

Abstract
During trans-endothelial migration (TEM), leukocytes use adhesion receptors such as intercellular adhesion molecule-1 (ICAM1) to adhere to the endothelium. In response to this interaction, the endothelium throws up dynamic membrane protrusions, forming a cup that partially surrounds the adherent leukocyte. Little is known about the signaling pathways that regulate cup formation. In this study, we show that RhoG is activated downstream from ICAM1 engagement. This activation requires the intracellular domain of ICAM1. ICAM1 colocalizes with RhoG and binds to the RhoG-specific SH3-containing guanine-nucleotide exchange factor (SGEF). The SH3 domain of SGEF mediates this interaction. Depletion of endothelial RhoG by small interfering RNA does not affect leukocyte adhesion but decreases cup formation and inhibits leukocyte TEM. Silencing SGEF also results in a substantial reduction in RhoG activity, cup formation, and TEM. Together, these results identify a new signaling pathway involving RhoG and its exchange factor SGEF downstream from ICAM1 that is critical for leukocyte TEM.
Introduction
Leukocyte trans-endothelial migration (TEM) is a key event in host defense. The passage of leukocytes across the vascular wall into the underlying tissues can be divided into distinct phases, including firm adhesion of leukocytes to the endothelium and subsequent diapedesis (Vestweber, 2002; Johnson-Leger and Imhof, 2003; van Buul and Hordijk, 2004; for review see Muller, 2003). Leukocyte adhesion to the endothelium initiates the formation of dynamic dorsal membrane protrusions, assembling a cuplike structure, which surrounds adherent leukocytes and contains the cell adhesion molecules intercellular adhesion molecule-1 (ICAM1) and VCAM1 (Barreiro et al., 2002; Carman et al., 2003; Carman and Springer, 2004). They have been referred to as docking structures (Barreiro et al., 2002) or trans-migratory cups (Carman and Springer, 2004). Little is known about the mechanisms that regulate their assembly, and their role in TEM remains uncertain.
During TEM, leukocytes adhere to ICAM1 on the endothelial cell surface, and this triggers diverse intracellular signals (Vestweber, 2002; Kluger, 2004). Engagement of ICAM1 can be mimicked by cross-linking ICAM1 with ICAM1-specific antibodies (Wojciak-Stothard et al., 1999; Etienne-Manneville et al., 2000; Thompson et al., 2002) or by beads coated with antibodies against ICAM1 (Tilghman and Hoover, 2002). Actin dynamics in endothelial cells are important for leukocyte TEM, which is prevented by inhibiting endothelial actin polymerization by cytochalasin D (Adamson et al. 1999; Carman and Springer, 2004). Cross-linking of ICAM1 stimulates the assembly of actin stress fibers (Wojciak-Stothard et al., 1999; Van Buul et al., 2002). In addition, actin polymerization is involved in assembly of the cups (Carman and Springer, 2004).
Actin membrane dynamics are controlled by small Rho-like GTPases. These proteins function as molecular switches and cycle between an inactive GDP-bound state and an active GTP-bound state. Blocking RhoA activity using Clostridium botulinum C3 transferase prevents the adhesion or migration of leukocytes across endothelial cell monolayers (Adamson et al., 1999; Wojciak-Stothard et al., 1999). However, the role of RhoA in the assembly of the cups is unclear. Barreiro et al. (2002) reported that assembly of these structures induced by VCAM1 is inhibited by Y27632, an inhibitor of Rho-associated coil-containing protein kinase (ROCK)/Rho kinase, which is a downstream effector of RhoA. In contrast, Carman and Springer (2004) found that treatment with Y27632 or C3 was unable to prevent cup formation downstream from ICAM1 engagement. The similarity of these apical cups to phagocytic cups (Barreiro et al., 2002; Carman et al., 2003) together with the role of RhoG in the phagocytosis of apoptotic cells (deBakker et al., 2004) has led us to examine whether RhoG may contribute to the formation of endothelial cups and participate in TEM.
In this study, we demonstrate that RhoG is a critical mediator of leukocyte TEM. RhoG and a guanine-nucleotide exchange factor (GEF) for RhoG, SH3-containing GEF (SGEF), are recruited to sites of ICAM1 engagement, where RhoG becomes activated. We find that ICAM1 interacts with SGEF through its SH3 domain. Finally, reduction of RhoG or SGEF expression in endothelial cells using siRNA decreases the assembly of the cups as well as the migration of leukocytes across endothelial cell monolayers.
Results
Endothelial cells form apical cups around leukocytes
Adhesion of myeloid leukemia HL60 cells to TNF-α–activated endothelial cells induced not only the recruitment of ICAM1 to sites of adhesion (Fig. 1 A) but also ICAM1-positive membrane protrusions that surrounded the adhered leukocyte (Fig. 1 B), which is consistent with previously reported findings (Barreiro et al., 2002; Carman et al., 2003). Also, GFP-actin, which is transiently expressed in endothelial cells, distributed to sites of leukocyte binding and colocalized with ICAM1 (Fig. 1 C). Of note, the endothelial cell–cell junctional marker vascular endothelial (VE) cadherin did not localize to these membrane protrusions (Fig. 1 A). Three-dimensional projections showed that ICAM1-positive protrusions arose from the apical plane of the endothelial cells but did not fully cover the leukocyte (Fig. 1 B). These protrusions resembled cuplike structures that extended ∼6–7 μm above the baso-lateral membrane (Fig. 1 B, d). To determine whether these ICAM1-rich cups formed around cells that were transmigrating, HL60 cells were plated on endothelial monolayers growing on transwell filters. Confocal analysis of fixed and stained preparations revealed rings of ICAM1 staining at the apical surface (i.e., cups) surrounding cells that were traversing the monolayer (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200612053/DC1). Scanning EM confirmed the presence of endothelial cuplike protrusions surrounding but not fully covering leukocytes 30 min after leukocyte adhesion (Fig. 1 D).
Figure 1.

Endothelial cells protrude ICAM1-expressing membrane ruffles around an adhered HL60 cell. (A) TNF-α–treated endothelial cells were incubated with HL60 cells for 30 min, processed, and stained for ICAM1 in green and for VE-cadherin in red. Confocal imaging shows that ICAM1 is recruited to sites of leukocyte adhesion at the baso-lateral focal plane (a) and as a ring structure surrounding the leukocyte at the apical focal plane (b). (B) Z-stack imaging shows ICAM1 staining in green (a) surrounding a leukocyte, which is stained for F-actin in red (asterisks; b). Image c shows the merge. Reconstruction of the Z-stack imaging shows ICAM1 surrounding a leukocyte in a cup-like structure (green, d). Vertical bar at the right shows the height of the protrusions (6 μm). (C) TNF-α–treated endothelial cells transiently transfected with GFP-actin were incubated with HL60 cells for 30 min, processed, and imaged for GFP-actin in green (a and e), ICAM1 in red (b and f), merge of GFP-actin and ICAM1 in yellow (c and g), and F-actin using phalloidin in white to visualize the adhered leukocyte (d and h). Confocal imaging revealed that actin and ICAM1 are recruited to sites of leukocyte adhesion, surrounding the leukocyte at the apical plane (e–h). (D) Scanning EM images show protruding endothelial membrane sheets (arrowheads) around adhered HL60 cells (asterisks). Bars (A), 10 μm; (B and C) 5 μm; (D) 1 μm.
RhoG and SGEF are enriched in dorsal membrane ruffles
The small GTPase RhoG and its specific GEF, SGEF, are known to induce dorsal ruffles (Ellerbroek et al., 2004). RhoG and SGEF are endogenously expressed in endothelial cells as well as in COS7 and HeLa cells (Fig. 2 A). Overexpression of the constitutively active mutant RhoG-Q61L or SGEF in endothelial cells induced ruffles on the apical surface (Fig. 2 B).
Figure 2.

RhoG and SGEF are expressed endogenously in endothelial cells and are localized to dorsal endothelial membrane protrusions. (A) Western blot analysis of tissue lysates of mouse brain (a positive control for SGEF), HUVECs, HeLa, and COS7 cells show the endogenous expression of SGEF (100 kD; top blot) and RhoG (18 kD; bottom blot). (B) Endothelial cells were transiently transfected with GFP–RhoG-Q61L (a) or GFP-SGEF (b) and stained for F-actin in red. Images represent the merge. Arrowheads show membrane ruffles.
To study the involvement of ICAM1 in the regulation of dorsal ruffles, COS7 cells that lack endogenous ICAM1 were used. The expression of ICAM1 tagged with GFP or the V5 epitope in COS7 cells showed distributions similar to ICAM1 in endothelial cells (Fig. 3 A). Interestingly, cotransfection of RhoG-Q61L or SGEF not only induced dorsal ruffles but also induced a redistribution of ICAM1 to these ruffles (Fig. 3, A and B; and Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200612053/DC1). ICAM1 colocalized with RhoG-Q61L or SGEF (Fig. 3, A and B). The localization of ICAM1 to ruffles required active RhoG because neither wild-type (wt) RhoG nor a dominant-negative mutant, T17N, colocalized with ICAM1 (unpublished data). As a control, transmembrane protein PECAM-1 was expressed together with RhoG-Q61L or SGEF and showed no colocalization (unpublished data). These data suggested a role for RhoG and SGEF in the formation of endothelial apical cup structures; therefore, we next tested the involvement of RhoG and SGEF in ICAM1 signaling and cup formation.
Figure 3.

SGEF and RhoG-Q61L colocalized with ICAM1. (A) COS7 cells were transiently cotransfected with ICAM1-GFP (a and g) or ICAM1-V5 (d) and with GFP–RhoG-Q61L (e) or myc-SGEF (h). Image b shows F-actin. Images c, f, and i represent the merge. ICAM1 colocalizes with RhoG-Q61L and SGEF. Moreover, RhoG-Q61L and SGEF induce a change in ICAM1 distribution from spikes (a) to ruffles (arrowheads; d and g). (B) COS7 cells were transiently cotransfected with ICAM1-GFP (a and d), myc-SGEF (b), or myc-RhoG-Q61L (e). Panels c and f show merged images. Confocal x-z section images show colocalization between ICAM1 and SGEF (a–c) and ICAM1 and RhoG-Q61L (d–f) in dorsal membrane ruffles (arrowheads). Bars (A), 20 μm; (B) 10 μm.
Recruitment of ICAM1-GFP to sites of adhesion
COS7 cells lacking endogenous ICAM1 were used to express ICAM1-GFP. Incubation of these COS7 cells with HL60 cells resulted in the majority of HL60 cells adhering to the ICAM1-GFP–transfected cells (Fig. 4 A). Three-dimensional projections showed that ICAM1-positive protrusions surrounded the adhered HL60 cells (Fig. 4 A, d), similar to those observed with endothelial cells (Fig. 1 B). To specifically study ICAM1 engagement and downstream signaling that would mimic leukocyte binding to ICAM1, beads coated with antibodies against ICAM1 were used as described in Materials and methods (see Bead adhesion assay section; Tilghman and Hoover, 2002). These beads, which are hereafter referred to as αICAM1 beads, specifically adhered to ICAM1 and recruited ICAM1-GFP within 30 min (Fig. 4 B and Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200612053/DC1). X-Z projections showed that ICAM1-GFP protruded around adhered αICAM1 beads (Fig. 4 B, d). Additionally, scanning EM images revealed that adhesion of αICAM1 beads induced dorsal ruffles comparable with those induced by leukocytes (Fig. 4 C). The αICAM1 beads did not bind to VCAM1-GFP–transfected cells or to nontransfected cells (unpublished data). In addition, blocking antibodies to ICAM1 completely inhibited binding of the αICAM1 beads to ICAM1 (unpublished data).
Figure 4.

HL60 cells and beads coated with ICAM1 antibodies recruited ICAM1-GFP. (A) COS7 cells were transiently transfected with ICAM1-GFP (a). HL60 cells were allowed to adhere for 30 min. Samples were fixed, permeabilized, and stained for F-actin (b). Image c shows the merge of images a and b. Note that the majority of the HL60 cells adhere to the ICAM1-GFP–expressing cells. Image d shows an x-z projection of ICAM1-GFP in green (arrowheads) that surrounds adhered HL60 cells (asterisks) stained with F-actin in red. (B) αICAM1 beads added for 30 min to ICAM1-GFP– expressing COS7 cells. Beads were stained with secondary AlexaFluor594 anti–mouse antibodies in red (b). Image c shows merge of images a and b, with additional F-actin distribution in white. Note that the majority of the αICAM1 beads adhere to the ICAM1-GFP–expressing cells. Image d shows x-z projection of ICAM1-GFP in green (arrowheads) that surrounds adhered αICAM1 beads (asterisks). (C) Scanning EM image shows endothelial membrane sheets (arrowheads) that surround an αICAM1 bead (asterisk). Bars, (A and B, panel c), 20 μm; (A and B, panel d) 5 μm; (C) 1 μm.
RhoG and SGEF are recruited to sites of ICAM1 engagement
To show that ICAM1-GFP was recruited specifically to the beads, cotransfections with ICAM1-V5 and GFP as a control were performed and revealed that GFP alone was not recruited to sites of adhesion (Fig. 5 A). Also, neither β-catenin–GFP nor VE-cadherin–GFP was recruited to sites of adhesion (Fig. 5 B). In contrast, GFP-SGEF and GFP–RhoG-Q61L were recruited to sites of ICAM1 engagement (Fig. 5, A and B). Additionally, as a control, beads coated with major histocompatibility complex (MHC) antibodies were incubated on human umbilical vein endothelial cells (HUVECs), and z-stack analysis was performed to measure actin-rich protrusions around adhered beads. The results revealed that αICAM1 beads induced substantially more F-actin–rich protrusions than the αMHC class I beads, whereas the total number of beads that adhered to the endothelium was equivalent (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200612053/DC1). Expression of GFP–RhoG-Q61L in HUVECs showed that RhoG is recruited by αICAM1 beads but not by αMHC class I beads (Fig. S2 B). Previous work has indicated that actin is a major component of the ICAM1- positive cup structures (Barreiro et al., 2002; Carman et al., 2003; Carman and Springer, 2004). Using GFP-actin, which is transiently expressed in endothelial cells, we confirmed that αICAM1 beads efficiently recruited actin to sites of adhesion (Fig. S2 C). These data indicate that ICAM1 specifically induces these protrusions and recruits RhoG to sites of adhesion.
Figure 5.

Beads coated with ICAM1 antibodies recruited SGEF and active RhoG to sites of adhesion. (A) As a control, cells expressing GFP alone and ICAM1-V5 were incubated with αICAM1 beads. ICAM1 mAbs are used to visualize ICAM1-V5. αICAM1 beads are stained with secondary AlexaFluor antibodies that also stained ICAM1 in red (a). Images show that GFP alone (green; b) is not recruited around αICAM1 beads that adhered to ICAM1-V5 (arrowheads; a–c). Image c shows the merge. Image d shows a magnification of image c. GFP-SGEF (f) and GFP–RhoG-Q61L (j) are recruited (arrowheads) to αICAM1 bead adhesion sites on ICAM1-V5–expressing COS7 cells (e and i). Merged images are shown in g and k. Images h and l show magnifications of g and k, respectively. (B) Quantification of GFP-expressing proteins that are recruited to adhesion sites induced by αICAM1 beads. All cells were transfected with ICAM1-V5 and subsequently cotransfected with GFP-tagged proteins except for single-transfected ICAM1-GFP. αICAM1 beads recruit ICAM1-GFP, GFP-SGEF, and GFP–RhoG-Q61L in 55–80% of the cases to sites of adhesion. In contrast, GFP, β-catenin–GFP, and VE-cadherin–GFP are not recruited. Data are means ± SEM (error bars) of at least four independent experiments. Bars (A, panel c), 10 μm; (A, panels d, h, and l) 5 μm; (A, g and k) 20 μm.
ICAM1 engagement activates RhoG
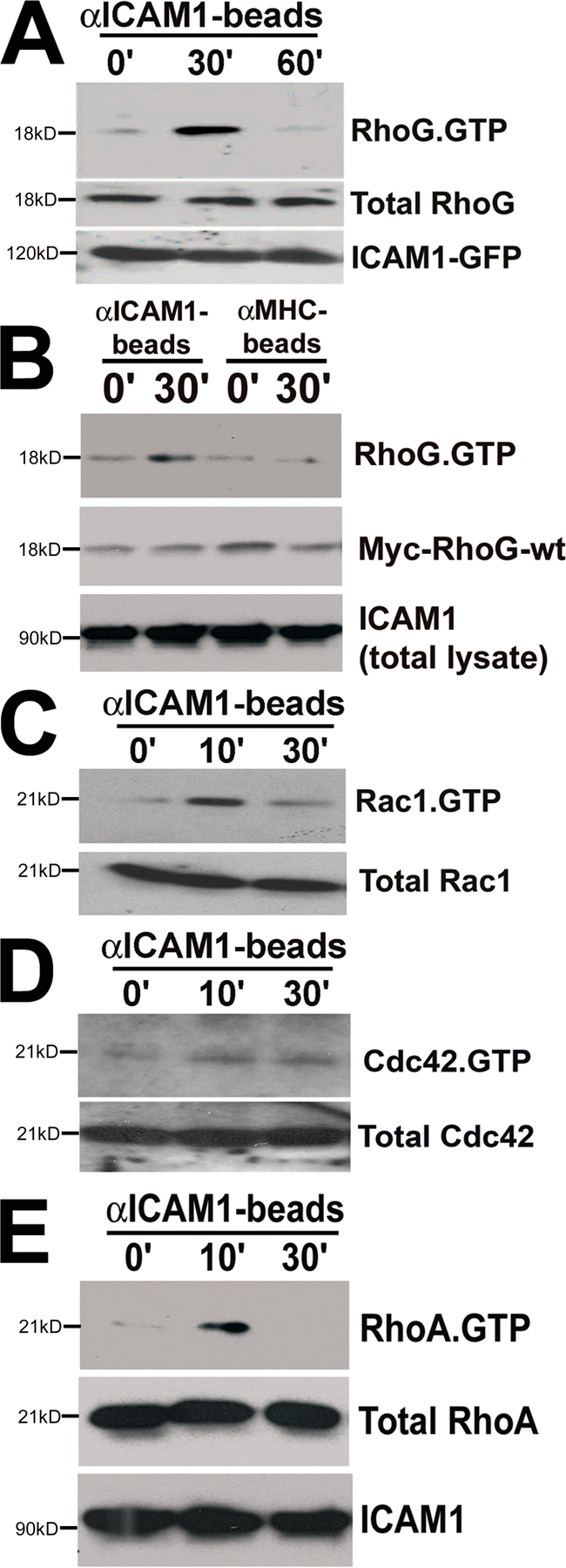
We next performed RhoG activation assays to determine RhoG activity downstream from ICAM1 engagement. We made use of the RhoG downstream effector ELMO (engulfment and cell motility), which specifically binds GTP-bound RhoG (Katoh and Negishi, 2003; Ellerbroek et al., 2004). In our initial experiments, we used an adenoviral vector to deliver myc-tagged RhoG to HUVECs and found that engagement of ICAM1 with αICAM1 beads induced RhoG activation (Fig. 6 A). Examining the activation of endogenous RhoG using a monoclonal antibody revealed that ICAM1 engagement showed a similar response (Fig. 6 B). It should be noted that TNF-α pretreatment did not change the activity of RhoG in endothelial cells, although overnight treatment slightly diminished RhoG expression (unpublished data). To delineate the pathway downstream from ICAM1, myc-tagged RhoG-wt together with ICAM1-GFP were expressed in COS7 cells as described in Materials and methods (see RhoG, RhoA, and Rac1 activation assay section). Treatment with αICAM1 beads induced RhoG activation after 10 and 30 min (Fig. 6 C). This activation was transient because the induced activity of RhoG declined after 60 min (Fig. 6 B and Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200612053/DC1). Beads coated with MHC class I antibodies did not induce any RhoG activation (Fig. S3 B). To examine whether leukocytes could activate RhoG through ICAM1, we added HL60 cells to myc–RhoG-wt and ICAM1-GFP–expressing COS7 cells. RhoG activation was stimulated by the adhesion of HL60 cells (Fig. 6 D). To study whether closely related GTPases Rac1 and Cdc42 are activated downstream from ICAM1 engagement, pull-down assays using the p21-activated kinase–binding domain (PBD) as bait were performed. Interestingly, Rac1 and Cdc42 were transiently activated downstream from ICAM1 engagement as well, although Rac1 activation peaked at 10 min (Fig. S3 C). RhoA activity measurements confirmed that RhoA became activated after ICAM1 engagement (Adamson et al. 1999; Wojciak-Stothard et al., 1999), and this was maximal after 10 min (Fig. S3 E).
Figure 6.

RhoG is activated downstream from ICAM1. (A) HUVECs were transiently transduced with myc-RhoG-wt using adenovirus. αICAM1 beads were added as described in Materials and methods. Using GST-ELMO, activated RhoG was pulled down from the lysate and detected by Western blot analysis using anti-myc antibodies. The middle blot shows protein expression levels of myc-RhoG-wt in endothelial cell lysates. The graph on the right shows quantification of two independent experiments. (B) αICAM1 beads were added to TNF-α–stimulated endothelial cells as described in Materials and methods. Using GST-ELMO, activated endogenous RhoG was isolated and detected by Western blot analysis using anti-RhoG mAbs. (A and B) αICAM1 beads increased RhoG activity in endothelial cells within 30 min (top). The bottom panel shows endogenous ICAM1 expression in endothelial cell lysates. (B) The middle panel shows expression levels of endogenous RhoG in endothelial cell lysates. (C) COS7 cells were transiently transfected with myc-RhoG-wt and ICAM1-GFP. αICAM1 beads were added as described in Materials and methods. Using GST-ELMO, activated RhoG was isolated and detected by Western blot analysis using anti-myc antibodies. αICAM1 beads increased RhoG activity up to 30 min (top). The middle panel shows expression levels of myc-RhoG-wt in cell lysates. The bottom panel shows ICAM1-GFP expression detected by anti-GFP antibodies in cell lysates. (D) Experiments were performed as in B, but with HL60 cells (5 × 105 cells per six wells). Single-transfected myc-RhoG-Q61L-COS7 cells were used as a positive control. The top panel shows that HL60 cell adhesion increased RhoG-GTP levels. The middle panel shows levels of transfected myc-RhoG-wt, and the bottom panel shows levels of ICAM1-GFP. (B–D) The graph on the right shows quantification of three independent experiments. Data are means ± SEM (error bars). *, P < 0.05; **, P < 0.01.
ICAM1–intracellular domain is required for RhoG activation
Previously, it has been shown that the intracellular domain of ICAM1 is required for leukocyte passage across the endothelium but is dispensable for the initial adhesion (Lyck et al., 2003; Sans et al., 2001). To investigate whether the intracellular domain of ICAM1 is required to transmit the signal that triggers RhoG activation, a C-terminal deletion mutant of ICAM1 lacking the intracellular domain and tagged to a V5 epitope (ICAM1-ΔC-V5) was generated and expressed in COS7 cells. The overexpression of ICAM1-ΔC-V5 together with GFP–RhoG-Q61L showed that ICAM1 required its intracellular domain to localize to RhoG-induced dorsal ruffles (Fig. 7 A). No difference in the adhesion of αICAM1 beads to either full-length or ICAM1-ΔC was observed (unpublished data). However, the αICAM1 beads were unable to activate RhoG in cells expressing ICAM1-ΔC (Fig. 7 B). Additionally, cells that expressed ICAM1-ΔC induced substantially less ICAM1-positive protrusions around adhered leukocytes than ICAM1-wt (Fig. 7 C). Together, these data show that ICAM1 engagement induces RhoG activation and subsequent membrane protrusions in a pathway that is dependent on its intracellular domain.
Figure 7.

ICAM1 intracellular domain was required for RhoG localization and activation. (A) COS7 cells were transiently cotransfected with full-length ICAM1 tagged with V5 (b) or a C-terminal deletion mutant of ICAM1-V5 (ICAM1-ΔC-V5; e) and with GFP–RhoG-Q61L (a and d). Panels c and f show the corresponding merged images. ICAM1-ΔC-V5 does not colocalize with RhoG-Q61L. (B) Experiments were performed as described in Fig. 6 C except ICAM1-GFP has been replaced with V5-tagged ICAM1-wt (ICAM1-wt) or the V5-tagged C-terminal truncated ICAM1 mutant (ICAM1-ΔC). The top panel shows that ICAM1 engagement increases RhoG-GTP levels in cells expressing ICAM1-wt but not in cells expressing ICAM1-ΔC. The bottom panel shows levels of myc-RhoG-wt in total cell lysates. The graph on the right shows quantification of three independent experiments. *, P < 0.05. (C) ICAM1 intracellular tail was required for efficient recruitment around an adhered leukocyte. Confocal imaging was used to visualize the apical and the baso- lateral plane of the COS7 cells that were expressing either ICAM1-wt or ICAM1-ΔC. Quantification of ICAM1-rich rings around HL60 cells that were allowed to adhere for 30 min showed a requirement of the ICAM1 tail for proper cup formation. The experiment was repeated three times. *, P < 0.01. (B and C) Data are means ± SEM (error bars). Bars, 20 μm.
ICAM1 associates with SGEF through its SH3 domain
The finding that RhoG is activated downstream from ICAM1 engagement coupled with the observation that SGEF and RhoG colocalized with ICAM1 led us to investigate whether ICAM1 and SGEF physically interact. Immunoprecipitation experiments showed that endogenous ICAM1 was precipitated with endogenous SGEF from TNF-α–treated endothelial cells (Fig. 8 A). To study this interaction in more detail, pull-down experiments were performed using biotinylated peptides. A peptide corresponding to the cytoplasmic domain of ICAM1 bound myc-tagged SGEF as well as endogenous SGEF (Fig. 8, B and C, respectively). Interestingly, the intracellular domain of ICAM1 comprises only 28 amino acids, and its C terminus contains four prolines in close proximity. We examined whether the SH3 domain of SGEF could directly associate with the cytoplasmic domain of ICAM1. Biotinylated ICAM1–intracellular domain peptide sedimented the SH3 domain of SGEF, which was fused to GST (GST-SH3SGEF) in vitro (Fig. 8 D, a). To further explore the interaction of SGEF with ICAM1, we used a myc-tagged mutant of SGEF lacking the SH3 domain (SGEF-ΔSH3). This mutant SGEF failed to coimmunoprecipitate with ICAM1-GFP (Fig. 8 D, b). Interestingly, the association between SGEF and ICAM1 did not depend on the GEF activity of SGEF; ICAM1 still associated with a catalytically dead mutant of SGEF (SGEF-ΔDH) that contained the SH3 domain (Fig. 8 D, b). An inactivating point mutant in the SH3 domain of SGEF (myc– SGEF-W826R) was previously generated in which the catalytic activity of SGEF remained intact (Ellerbroek et al., 2004). This construct and SGEF-wt were overexpressed in COS7 cells together with ICAM1-GFP. Immunoprecipitation assays confirmed that SGEF-wt interacted with ICAM1, but SGEF-W826R revealed decreased binding (Fig. 8 E). These data indicated that the ICAM1–SGEF interaction requires an intact SGEF-SH3 domain. To test whether ICAM1 associates through its proline-rich sequence to SGEF, we deleted this proline-rich sequence from the cytoplasmic domain of ICAM1. Immunoprecipitation studies revealed that ICAM1 lacking the proline-rich sequence failed to bind to SGEF (Fig. 8 F).
Figure 8.

SGEF interacts through its SH3 domain with the C-terminal domain of ICAM1. (A) ICAM1 was immunoprecipitated (IP) using anti-ICAM1 antibodies, and IgG was used as control. Immunoblotting with anti-SGEF antibodies showed that endogenous ICAM1 and SGEF interacted in endothelial cells, whereas IgG did not show any interaction with SGEF (top). The second panel shows that ICAM1 was efficiently immunoprecipitated by anti-ICAM1 antibodies (left lane) but not by IgG (right lane). The two bottom panels show levels of endogenous SGEF and ICAM1 in endothelial cell lysates. (B) COS7 cells were transfected with myc-SGEF-wt, lysed, and subsequently incubated with biotinylated peptides that correspond to the intracellular domain of ICAM1 (ICAM1-C-term.) or to the intracellular domain of αv-integrin, which was used as control (αv-C-term). Streptavidin-based pull-downs show that the intracellular domain of ICAM1 binds myc-SGEF. The right lane shows myc-SGEF expression in one tenth of the total cell lysate using anti-myc antibodies. (C) Endothelial cells were lysed and subsequently incubated with biotinylated peptides as described in B. The top panel shows that ICAM1-peptide bound endogenous SGEF, whereas the αv-C-term-peptide did not. (D, a) GST-SH3SGEF (amino acids 789–850) was purified and incubated with the peptides described in B. Pull-down experiments using streptavidin-coated Sepharose beads were performed, and GST-SH3SGEF was detected using anti-GST antibodies. SH3SGEF interacted with the ICAM1 tail but not with the tail of αv-integrin (αv-C-term). (D, b) COS7 cells were transfected with ICAM1-GFP and myc-SGEF-ΔDH or myc-SGEF-ΔSH3 and were processed for immunoprecipitation using anti-GFP antibodies. Western blot analysis revealed that ICAM1-GFP binds to myc-SGEF that contains the SH3 domain independent of the DH domain. (E) COS7 cells were transiently cotransfected with ICAM1-GFP and myc-SGEF-wt or myc-SGEF-W826R. ICAM1-GFP was immunoprecipitated using anti-GFP antibodies. Immunoblotting with anti-myc antibodies showed the binding of SGEF-wt but reduced binding of SGEF-W826R to ICAM1-GFP (top). The bottom blots show levels of immunoprecipitated ICAM1-GFP and myc constructs in total cell lysates as indicated. (F) COS7 cells were transiently cotransfected with myc-SGEF-wt and ICAM1-wt-GFP or ICAM1-ΔProline (Pro). GFP was immunoprecipitated using anti-GFP antibodies. Immunoblotting with anti-myc antibodies showed the binding of SGEF-wt but reduced binding of SGEF-wt to ICAM1-ΔPro-GFP (top). The bottom blots show levels of immunoprecipitated ICAM1-wt-GFP or ICAM1-ΔPro-GFP and GFP and myc constructs in total cell lysates as indicated. Note that ICAM1-ΔPro-GFP runs a little bit lower than wt.
RhoG is required for leukocyte TEM
To study RhoG involvement in TEM, siRNA was used to reduce RhoG expression in primary endothelial cells. Western blot analysis revealed that the relevant siRNA reduced RhoG protein expression in endothelial cells but did not affect other proteins known to be present in cup structures or involved in transmigration, such as moesin and ICAM1 (Barreiro et al., 2002; Carman et al., 2003; Millán et al., 2006). Also, the expression levels of other closely related small GTPases such as Rac1, Cdc42, and RhoA were unaffected (Fig. 9 A). Adhesion of leukocytes to endothelial monolayers that showed reduced RhoG expression was not affected. Similarly, expression of dominant-negative RhoG did not affect leukocyte adhesion (unpublished data). However, the formation of cup structures, which was quantified as ICAM1-positive ringlike structures that surrounded adhered leukocytes, was decreased compared with control cells (Fig. 9 B). Transmigration of HL60 cells across endothelial cell monolayers was also substantially attenuated by the knockdown of RhoG expression (Fig. 9 C).
Figure 9.
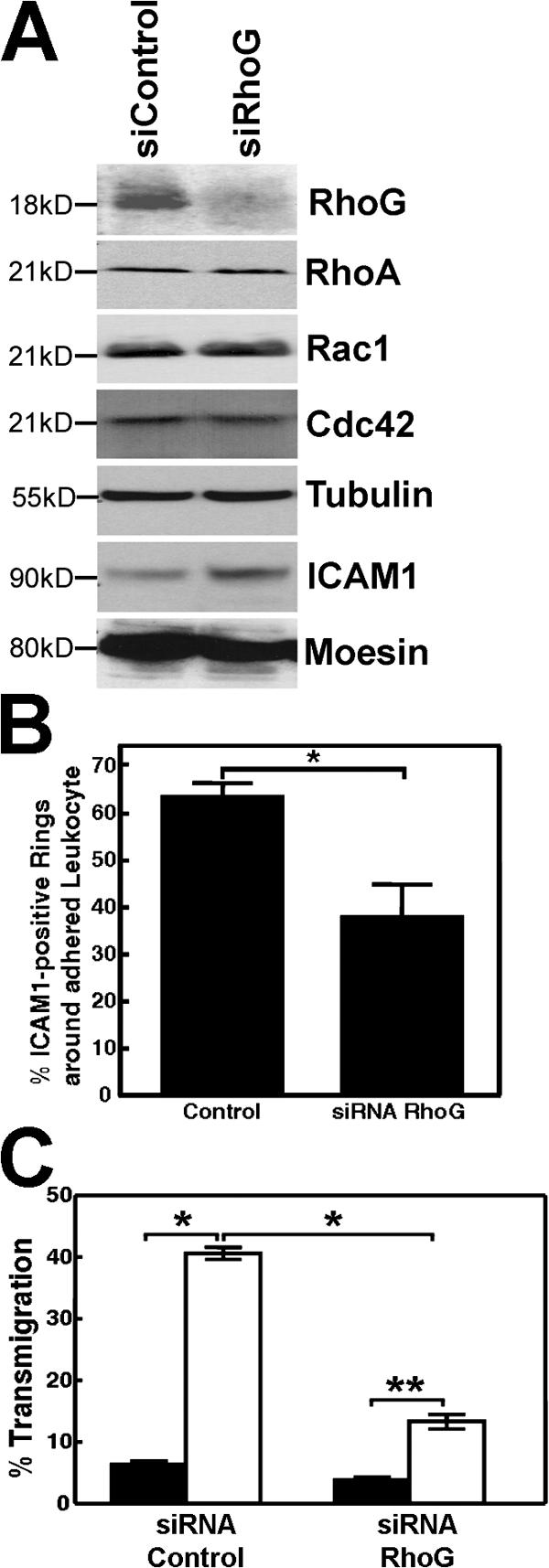
Transmigration of HL60 cells and formation of cups depended on RhoG expression levels. (A) siRNA against RhoG efficiently reduced the expression of endogenous RhoG in endothelial cells. Longer exposure did show some expression of RhoG. The siRNA did not affect the expression levels of RhoA, Rac1, Cdc42, tubulin, ICAM1, or moesin. (B) Quantification of ICAM1-positive rings around adhered HL60 cells, which was measured as described in Materials and methods, showed that cup formation was significantly decreased when RhoG expression was reduced. *, P < 0.01. (C) Endothelial cells were cultured on transwell filters and transfected with RhoG siRNA as described in Materials and methods. 48 h later, differentiated HL60 cells were allowed to transmigrate for 4 h under spontaneous conditions (black bars) or to 50 ng/ml SDF-1 (white bars). Reduced endothelial RhoG expression resulted in decreased spontaneous and SDF-1– induced transmigration. *, P < 0.001; **, P < 0.01. (B and C) The experiment was repeated four times. Data are means ± SEM (error bars).
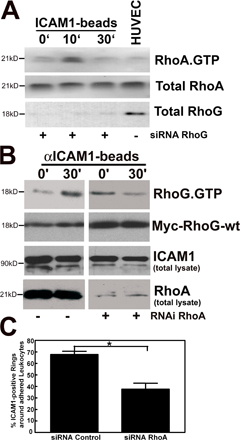
Several previous studies have addressed the role of RhoA in endothelial cells during leukocyte TEM, demonstrating that it is required for TEM (Adamson et al., 1999; Wojciak-Stothard et al., 1999) and showing that it becomes activated downstream from ICAM1 cross-linking (Wojciak-Stothard et al., 1999; Etienne-Manneville et al., 2000; Thompson et al., 2002). We were interested to relate our RhoG results to this previous body of work on RhoA. Reducing RhoG expression by siRNA did not affect RhoA activation downstream of ICAM1 engagement (Fig. S4 A, available at http://www.jcb.org/cgi/content/full/jcb.200612053/DC1), which is consistent with the activation of RhoA occurring faster than the activation of RhoG (Fig. S4, A and E). Interestingly, reducing RhoA expression by siRNA depressed ICAM1-induced RhoG activation (Fig. S4 B). This suggested that RhoA acts upstream of RhoG activation in the pathway from ICAM1 engagement. Whether RhoA has a role in cup formation has been controversial. Barreiro et al. (2002) found that inhibiting the RhoA effector ROCK/Rho kinase with Y27632 diminished cup formation. However, this was not found by Carman and Springer (2004), who also were unable to block cup formation by treating endothelial cells with C3 or Y27632 (Carman et al., 2003). Our finding that RhoA is required upstream of RhoG activation suggested that RhoA may be necessary for cup formation. Consequently, we investigated this directly using micro-RNA (miRNA) of RhoA to depress its expression. We have found that the depletion of RhoA reduced the formation of cups induced by αICAM1 beads (Fig. S4 C).
SGEF and leukocyte TEM
We wished to explore whether SGEF has a role in leukocyte TEM and, thus, have used siRNA to knockdown SGEF expression in endothelial cells. We confirmed that the siRNA decreased SGEF expression and that it did not affect the expression of RhoG, Rac1, or other proteins involved in cups, such as ICAM1 or moesin (Fig. 10 A). Importantly, SGEF knockdown did impair the activation of RhoG downstream from ICAM1 engagement (Fig. 10 B), and, consistent with this, it also resulted in decreased cup formation, as judged by the number of ICAM1-positive rings surrounding adherent leukocytes (Fig. 10 C). Together, these data indicate a pathway from ICAM1 clustering to SGEF to RhoG activation resulting in the formation of cups. Finally, we examined the effect of SGEF knockdown on TEM and found that it caused a decrease in the migration of HL60 cells across endothelial monolayers by up to 50% (Fig. 10 D).
Figure 10.

Reducing SGEF expression in HUVECs blocked RhoG activation and TEM. (A) siRNA against SGEF reduces the expression of endogenous SGEF in endothelial cells. The siRNA did not affect the expression levels of endogenous RhoG, Rac1, ICAM1, or moesin. (B) Knockdown of SGEF expression by siRNA inhibits the activation of RhoG downstream from ICAM1. HUVECs were transiently transduced with myc-RhoG-wt and treated with TNF-α. SGEF expression was reduced by siRNA in HUVECs, and RhoG activity, which was induced by αICAM1 beads, was measured using GST-ELMO. The top panel shows RhoG activity after 30 min, which was depressed when SGEF expression was reduced (bottom panel). The second panel shows equal expression for myc-RhoG-wt in HUVECs. The third panel shows equal levels of ICAM1 in cell lysates. The bottom panel shows reduced SGEF expression after siRNA treatment in HUVECs. The experiment was performed two times. (C) Knockdown of SGEF expression decreased ICAM1 cup formation. Endothelial cells transfected with SGEF siRNA were incubated with HL60 cells. Quantification of ICAM1-positive rings around adhered leukocytes, which was measured as described in Materials and methods, shows that cup formation was significantly decreased when SGEF levels were reduced. *, P < 0.05. (D) Knockdown of SGEF expression inhibited transmigration. Endothelial cells were cultured on transwell filters and transfected with the appropriate siRNA as described in Materials and methods. 48 h later, differentiated HL60 cells were allowed to transmigrate for 4 h under spontaneous conditions (black bars) or toward 50 ng/ml SDF-1 in the lower chamber (white bars). Reduced SGEF levels diminish SDF-1–induced transmigration significantly. *, P < 0.001; **, P < 0.05. (C and D) The experiment was repeated nine times. Data are means ± SEM (error bars).
Discussion
During the last decade, it has become increasingly clear that endothelial cells, rather than being a passive barrier, actively participate in the process of leukocyte TEM. This study focuses on a recently discovered phenomenon that occurs during TEM in which the endothelial cell extends sheets of membrane to form a cuplike structure that surrounds adherent leukocytes (Barreiro et al., 2002; Carman et al., 2003; Carman and Springer, 2004; Doulet et al., 2006). Although their precise function is unclear, evidence has been presented that these structures assist leukocytes on their way through the endothelium (Carman and Springer, 2004).
Our data reveal a new signaling pathway downstream from leukocyte adhesion that involves the small GTPase RhoG. We show here that RhoG activation is triggered through the engagement of ICAM1 and is critical for formation of the apical cups. Additionally, RhoG expression is needed for optimal leukocyte passage across the endothelium. Our data show a strong correlation between formation of the cups and TEM. The endothelial apical cups resemble phagocytic cups, and it is notable that RhoG has been implicated previously in the phagocytosis of apoptotic cells in Caenorhabditis elegans (deBakker et al., 2004). Recent work has also implicated RhoG as well as its exchange factor SGEF in the uptake of Salmonella by epithelial cells (Patel and Galan, 2006). Engulfment of Salmonella is promoted by several bacterial proteins that function to activate multiple Rho family GTPases. Interestingly, the Salmonella protein SopB was found to activate SGEF and RhoG, thereby stimulating the formation of phagocytic cups on the surfaces of epithelial cells (Patel and Galan, 2006). Together, these results suggest that SGEF and RhoG may function in a variety of physiological and pathological situations in which phagocytosis or the uptake of particulate material is involved.
The route by which leukocytes pass through the endothelium, whether it is paracellular or transcellular, has generated considerable debate for many years. In tissue culture models, it has been estimated that only 10–25% of all leukocytes use the transcellular route, with the majority migrating through cell–cell junctions (Carman and Springer, 2004). Millán et al. (2006) have shown that the redistribution of ICAM1 to caveolin-rich membrane domains in response to engagement is followed by transcytosis to the baso-lateral side of the endothelium. The induction of apical cups by RhoG as well as the similarity of these structures to phagocytic cups might lead to the idea that RhoG would function primarily in transcellular rather than paracellular migration. However, our data show that silencing RhoG results in >70% inhibition of leukocyte TEM. Although our work does not discriminate between the para- and trans-cellular migration routes, this decrease in TEM cannot be explained by blocking the trans-cellular pathway only. Consistent with this, the work of Carman and Springer (2004) suggests that trans-migratory cups are not restricted to the trans-cellular route but may function to facilitate and guide leukocyte TEM in general. Alternatively, RhoG may have additional functions in TEM besides mediating cup formation.
The role of Rho family GTPases in formation of the cup structures has begun to be investigated. Barreiro et al. (2002) found that Y27632, which inhibits ROCK/Rho kinase downstream of RhoA, decreased the assembly of these structures induced by VCAM1 engagement. However, Carman and Springer reported that neither C3 nor Y27632 inhibited the assembly of the cups induced by ICAM1 cross-linking (Carman et al., 2003; Carman and Springer, 2004). In our hands, we have observed the partial inhibition of cup formation by Y27632 (our unpublished data) and have found that knockdown of RhoA also inhibits cup formation (Fig. S4 C). The depression of cup formation may, in part, be caused by the inhibition of RhoG activation in cells in which RhoA has been knocked down (Fig. S4 B). How RhoA regulates RhoG activation remains to be determined. In addition, RhoA may play other roles in the assembly of endothelial apical cups.
In this study, we have focused on RhoG, a close relative of Rac1 (Wennerberg et al., 2002), because it induces dorsal membrane ruffles and has been implicated in phagocytosis (deBakker et al., 2004). However, we have observed that ICAM1 engagement leads to the activation of not only RhoG and RhoA but also Rac1 and Cdc42 (Figs. 6 and S3). It is notable that RhoG can activate Rac1 through the DOCK180-binding protein ELMO (Katoh and Negishi, 2003), raising the possibility that the activation of RhoG we observe stimulates Rac1 activation. However, the time course of the activation of Rac1 and RhoG is not consistent with this idea. In future work, it will be interesting to identify the pathways leading to the activation of these other Rho family members.
The intracellular domain of ICAM1 is a prerequisite for optimal TEM of leukocytes (Sans et al., 2001; Lyck et al., 2003). ICAM1 lacking its intracellular domain (ICAM1-ΔC) fails to promote leukocyte TEM, although leukocyte adhesion to ICAM1-ΔC is unaffected. Engagement of ICAM1-ΔC by αICAM1 beads also fails to activate RhoG. The fact that ICAM1-ΔC cannot activate RhoG is likely the result of its inability to bind SGEF. We found that the proline-rich region of the intracellular domain of ICAM1 binds the SH3 domain of SGEF. This interaction is independent of SGEF activation because catalytically inactive mutants of SGEF that express the SH3 domain still bind ICAM1. Engagement of ICAM1 does not promote the association between SGEF and ICAM1 but does increase the activation of SGEF, as judged by the increased binding of SGEF to nucleotide-free RhoG (unpublished data). Thus, SGEF and ICAM1 likely form a stable interacting pair.
Additional signals such as tyrosine phosphorylation may be necessary to trigger SGEF activation, as has been shown for other GEFs (Rossman et al., 2005). One such signal may depend on Src-kinase activity. Src-kinase is rapidly activated after ICAM1 engagement and is required for optimal leukocyte TEM but also does not affect leukocyte adhesion (Etienne-Manneville et al., 2000; Tilghman and Hoover, 2002; Wang et al., 2003; Yang et al., 2006a). Our preliminary results show that inhibiting Src family kinases using PP2 prevented RhoG activation downstream from ICAM1 engagement (unpublished data). These data support the idea that additional signals such as tyrosine phosphorylation are needed to activate SGEF. It is likely that there are multiple targets for Src downstream from ICAM1. One Src substrate that has been implicated in TEM is cortactin (Yang et al., 2006b). Cortactin is a regulator of the actin cytoskeleton that is notably prominent in structures like membrane ruffles and phagocytic cups (Weed and Parsons, 2001).
The passage of leukocytes across the endothelium is a critical event in immune surveillance and in inflammation. Although inflammation is physiologically important, it also underlies many pathological conditions. Consequently, there is considerable interest in understanding the pathways by which leukocytes cross the endothelial barrier so that inappropriate inflammation can be controlled. Much remains to be learned about TEM, including the role of the cups that are formed in response to ICAM1 engagement. Different leukocyte types may induce different effects on the kinetics of ICAM1 signaling and subsequent apical cup formation. In this study, we have identified a pathway downstream from ICAM1 involving RhoG and its exchange factor SGEF that leads to endothelial apical cup formation. Inhibition of either RhoG or SGEF not only inhibits apical cup formation but also depresses TEM, which is consistent with, although does not prove, a role for the cups in TEM.
Materials and methods
Reagents and antibodies
pAbs against ICAM1 (for Western blotting) and mAb against RhoA were obtained from Santa Cruz Biotechnology, Inc. mAbs against Rac1, Cdc42, and MHC class I (MHC-A, -B, and -C) were purchased from BD Biosciences. Recombinant TNF-α and a mAb against ICAM1 were purchased from R&D Systems. The GFP and myc (clone 9E10) mAbs were purchased from Invitrogen. Polyclonal rabbit antibody against VE-cadherin was purchased from Cayman Chemical. The SGEF rabbit pAb was generated in our laboratory as described previously (Ellerbroek et al., 2004). The mAb against RhoG (clone IF-3-B3-E5) was raised in the laboratory of M.A. Schwartz (Robert M. Berne Cardiovascular Research Center, University of Virginia, Charlottesville, VA) against the C-terminal RhoG peptide (AA162-180) of the sequence QQDGVKEVFAEAVRAVLNPT. Dot blots showed that the mAb did not cross react with bacterially expressed Rac1, Cdc42, and RhoA. Western blotting analysis showed that the RhoG antibody did recognize GFP–RhoG-wt but not GFP–Rac1-wt expressed in COS7 cells.
Expression vectors
SGEF cDNA was subcloned using BamHI–EcoRI restriction sites into pCMV6M, an N-terminal myc epitope–tagged eukaryotic expression vector, as described previously (Ellerbroek et al., 2004). SGEF deletion mutants were generated using the QuikChange Site-Directed Mutagenesis kit (Stratagene) and were subcloned into pCMV6M. pGEX-4T2-ELMO was a gift from K. Ravichandran (University of Virginia, Charlottesville, VA). Generation of eukaryotic expression vectors pCMV-myc-Rac(Q61L), pCMV-myc–Rac-wt, pCMV-myc-Rac(T17N), pCMV-myc-RhoG(Q61L), pCMV-myc–RhoG-wt, and pCMV-myc-RhoG(T17N) was described previously by our laboratory (Wennerberg et al., 2002). wt and mutant Rac1 and RhoG constructs were subsequently subcloned into pEGFP-C3 (CLONTECH Laboratories, Inc.) as described previously (Wennerberg et al., 2002). SGEF was subcloned into pEGFP-C2. ICAM1-GFP was a gift from F. Sanchez-Madrid (Hospital de la Princesa, Universidad Autónoma de Madrid, Madrid, Spain). For ICAM1-ΔPro-GFP, the last 11 amino acids of the intracellular tail of ICAM1 were deleted. ICAM1-wt and C-terminal deletion mutant (lacking the last 28 amino acids) cDNA was subcloned into the pAdCMV-V5-DEST vector using the Gateway expression system (Invitrogen).
Cell cultures, treatments, and transfections
HUVECs were obtained from Cambrex and cultured as described previously (Worthylake et al., 2001). Endothelial cells were activated with 10 ng/ml TNF-α overnight as indicated to mimic inflammation. All cell lines were cultured or incubated at 37°C at 10% CO2. The HL60 promyelocytic cell line was obtained from the University of North Carolina's Lineberger Comprehensive Cancer Center Tissue Culture Facility and grown in Optimem plus 5% FBS. In all experiments described, differentiated HL60 cells were used. Differentiation to a neutrophil-like lineage was achieved by adding 1.3% DMSO for 3–5 d (Back et al., 1992). COS7 cells were maintained in growth medium (Iscove's modified Dulbecco's medium with 10% FCS; Sigma-Aldrich). Cells were transiently transfected with the expression vectors indicated in each experiment according to the manufacturer's protocol using LipofectAMINE PLUS (Invitrogen) or Fugene 6 (Roche). Myc–RhoG-wt cDNA was transferred to an AdV expression vector and transfected into 293 cells, and high titer virus stocks were produced. Subsequently, myc–RhoG-wt was transiently delivered into HUVECs by adenovirus transduction.
Immunofluorescence
Cells were cultured on glass coverslips, fixed, and immunostained with the indicated primary antibodies for 60 min at RT as described previously (van Buul et al., 2002). Subsequent visualization was performed with AlexaFluor-conjugated secondary antibodies for 30 min (Invitrogen). F-actin was visualized with fluorescently labeled phalloidin (Invitrogen). Glass coverslips were mounted in MOWIOL at RT. Images were collected with a confocal microscope (LSM510; Carl Zeiss MicroImaging, Inc.) equipped with a microscope (Axiovert 100M; Carl Zeiss MicroImaging, Inc.) and an oil immersion plan-Neofluar 63× NA 1.3 oil lens (Carl Zeiss MicroImaging, Inc.). Cross talk between the different channels was avoided by the use of sequential scanning. Images were processed using imaging examiner software (Carl Zeiss MicroImaging, Inc.) and Photoshop CS (Adobe).
Scanning EM
Transfected cells were grown on glass coverslips, fixed in 2.5% glutaraldehyde/PBS for 30 min at room temperature, and processed for scanning EM as described previously (Ellerbroek et al., 2004). In brief, samples were incubated with 2% aqueous osmium tetroxide for 45 min, dehydrated in a graded ethanol series, and critical point dried in liquid CO2 using a drying apparatus (CPD 010; Balzers Instruments). Samples were mounted on aluminum stubs (Ted Pella, Inc.) and sputter coated with gold/palladium using Polaron scanning EM. Cells were examined on a scanning electron microscope (model 820; JEOL) at 15 kV.
TEM assay
Migration assays were performed in transwell plates (Corning) of 6.5-mm diameter with 8-μm pore filters. Approximately 20,000 endothelial cells were plated on matrigel-coated transwell filters, which were treated the next day with siRNA as indicated. The following day, endothelial cells were treated with siRNA again and with 10 ng/ml TNF-α overnight at 37°C and 10% CO2. 100,000 differentiated HL60 cells were added to the upper compartment, and HL60 cells were allowed to migrate to 50 ng/ml stromal cell–derived factor-1 (SDF-1; placed in the lower chamber to generate a chemotactic gradient; R&D Systems) for 4 h at 37°C and 10% CO2. An input control (i.e., 100,000 HL60 cells) was set as 100%. After collecting the migrated HL60 cells, filters were inspected by confocal laser-scanning microscopy using fluorescently labeled phalloidin to stain F-actin; coating of matrigel on the transwell filter did not affect the formation of a confluent endothelial monolayer. Migrated HL60 cells were counted and compared with 100% input, and the percent migration of HL60 cells was calculated. To confirm efficient knockdown of the protein by siRNA, cells were simultaneously grown in six-well plates and equally treated with siRNA constructs and were analyzed by Western blotting.
Immunoprecipitation and Western blotting
Cells were grown to confluency, washed twice gently with ice-cold Ca2+- and Mg2+-containing PBS, and lysed in 300 μl lysis buffer (25 mM Tris, 150 mM NaCl, 10 mM MgCl2, and 1% Triton X-100 with the addition of fresh protease inhibitors, pH 7.4). Immunoprecipitation was performed as previously described (Barreiro et al., 2002) and analyzed by Western blotting using an enhanced ECL detection system (GE Healthcare). The intensity of the bands was quantified by using ImageJ version 1.36 (National Institutes of Health, Bethesda, MD).
Apical cup quantification
Using confocal laser-scanning microscopy, z-stacks were taken to confirm the formation of a cup around an adhered leukocyte. The length of the protrusion was ∼6–7μm above the baso-lateral plane of the substrate (Fig. 1 B, d). The apical plane was set to 4 μm from the baso-lateral plane (Fig. 1 A). ICAM1-positive rings in the apical plane were counted as positive cups.
RhoG, RhoA, and Rac1 activation assay
For RhoG activation assays, a transient coexpression of myc-tagged RhoG was used because of the lack of a high affinity antibody that is appropriate for these assays (according to Katoh and Negishi [2003]). Transfected cells were lysed in 300 μl of 50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, 1 mM PMSF, and 10 μg/ml each of aprotinin and leupeptin. Lysates were cleared at 14,000 g for 10 min. Supernatants were rotated for 30 min with 60–90 μg GST-ELMO (GST fusion protein containing the full-length RhoG effector ELMO) conjugated to glutathione–Sepharose beads (GE Healthcare). Beads were washed in 50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, and protease inhibitors. Pull-downs and lysates were then immunoblotted for the myc epitope tag. For RhoA and Rac1, GST-Rhotekin and GST-PBD were used as baits, respectively, and used as described for GST-ELMO.
Fusion proteins
GST-ELMO, GST-SH3SGEF (SGEF789–850), GST-Rhotekin, and GST-PBD fusion proteins were purified from BL21 Escherichia coli cells (Stratagene) using glutathione–Sepharose 4B as previously described (Ellerbroek et al., 2004). GST fusion proteins were eluted with free, reduced glutathione in TBS medium (50 mM Tris, 150 mM NaCl, 5 mM MgCl2, pH 7.4, and 1 mM DTT) and stored in 30% glycerol at −80°C.
Antibody-coated beads
3 μm polystyrene beads (Polysciences, Inc.) were pretreated with 8% glutaraldehyde overnight, washed five times with PBS, and were incubated with 300 μg/ml ICAM1/MHC mAb according to the manufacturer's protocol.
Bead adhesion assay
For immunofluorescence or scanning EM, 1 μg/ml of antibody-containing beads was washed and resuspended in culture medium. 1 μg/ml of antibody-coated beads was incubated in wells of 24-well dishes containing glass coverslips, on which TNF-α–pretreated HUVECs or COS7 cells were cultured. After the appropriate time, unbound beads were removed, and coverslips were put on ice, gently washed three times with ice-cold PBS containing 1 mM Ca2+/Mg2+, and subsequently processed for immunofluorescence. For biochemistry, 10 μg/ml of antibody-coated beads were incubated on the cells, after which cells were washed as described above (see Bead adhesion assay section) and subsequently lysed and processed as described (see Immunoprecipitation and Western blotting and RhoG, RhoA, and Rac1 activation assay sections).
Knockdown using siRNA
siRNA duplexes against human RhoG (sense, GCAACAGGAUGGUGUCAAGUU; antisense, 5′-P-UCGUCCAAGAUCGACAUCC UU) and SGEF mRNA (sense, CAAAUGGCCUUGCCGCUAAUU; antisense, 5′-P-UUAGCGGCAAGGCCAUUUGUU) and siControl nontargeting siRNA were obtained from the Dharmacon siRNA collection. HUVECs were transfected twice with 50 nmol/l siRNA using RNAifect transfection reagent (QIAGEN). After 48 h, cells were processed as described in the previous paragraph.
Knockdown using miRNA adenovirus
miRNA adenoviral constructs were engineered according to the manufacturer's protocol (Invitrogen). In brief, two sets of DNA oligonucleotides were designed to target human RhoA mRNA and were named RhoA#1 and RhoA#2: TGCTGAAGACTATGAGCAAGCATGTCGTTTTGGCCACTGACTGACGACATGCTCTCATAGTCTT and CCTGAAGACTATGAGAGCATGTCGTCAGTCAGTGGCCAAAACGACATGCTTGCTCATAGTCTTC (RhoA#1) and GCTGTTTCCATCCACCTCGATATCTGTTTTGGCCACTGACTGACAGATATCGGTGGATGGAAA and CCTGTTTCCATCCACCGATATCTGTCAGTCAGTGGCCAAAACAGATATCGAGGTGATGGAAAC (RhoA#2). The oligonucleotides were annealed and ligated into pcDNA6 EmGFP. The EmGFP MiR RNA cassette was subsequently transferred to pDONR221 and finally to pAd by two sequential Gateway BP and LR recombinations. Each construct was sequence verified, and viral particles were produced by transfection in 293A cells.
Biotinylated peptides
Peptides were synthesized with the following sequence: ICAM1–intracellular tail peptide; biotin-GRQRKIKKYRLQQAQKGTPMKPNTQATPP-OH; αv peptide; and biotin-GHENGEGNSET-OH.
Live cell imaging
COS7 cells were cultured on glass coverslips and transfected with cDNA as indicated. After 24 h, cells were placed in a heating chamber at 37°C and recorded with a confocal microscope (LSM510; Carl Zeiss MicroImaging, Inc.) equipped with a microscope (Axiovert 100M; Carl Zeiss MicroImaging, Inc.) and an oil immersion plan-Neofluar 63× NA 1.3 oil lens (Carl Zeiss MicroImaging, Inc.).
Online supplemental material
Fig. S1 shows the recruitment of endogenous ICAM1 around a migrating HL60 cell. Fig. S2 shows the recruitment of GFP–RhoG-Q61L and F-actin to αICAM1 beads but not to αMHC class I beads. Fig. S3 shows activation of the small GTPases RhoG, Rac1, Cdc42, and RhoA downstream of ICAM1 engagement. Fig. S4 shows that reduced RhoG expression does not affect ICAM1-mediated RhoA activation but that the reduced expression of RhoA does influence RhoG activity downstream from ICAM1 engagement, which is induced by αICAM1 beads. Video 1 shows a real-time recording of 10 min of GFP–RhoG-Q61L expression in COS7 cells. Video 2 shows a real-time recording of 10 min of GFP-SGEF expression in COS7 cells. Video 3 shows a real-time recording of ICAM1-GFP expressed in COS7 cells and incubated with αICAM1 beads for 30 min. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200612053/DC1.
Supplementary Material
Acknowledgments
We thank Wendy Salmon and Dr. Michael Chua (Michael Hooker Microscopy Facility, University of North Carolina, Chapel Hill, NC) as well as Hal Mekeel (University of North Carolina, Chapel Hill, NC) for their technical assistance with confocal and electron microscopy. We thank Lisa Sharek for outstanding technical assistance and members of the Burridge laboratory for their encouragement. We thank Dr. Peter Hordijk for critically reading the manuscript. We also thank Jos van Rijssel and Floris van Alphen for their technical assistance.
This work was supported, in part, by National Institutes of Health grants HL45100 and HL080166 and a Kenan Distinguished Professorship to K. Burridge. J.D. van Buul was supported by the Ter Meulen Fund, Royal Netherlands Academy of Arts and Sciences, and an E. Dekker stipendium from the Netherlands Heart Foundation. R. Garcia-Mata was supported by a postdoctoral fellowship from the Susan Komen Foundation. T. Samsom was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (grant Sa 1636/1-1).
J.D. van Buul's present address is Dept. of Molecular Cell Biology, Sanquin Research and Landsteiner Laboratory, Academic Medical Center, University of Amsterdam, 1012 ZA Amsterdam, Netherlands.
Abbreviations used in this paper: GEF, guanine-nucleotide exchange factor; HUVEC, human umbilical vein endothelial cell; ICAM1, intercellular adhesion molecule-1; MHC, major histocompatibility complex; miRNA, micro-RNA; PBD, p21-activated kinase–binding domain; ROCK, Rho-associated coil-containing protein kinase; SDF-1, stromal cell–derived factor-1; SGEF, SH3-containing GEF; TEM, trans-endothelial migration; VE, vascular endothelial; wt, wild type.
References
- Adamson, P., S. Etienne, P.O. Couraud, V. Calder, and J. Greenwood. 1999. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a Rho-dependent pathway. J. Immunol. 162:2964–2973. [PubMed] [Google Scholar]
- Back, A.L., K.A. Gollahon, and D.D. Hickstein. 1992. Regulation of expression of the leukocyte integrin CD11a (LFA-1) molecule during differentiation of HL-60 cells along the monocyte/macrophage pathway. J. Immunol. 148:710–714. [PubMed] [Google Scholar]
- Barreiro, O., M. Yanez-Mo, J.M. Serrador, M.C. Montoya, M. Vicente-Manzanares, R. Tejedor, H. Furthmayr, and F. Sanchez-Madrid. 2002. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J. Cell Biol. 157:1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman, C.V., and T.A. Springer. 2004. The transcellular railway: insights into leukocyte diapedesis. J. Cell Biol. 167:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman, C.V., C.D. Jun, A. Salas, and T.A. Springer. 2003. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J. Immunol. 171:6135–6144. [DOI] [PubMed] [Google Scholar]
- deBakker, C.D., L.B. Haney, J.M. Kinchen, C. Grimsley, M. Lu, D. Klingele, P.K. Hsu, B.K. Chou, L.C. Cheng, A. Blangy, et al. 2004. Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr. Biol. 14:2208–2216. [DOI] [PubMed] [Google Scholar]
- Doulet, N., E. Donnadieu, M.P. Laran-Chich, F. Niedergang, X. Nassif, P.O. Couraud, and S. Bourdoulous. 2006. Neisseria meningitidis infection of human endothelial cells interferes with leukocyte transmigration by preventing the formation of endothelial docking structures. J. Cell Biol. 173:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerbroek, S.M., K. Wennerberg, W.T. Arthur, J.M. Dunty, D.R. Bowman, K.A. DeMali, C.J. Der, and K. Burridge. 2004. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol. Biol. Cell. 15:3309–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S., J.B. Manneville, P. Adamson, B. Wilbourn, J. Greenwood, and P.O. Couraud. 2000. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J. Immunol. 165:3375–3383. [DOI] [PubMed] [Google Scholar]
- Johnson-Leger, C., and B.A. Imhof. 2003. Forging the endothelium during inflammation: pushing at a half-open door? Cell Tissue Res. 314:93–105. [DOI] [PubMed] [Google Scholar]
- Katoh, H., and M. Negishi. 2003. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature. 424:461–464. [DOI] [PubMed] [Google Scholar]
- Kluger, M.S. 2004. Vascular endothelial cell adhesion and signaling during leukocyte recruitment. Adv. Dermatol. 20:163–201. [PubMed] [Google Scholar]
- Lyck, R., Y. Reiss, N. Gerwin, J. Greenwood, P. Adamson, and B. Engelhardt. 2003. T-cell interaction with ICAM-1/ICAM-2 double-deficient brain endothelium in vitro: the cytoplasmic tail of endothelial ICAM-1 is necessary for transendothelial migration of T cells. Blood. 102:3675–3683. [DOI] [PubMed] [Google Scholar]
- Millán, J., L. Hewlett, M. Glyn, D. Toomre, P. Clark, and A.J. Ridley. 2006. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat. Cell Biol. 8:113–123. [DOI] [PubMed] [Google Scholar]
- Muller, W.A. 2003. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 24:327–334. [DOI] [PubMed] [Google Scholar]
- Patel, J.C., and J.E. Galan. 2006. Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J. Cell Biol. 175:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman, K.L., C.J. Der, and J. Sondek. 2005. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6:167–180. [DOI] [PubMed] [Google Scholar]
- Sans, E., E. Delachanal, and A. Duperray. 2001. Analysis of the roles of ICAM1 in neutrophil transmigration using a reconstituted mammalian cell expression model: implication of ICAM1 cytoplasmic domain and Rho-dependent signaling pathway. J. Immunol. 166:544–551. [DOI] [PubMed] [Google Scholar]
- Thompson, P.W., A.M. Randi, and A.J. Ridley. 2002. Intercellular adhesion molecule (ICAM)-1, but not ICAM-2, activates RhoA and stimulates c-fos and rhoA transcription in endothelial cells. J. Immunol. 169:1007–1013. [DOI] [PubMed] [Google Scholar]
- Tilghman, R.W., and R.L. Hoover. 2002. The Src-cortactin pathway is required for clustering of E-selectin and ICAM-1 in endothelial cells. FASEB J. 16:1257–1259. [DOI] [PubMed] [Google Scholar]
- van Buul, J.D., and P.L. Hordijk. 2004. Signaling in leukocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol. 24:824–833. [DOI] [PubMed] [Google Scholar]
- van Buul, J.D., C. Voermans, V. van den Berg, E.C. Anthony, F.P. Mul, S. van Wetering, C.E. van der Schoot, and P.L. Hordijk. 2002. Migration of human hematopoietic progenitor cells across bone marrow endothelium is regulated by vascular endothelial cadherin. J. Immunol. 168:588–596. [DOI] [PubMed] [Google Scholar]
- Vestweber, D. 2002. Regulation of endothelial cell contacts during leukocyte extravasation. Curr. Opin. Cell Biol. 14:587–593. [DOI] [PubMed] [Google Scholar]
- Wang, Q., G.R. Pfeiffer II, and W.A. Gaarde. 2003. Activation of SRC tyrosine kinases in response to ICAM-1 ligation in pulmonary microvascular endothelial cells. J. Biol. Chem. 278:47731–47743. [DOI] [PubMed] [Google Scholar]
- Weed, S.A., and J.T. Parsons. 2001. Cortactin: coupling membrane dynamics to cortical actin assembly. Oncogene. 20:6418–6434. [DOI] [PubMed] [Google Scholar]
- Wennerberg, K., S.M. Ellerbroek, R.Y. Liu, A.E. Karnoub, K. Burridge, and C.J. Der. 2002. RhoG signals in parallel with Rac1 and Cdc42. J. Biol. Chem. 277:47810–47817. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard, B., L. Williams, and A.J. Ridley. 1999. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J. Cell Biol. 145:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthylake, R.A., S. Lemoine, J.M. Watson, and K. Burridge. 2001. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 154:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L., J.R. Kowalski, X. Zhan, S.M. Thomas, and F.W. Luscinskas. 2006. a. Endothelial cell cortactin phosphorylation by Src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ. Res. 98:394–402. [DOI] [PubMed] [Google Scholar]
- Yang, L., J.R. Kowalski, P. Yacono, M. Bajmoczi, S.K. Shaw, R.M. Froio, D.E. Golan, S.M. Thomas, and F.W. Luscinskas. 2006. b. Endothelial cell cortactin coordinates intercellular adhesion molecule-1 clustering and actin cytoskeleton remodeling during polymorphonuclear leukocyte adhesion and transmigration. J. Immunol. 177:6440–6449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}