

Abstract
Recent advances in computational biology have made it possible to map the complete network and energy profile of gas migration pathways inside proteins. Although networks of O2 pathways have already been characterized for a small number of proteins, the general properties and locations of these pathways have not been previously compared between proteins. In this study, maps of the O2 pathways inside 12 monomeric globins were computed. It is found that, despite the conserved tertiary structure fold of the studied globins, the shape and topology of O2 pathway networks exhibit a large variability between different globins, except when two globins are nearly identical. The locations of the O2 pathways are, however, found to be correlated with the location of large hydrophobic residues, and a similar correlation is observed in two unrelated protein families: monomeric globins and copper-containing amine oxidases. The results have implications for the evolution of gas pathways in proteins and for protein engineering applications involving modifications of these pathways.
INTRODUCTION
For many classes of proteins, enzymatic reaction with, or binding to, gas molecules is an essential component of their function. Such proteins often bind gas molecules by means of buried active sites consisting of metal ions or metal-containing compounds. Gas molecules such as O2 must make their way across the protein's interior to reach these active sites. In the majority of cases, permanent gas channels can neither be detected nor found present in the protein's static structure; instead, the migrating gas molecules take advantage of a network of transient pathways, which encompass a number of highly favorable gas-holding sites connected by less favorable regions which are used by gas molecules to hop between the holding sites. These pathways are due to thermal fluctuations of the protein (1). By monitoring the occurrence of these transient pathways over time and making use of recent methodological advances such as volumetric gas accessibility maps (2) and implicit ligand sampling (3), one can comprehensively map and describe networks of gas migration pathways inside proteins.
For a large number of proteins that interact with O2, finding the location of O2 migration pathways has important implications. For example, locating the O2 pathways in oxygenases and O2-consuming oxidases provides important clues regarding their enzymatic activity and operating mechanism. Also, the elucidation of O2 pathways in hydrogenases is assisting efforts aiming to block O2 access to the hydrogenase active sites, thus making this protein useful for commercial hydrogen gas production (4–6). To date, complete maps of O2 pathway networks have only been computed for a small number of proteins, including CpI hydrogenase (2), sperm whale myoglobin (Mb) (3), AQP1 aquaporin (7), and a set of copper amine oxidases (8). As more proteins are scrutinized in terms of their O2 migration pathways, one will gain a better grasp of how gases are transported inside proteins, discern patterns of how such pathways are conserved across protein families, and develop rules of thumb for quickly identifying these pathways. In this article, we address the question of O2 pathway conservancy within a given protein fold by computing and comparing maps of the O2 pathway networks across a range of proteins from the globin superfamily. The comprehensive comparison provides a basic understanding as to how O2 pathways are formed inside proteins and, in turn, provides clues as to how to engineer gas accessibility in proteins.
Globins are a large and ancient family of proteins for which all members, with few exceptions, share an exceptionally well-conserved tertiary structure: the globin fold. At the heart of this fold lies a prosthetic group, the heme, universally used by globins to reversibly bind to, and to temporarily hold, O2 and other gas ligands. This investigation focuses on three types of pentacoordinated globins: monomeric hemoglobins (Hbs), which transport O2 throughout entire organisms, Mbs, which store and transport O2 within muscle cells, and leghemoglobins (Lbs), which store, transport, and/or scavenge O2 to maintain a population of symbiotic bacteroids in the root nodules of symbiotic plants (9). We stay clear of hexacoordinated globins such as neuroglobin and cytoglobin, which exhibit poorly understood behavior regarding how O2 binds to their heme.
In addition to binding O2, many globins have other physiological functions. For example, although the role of Mb has been long considered to be well established (10–12), recent studies have shown Mb to also be involved in secondary roles (12,13) like scavenging and inactivating nitric oxide (NO) (14) and weak peroxidase activity (15). Many invertebrate Hbs also possess a number of interesting characteristics, such as the ability to react with sulfide (16) and the ability to tune their affinities to O2 depending on their environment. By forming multimeric assemblies (16–18), many Hb monomers can bind gases such as O2 and CO2 cooperatively by making their affinity for O2 depend on the O2 occupancy of the neighboring monomers, either through intermonomer conformational changes or through multimeric association/dissociation. It is clear that despite the well-studied nature of the globin family, there remains a large number of globin properties relating to their structure and gas-conduction properties that is still poorly understood and possibly not even known.
In this article, we focus on the O2-transporting role of the globin protein matrix. A globin's main function is performed by its heme, which binds gas ligands for extended periods of time. Consequently, one can regard the conserved protein fold surrounding the heme as merely a shell. Nevertheless, this “shell” provides important functionality. First, the protein shell protects the heme from oxidizing into an inactive ferric state, which would happen if the heme were to float freely in solution. Second, the protein component strongly modulates the environment of the heme-bound ligand and, thus, influences its binding affinity, its binding rates, and the globin's relative selectivity for various ligands. Finally, the protein matrix provides cavities and pathways for gas ligands to travel from the exterior solution to the heme and vice versa. Since all globins have an identical heme, the protein shell is what differentiates globins in regard to globin behavior and properties.
A large body of work, both experimental and theoretical, has focused on finding gas ligand pathways inside globins, particularly sperm whale Mb and certain Hbs. In particular, x-ray crystallography in the presence of xenon (19), x-ray crystallography of intermediate states (20), time-resolved x-ray crystallography (21–25), spectroscopy of the geminate recombination process (26–32), and molecular dynamics simulation (33–37) all provide important clues as to how gas molecules make their way into proteins. In terms of the pathways' locations, only the areas of the pathways that are most favorable to O2, often termed “cavities”, “xenon-binding sites”, or “holding regions”, can be observed experimentally since only in these regions is O2 concentrated enough to be observed. The effects of point mutations on overall O2 association and dissociation rates in globins can also be observed experimentally. We complement this work by computing the complete map of O2 pathways inside a broad range of monomeric globins.
METHOD
Simulations were run for a selected set of monomeric globins of known structure, taken from the Protein Data Bank (PDB). In every case, a deoxy form of the globin was created from the PDB coordinates, and any water or gas ligand present in the distal pocket (DP) was removed. For the cases in which only the ferric state of the globin is available (whether unbound or bound to small compounds), the coordinates of the ferric heme were used as starting points, but the hemes were modeled using parameters for the ferrous state.
For every globin, the simulation system was built from the PDB coordinates by adding hydrogen atoms, binding the globin's proximal histidine to the hemes, and picking an appropriate titration state for every histidine based on its immediate environment. The Dowser water placement program (38) was then used to internally solvate the globin atomic structures, though rarely resulting in the placement of additional water molecules. The heme-protein complex was solvated using a water box whose sides exceeded those of the protein by at least 20 Å in all dimensions. A total of 50 mM of NaCl was added, adjusting the relative concentrations of Na+ and Cl− to make the whole system chargeless.
The equilibration protocol used two preequilibration stages: an initial 30 ps simulation stage had the protein and heme fixed and allowed the solvent to relax at constant temperature (300 K) and pressure (1 atm); a 50 ps stage then allowed both protein side chains and solvent to equilibrate while constraining the protein backbone. The entire protein-solvent systems were equilibrated for another 950 ps. Finally, an additional 10 ns of simulation at the same NPT conditions was performed for analysis. The 10 ns simulations were processed using the implicit ligand sampling method, reviewed in Cohen et al. (3) and included with the VMD visualization program (39). The implicit ligand sampling method takes advantage of the fact that weakly interacting particles such as gas molecules embedded in a protein do not distort the protein much. In this approximation, it is possible to estimate the free energy of placing a gas molecule at any point inside the protein by treating the gas ligand as a perturbation and by using a one-step free energy perturbation scheme. This allows us to infer the free energy of placing a gas molecule at any given point in space from the energies of placing the molecule directly into protein conformations taken from the frames of an equilibrium simulation trajectory of the dynamics of the protein without gas. The end result is a three-dimensional potential of mean force (PMF) map of the complete network of O2 migration pathways for each globin.
All simulations were performed using the molecular dynamics program NAMD (40) in combination with the NAMD-G simulation automation engine (41). Simulation parameters were taken from the CHARMM22 force field (42). Particle mesh Ewald, with a resolution of at least 1 Å, was used everywhere for long-range electrostatics. Langevin dynamics and a Langevin piston were used to maintain constant temperature and pressure, respectively. Finally, integration time steps of 1, 2, and 4 ps, respectively, were used for bonded, nonbonded, and long-range electrostatics interactions.
To determine which residue types promote the formation of O2 cavities and pathways, we performed a scan of the O2 PMF profile maps to assess residue proximity to O2 cavities. This was done by computing, for each location on the O2 PMF grid, the number of residues of each type whose side chains lie within 2 Å of that grid point. This quantity is then averaged over all PMF grid points, using the O2 occupancy at each grid point as the weight. Finally, this average is divided by the proportional population of each given residue type to remove the bias caused by having certain residue types being more abundant than others. The propensity 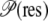 for each residue type to lie near an O2 pathways can thus be evaluated as
for each residue type to lie near an O2 pathways can thus be evaluated as
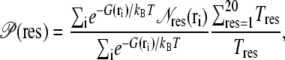 |
(1) |
where the sum over i is over every PMF grid point within a specified region (corresponding to either the interior or the surface of the protein); G(ri) is the PMF at i; 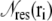 is the number of residues of type “res” whose side chains lie within 2 Å of i; and Tres is the total population of residues of type “res” which lie within 2 Å of any PMF grid point in the region of interest.
is the number of residues of type “res” whose side chains lie within 2 Å of i; and Tres is the total population of residues of type “res” which lie within 2 Å of any PMF grid point in the region of interest.
Using residue locations from the crystal structure, as opposed to trajectory snapshots, produced no discernible change to 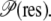 The relative propensities are also not significantly changed when compensated to account for residue size (except for the case of Gly) or when using a different value for the 2 Å distance threshold (tested up to 4 Å).
The relative propensities are also not significantly changed when compensated to account for residue size (except for the case of Gly) or when using a different value for the 2 Å distance threshold (tested up to 4 Å).
RESULTS AND DISCUSSION
O2 pathways in monomeric globins
Although the O2 pathways and cavities in Mbs (particularly sperm whale Mb) have been studied extensively, those in most other globins remain unknown. We investigated the networks of O2 pathways in a broad set of pentacoordinated monomeric globins, listed in Table 1, for which atomic coordinates are available. For each protein, its O2 PMF map was computed using the implicit ligand sampling method (3) (see Methods). These maps provided the locations and energy barriers of the complete network of O2 pathways inside every simulated globin.
TABLE 1.
List of pentacoordinated monomeric globins investigated in this study
| Globin | Species | PDB code |
|---|---|---|
| Myoglobins | ||
| Sperm whale Mb | Physeter catodon | 1A6M, 1A6N (60) |
| Sperm whale Mb (YQR mutant) | Physeter catodon | 1MYZ (24) |
| Horse heart Mb | Equus caballus | 1WLA (61) |
| Sea hare Mb | Aplysia limacina | 1MBA (62) |
| Invertebrate hemoglobins | ||
| Pig roundworm Hb domain I | Ascaris suum | 1ASH (63) |
| Trematode Hb | Paramphistomum epiclitum | 1H97 (64) |
| Marine bloodworm Hb component III | Glycera dibranchiata | 1JF3 (65) |
| Midge HbIII | Chironomus thummi thummi | 1ECO (66) |
| Clam HbI | Lucina pectinata | 1FLP (67) |
| Leghemoglobins | ||
| Yellow lupin Lb II | Lupinus luteus | 1GDJ (68) |
| Soybean Lb A | Glycine max | 1BIN (69) |
For the case of Mbs, we computed the O2 PMFs for sperm whale, horse heart, and sea hare Mb. In the case of sperm whale Mb, we also looked at two additional variants: deoxy-Mb, in which the distal pocket (DP) contains a water molecule, and the sperm whale (L29Y, H64Q, T67R) “YQR-Mb” mutant, designed to mimic the slower association/dissociation rates observed in the Ascaris nematode Hb (43).
The PMF maps for sperm whale oxy-Mb, deoxy-Mb, and YQR-Mb were considered to test the reproducibility of the implicit ligand sampling approach, as well as to observe the magnitude of the changes caused by the presence of water in the DP and by point mutations. The O2 PMF maps for sperm whale oxy-Mb and deoxy-Mbs matched particularly well: every favorable O2 holding region (red, in Fig. 1, a and b) and the O2 pathway interconnections (blue, in Fig. 1, a and b) exhibit an excellent correspondence between the two maps, both in shape and in size, as they should. The YQR-Mb mutant, also, exhibits strong similarities with the other sperm whale Mb, except for the shape of the DP, as expected, which is where the three “YQR” point mutations are located. The variation in shape of the O2 pathways near the Xe1 binding site in YQR-Mb is due to the presence of a crystal water molecule at that location which is not present in the other Mbs. Also, a reduction in the size of the pathways far away from the heme (bottom of Mb in Fig. 1 c) appears to be due to statistical variations in the presence of water molecules inside all Mbs near those locations over the course of the simulations. All in all, despite the effect of trapped water molecules inside the proteins, the maps for all three sperm whale Mbs were the most similar to each other of all globin maps, and the details of these maps were remarkably well reproduced between each other (as well as with the independently computed CO PMF map for sperm whale presented in Cohen et al. (3)). These results lend further credibility to the reproducibility of the implicit ligand sampling approach for mapping gas migration pathways.
FIGURE 1.

O2 PMF maps for various monomeric globins. Shown are the 0 kcal/mol (red) and 1.6 kcal/mol (blue) O2 free energy contours, along with the four sperm whale Mb xenon binding sites as green spheres. The globins are sperm whale (a) oxyMb, (b) deoxyMb, and (c) YQR mutant Mb, (d) horse, and (e) sea hare Mbs, (f) soy, and (i) lupin Lbs, (g) roundworm, (h) trematode, (j) bloodworm, (k) clam, and (l) midge Hbs. The Xe binding sites of sperm whale Mb are shown as green spheres, and the proteins' α-helices are displayed as black lines.
When comparing Mb O2 PMF maps across species, we again see a good agreement between sperm whale Mb and horse Mb as we did between the various sperm whale Mbs, reflecting the fact that these globins are all, to a practical extent, almost the same protein. The comparison becomes interesting, however, when one looks at the O2 PMF for sea hare Mb (Fig. 1 d). Despite the very strong similarities in both function and structure between sperm whale and sea hare Mb, the location of O2 pathways is, surprisingly, very different for these two Mbs.
The mapped O2 pathways provide new insights into the behavior of Mbs. A closer examination of the YQR-Mb O2 PMF map reveals that its DP is much more unfavorable to O2 than the DP of sperm whale oxy-Mb, namely, by ∼3 kcal/mol. Paradoxically, the shape and energy features of the DP do not resemble those of the Ascaris roundworm Hb, which served as a template for YQR-Mb. According to the O2 maps, YQR-Mb's low association/dissociation constants are due to a DP which is unfavorable to O2, resulting in a lower probability of O2 occupation in the DP and lower chance of binding to the heme, rather than having higher energy barriers to reach the DP through the protein matrix, given that the latter is not observed here.
In particular, we find it interesting that the minima of the three-dimensional energy maps occur at the sperm whale Mb's DP, both in the presence (deoxy-Mb) and absence (oxy-Mb) of a water molecule inside it, implying that water does not prevent O2 from reaching the DP. The deoxy-Mb DP is, however, measured to be less favorable to O2 by 3 kcal/mol. Surprisingly, we did not observe any opening of the distal channel (defined as the pathways going through the swinging His-64 “gate”) during any of the 10 ns simulations of sperm whale Mb (which we extended to 25 ns for the case of oxy-Mb) or of horse Mb, resulting in the absence of this pathway in the maps presented in this article. The distal pathway was observed in a previous PMF map of Mb (3); however the initial structure of Mb used in that study contained large crystal deformations (with respect to the other known structures of whale Mb) that may have contributed to this discrepancy in the distal pathway behavior. Other computational studies have, however, reported observing the spontaneous swinging of the distal histidine “gate” in Mb on 10–100 ns timescales (44).
When we extend the comparison to include the O2 pathways for various monomeric invertebrate Hbs, we note a surprising fact. Fig. 1 illustrates the O2 pathways for the 12 simulated globins. Aside from a prominent DP, the various monomeric globins exhibit O2 pathway and cavity locations which are completely different from one Hb to another. These variations are significant and reproducible and cannot at all be attributed to errors in the evaluation of the PMF. Since the globin fold is well conserved among the studied globins and their tertiary structure is near identical (see Fig. 2), our results suggest that the locations of O2 pathways (which in general are the same as those for CO and NO (3)) are not determined by the protein's secondary and tertiary structures, which are conserved.
FIGURE 2.

Structure of 10 monomeric globins are aligned and superimposed, demonstrating the very strong conservation of their secondary structure globin fold. The structures are sperm whale (blue), horse (green), and sea hare (cyan) Mbs, soy (black) and lupin (white) Lbs, roundworm (yellow), trematode (red), and bloodworm (orange) Hbs, clam (pink), and midge (purple) Hbs. The Xe binding sites of sperm whale Mb are shown as green spheres, and the proteins' α-helices are displayed as black lines.
A second notable observation arising from the O2 PMF maps for the invertebrate monomeric Hbs is the large number of exits and pathways present in each globin. Our results suggest that multiple exits and an overall porousness to O2 might be the norm in globins. This is surprising and contrasts with the common assumption of a single O2 entryway in many kinetic models of O2 migration in Mb (22,30), as well as with the O2 PMF maps of the other proteins for which O2 pathways have been mapped: CpI hydrogenase, which was found to be largely impermeable to O2 except along two well-defined and very localized pathways (2), and AQP1 aquaporin (7), for which O2 pathways are only found at the interface of the protein's constituent monomers.
Finally, we studied two Lbs, from lupin and soy, both of which exhibit very similar O2 PMF maps (see Fig. 3 b). The O2 PMF maps for both Lbs show them to be mostly inaccessible to O2, except for a very short and direct exit between the DP and the external solution. The main exit seen in both lupin and soy Lbs (in lupin Lb: Ala-37, Leu-43, His-106, Val-109; in soy Lb: Pro-38, Leu-43, Gln-101, Val-104) is the same as the one reported in lupin Lb by Czerminski and Elber (45) using locally enhanced sampling simulation. Despite the presence of a distal histidine in Lb at the same location as the gating histidine in Mbs, the distal pathway is not present at all in the Lb O2 PMF maps (the distal pathway was in fact observed here for every Mb, even though it was in some cases seen to be blocked by a closed histidine “gate”).
FIGURE 3.

Comparisons of the 1.6 kcal/mol O2 PMF surfaces for similar monomeric globins. The globins are (a) sperm whale Mb (blue) versus horse heart Mb (red), and (b) lupin (blue) versus soy Lb (red).
In comparison with the other monomeric globins from Fig. 1, both Lbs are exceptional in having a single, and conserved, dominant exit next to the heme. The only possible other exit, according to the O2 PMF maps, is an energetically less accessible secondary exit that is still located right next to the primary exit. This is in stark contrast to all the other globins in this study, which are very porous to O2, offering it multiple exits. Both soybean and lupin Lbs must transport O2 to symbiotic Rhizobium bacteroids inside root nodules in the plant, while simultaneously ensuring that as little as possible of this O2 reaches the Rhizobia's nitrogenase enzymes, which are essential to the plant host and which are intolerant to O2. By having a unique exit, the possibility is raised that this exit could be blocked during transport and/or that Lb could deliver O2 to a precise target while ensuring minimal O2 leakage. Our results do not prove such a conclusion; nevertheless, they do show that Lbs have a peculiar O2 pathway arrangement which is compatible with the possibility of their role in sequestering O2 in a way that is not realized by any other globin examined in this study.
Specific residues promoting O2 pathways
From the comparison of the various globin O2 pathway maps performed in this study, it is evident that O2 pathways are not defined by a protein's overall fold, which is conserved across all globins; instead, O2 pathways are found to correlate with the location of certain residue types. Although there is no guarantee that specific residue types have any individual effect on the location of O2 pathways in the protein, we find that certain residues, on average, are more likely than others to be found near O2 pathways. By collecting statistical information regarding the propensity of different residue types to form or not form O2 pathways, one can guide future efforts to manipulate O2 pathways inside proteins (46–48).
Fig. 4 shows the propensity of residues to form O2 pathways and cavities normalized by residue abundance (using Eq. 1), sorted by residue type. The analysis was repeated for two different sets of proteins: a set of nine monomeric globins and a set of four copper-containing amine oxidases from yeast (Hansenula polymorpha, Pichia pastoris) (8,49), bacteria (Arthrobacter globiformis), and plant (Pisum sativum) (50). As can be seen, the propensity of given residue types to be near an O2 pocket is loosely correlated with its hydrophobicity. Large flexible hydrophobic residues and those possessing aromatic rings are most often seen near O2 pathways, indicating that the large size and mobility of these residues most likely promote the formation of cavities, rather than fill them up as is often assumed.
FIGURE 4.

Relative propensities of residue types to form pathways, computed using Eq. 1, for O2 pathways (a) inside and (b) on the surface of proteins. The data were collected for a set of nine monomeric globins (excluding whale deoxy-Mb, YQR-Mb, and soy Lb, which are redundant), and four copper amine oxidases. The relative (%) (a) internal and (b) surface residue composition of the combined proteins is displayed as a dotted line (correspondingly, residues which are less abundant are also less well sampled).
It is illuminating that the propensities plotted in Fig. 4 are similar between protein families, which suggests that we are observing a universal behavior. Furthermore, the propensities are not correlated with residue populations in the protein. These results have meaningful relevance to protein engineering applications since they state that to alter the O2 pathway and cavity networks in proteins, one should focus on the residue side chains near that location, rather than altering the protein's fold.
In the proteins' interiors, the residues which form O2 pathways are bulky and hydrophobic, whereas residues that are unlikely to form pathways are polar and/or charged (Fig. 4 a). These results are independent of the fact that hydrophobic residues form the core of globular proteins. A priori, it is not obvious whether large mobile residues with nonspecific interactions predominantly create pathways through suboptimal protein packing or whether they fill pathways by occupying volume inside the protein. Based on our results, it appears that the former behavior is the dominant one. However, the correlation between propensities and residue type observed inside the proteins is not readily visible on the proteins' surface, where the solvation energy of O2 in water dominates the O2 PMF (Fig. 4 a).
Significance of the O2 pathway networks
There has been, over the course of decades, a large body of work dedicated to understanding and characterizing O2 migration kinetics inside a small number of representative globins. Our results clearly show that the shapes of the networks of O2 pathways inside globins vary greatly from one globin to the next and that whatever conclusion can be experimentally drawn for one specific globin is likely to be only applicable to that specific globin. One might even wonder if the actual location of pathways and cavities inside a globin bear much relevance to its function. As long as the global properties of the O2 pathways match the desired function of the protein, there may well be no incentive for the protein to conserve or even tune O2 pathway locations, especially given that most globins appear to possess a large number of O2 access routes. It is not clear to us that O2 pathways are critically important. A discussion of the role of O2 pathways should therefore start with what they are not.
O2 pathway networks do not affect O2 binding affinities. A differentiating property of individual globins is their affinity to gas ligands such as O2. Maintaining fine-tuned affinities to O2 is crucial to organism function. For example, most Hbs undergo conformational changes to vary their O2 affinity through cooperative binding: they require high affinities near an O2 source such as the lungs along with a decreased affinity in low O2 environments. Similarly, vertebrate Mb as well as secondary Hbs in invertebrates require a relatively high O2 affinity to uptake O2 from the primary Hb carrier. Globin O2 affinity, however, is not a property of the O2 pathway network; it only depends on the free energy of O2 binding at the heme, which is almost exclusively influenced by interaction with the residues located in the DP, as evidenced by the high sensitivity of globin-gas affinities to mutations of DP residues (51–53). The pathway taken by O2 to reach the DP are themselves not expected to have an influence on binding affinities but only on binding rates.
The properties of pathways in the protein matrix, in principle, affect the O2 on/off rates between heme and exterior. Given a fixed O2 affinity (which is related to the ratio of on and off rates), altering the energy barriers along, and shapes of, the O2 pathways could slow or hasten O2's migration speeds. Although most globins bind to O2 for short times (with a 1–100 ms half-life), the Hbs of the Paramphistomum trematode and Ascaris roundworm both display exceptionally long O2 binding half-lives (21 s and 175 s, respectively) (16). Despite the fact that both the roundworm and trematode Hbs exhibit the two highest free energy barriers for O2 exit of all the globins studied here, the barriers that we measure are too low to explain by themselves the 4–5 orders of magnitude difference in O2 binding times for these proteins compared to that of the average globin. Assuming an Arrhenius process, such a difference in binding times would require an energy barrier of at least 9 kBT, which would have been readily measured by the implicit ligand sampling calculation. Both roundworm and trematode O2 dissociation rates are well explained by the O2 affinities of their hemes (and DP), and the details of their O2 pathway networks have very little effect on these rates. In practice, the effect of the O2 pathway shapes and locations thus appears to be of minor importance relative to the effect of the bound ligand's environment at the heme, even for extreme cases.
The different pathways in globins may, however, have important roles not yet appreciated or understood. For one, these pathways may have enzymatic functions such as kinetic proofreading as a means to increase the selectivity of Mb binding to various gases, as suggested by Radding and Phillips (54), or as a means to promote the NO to 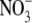 reaction catalyzed by some oxyHbs and oxyMbs (55). Most obviously, such pathways would provide many ways for O2 to enter and escape globins, and many entrances in the globin surface would also increase the capture (and release) rates of gas molecules by globins. Ever since the hypothesis by Perutz and Mathews (56) that O2 enters Mb and some Hbs through a conserved swinging histidine gate, this pathway has often been considered the dominant pathway for O2 to enter these globins. This hypothesis has remained popular because the His gate is and has been the only visible O2 pathway in static crystal structures of Mbs/Hbs. However, when thermal fluctuations and protein dynamics are accounted for, numerous other possible O2 pathways are revealed, and it is likely that the His gate is just one pathway among many (3,34,57). In all likelihood, the swinging of the His gate (as opposed to the direct interaction of the gating His with the bound ligand in the DP) is not a critical factor in the regulation of O2 entry or exit from the protein. We, furthermore, postulate that the conserved His gate appears to gate a water channel, which could permit, for example, the escape of the charged
reaction catalyzed by some oxyHbs and oxyMbs (55). Most obviously, such pathways would provide many ways for O2 to enter and escape globins, and many entrances in the globin surface would also increase the capture (and release) rates of gas molecules by globins. Ever since the hypothesis by Perutz and Mathews (56) that O2 enters Mb and some Hbs through a conserved swinging histidine gate, this pathway has often been considered the dominant pathway for O2 to enter these globins. This hypothesis has remained popular because the His gate is and has been the only visible O2 pathway in static crystal structures of Mbs/Hbs. However, when thermal fluctuations and protein dynamics are accounted for, numerous other possible O2 pathways are revealed, and it is likely that the His gate is just one pathway among many (3,34,57). In all likelihood, the swinging of the His gate (as opposed to the direct interaction of the gating His with the bound ligand in the DP) is not a critical factor in the regulation of O2 entry or exit from the protein. We, furthermore, postulate that the conserved His gate appears to gate a water channel, which could permit, for example, the escape of the charged 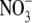 product from the DP. At this point, the roles of such pathways in Mb and other globins, including the histidine gate, are still partially speculative, but in light of new developments in the localization of the O2 pathways in globins, it is now appropriate to carefully reconsider the assumptions made in the past.
product from the DP. At this point, the roles of such pathways in Mb and other globins, including the histidine gate, are still partially speculative, but in light of new developments in the localization of the O2 pathways in globins, it is now appropriate to carefully reconsider the assumptions made in the past.
CONCLUSION
Although there has been a large amount of progress in recent years in first identifying major gas-holding packing defects (19,55) and later complete maps of O2 pathways inside many proteins (2,3,36,44,58,59), a more fundamental understanding of how these pathways occur in general has been lacking. In this study, we characterized the network of O2 pathways inside a large range of proteins from the globin superfamily. Despite the fact that our results are reproducible and similar for very closely related proteins, we find a complete lack of conservancy of the location, topology, or sizes of the O2 pathway networks from one individual monomeric globin to the next, despite the similar folds of the proteins. On the one hand, this suggests that the specific details and locations of the O2 pathways in proteins do not matter much for the protein's function, as long as the pathways are present and provide adequate transport to gas molecules across the protein matrix. On the other hand, this implies that although these pathways are rather independent of the protein's secondary and tertiary structural features, they may actually be dependent on the specific composition of residues inside the protein. This hypothesis was tested, and it was found that the propensity of certain residues to be adjacent or not to O2 pathways is well reproduced across two protein families: monomeric globins and copper-containing amine oxidases. Such results can be used to plan gas migration pathway-altering mutations inside proteins by substituting residues which have a predisposition to create O2 favorable regions with those that do not and vice versa.
Acknowledgments
We thank Ken Olsen and Yi Wang for insightful discussion as well as Carrie Wilmot and Bryan Johnson for providing the Hansenula polymorpha copper-containing amine oxidase atomic structure ahead of publication. All molecular graphics were made using VMD (39).
This work was supported by grants from the National Institutes of Health PHS-5-P41-RR05969, the National Science Foundation SCI04-38712, and the Department of Energy. Supercomputer time was provided by the National Center for Supercomputing Applications via National Resources Allocation Committee grant MCA93S028.
Editor: Ron Elber.
References
- 1.Carlson, M. L., R. M. Regan, and Q. H. Gibson. 1996. Distal cavity fluctuations in myoglobin: protein motion and ligand diffusion. Biochemistry. 35:1125–1136. [DOI] [PubMed] [Google Scholar]
- 2.Cohen, J., K. Kim, P. King, M. Seibert, and K. Schulten. 2005. Finding gas diffusion pathways in proteins: application to O2 and H2 transport in CpI [FeFe]-hydrogenase and the role of packing defects. Structure. 13:1321–1329. [DOI] [PubMed] [Google Scholar]
- 3.Cohen, J., A. Arkhipov, R. Braun, and K. Schulten. 2006. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys. J. 91:1844–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boichenko, V. A., E. Greenbaum, and M. Seibert. 2004. Hydrogen production by photosynthetic microorganisms. In Photoconversion of Solar Energy: Molecular to Global Photosynthesis. M. D. Archer and J. Barber, editors. Imperial College Press, London. 397–452.
- 5.Mertens, R., and A. Liese. 2004. Biotechnological applications of hydrogenases. Curr. Opin. Biotechnol. 15:343–348. [DOI] [PubMed] [Google Scholar]
- 6.Ghirardi, M. L., L. Zhang, J. W. Lee, T. Flynn, M. Seibert, E. Greenbaum, and A. Melis. 2000. Microalgae: a green source of renewable H2. Trends Biotechnol. 18:506–511. [DOI] [PubMed] [Google Scholar]
- 7.Wang, Y., J. Cohen, W. F. Boron, K. Schulten, and E. Tajkhorshid. 2007. Exploring gas permeability of cellular membranes and membrane channels with molecular dynamics. J. Struct. Biol. 157:534–544. [DOI] [PubMed] [Google Scholar]
- 8.Johnson, B. J., J. Cohen, R. W. Welford, A. R. Pearson, K. Schulten, J. P. Klinman, and C. M. Wilmot. 2007. Exploring molecular oxygen pathways in Hanseluna polymorpha copper-containing amine oxidase. J. Biol. Chem. 282:17767–17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Appleby, C. A. 1984. Leghemoglobin and rhizobium respiration. Ann. Rev. Plant Physiol. 35:443–478. [Google Scholar]
- 10.Frauenfelder, H., B. H. McMahon, R. H. Austin, K. Chu, and J. T. Groves. 2001. The role of structure, energy landscape, dynamics, and allostery in the enzymatic function of myoglobin. Proc. Natl. Acad. Sci. USA. 98:2370–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunori, M., D. Bourgeois, and B. Vallone. 2004. The structural dynamics of myoglobin. J. Struct. Biol. 147:223–234. [DOI] [PubMed] [Google Scholar]
- 12.Wittenberg, J. B., and B. A. Wittenberg. 2003. Myoglobin function reassessed. J. Exp. Biol. 206:2011–2020. [DOI] [PubMed] [Google Scholar]
- 13.Garry, D. J., S. B. Kanatous, and P. P. A. Mammen. 2003. Emerging roles for myoglobin in the heart. Trends Cardiovasc. Med. 13:111–116. [DOI] [PubMed] [Google Scholar]
- 14.Flögel, U., M. W. Merx, A. Gödecke, U. K. M. Decking, and J. Schrader. 2001. Myoglobin: a scavenger of bioactive NO. Proc. Natl. Acad. Sci. USA. 98:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan, L., M. B. Twitchett, L. D. Eltis, A. G. Mauk, and M. Smith. 1998. In vitro evolution of horse heart myoglobin to increase peroxidase activity. Proc. Natl. Acad. Sci. USA. 95:12825–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber, R. E., and S. N. Vinogradov. 2001. Non-vertebrate hemoglobins: functions and molecular adaptations. Physiol. Rev. 81:569–627. [DOI] [PubMed] [Google Scholar]
- 17.Perutz, M. F. 1979. Regulation of oxygen affinity of hemoglobin: influence of structure of the globin on the heme iron. Annu. Rev. Biochem. 48:327–386. [DOI] [PubMed] [Google Scholar]
- 18.Royer, W. E. Jr., H. Zhu, T. A. Gorr, J. F. Flores, and J. E. Knapp. 2005. Allosteric hemoglobin assembly: diversity and similarity. J. Biol. Chem. 39:27477–27480. [DOI] [PubMed] [Google Scholar]
- 19.Tilton, R. F., I. D. Kuntz, and G. A. Petsko. 1984. Cavities in proteins: structure of a metmyoglobin-xenon complex solved to 1.9 Å. Biochemistry. 23:2849–2857. [DOI] [PubMed] [Google Scholar]
- 20.Brunori, M., B. Vallone, F. Cutruzzola, C. Travaglini-Allocatelli, J. Berendzen, K. Chu, R. M. Sweeti, and I. Schlichting. 2000. The role of cavities in protein dynamics: crystal structure of a photolytic intermediate of a mutant myoglobin. Proc. Natl. Acad. Sci. USA. 97:2058–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schotte, F., M. Lim, T. A. Jackson, A. V. Smirnov, J. Soman, J. S. Olson, G. N. Phillips Jr., M. Wulff, and P. A. Anfinrud. 2003. Watching a protein as it functions with 150-ps time-resolved x-ray crystallography. Science. 300:1944–1947. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt, M., K. Nienhaus, R. Pahl, A. Krasselt, S. Anderson, F. Parak, G. U. Nienhaus, and V. Šrajer. 2005. Ligand migration pathway and protein dynamics in myoglobin: a time-resolved crystallographic study on L29W MbCO. Proc. Natl. Acad. Sci. USA. 102:11704–11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Šrajer, V., Z. Ren, T. Y. Teng, M. Schmidt, T. Ursby, D. Bourgeois, C. Pradervand, W. Schildkamp, M. Wulff, and K. Moffat. 2001. Protein conformational relaxation and ligand migration in myoglobin: a nanosecond to millisecond molecular movie from time-resolved Laue x-ray diffraction. Biochemistry. 40:13802–13815. [DOI] [PubMed] [Google Scholar]
- 24.Bourgeois, D., B. Vallone, F. Schotte, A. Arcovito, A. E. Miele, G. Sciara, M. Wulff, P. Anfinrud, and M. Brunori. 2003. Complex landscape of protein structural dynamics unveiled by nanosecond Laue crystallography. Proc. Natl. Acad. Sci. USA. 100:8704–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Šrajer, V., T. Y. Teng, T. Ursby, C. Pradervand, Z. Ren, S. Adachi, W. Schildkamp, D. Bourgeois, M. Wulff, and K. Moffat. 1996. Photolysis of the carbon monoxide complex of myoglobin: nanosecond time-resolved crystallography. Science. 274:1726–1729. [DOI] [PubMed] [Google Scholar]
- 26.Austin, R. H., K. W. Beeson, L. Eisenstein, H. Frauenfelder, and I. C. Gunsalus. 1975. Dynamics of ligand binding to myoglobin. Biochemistry. 14:5355–5373. [DOI] [PubMed] [Google Scholar]
- 27.Scott, E. E., Q. H. Gibson, and J. S. Olson. 2001. Mapping the pathways for O2 entry into and exit from myoglobin. J. Biol. Chem. 276:5177–5188. [DOI] [PubMed] [Google Scholar]
- 28.Ostermann, A., R. Waschipky, F. G. Parak, and G. U. Nienhaus. 2000. Ligand binding and conformational motions in myoglobin. Nature. 404:205–208. [DOI] [PubMed] [Google Scholar]
- 29.Dantsker, D., C. Roche, U. Samuni, G. Blouin, J. S. Olson, and J. M. Friedman. 2005. The position 68(E11) side chain in myoglobin regulates ligand capture, bond formation with heme iron, and internal movement into the xenon cavities. J. Biol. Chem. 280:38740–38755. [DOI] [PubMed] [Google Scholar]
- 30.Scott, E. E., and Q. H. Gibson. 1997. Ligand migration in sperm whale myoglobin. Biochemistry. 36:11909–11917. [DOI] [PubMed] [Google Scholar]
- 31.Gibson, Q. H., R. Regan, R. Elber, J. S. Olson, and T. E. Carver. 1992. Distal pocket residues affect picosecond ligand recombination in myoglobin. J. Biol. Chem. 267:22022–22034. [PubMed] [Google Scholar]
- 32.Rohlfs, R. J., J. S. Olson, and Q. H. Gibson. 1988. A comparison of the geminate recombination kinetics of several monomeric heme proteins. J. Biol. Chem. 263:1803–1813. [PubMed] [Google Scholar]
- 33.Case, D. A., and M. Karplus. 1979. Ligands binding to heme proteins. J. Mol. Biol. 132:353–368. [DOI] [PubMed] [Google Scholar]
- 34.Elber, R., and M. Karplus. 1990. Enhanced sampling in molecular dynamics: use of the time-dependent Hartree approximation for a simulation of carbon monoxide diffusion through myoglobin. J. Am. Chem. Soc. 112:9161–9175. [Google Scholar]
- 35.Bossa, C., M. Anselmi, D. Roccatano, A. Amadei, B. Vallone, M. Brunori, and A. D. Nola. 2004. Extended molecular dynamics simulation of the carbon monoxide migration in sperm whale myoglobin. Biophys. J. 86:3855–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hummer, G., F. Schotte, and P. A. Anfinrud. 2004. Unveiling functional protein motions with picosecond x-ray crystallography and molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 101:15330–15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nutt, D. R., and M. Meuwly. 2004. CO migration in native and mutant myoglobin: atomistic simulations for the understanding of protein function. Proc. Natl. Acad. Sci. USA. 101:5998–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang, L., and J. Hermans. 1996. Hydrophilicity of cavities in proteins. Proteins. 24:433–438. [DOI] [PubMed] [Google Scholar]
- 39.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—Visual Molecular Dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 40.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gower, M., J. Cohen, J. Phillips, R. Kufrin, and K. Schulten. 2006. Managing biomolecular simulations in a grid environment with NAMD-G. Proc. 2006 TeraGrid Conf. 7 pp.
- 42.MacKerell, A. Jr., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 43.Allocatelli, C. T., F. Cutruzzolà, A. Brancaccio, B. Vallone, and M. Brunori. 1994. Engineering Ascaris hemoglobin oxygen affinity in sperm whale myoglobin: role of tyrosine B10. FEBS Lett. 352:63–66. [DOI] [PubMed] [Google Scholar]
- 44.Bossa, C., A. Amadei, I. Daidone, M. Anselmi, B. Vallone, M. Brunori, and A. D. Nola. 2005. Molecular dynamics simulation of sperm whale myoglobin: effects of mutations and trapped CO on the structure and dynamics of cavities. Biophys. J. 89:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Czerminski, R., and R. Elber. 1991. Computational studies of ligand diffusion in globins: I. Leghemoglobin. Proteins. 10:70–80. [DOI] [PubMed] [Google Scholar]
- 46.Salomonsson, L., A. Lee, R. B. Gennis, and P. Brzezinski. 2004. A single-amino-acid lid renders a gas-tight compartment within a membrane-bound transporter. Proc. Natl. Acad. Sci. USA. 101:11617–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buhrke, T., O. Lenz, N. Krauss, and B. Friedrich. 2005. Oxygen tolerance of the H2-sensing [NiFe] hydrogenase from Ralstonia eutropha H16 is based on limited access of oxygen to the active site. J. Biol. Chem. 280:23791–23796. [DOI] [PubMed] [Google Scholar]
- 48.Ghirardi, M. L., J. Cohen, P. King, K. Schulten, K. Kim, and M. Seibert. 2006. [FeFe]-hydrogenases and photobiological hydrogen production. Proc. SPIE: Solar Hydrogen and Nanotechnology. L. Vayssieres, editor. 6340:253–258. [Google Scholar]
- 49.Duff, A., A. E. Cohen, P. J. Ellis, J. A. Kuchar, D. B. Langley, E. M. Shepard, D. M. Dooley, H. C. Freeman, and J. M. Guss. 2003. The crystal structure of Pichia pastoris lysyl oxidase. Biochemistry. 42:15148–15157. [DOI] [PubMed] [Google Scholar]
- 50.Duff, A., D. Trambaiolo, A. Cohen, P. Ellis, G. Juda, E. Shepard, D. Langley, D. Dooley, H. Freeman, and J. Guss. 2004. Using xenon as a probe for dioxygen-binding sites in copper amine oxidases. J. Mol. Biol. 344:599–607. [DOI] [PubMed] [Google Scholar]
- 51.Springer, B. A., S. G. Sligar, J. S. Olson, and G. N. Phillips Jr. 1994. Mechanisms of ligand recognition in myoglobin. Chem. Rev. 94:699–714. [Google Scholar]
- 52.Olson, J. S., and G. N. Phillips Jr. 1997. Myoglobin discriminates between O2, NO, and CO by electrostatic interactions with the bound ligand. J. Biol. Inorg. Chem. 2:544–552. [Google Scholar]
- 53.Liong, E. C., Y. Dou, E. E. Scott, J. S. Olson, and G. N. Phillips. 2001. Waterproofing the heme pocket. J. Biol. Chem. 276:9093–9100. [DOI] [PubMed] [Google Scholar]
- 54.Radding, W., and G. N. Phillips Jr. 2004. Kinetic proofreading by the cavity system of myoglobin: protection from poisoning. Bioessays. 26:422–433. [DOI] [PubMed] [Google Scholar]
- 55.Brunori, M., and Q. H. Gibson. 2001. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2:676–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perutz, M. F., and F. S. Mathews. 1966. An x-ray study of azide methaemoglobin. J. Mol. Biol. 21:199–202. [DOI] [PubMed] [Google Scholar]
- 57.Huang, X., and S. G. Boxer. 1994. Discovery of new ligand binding pathways in myoglobin by random mutagenesis. Nat. Struct. Biol. 1:226–229. [DOI] [PubMed] [Google Scholar]
- 58.Amara, P., P. Andreoletti, H. M. Jouve, and M. J. Field. 2001. Ligand diffusion in the catalase from Proteus mirabilis: a molecular dynamics study. Prot. Sci. 10:1927–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Montet, Y., P. Amara, A. Volbeda, X. Vernede, E. C. Hatchikian, M. J. Field, M. Frey, and J. C. Fontecilla-Camps. 1997. Gas access to the active site of Ni-Fe hydrogenase probed by x-ray crystallography and molecular dynamics. Nat. Struct. Biol. 4:523–526. [DOI] [PubMed] [Google Scholar]
- 60.Vojtechovsky, J., K. Chu, J. Berendzen, R. Sweet, and I. Schlichting. 1999. Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys. J. 77:2153–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maurus, R., C. Overall, R. Bogumil, Y. Luo, A. Mauk, M. Smith, and G. Brayer. 1997. A myoglobin variant with a polar substitution in a conserved hydrophobic cluster in the heme binding pocket. Biochim. Biophys. Acta. 1341:1–13. [DOI] [PubMed] [Google Scholar]
- 62.Bolognesi, M., S. Onesti, G. Gatti, A. Coda, P. Ascenzi, and M. Brunori. 1989. Aplysia limacina myoglobin. Crystallographic analysis at 1.6 Å resolution. J. Mol. Biol. 205:529–544. [DOI] [PubMed] [Google Scholar]
- 63.Yang, J., A. P. Kloek, D. E. Goldberg, and F. S. Mathews. 1995. The structure of Ascaris hemoglobin domain I at 2.2 Å resolution: molecular features of oxygen avidity. Proc. Natl. Acad. Sci. USA. 92:4224–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pesce, A., S. Dewilde, L. Kiger, M. Milani, P. Ascenzi, M. C. Marden, M. L. V. Hauwaert, J. Vanfleteren, L. Moens, and M. Bolognesi. 2001. Very high resolution structure of a trematode hemoglobin displaying a TyrB10-TyrE7 heme distal residue pair and high oxygen affinity. J. Mol. Biol. 309:1153–1164. [DOI] [PubMed] [Google Scholar]
- 65.Park, H. J., C. Yang, N. Treff, J. D. Satterlee, and C. Kang. 2002. Crystal structures of unligated and CN-ligated Glycera dibranchiata monomer ferric hemoglobin components III and IV. Proteins. 49:49–60. [DOI] [PubMed] [Google Scholar]
- 66.Steigemann, W., and E. Weber. 1979. Structure of erythrocruorin in different ligand states refined at 1.4 Å resolution. J. Mol. Biol. 127:309–338. [DOI] [PubMed] [Google Scholar]
- 67.Rizzi, M., J. B. Wittenberg, A. Coda, M. Fasano, P. Ascenzi, and M. Bolognesi. 1994. Structure of the sulfide-reactive hemoglobin from the clam Lucina pectinata. Crystallographic analysis at 1.5 Å resolution. J. Mol. Biol. 244:86–99. [DOI] [PubMed] [Google Scholar]
- 68.Harutyunyan, H. E., T. N. Safonova, I. P. Kuranova, A. N. Popov, A. V. Teplyakov, G. V. Obmolova, A. A. Rusakov, B. K. Vainshtein, G. G. Dodson, J. C. Wilson, and M. F. Perutz. 1995. The structure of deoxy- and oxy-leghaemoglobin from lupin. J. Mol. Biol. 251:104–115. [DOI] [PubMed] [Google Scholar]
- 69.Hargrove, M., J. Barry, E. Brucker, M. Berry, G. Phillips Jr., J. Olson, R. Arredondo-Peter, J. Dean, R. Klucas, and G. Sarath. 1997. Characterization of recombinant soybean leghemoglobin a and apolar distal histidine mutants. J. Mol. Biol. 266:1032–1042. [DOI] [PubMed] [Google Scholar]