ohno. 2006. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 20:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N., G. Stoecklin, M. Ayodele, P. Yacono, J. Lykke-Andersen, M.J. Fitzler, D. Scheuner, R.J. Kaufman, D.E. Golan, and P. Anderson. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169:871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y.K., L. Furic, L. Desgroseillers, and L.E. Maquat. 2005. Mammalian Staufen1 recruits Upf1 to specific mRNA 3′UTRs so as to elicit mRNA decay. Cell. 120:195–208. [DOI] [PubMed] [Google Scholar]
- Kuzmiak, H.A., and L.E. Maquat. 2006. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol. Med. 12:306–316. [DOI] [PubMed] [Google Scholar]
- Lejeune, F., and L.E. Maquat. 2004. Immunopurification and analysis of protein and RNA components of mRNP in mammalian cells. Methods Mol. Biol. 257:115–124. [DOI] [PubMed] [Google Scholar]
- Lejeune, F., and L.E. Maquat. 2005. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr. Opin. Cell Biol. 17:309–315. [DOI] [PubMed] [Google Scholar]
- Lejeune, F., X. Li, and L.E. Maquat. 2003. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol. Cell. 12:675–687. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen, J., M.D. Shu, and J.A. Steitz. 2000. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell. 103:1121–1131. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen, J., M.D. Shu, and J.A. Steitz. 2001. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science. 293:1836–1839. [DOI] [PubMed] [Google Scholar]
- Maquat, L.E. 2004. a. Nonsense-mediated mRNA decay: a comparative analysis of different species. Curr. Genomics. 5:175–190. [Google Scholar]
- Maquat, L.E. 2004. b. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5:89–99. [DOI] [PubMed] [Google Scholar]
- Mendell, J.T., C.M. ap Rhys, and H.C. Dietz. 2002. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science. 298:419–422. [DOI] [PubMed] [Google Scholar]
- Mendell, J.T., N.A. Sharifi, J.L. Meyers, F. Martinez-Murillo, and H.C. Dietz. 2004. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 36:1073–1078. [DOI] [PubMed] [Google Scholar]
- Moriarty, P.M., C.C. Reddy, and L.E. Maquat. 1998. Selenium deficiency reduces the abundance of mRNA for Se-dependent glutathione peroxidase 1 by a UGA-dependent mechanism likely to be nonsense codon-mediated decay of cytoplasmic mRNA. Mol. Cell. Biol. 18:2932–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi, T., A. Yamashita, I. Kashima, T. Schell, K.R. Anders, A. Grimson, T. Hachiya, M.W. Hentze, P. Anderson, and S. Ohno. 2003. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell. 12:1187–1200. [DOI] [PubMed] [Google Scholar]
- Page, M.F., B. Carr, K.R. Anders, A. Grimson, and P. Anderson. 1999. SMG-2 is a phosphorylated protein required for mRNA surveillance in Caenorhabditis elegans and related to Upf1p of yeast. Mol. Cell. Biol. 19:5943–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal, M., Y. Ishigaki, E. Nagy, and L.E. Maquat. 2001. Evidence that phosphorylation of human Upfl protein varies with intracellular location and is mediated by a wortmannin-sensitive and rapamycin-sensitive PI 3-kinase-related kinase signaling pathway. RNA. 7:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai, R.S., S.N. Bhattacharyya, C.G. Artus, T. Zoller, N. Cougot, E. Basyuk, E. Bertrand, and W. Filipowicz. 2005. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 309:1573–1576. [DOI] [PubMed] [Google Scholar]
- Rehwinkel, J., I. Letunic, J. Raes, P. Bork, and E. Izaurralde. 2005. Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA. 11:1530–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivalle, C., C. Ducrocq, J.M. Lhoste, F. Wendling, E. Bisagni, and J.C. Chermann. 1981. 11H-Pyrido [3′,2′:4,5]pyrrolo[2,3-g]isoquinoléines (aza-7 ellipticines) substituées sur leur position 6. Tetrahedron. 37:2097–2103. [Google Scholar]
- Serin, G., A. Gersappe, J.D. Black, R. Aronoff, and L.E. Maquat. 2001. Identification and characterization of human orthologues to Saccharomyces cerevisiae Upf2 protein and Upf3 protein (Caenorhabditis elegans SMG-4). Mol. Cell. Biol. 21:209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth, U., and R. Parker. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 300:805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth, U., and R. Parker. 2006. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell. 125:1095–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soret, J., N. Bakkour, S. Maire, S. Durand, L. Zekri, M. Gabut, W. Fic, G. Divita, C. Rivalle, D. Dauzonne, et al. 2005. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc. Natl. Acad. Sci. USA. 102:8764–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X., H.A. Perlick, H.C. Dietz, and L.E. Maquat. 1998. A mutated human homologue to yeast Upf1 protein has a dominant-negative effect on the decay of nonsense-containing mRNAs in mammalian cells. Proc. Natl. Acad. Sci. USA. 95:10009–10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau, A., R. Gattoni, Y. Dooghe, J. Stevenin, and J. Soret. 2001. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 20:1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira, D., U. Sheth, M.A. Valencia-Sanchez, M. Brengues, and R. Parker. 2005. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA. 11:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann, R., G. Neu-Yilik, A. Deters, U. Frede, K. Wehr, C. Hagemeier, M.W. Hentze, and A.E. Kulozik. 1998. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 17:3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourriere, H., K. Chebli, L. Zekri, B. Courselaud, J.M. Blanchard, E. Bertrand, and J. Tazi. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Unterholzner, L., and E. Izaurralde. 2004. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell. 16:587–596. [DOI] [PubMed] [Google Scholar]
- van Dijk, E., N. Cougot, S. Meyer, S. Babajko, E. Wahle, and B. Seraphin. 2002. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 21:6915–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollerton, M.C., C. Gooding, E.J. Wagner, M.A. Garcia-Blanco, and C.W. Smith. 2004. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol. Cell. 13:91–100. [DOI] [PubMed] [Google Scholar]
- Yamashita, A., T. Ohnishi, I. Kashima, Y. Taya, and S. Ohno. 2001. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 15:2215–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J.H., W.H. Yang, T. Gulick, K.D. Bloch, and D.B. Bloch. 2005. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA. 11:1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J., X. Sun, Y. Qian, and L.E. Maquat. 1998. Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA. 4:801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}