

Abstract
hSgo2 (previously annotated as Tripin) was recently reported to be a new inner centromere protein that is essential for centromere cohesion (Kitajima et al., 2006). In this study, we show that hSgo2 exhibits a dynamic distribution pattern, and that its localization depends on the BUB1 and Aurora B kinases. hSgo2 is concentrated at the inner centromere of unattached kinetochores, but extends toward the kinetochores that are under tension. This localization pattern is reminiscent of MCAK, which is a microtubule depolymerase that is believed to be a key component of the error correction mechanism at kinetochores. Indeed, we found that hSgo2 is essential for MCAK to localize to the centromere. Delocalization of MCAK accounts for why cells depleted of hSgo2 exhibit kinetochore attachment defects that go uncorrected, despite a transient delay in the onset of anaphase. Consequently, these cells exhibit a high frequency of lagging chromosomes when they enter anaphase. We confirmed that hSgo2 is associated with PP2A, and we propose that it contributes to the spatial regulation of MCAK activity within inner centromere and kinetochore.
Introduction
Tripin is a mammalian protein of unknown function that was reported to contain a domain that is conserved amongst Shugoshin (Sgo) family members (Kitajima et al., 2004). Sgo1 is a family of evolutionarily conserved proteins that was first identified in yeast (Katis et al., 2004; Kitajima et al., 2004; Marston et al., 2004; Rabitsch et al., 2004) and Drosophila melanogaster (MEI-S332; Kerrebrock et al., 1995; Moore et al., 1998) as mutants that failed to maintain centromeric cohesion during meiosis I. Although Sgo1/MEI-S332 are essential for maintaining centromere cohesion during meiosis I in yeast and flies, they are not essential for mitotic chromosome segregation in both species. Sgo2 is a paralogue of Sgo1 fission yeast, and studies in have shown that it acts both in meiosis and mitosis (Kitajima et al., 2004; Rabitsch et al., 2004). During meiosis I, Sgo2 specifies monopolar attachments of paired chromatids, as opposed to a role in centromere cohesion (Rabitsch et al., 2004; Vaur et al., 2005). Recent studies in fission yeast showed that Sgo2 is important for bipolar attachments of chromosomes in mitosis, and it specifies the centromeric localization of the chromosome passenger proteins Bir1/survivin, Pic1/INCENP, and Ark1/Aurora B kinase (Kawashima et al., 2007; Vanoosthuyse et al., 2007). As Aurora B kinase is a critical component of the error correction machinery at kinetochores that monitors defective attachments (Tanaka et al., 2002; Cimini et al., 2006; Pinsky et al., 2006), its loss from centromeres in Sgo2 mutants explains the defects in establishing stable bipolar attachments.
Comparison of the primary sequences of Sgo1 and Sgo2 amongst different species of fungi revealed a common coiled-coil domain near their N termini and a conserved basic region of ∼30 residues near their C termini (Kitajima et al., 2004; Rabitsch et al., 2004). Identification of mutations within the conserved elements in D. melanogaster MEI-S322 (Suzuki et al., 2006) established that it was related to Sgo1 both in structure and in function. Although vertebrate proteins with these conserved elements were also identified, the first vertebrate Sgo1 was identified in a biochemical screen for microtubule-binding proteins in Xenopus laevis egg extracts (Salic et al., 2004). Consistent with the microtubule-binding activity, both the frog and human Sgo1 were found to be essential for establishing kinetochore–microtubule attachments (Salic et al., 2004; Tang et al., 2004; Suzuki et al., 2006). These proteins were also essential for maintaining chromatid cohesion during mitosis, as cells depleted of Sgo1 were delayed in mitosis with unattached kinetochores and separated chromatids (Salic et al., 2004; Tang et al., 2004; McGuinness et al., 2005). The vertebrate Sgo1 was concentrated near the outer kinetochore, which is where one would expect to find a microtubule-binding protein (Salic et al., 2004). Others reported that hSgo1 was concentrated at the inner centromere (Tang et al., 2004; McGuinness et al., 2005), which is where one would expect to find a protein that is responsible for centromeric cohesion.
The annotation of Tripin as Shugoshin 2 (Sgo2) in the database was based on the presence of the conserved C-terminal basic region that is shared amongst Sgo family members. Both the mouse and human proteins lack the conserved coiled-coil domain that is present within the N terminus of Sgo1. Additionally, Tripin is significantly larger than Sgo1 (∼1,200 vs. 480 residues) and yeast Sgo2 (647 residues). A recent study linking hSgo1 to PP2A phosphatase also reported that Tripin/hSgo2 is localized at the inner centromere, where it is important for centromere cohesion (Kitajima et al., 2006). Mechanistically, hSgo2 was proposed to maintain cohesion in a manner that is similar to that of hSgo1. Namely, hSgo1 and hSgo2 recruit PP2A to the centromere, where they can neutralize Plk1's ability to phosphorylate and release cohesin complexes (McGuinness et al., 2005; Kitajima et al., 2006; Riedel et al., 2006; Rivera and Losada, 2006; Tang et al., 2006).
Our studies show that hSgo2 is, indeed, a component of the inner centromere and that it exhibits a dynamic localization pattern where it is concentrated in between sister kinetochores during prometaphase, but extends toward the kinetochore by metaphase. We show that hSgo2 is released from the inner centromere shortly after the onset of anaphase and does not reappear there until late G2/prophase. This pattern is similar to that reported for the localization of Sgo2 during meiosis II in mouse spermatocytes (Gomez et al., 2007).
Functionally, cells quantitatively depleted of hSgo2 exhibited kinetochore attachment defects that transiently delayed cells at metaphase. When the cells entered anaphase, they invariably contained lagging chromosomes, which suggested that the defective attachments were never corrected. We ascribe the attachment defects to the delocalization of the microtubule depolymerase MCAK. Although we have no evidence to indicate that hSgo2 is essential for centromere cohesion, we confirmed that hSgo2 is, indeed, associated with PP2A (Kitajima et al., 2006). We speculate that this subpopulation of PP2A may regulate the targeting or activity of MCAK at the inner centromere and kinetochore.
Results
Antibodies to Tripin/hSgo2 were generated to characterize its localization and expression patterns in HeLa cells. Consistent with its predicted size of 1,265 aa, the antibodies identified an ∼150-kD protein in HeLa lysates that is depleted by Tripin/hSgo2 siRNA (Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200701122/DC1). Costaining with hBUB1 antibodies showed that hSgo2 is concentrated at the inner centromere (Fig. 1 A). hSgo2 is diffusely distributed in the nucleus during interphase (unpublished data). In cells whose chromosomes have begun to condense (mid to late G2), hSgo2 accumulated at foci that were coincident with hBUB1. At a later stage of G2, when the nascent kinetochore pairs have resolved, pairs of hSgo2 foci that were positioned internal to hBUB1 were clearly evident. After nuclear envelope breakdown, hSgo2 staining appeared as a single focus that was positioned in between the sister kinetochores as defined by hBUB1 staining. At metaphase, hSgo2 was distributed across the width of the centromere and extended toward, and sometimes overlapped, the kinetochores. In early anaphase cells, hSgo2 and hBUB1 remain colocalized, but kinetochores exhibiting only hBUB1 staining in the same cells were also observed. Thus, the release of hSgo2 from kinetochores does not appear to be regulated solely by mitotic timing. By late anaphase, neither hSgo2 nor hBUB1 were detected at kinetochores. The dynamic properties of hSgo2 were confirmed in real time by tracking cells expressing GFP/hSgo2 (unpublished data).
Figure 1.

hSgo2 localization and expression patterns. (A) HeLa cells released from a double thymidine block were costained with Bub1 (b, f, j, n, r, z) and hSgo2 (c, g, k, o, s, 1). Shown are cells from G2 through anaphase A. Insets show enlarged images of kinetochore pairs. Color images show the merged signals of Bub1 (green) and hSgo2 (red) staining. (B) Western blots of lysates prepared from cells released from a mitotic block. Lane 1, asynchronous cells; lanes 2–7, cells at 0, 30, 60, 90, 120, and 150 min after nocodazole washout; lane 8, cells released into medium with MG132 and harvested at 150 min. Bub3 is used as a loading control. (C) Costaining of hSgo2, MCAK, and ACA in prophase (Pro) and prometaphase (Prometa) HeLa cells. (D) Intensity profiles of hSgo2 (red), MCAK (green), and ACA (blue) of a representative kinetochore (C, arrow) in prophase (left) and prometaphase (right). Intensity values were obtained from the separate channels and plotted as a function of distance (inset, white dotted line). 25 kinetochores with discrete signals in all three channels were measured.
We monitored the stability of hSgo2 as a function of mitotic exit by probing lysates prepared from cells that were released from a nocodazole block (Fig. 1 B). Between 60 and 90 min after release, the majority of cells entered anaphase, as cyclin B levels abruptly declined. hSgo2, along with CENP-E, also began to decline at this time, although their kinetics of degradation appeared to lag behind cyclin B. Loss of hSgo2 (as with cyclin B and CENP-E) was dependent on the proteosome as it was stabilized when cells were treated with a proteosome inhibitor.
The localization pattern of hSgo2 is reminiscent of MCAK, which is a microtubule depolymerase (Desai et al., 1999; Kinoshita et al., 2006) that is concentrated at the inner centromere, but is redistributed toward the kinetochores in response to microtubule attachments (Kline-Smith et al., 2004). Indeed, hSgo2 localization was found to be coincident with MCAK (Fig. 1 C), as recently reported in mouse spermatocytes (Gomez et al., 2007). In late prophase, hSgo2 and MCAK were colocalized at a single focus in between kinetochore pairs that were stained with ACA (Fig. 1 D). In prometaphase, we found examples where hSgo2 and MCAK staining were skewed toward one of the sister kinetochores (Fig. 1 D). For MCAK, this was shown to reflect its redistribution toward the leading kinetochore of a congressing chromosome (Kline-Smith et al., 2004).
The relocalization of hSgo2 from the inner centromere to the kinetochore at metaphase led us to test whether its localization pattern was sensitive to microtubule attachments or kinetochore tension. HeLa cells were treated with a dose of nocodazole that suppressed microtubule dynamics, and thus reduced kinetochore tension without affecting attachment. The reduction in the interkinetochore distance of bipolar attached kinetochores in the drug-treated samples relative to controls (2.2 vs. 1.3 μm, respectively) confirmed that nocodazole reduced kinetochore tension. Despite the reduction in tension, hSgo2 was still able to redistribute from a single dot, seen at unattached kinetochores (Fig. 2 A, inset 1), to a bar that stretched between the bipolar attached kinetochore (Fig. 2 A, insets 2 and 3). However, when tension was reduced, the peaks of hSgo2 staining did not overlap with hBUB1 to the same extent as seen in kinetochores that are under maximal tension (Fig. 2 B, insets 1 and 2). Thus, the extent of hSgo2's redistribution from the centromere to the kinetochore is sensitive to tension, as previously suggested (Gomez et al., 2007).
Figure 2.

hSgo2 localization is affected by kinetochore tension. (A) Cells treated with a low-dose nocodazole to suppress microtubule dynamics, and thus reduce tension between bipolar attached kinetochores, were costained with Bub1 (red), hSgo2 (green), and DAPI (blue). Insets depict enlarged images of single kinetochore pairs highlighted by the dashed boxes. 1, unattached kinetochore pair; 2 and 3, bipolar attached kinetochores of aligned chromosomes. (B) Control metaphase cells. 1 and 2, bipolar attached kinetochores under full tension. Distances were measured from the centers of Bub1 and hSgo2 intensity profiles. Scale, 1 pixel = 0.117 μm. All images were captured and processed identically.
Next, we used FRAP to compare the turnover rates of hSgo2 at kinetochores of different microtubule-binding status. Kinetochores expressing GFP/hSgo2 were photobleached, and the rate of recovery of the GFP signal was monitored (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200701122/DC1). The t 1/2 during G2 phase, when hSgo2 is first recruited to the nascent kinetochore, is 5.25 ± 2.1 s. The t 1/2 increased to 10.17 ± 5.93 and 9.17 ± 3.39 s in prometaphase and metaphase, respectively. We then compared the turnover rates in mitotic cells that were exposed to vinblastine or taxol. In vinblastine-treated cells whose kinetochores lacked attachments, the t 1/2 was 13.22 ± 4.89 s, as opposed to 8.87 ± 1.67 s kinetochores in taxol-treated cells. Regardless of the microtubule-binding status or phase of the cell cycle, all of the kinetochores examined (n = 38) were able to recover >94% of the prebleached level of GFP/hSgo2. Thus, the slightly faster turnover rate of hSgo2 at metaphase kinetochores may be affected by increased microtubule attachments. This explanation cannot account for the more rapid turnover rates in G2, which may be governed by determinants that specify kinetochore assembly.
hSgo2 specifies kinetochore microtubule attachments
We next used siRNA to examine the role of hSgo2 during mitosis. We first tested the efficiency of depletion by different siRNAs that were targeted against hSgo2. Quantitative immunoblots of lysates prepared from transfected cells showed that hSgo2 siRNAs 2, 3, 4 and the pooled depleted hSgo2 by >90% (Fig. 3 A), as compared with an unrelated siRNA. Further analysis using immunofluorescence staining showed that in cells transfected with the pooled siRNAs and siRNA 3, hSgo2 levels were reduced by 94 and 98%, respectively (Fig. S1 B). Functional studies were thus conducted with the pooled siRNAs and siRNA 3. In all of our functional studies, we confirmed in parallel samples that the staining intensity of the siRNA target was depleted by >95%.
Figure 3.

Characterization of cells depleted of hSgo2 by siRNA. (A) Efficiency of depletion of hSgo2 by different siRNAs. Lysates were harvested 48 h after transfection and probed for hSgo2 and CENP-F, which served as a loading control. Lanes 1–3, 50, 10, and 5 μg of lysates from cells transfected with control siRNA; lanes 4–7, 50 μg of lysates from cells transfected with pooled, 1, 2, 3, and 4 hSgo2 siRNAs. The hSgo2 signal intensity from cells transfected with oligos 2, 3, and the pooled siRNAs was less than or equal to the signal obtained from control samples containing 10-fold less protein (>90% depletion). (B) Cells transfected with hSgo1 exhibit attachment (top left) and cohesion defects (bottom left). Cells depleted of hSgo2 by the pooled siRNA (middle) can achieve metaphase alignment, as with controls (right). (C) Select frames from videos of HeLa (GFP/H2B) cells transfected with the different siRNAs. Two videos of hSgo2-depleted cells are shown to document different spindle orientations relative to the slide. (D) Fraction of cells from time-lapse studies that exhibited mitotic errors in anaphase. Orange, morphologically normal anaphase (nondisjunction would not be scored); green, lagging chromosomes; blue, died in mitosis. (E) HeLa (GFP/H2B) cells transfected with control, hSgo2, and MCAK siRNAs were examined by time-lapse videomicroscopy. The time of anaphase onset was determined as the time from NEBD to chromosome separation. Anaphase times for cells (n > 50 for each sample) were determined in each experiment (n = 6) and plotted as the frequency of all mitotic cells at each of the recorded times. Black, control siRNA; pink, hSgo2 siRNA smartpool; green, hSgo2 siRNA 3; red, MCAK siRNA. Error bars represent the SEM from six independent experiments.
We first compared cells that were transfected with hSgo1 and hSgo2 siRNAs to test their relative contributions to cohesion. As previously reported (Salic et al., 2004; Tang et al., 2004; Kitajima et al., 2005; McGuinness et al., 2005), cells transfected with hSgo1 siRNA blocked in mitosis with large numbers of unattached chromosomes and separated sister chromatids (Fig. 3 B). In contrast, chromosomes in mitotic cells depleted of hSgo2 did not exhibit obvious defects in cohesion. Many cells depleted of hSgo2 were able to align their chromosomes, as with control metaphase cells (Fig. 3 B). However, kinetochore attachment in hSgo2-depleted cells were likely defective, as anaphase cells exhibited a high frequency of lagging chromosomes compared with control samples (see the following paragraph).
We next examined chromosome behavior in synchronized HeLa (GFP/H2B) cells that were transfected with control or the pooled siRNAs and hSgo2 siRNA 3 (Fig. 3 C). Control cells took 25–35 min from the onset of mitosis (nuclear envelope breakdown) to align their chromosomes at the metaphase plate (Fig. 3 C, top row). Anaphase onset ensued within 10 min after all the chromosomes achieved metaphase alignment, and ∼45 min after NEBD (Fig. 3 E). Cells depleted of hSgo2 took more time to align their chromosomes (∼30–90 min), as they were delayed in mitosis with their chromosomes organized in a ring (Fig. 3 C). The chromosomes eventually reached the spindle equator, but took an additional >20–45 min before entering anaphase (Fig. 3 C, middle). The metaphase delay is likely also caused by defective kinetochore attachments, as cells that eventually entered anaphase invariably contained lagging chromosomes (Fig. 3, C and D; and Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200701122/DC1). In addition to lagging chromosomes, some anaphase cells showed two separated rings of chromosomes (Fig. 3 C, bottom, times 4:00–4:12; Fig. S2 A). These examples reflect a rotation of the spindle axis out of the plane of the slide such that the aligned chromosomes appear as a disc (Fig. 3 C, bottom, times 2:27–3:57) and the separated chromatids appear as two rings.
MCAK localization depends on hSgo2
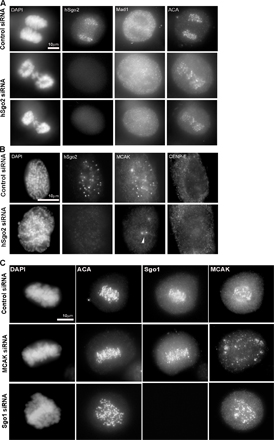
Given that the localization pattern of hSgo2 was similar to MCAK, and the loss of hSgo2 resulted in defective attachments that are proposed to be resolved by MCAK, we examined if there was a connection between these two proteins. In cells transfected with a control siRNA, MCAK and hSgo2 were colocalized during prophase and prometaphase (Fig. 4 A and Fig. S3 B). In contrast, MCAK was delocalized from the inner centromeres in prophase and prometaphase cells that were depleted of hSgo2 (Fig. 4 A and Fig. S3 B). Delocalization of MCAK from the centromere was specific, as it was still detected at centrosomes (Fig. S3 B). Quantitative analysis showed that depletion of hSgo2 resulted in a >30-fold reduction in MCAK staining intensity at the inner centromere. The same magnitude of reduction was also observed when the intensity level of MCAK was normalized to ACA, whose staining was unaffected by hSgo2 (Fig. 4 B). Western blots showed that MCAK levels were unaffected by the depletion of hSgo2 (Fig. 4 C), and the relative amounts of phosphorylated MCAK (based on slower migration; Fig. S4 A) was not grossly different between control and hSgo2-depleted cells. To demonstrate that the delocalization of MCAK was caused by a failure to assemble onto the inner centromere, as opposed to some indirect affect by microtubule attachments, we repeated the analysis in the presence of nocodazole. In the absence of microtubules, depletion of hSgo2 also prevented recruitment of MCAK to the inner centromere (Fig. 4 D). In contrast to hSgo2, MCAK localization was unaffected when cells were depleted of hSgo1 (Fig. S3 C).
Figure 4.

hSgo2 specifies localization of MCAK to the inner centromere. (A) Prometaphase cells after transfection with control and hSgo2 siRNAs were costained for hSgo2, MCAK, and ACA. Chromosomes were stained with DAPI. (B) Comparison of the intensity ratios of MCAK (left) and MCAK normalized to ACA (right) between control and hSgo2-depleted cells (n > 30). (left) The depletion efficiency of MCAK siRNA. (right) Ratios of the intensity of MCAK to ACA at kinetochores (n > 30) in cells treated with nocodazole. (C) Mitotic cells harvested after transfection with control and hSgo2 siRNAs were probed with anti–CENP-F, anti-hSgo2, and anti-MCAK antibodies. The slower migrating MCAK is hyperphosphorylated (p-MCAK). (D) Cells transfected with control, MCAK, and hSgo2 siRNAs were treated with a high dose of nocodazole to depolymerize microtubules, and then costained for ACA, hSgo2, and MCAK.
Functionally, cells depleted of MCAK exhibited chromosome attachment defects (Fig. S4, B and C) that delayed mitotic exit (Fig. 3 E) in a manner that was similar to when hSgo2 was depleted from cells. We next tested whether MCAK might be physically associated with hSgo2 in mitotic HeLa cells. MCAK antibodies immunoprecipitated detectable amounts of hSgo2, but hSgo2 immunoprecipitates did not contain detectable MCAK (Fig. S4 D). We found that hSgo2 was associated with PP2A, as previously reported (Fig. S4 D; Kitajima et al., 2006). This interaction was confirmed by the fact that PP2A copurified with a transfected GST/hSgo2, but not with GST alone or GST/BubR1 (Fig. S4 E). As interaction between MCAK and hSgo2 was not detected by yeast two-hybrid assay (unpublished data), this, along with the immunoprecipitation results, suggest that MCAK is unlikely to form a stable complex with hSgo2.
hSgo2 is not essential for chromatid cohesion
As cohesion defects were not observed in the experiments described above, we prepared metaphase spreads to directly assess cohesion (Fig. S5 A, available at http://www.jcb.org/cgi/content/full/jcb.200701122/DC1). Cells transfected with control, and hSgo1, hSgo2, and MCAK siRNAs were blocked in mitosis with nocodazole before harvesting. Consistent with the ∼33-fold increase in the frequency of separated chromatids seen in hSgo1-depleted cells over controls (Kitajima et al., 2006), we observed a 37.4-fold increase in separated chromatids (86 vs. 2.3% for hSgo1 and control siRNAs, respectively) in cells depleted of hSgo1 as compared with controls. Depletion of hSgo2 increased the frequency of separated chromatids by 5.3-fold over controls (12.3 vs. 2.3%). This contrasts with the ∼15-fold increase in separated chromatids reported for cells depleted of Tripin/hSgo2 (Kitajima et al., 2006). Depletion of MCAK increased the frequency of separated chromatids by 3.5-fold over controls (8 vs. 2.3%). In our hands, the frequency of premature chromatid separation in cells depleted of hSgo2 (and MCAK) is over sevenfold lower than seen in cells depleted of hSgo1.
hSgo2 is essential for correcting aberrant attachments
The time lapse studies showed that chromosomes in hSgo2-depleted cells were consistently arranged in a ring before they reached the spindle equator. These rings are not a consequence of unseparated spindle poles, as tubulin staining revealed that the chromosomes were positioned in between a bipolar spindle (unpublished data). We next compared the microtubule attachments at kinetochores of control, hSgo2-, and MCAK-depleted cells (Fig. 5 A). Cells were first briefly exposed to the cold to enrich for stable kinetochore microtubules. In control metaphase cells, sister kinetochores established end-on attachments to microtubules from opposite poles. Cold treatment reduced the density of microtubules in hSgo2-depleted cells, suggesting fewer stable kinetochore attachments. The attachments that were observed included merotelic connections (one kinetochore attached to microtubules from opposite poles), and some with syntelic connections (both kinetochores attached to one pole; Fig. 5 A). Likewise, cells depleted of MCAK exhibited similar attachment defects seen in cells depleted of hSgo2 (Fig. 5 A). These defects, if unresolved by the time the cell enters anaphase, would contribute to lagging chromosomes.
Figure 5.

Kinetochores depleted of hSgo2 exhibit attachment defects. (A) Metaphase cells transfected with control, hSgo2, and MCAK siRNAs were chilled as previously described (Lampson and Kapoor, 2005) before fixing and staining for ACA and tubulin. Images of whole cells are from maximum projections. Insets are images from a single optical slice. (B) Control (top row) and hSgo2 siRNA-transfected cells (middle and bottom rows) were costained with hSgo2 and Mad1 to monitor microtubule attachment status at aligned (middle row) and unaligned (bottom row) kinetochores. ACA was used to identify kinetochores. (C) To measure the interkinetochore distance, sister kinetochores were identified by pairs of Bub1 foci that flanked Aurora B (not depicted). Interkinetochore distances (n > 40) of aligned chromosomes in control metaphase- (1), hSgo2- (2), and MCAK-depleted (3) cells and in cells treated with a low dose of nocodazole (4). Interkinetochore distances of unattached kinetochores at low dose (5), high dose nocodazole (6), and in hSgo2-depleted cells (7). Black bars represent the mean.
We next examined Mad1 localization at kinetochores to assess the microtubule attachment status in cells depleted of hSgo2. Mad1 was clearly detectable at kinetochores that did not have microtubule attachments (Fig. 5 B, bottom). In cells that have reached metaphase, Mad1 was detected at some, but not at the majority of kinetochores (unpublished data). Thus, the metaphase delay may be caused by the few remaining kinetochores that have not fully attached to the spindle and were generating the “wait for anaphase” signal. Indeed, we were able to identify some metaphase cells that lacked any detectable Mad1 staining (Fig. 5 B, middle). In the hSgo2-depleted cells, we observed a 25-fold difference in Mad1 staining intensity between unattached and attached kinetochores. This difference is similar to the 30-fold difference that was seen in control cells (Fig. S5 B). Thus, the magnitude of Mad1 reduction at the attached kinetochores in hSgo2-depleted cells is similar to that seen in control metaphase cells that are ready to exit mitosis.
The absence of Mad1 from the bipolar attached kinetochores in the hSgo2-depleted cells suggested that the kinetochores were saturated with microtubules. We then measured the interkinetochore distance to assess the level of tension (Fig. 5 C). The mean interkinetochore distance of the attached kinetochores in hSgo2-depleted cells was 1.9 μm, as compared with 2.2 μm seen in normal bipolar attached kinetochores (P = 5.6 × 10−5). Consistent with the dependence of MCAK localization on hSgo2, the mean interkinetochore distance in MCAK-depleted cells was 1.8 μm. The magnitude of reduction, however, was not as great as when microtubule dynamics was suppressed by nocodazole (from 2.2 to 1.5 μm). The mean interkinetochore distance of unattached kinetochores in cells depleted of hSgo2 (1.0 μm) was virtually identical to the unattached kinetochores in cells treated with low (1.1 μm) and high (1.0 μm) concentrations of nocodazole. Consistent with the loss of MCAK (Andrews et al., 2004; Kline-Smith et al., 2004), depletion of hSgo2 reduced but did not abolish, tension between attached kinetochores. However, the reduction in interkinetochore distance may not solely be ascribed to reduced tension as it may also result from structural defects that were observed at the EM level (see Fig. 7).
Figure 7.

Kinetochore ultrastructure is altered in cells depleted of hSgo2 and Aurora B. EM images of thin sections of mitotic cells that were transfected with control (A), hSgo2 (B and C), and Aurora B (D) siRNAs. Insets show enlarged views of kinetochores indicated by the white and black arrows. (A) Normal kinetochores (n = 32) with discrete inner (small white arrow) and outer plates (large white arrow). (B) hSgo2-depleted kinetochores (n = 12) reveal an outer plate, but lack a discernable inner plate, and the subjacent chromatin appears undercondensed (bracket). (C) Example of a C-shaped kinetochore with a prominent fibrous corona. (D) Cells depleted of Aurora B (n = 24) exhibit C-shaped kinetochores.
Recruitment of hSgo2 to the inner centromere depends on Bub1 and Aurora B
To understand how hSgo2 is recruited to the inner centromere, we tested its dependence on hBUB1, as both proteins appeared at this location at about the same time (Fig. 1 A). Cells depleted of hBUB1 by siRNA failed to recruit hSgo2 to the inner centromere (Fig. 6). However, the assembly of hBUB1 at kinetochores was not dependent on hSgo2 (unpublished data). Although hBUBR1 localization depends on hBUB1(Johnson et al., 2004), hSgo2 localization was not dependent on hBUBR1. Consistent with the dependence of hSgo2 localization on hBUB1, we confirmed that MCAK localization was also dependent on hBUB1 (Fig. 6; Liu et al., 2006). In addition, we found that hSgo2 localization was also dependent on Aurora B (Fig. 6). This finding is consistent with studies that showed MCAK localization was also dependent on Aurora B (Andrews et al., 2004; Lan et al., 2004). The localization of Aurora B kinase, however, was not dependent on hSgo2. Depletion of hSgo2 also did not affect other chromosome passengers, such as INCENP and survivin (Fig. S5 C). Thus, the dependence of MCAK localization on hBUB1 and Aurora B is likely mediated through hSgo2.
Figure 6.

hSgo2 localization depends on hBuB1 and Aurora B. Cells were transfected with control, hBuB1, hBuBR1, and AuroraB siRNAs and stained with the indicated antibodies. Samples were also costained with hSgo2, MCAK, or ACA. Exposure times were identical between control and siRNA samples.
Kinetochore ultrastructure is disrupted by the loss of hSgo2
We next conducted EM studies to evaluate the contribution of hSgo2 to the organization of the kinetochore at the ultrastructure level. A trilaminar kinetochore with discernable outer and inner plates was clearly visible in control mitotic cells (Fig. 7). The majority of mitotic cells from hSgo2 siRNA-treated cultures contained clusters of chromosomes (Fig. 7) that were likely fixed at the “ring” stages shown in Fig. 3 C. Short microtubules could be seen to extend from the centrosome region to the chromosomes. The chromosomes appeared uniformly condensed and kinetochores possessed a clearly defined outer plate. However, the inner plate was not discernable as the region between the outer plate and the subadjacent heterochromatin, appeared undercondensed or expanded (Fig. 7, inset). Cells whose kinetochore assumed a C-shaped morphology and a very prominent fibrous corona were identified (Fig. 7). Microtubules were generally not found in association with these kinetochores and probably corresponded to cells that had just entered mitosis. Consistent with the fact that hSgo2 localization depends on Aurora B, cells depleted of Aurora B also contained C-shaped kinetochores (Fig. 7). As C-shaped kinetochores were not seen in control samples, the EM data suggest that hSgo2 contributes to the higher organization of the kinetochore.
Discussion
Consistent with recent findings, we found that hSgo2 exhibits a dynamic localization pattern (Kitajima et al., 2006). hSgo2 accumulates at an early stage of kinetochore assembly when hBUB1 is first detected at the nascent kinetochore. At this time, kinetochores have not resolved into discrete pairs, as hBUB1 appears as single spot instead of pairs of foci. Once kinetochore pairs were resolved, hSgo2 was concentrated at the inner centromere. hSgo2 localization was not always centered between kinetochores because it tended to skew toward the leading kinetochore, as was reported for MCAK. Once stable bipolar attachments are made, hSgo2 was found to span the width of the centromere and partially overlap with the kinetochore, as was shown for MCAK. Consistent with the recent finding that the localization of Sgo2 in mouse was sensitive to tension (Gomez et al., 2007), we found that the extent of the overlap between hSgo2 and kinetochores (based on hBUB1 colocalization) was reduced when tension was reduced by nocodazole treatment. Thus, some aspects of the dynamic localization pattern of hSgo2 within the centromere–kinetochore complex appear to be sensitive to tension. However, we cannot rule out the role of microtubule attachments in the redistribution of hSgo2 from the inner centromere toward the kinetochore.
Functionally, we established that hSgo2 is essential for recruiting MCAK to the inner centromere. Cells depleted of hSgo2 exhibited a quantitative depletion of MCAK from centromeres by >95%. Although we detected some hSgo2 in immunoprecipitates obtained with MCAK antibodies, hSgo2 immunoprecipitates did not contain MCAK. As neither gel-filtration analysis of HeLa lysates nor yeast two-hybrid assays indicated that MCAK associated with hSgo2, MCAK is unlikely to be recruited to the centromere in a stable complex with hSgo2. Our studies also showed that hSgo2 localization is dependent on Aurora B. As Aurora B has been shown to specify the recruitment of MCAK to the centromere (Andrews et al., 2004; Lan et al., 2004), the combined data suggests the following linear assembly pathway: Aurora B → hSgo2 → MCAK. The relationship may be more complex, given how Aurora B is thought to specify MCAK localization. MCAK has been shown to be a substrate of Aurora B kinase in vitro and in vivo (Andrews et al., 2004; Lan et al., 2004), but it is not entirely clear whether recruitment of MCAK depends on these phosphorylation sites. This is based on the finding that mutating all five of the MCAK phosphorylation sites to phosphodefective and phosphomimic mutants did not prevent their assembly to the centromere (Andrews et al., 2004). Instead, the distribution of MCAK between the inner centromere and the kinetochore seemed to be affected by its phosphorylations status (Andrews et al., 2004; Lan et al., 2004). This suggests that the role of Aurora B in recruiting MCAK to the inner centromere may differ from its role in regulating the dynamic distribution of MCAK within the centromere and kinetochore. Recruitment of MCAK may depend on other proteins, such as hSgo2, that are also targets of Aurora B. One role for the PP2A that is associated with hSgo2 might be to locally regulate the spatial distribution of MCAK within the centromere and kinetochore. Finally, we showed that hBub1 is also required by hSgo2 and MCAK to localize to centromeres. As hBUB1 and Aurora B do not depend on each other for their localization to kinetochores, the assembly of hSgo2 and MCAK appears to depend on two parallel pathways. The significance of this is unclear, but the use of multiple pathways appears to be a common feature used for kinetochore assembly (Liu et al., 2006).
The importance of hSgo2 in recruiting MCAK to centromeres also provides a mechanistic explanation for the kinetochore attachment defects seen in cells depleted of hSgo2. Time-lapse studies of chromosome dynamics in cells depleted of hSgo2 showed a delay in congression to the spindle equator. Whereas virtually all chromosomes eventually achieved alignment, kinetochores with merotelic and syntelic attachments were identified. These defective attachments accumulated because no MCAK was present to sever them. This interpretation is supported by the fact that similar defects were observed when MCAK was directly depleted from cells. Failure to resolve these defective attachments in the hSgo2-depleted cells before anaphase onset explains the high incidence of lagging chromosomes once cells enter anaphase.
Given that the microtubule depolymerase activity of MCAK has been shown to be negatively regulated by phosphorylations mediated by Aurora B kinase (Andrews et al., 2004; Lan et al., 2004), there may be another role for the PP2A that is associated with hSgo2. PP2A may provide a way to locally control MCAK activity so that only defective microtubule attachments are severed, while productive attachments are preserved. In this scenario, PP2A associated with hSgo2 may dephosphorylate and activate MCAK depolymerase activity. This model implies that hSgo2/PP2A, MCAK, and Aurora B activities must be highly regulated so that they can spatially restrict their actions to just a single defective attachment.
Our interpretation of the PP2A–hSgo2 interaction differs from that proposed for how the PP2A–hSgo1 interaction maintains centromeric cohesion (Kitajima et al., 2006; Riedel et al., 2006; Tang et al., 2006). As with hSgo1, hSgo2 is thought to target PP2A to the inner centromere, where it can neutralize the phosphorylation of cohesin subunit Scc3 introduced by Polo kinase 1 (Kitajima et al., 2006). This is supported by their finding that depletion of hSgo2 resulted in a high incidence (15-fold increase) of prematurely separated chromatids. However, we found that cells depleted of ∼95% of hSgo2 only exhibited a small increase in chromatid separation, which was also seen in cells depleted of MCAK. We believe that the loss of hSgo2 and MCAK from the inner centromere affects the higher-order organization of this region in a way that indirectly weakens centromeric cohesion. This is partially confirmed at the EM level, which showed that depletion of hSgo2 and Aurora B compromised the organization of the inner kinetochore and the subjacent chromatin. Given that Aurora B lies upstream of hSgo2, we would expect that its removal should lead to a dramatic increase in the frequency of separated chromatids if hSgo2 is, indeed, essential for cohesion. On the contrary, inhibition of Aurora B kinase has been reported to not affect chromatid cohesion (Hauf et al., 2003; McGuinness et al., 2005). At present, we cannot provide a satisfactory explanation for the discrepancy in the functional roles of hSgo2 presented by the two studies. The microtubule attachment defects we identified in cells depleted of hSgo2 is fully consistent with its role in recruiting MCAK. It is formally possible that we failed to see cohesion defects because depletion of ∼95% of hSgo2 was insufficient to manifest the phenotype. It is noteworthy that a recent study showed that Sgo2 in fission yeast is not essential for cohesion (Kawashima et al., 2007; Vanoosthuyse et al., 2007). Instead, both studies showed that Sgo2 facilitated chromosome biorientation, most likely via its role in recruiting the Aurora kinase complex to the centromere. Although our results differ in respect to the fact that hSgo2 was not important for recruiting chromosome passenger complexes to the centromere, its role in kinetochore attachments is consistent with those reported for the fission yeast Sgo2.
Materials and methods
DNA cloning and antibodies
hSgo2 was PCR amplified from a cDNA library (Marathon-ready cDNA; CLONTECH Laboratories, Inc.) and confirmed by sequence analysis. The full-length cDNA or fragments were cloned into pENTR (Gateway) to facilitate transfer into mammalian and bacterial expression vectors by in vitro recombination reactions. The cDNA encoding N-terminal 469 aa of hSgo2 was inserted into the bacterial expression vector pDEST17 (Gateway) and recombinant protein was purified by Ni-beads under denaturing condition. Purified protein was used to immunize animals and coupled to Affigel-10 (BioRad Laboratories). The affinity column was used to purify antibodies from rabbit and rat sera.
Cell culture and RNA interference
HeLa cells were grown in DME + 10% FBS in a humidified incubator at 37°C. Nocodazole was used at 20 nM (low) and 60 nM (high) final concentrations, respectively.
SMARTpool and single siRNAs targeting hSgo1 (Salic et al., 2004) and hSgo2 (siRNA 1, 2, 3, and 4 sense sequences were as follows: UCAAAGACAUUACCUGAUAUU, GAACACAUUUCUUCGCCUAUU, UCGGAAGUGUUAUUUCUUAUU, and GAGAAACGCCCAGUCUAUUUU) were obtained from Dharmacon. siRNAs were diluted in serum-free OptiMEM and HiPerfect (QIAGEN) as per the manufacturer's instructions and added to cells so that the final concentration of siRNA was 20 nM. 24–36 h after transfection, cells were fixed and stained or lysed in SDS sample buffer.
Microscopy
Cells were fixed for 7 min in freshly prepared 3.5% paraformaldehyde/PBS, pH 6.9, extracted in KB (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, and 0.1% BSA) plus 0.2% Triton X-100 for 4 min at room temperature, and then rinsed in KB. In some cases, cells were preextracted for 2 min before fixing. Primary and secondary antibodies were diluted in KB and added to coverslips for 30 min at 37°C in a humidified chamber. Antibodies to tubulin (Sigma-Aldrich), Aurora B (BD Biosciences), and survivin (Novus Biologicals) were obtained commercially. Human ACA, INCENP, and MCAK antibodies were gifts from J.B. Rattner (University of Alberta, Calgary, Canada), W. Earnshaw (Edinburgh University, Edinburgh, UK), and L. Wordeman (University of Washington, Seattle, WA), respectively. Antibodies to hBUB1, hBUBR1, hBUB3, and Mad1 were obtained from our laboratory (Chan et al., 1998; Jablonski et al., 1998; Campbell et al., 2001). Antibodies were used at a final concentration of 0.5–1.0 μg/ml. Secondary antibodies conjugated to Alexa Fluor 488, 555, and 647 (Invitrogen) were used at 1 μg/ml. Images were visualized with a 100×/1.4 NA objective on a microscope (Eclipse TE2000S; Nikon) and 0.5-μm image stacks were captured with a charge-coupled device camera (Roper Scientific). Images are presented as maximum projections and quantitated as previously described (Hoffman et al., 2001). Deconvolution was conducted with AutoQuant (Media Cybernetics).
For time-lapse studies, HeLa (GFP/H2B) were plated onto glass-bottomed 35-mm dishes (MakTek) in Hepes-buffered, phenol red–free medium, transfected with siRNA, and imaged with an UltraView spinning disc confocal microscope. Images were captured every 3–5 min overnight at 37°C. For FRAP experiments, HeLa cells were transfected with Lipofectamine 2000 (Invitrogen). GFP-labeled kinetochores were imaged with a 63× objective on a multiphoton laser scanning microscope (NLO510; Carl Zeiss MicroImaging, Inc.) that is equipped with a heated stage and objective heater. FRAP was performed essentially as previously described (Howell et al., 2000). LSM software (Carl Zeiss MicroImaging, Inc.) was used to measure integrated fluorescence intensities of kinetochores. The intensities are normalized as the percentage of recovery. The normalized data was fit to the nonlinear regression curve in Prism (Graftpad Software).
Chromosome spreads were prepared as previously described (Henegariu et al., 2001). In brief, mitotic cells were removed by shake-off, pelleted, hypotonically swollen in 75 mM KCl at 37°C for 20 min. Cells were pelleted, fixed with methanol/glacial acetic acid (3:1) for 5–10 min, dropped onto clean glass slides, and allowed to air dry. Slides were rehydrated in an 80°C steam bath for several seconds, dried on a 70°C heatblock, and stained with DAPI.
For EM, HeLa cells transfected with siRNAs were fixed in 3% glutaraldehyde and 0.2% tannic acid in 200 mM Na cacodylate buffer for 1 h at room temperature. Postfixation was in 2% OsO4 for 20 min. The cells were dehydrated in ethanol, and then infiltrated with Polybed 812 resin (Polysciences). Polymerization was performed at 60°C for 24 h. Silver-gray sections were cut with an ultramicrotome (Leica) equipped with a Diamond knife, and sections were stained with uranyl acetate and lead citrate and examined in an electron microscope (H-7000; Hitachi).
Online supplemental material
Fig. S1 shows that the specificity of hSgo2 antibody and the efficiency of hSgo2 siRNAs depletion by Western blot and immunofluorescence. Fig. S2 shows the turnover rates of GFP/hSgo2 at kinetochores at different cell cycle phases as determined by FRAP. Fig. S3 shows that cells depleted of hSgo2 by siRNA exhibit anaphase bridges and delocalization of MCAK from the centromere. In contrast, depletion of Sgo1 does not affect the centromeric localization of MCAK. Fig. S4 shows the effects of hSgo2 depletion on the phosphorylation state of MCAK, and cells depleted of MCAK exhibit mitotic defects similar to depletion of hSgo2. Coimmunoprecipitation experiments reveal a weak interaction between hSgo2 and MCAK and a clear association with endogenous and transfected hSgo2 with PP2A-C. Fig. S5 shows that hSgo2 is neither essential for centromere cohesion nor centromeric localization of chromosomal passenger proteins. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200701122/DC1.
Supplementary Material
Acknowledgments
We are grateful for the expert technical services provided by J. Hittle, B. Conner, and the Lab Animal, Hybridoma, Oligo, and DNA synthesis facilities at FCCC. Special thanks to P. Lau for the yeast two-hybrid analysis. We also acknowledge the support of the Cross Cancer Institute Cell Imaging Facility, which is where the FRAP experiments were performed.
G.K.T. Chan is supported by a Canadian Institute of Health Research (CIHR) New Investigator Award, CIHR operating grant MOP-57723, and the Alberta Cancer Board (ACB). J. Famulski is supported by a studentship from the ACB. J.B. Rattner is supported by a grant from the National Science and Engineering Council of Canada. T.J. Yen, R. Muschel, and G.D. Kao are supported by grant PO1 CA75138. T.J. Yen is also supported by National Institutes of Health grants CA099423 and core grant CA06927, The Leukemia and Lymphoma Society, and an Appropriation from the Commonwealth of Pennsylvania.
Abbreviation used in this paper: ACA, anticentromere antibodies.
References
- Andrews, P.D., Y. Ovechkina, N. Morrice, M. Wagenbach, K. Duncan, L. Wordeman, and J.R. Swedlow. 2004. Aurora B regulates MCAK at the mitotic centromere. Dev. Cell. 6:253–268. [DOI] [PubMed] [Google Scholar]
- Campbell, M.S., G.K. Chan, and T.J. Yen. 2001. Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J. Cell Sci. 114:953–963. [DOI] [PubMed] [Google Scholar]
- Chan, G.K., B.T. Schaar, and T.J. Yen. 1998. Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J. Cell Biol. 143:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini, D., X. Wan, C.B. Hirel, and E.D. Salmon. 2006. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr. Biol. 16:1711–1718. [DOI] [PubMed] [Google Scholar]
- Desai, A., S. Verma, T.J. Mitchison, and C.E. Walczak. 1999. Kin I kinesins are microtubule-destabilizing enzymes. Cell. 96:69–78. [DOI] [PubMed] [Google Scholar]
- Gomez, R., A. Valdeolmillos, M.T. Parra, A. Viera, C. Carreiro, F. Roncal, J.S. Rufas, J.L. Barbero, and J.A. Suja. 2007. Mammalian SGO2 appears at the inner centromere domain and redistributes depending on tension across centromeres during meiosis II and mitosis. EMBO Rep. 8:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf, S., R.W. Cole, S. LaTerra, C. Zimmer, G. Schnapp, R. Walter, A. Heckel, J. van Meel, C.L. Rieder, and J.M. Peters. 2003. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henegariu, O., N.A. Heerema, L. Lowe Wright, P. Bray-Ward, D.C. Ward, and G.H. Vance. 2001. Improvements in cytogenetic slide preparation: controlled chromosome spreading, chemical aging and gradual denaturing. Cytometry. 43:101–109. [PubMed] [Google Scholar]
- Hoffman, D.B., C.G. Pearson, T.J. Yen, B.J. Howell, and E.D. Salmon. 2001. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell. 12:1995–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, B.J., D.B. Hoffman, G. Fang, A.W. Murray, and E.D. Salmon. 2000. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 150:1233–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski, S.A., G.K. Chan, C.A. Cooke, W.C. Earnshaw, and T.J. Yen. 1998. The hBUB1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma. 107:386–396. [DOI] [PubMed] [Google Scholar]
- Johnson, V.L., M.I. Scott, S.V. Holt, D. Hussein, and S.S. Taylor. 2004. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 117:1577–1589. [DOI] [PubMed] [Google Scholar]
- Katis, V.L., M. Galova, K.P. Rabitsch, J. Gregan, and K. Nasmyth. 2004. Maintenance of cohesin at centromeres after meiosis I in budding yeast requires a kinetochore-associated protein related to MEI-S332. Curr. Biol. 14:560–572. [DOI] [PubMed] [Google Scholar]
- Kawashima, S.A., T. Tsukahara, M. Langegger, S. Hauf, T.S. Kitajima, and Y. Watanabe. 2007. Shugoshin enables tension-generating attachment of kinetochores by loading Aurora to centromeres. Genes Dev. 21:420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrebrock, A.W., D.P. Moore, J.S. Wu, and T.L. Orr-Weaver. 1995. Mei-S332, a Drosophila protein required for sister-chromatid cohesion, can localize to meiotic centromere regions. Cell. 83:247–256. [DOI] [PubMed] [Google Scholar]
- Kinoshita, K., T.L. Noetzel, I. Arnal, D.N. Drechsel, and A.A. Hyman. 2006. Global and local control of microtubule destabilization promoted by a catastrophe kinesin MCAK/XKCM1. J. Muscle Res. Cell Motil. 27:107–114. [DOI] [PubMed] [Google Scholar]
- Kitajima, T.S., S.A. Kawashima, and Y. Watanabe. 2004. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature. 427:510–517. [DOI] [PubMed] [Google Scholar]
- Kitajima, T.S., S. Hauf, M. Ohsugi, T. Yamamoto, and Y. Watanabe. 2005. Human Bub1 defines the persistent cohesion site along the mitotic chromosome by affecting Shugoshin localization. Curr. Biol. 15:353–359. [DOI] [PubMed] [Google Scholar]
- Kitajima, T.S., T. Sakuno, K. Ishiguro, S. Iemura, T. Natsume, S.A. Kawashima, and Y. Watanabe. 2006. Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature. 441:46–52. [DOI] [PubMed] [Google Scholar]
- Kline-Smith, S.L., A. Khodjakov, P. Hergert, and C.E. Walczak. 2004. Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol. Biol. Cell. 15:1146–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson, M.A., and T.M. Kapoor. 2005. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat. Cell Biol. 7:93–98. [DOI] [PubMed] [Google Scholar]
- Lan, W., X. Zhang, S.L. Kline-Smith, S.E. Rosasco, G.A. Barrett-Wilt, J. Shabanowitz, D.F. Hunt, C.E. Walczak, and P.T. Stukenberg. 2004. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr. Biol. 14:273–286. [DOI] [PubMed] [Google Scholar]
- Liu, S.T., J.B. Rattner, S.A. Jablonski, and T.J. Yen. 2006. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J. Cell Biol. 175:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston, A.L., W.H. Tham, H. Shah, and A. Amon. 2004. A genome-wide screen identifies genes required for centromeric cohesion. Science. 303:1367–1370. [DOI] [PubMed] [Google Scholar]
- McGuinness, B.E., T. Hirota, N.R. Kudo, J.M. Peters, and K. Nasmyth. 2005. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol. 3:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, D.P., A.W. Page, T.T. Tang, A.W. Kerrebrock, and T.L. Orr-Weaver. 1998. The cohesion protein MEI-S332 localizes to condensed meiotic and mitotic centromeres until sister chromatids separate. J. Cell Biol. 140:1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsky, B.A., C. Kung, K.M. Shokat, and S. Biggins. 2006. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat. Cell Biol. 8:78–83. [DOI] [PubMed] [Google Scholar]
- Rabitsch, K.P., J. Gregan, A. Schleiffer, J.P. Javerzat, F. Eisenhaber, and K. Nasmyth. 2004. Two fission yeast homologs of Drosophila Mei-S332 are required for chromosome segregation during meiosis I and II. Curr. Biol. 14:287–301. [DOI] [PubMed] [Google Scholar]
- Riedel, C.G., V.L. Katis, Y. Katou, S. Mori, T. Itoh, W. Helmhart, M. Galova, M. Petronczki, J. Gregan, B. Cetin, et al. 2006. Protein phosphatase 2A protects centromeric sister chromatid cohesion during meiosis I. Nature. 441:53–61. [DOI] [PubMed] [Google Scholar]
- Rivera, T., and A. Losada. 2006. Shugoshin and PP2A, shared duties at the centromere. Bioessays. 28:775–779. [DOI] [PubMed] [Google Scholar]
- Salic, A., J.C. Waters, and T.J. Mitchison. 2004. Vertebrate shugoshin links sister centromere cohesion and kinetochore microtubule stability in mitosis. Cell. 118:567–578. [DOI] [PubMed] [Google Scholar]
- Suzuki, H., N. Akiyama, M. Tsuji, T. Ohashi, S. Saito, and Y. Eto. 2006. Human Shugoshin mediates kinetochore-driven formation of kinetochore microtubules. Cell Cycle. 5:1094–1101. [DOI] [PubMed] [Google Scholar]
- Tanaka, T.U., N. Rachidi, C. Janke, G. Pereira, M. Galova, E. Schiebel, M.J. Stark, and K. Nasmyth. 2002. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 108:317–329. [DOI] [PubMed] [Google Scholar]
- Tang, Z., Y. Sun, S.E. Harley, H. Zou, and H. Yu. 2004. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc. Natl. Acad. Sci. USA. 101:18012–18017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., H. Shu, W. Qi, N. Mahmood, M.C. Mumby, and H. Yu. 2006. PP2A is required for centromeric localization of Sgo1 and proper chromosome segregation. Dev. Cell. 10:575–585. [DOI] [PubMed] [Google Scholar]
- Vanoosthuyse, V., S. Prykhozhij, and K.G. Hardwick. 2007. Shugoshin2 regulates localization of the chromosomal passenger proteins in fission yeast mitosis. Mol. Biol. Cell. DOI: 10.1091/mbc.E06-10-0890. [DOI] [PMC free article] [PubMed]
- Vaur, S., F. Cubizolles, G. Plane, S. Genier, P.K. Rabitsch, J. Gregan, K. Nasmyth, V. Vanoosthuyse, K.G. Hardwick, and J.P. Javerzat. 2005. Control of Shugoshin function during fission-yeast meiosis. Curr. Biol. 15:2263–2270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}