

Abstract
The T cell receptor (TCR) expressed on most T cells is a protein complex consisting of TCRαβ heterodimers that bind antigen and cluster of differentiation (CD) 3εδ, εγ, and ζζ dimers that initiate signaling. A long-standing controversy concerns whether there is one, or more than one, αβ heterodimer per complex. We used a form of single-molecule spectroscopy to investigate this question on live T cell hybridomas. The method relies on detecting coincident fluorescence from single molecules labeled with two different fluorophores, as the molecules diffuse through a confocal volume. The fraction of events that are coincident above the statistical background is defined as the “association quotient,” Q. In control experiments, Q was significantly higher for cells incubated with wheat germ agglutinin dual-labeled with Alexa488 and Alexa647 than for cells incubated with singly labeled wheat germ agglutinin. Similarly, cells expressing the homodimer, CD28, gave larger values of Q than cells expressing the monomer, CD86, when incubated with mixtures of Alexa488- and Alexa647-labeled antibody Fab fragments. T cell hybridomas incubated with mixtures of anti-TCRβ Fab fragments labeled with each fluorophore gave a Q value indistinguishable from the Q value for CD86, indicating that the dominant form of the TCR comprises single αβ heterodimers. The values of Q obtained for CD86 and the TCR were low but nonzero, suggesting that there is transient or nonrandom confinement, or diffuse clustering of molecules at the T cell surface. This general method for analyzing the subunit composition of protein complexes could be extended to other cell surface or intracellular complexes, and other living cells.
Keywords: cell membrane, organization, protein, spectroscopy, TCR/CD3 complex
The cell surface, which has a central role in determining cellular function and fate, presents a particular challenge for the in situ analysis of protein organization because of the relatively low levels of expression of many of the molecules present there. Whereas the overall compositional complexity of the best characterized mammalian cell surface, that is, that of the T cell, is now largely known (1), the organizational properties of some of its most important constituents are poorly characterized. The outstanding example is the T cell receptor (TCR), which initiates T cell activation by binding antigenic peptides complexed with MHC molecules expressed on antigen-presenting cells. The TCR consists of the clonotypic, antigen-binding, disulfide-linked TCR α and β (or γ and δ) chains, which are noncovalently associated with the signaling subunits, CD3γ, δ, ε, and ζ. Precisely how these elements are assembled beyond the formation of TCRαβ (or γδ), εδ, and εγ heterodimers and ζζ homodimers (2) is not known. It has variously been proposed that the TCR is monovalent (i.e., consists of a single αβ (or γδ) heterodimer; see refs. 3 and 4), invariably multivalent (5), or a mixture of the two (6). When it is finally understood in detail, the structure of the TCR is likely to place important constraints on theories of antigen recognition and TCR triggering.
More generally, there is a paucity of methods for characterizing the subunit compositions of protein complexes that are useful in the context of the relatively low levels of protein expression observed in vivo. Resonance energy transfer has been used (7–9) but requires that two fluorophores must be close enough for energy transfer to occur (1–10 nm), precluding the analysis of large complexes. Immunoprecipitation followed by gel electrophoresis (4, 10, 11) suffers from the major drawback that it yields subunit information for detergent-solubilized, rather than native, complexes. One general approach is to use a single fluorophore to label the protein and to try to detect a doubling in fluorescence intensity on dimer or complex formation (12). A disadvantage of this method is that it requires all of the molecules to be labeled, which may not be possible in situations where there are high endogenous levels of proteins (e.g., the cytoplasm).
Two general types of ultrasensitive fluorescence-based methods have been used successfully to probe the cell membrane. In single-molecule spectroscopy (12–16) individual molecules are resolved by working with sufficiently small numbers of labeled proteins, whereas in fluorescence correlation spectroscopy single molecules are not resolvable and, instead, fluctuations in fluorescence intensity in both amplitude and time are the properties of interest (17–20). We have recently introduced a complementary method called two-color coincidence detection (TCCD), which is a single-molecule method based on the coincident detection of fluorescence from two different fluorophores on the same molecule or complex that are excited with focused, overlapped lasers. In TCCD experiments, we measure the “association quotient,” Q, which is the fraction of all events that are coincident above the random statistical background and is directly proportional to the fraction of associated molecules (21, 22). Solution experiments (23) have shown that the TCCD method greatly extends the sensitivity of the single-molecule approach by allowing detection of low levels of complex against a high background of monomer and by reducing the background from autofluorescent impurities.
We have now used TCCD to study the subunit composition of the TCR. Our results show that the dominant form of the TCR complex expressed on T cells is composed of a single αβ heterodimer. We consider the implications of our findings in the contexts of antigen recognition and TCR triggering.
Results
Principle of the Method.
The membrane proteins investigated in this study and their associated antibody fragments are depicted in Fig. 1A, whereas the underlying principle of TCCD experiments, as described in detail for solution experiments in ref. 23, is shown in Fig. 1B. A T cell hybridoma is incubated with the Fab fragments of receptor-specific antibodies labeled with either a fluorophore excitable with a blue laser (Alexa488) or a fluorophore excitable with a red laser (Alexa647). The two beams are overlapped and focused on the apical surface of the cell. For TCCD to be applicable, the fluorescence burst rate must be low enough for each event to be resolvable as individual, labeled membrane receptors diffuse through the laser beams. On the other hand, the fluorescence burst rate should not be so low that it becomes impossible to obtain enough data for the analysis. Under suitable experimental conditions, we reasoned that if the TCR complex consists of two or more αβ heterodimers, then by tagging the TCRβ chain with mixtures of Fab fragments carrying either fluorophore, coincident bursts of fluorescence would be detected as dual-color-labeled TCRs diffuse through the beam (Fig. 1B Left). Note that it is not possible to overlap the beams perfectly, so the efficiency of coincidence detection is less than unity. In contrast, if the TCR comprises a single αβ heterodimer, then only red- or blue-excited bursts would be detected (Fig. 1B Right), apart from occasional coincident events arising from the simultaneous, random diffusion of red- and blue-excited TCRs into the beams. These random events constitute a statistical background, the magnitude of which must be determined accurately to detect associated molecules (21). A schematic view of the experimental apparatus used for dual-color excitation and TCCD is shown in Fig. 1C.
Fig. 1.

Two-color coincidence detection microscopy. (A) Drawings, approximately to scale, of the proteins, that is, CD86, CD28, and the TCR complex, and the fragments of antibodies (red and blue), that is, BU63 (anti-CD86), 7.3B6 (anti-CD28), KT3 (anti-CD3ε), and F23.1 (anti-TCRβ) used in this study. A TCR complex composed of a single αβ heterodimer is shown. (B) Diagrammatic representation of two types of TCR complex, that is, TCRs comprising two αβ heterodimers (Left) or a single αβ heterodimer (Right), bound to Alexa488 (blue)- and Alexa647 (red)-labeled anti-TCRβ Fab fragments. On dual-color excitation, a fraction of the complexes in Left will produce fluorescence from both fluorophores, whereas those in Right will only ever exhibit single-color fluorescence. The expected avalanche photodiode (APD) output, against time, for each scenario is also shown. (C) Schematic view of the experimental apparatus used for dual-color excitation and two-color coincidence detection (LP/BP, long pass/band pass filters).
The method detects coincident fluorescence bursts from two different fluorophores attached to any oligomeric structure. There are no restrictions on the distance between these fluorophores, nor is there any dependence of the signal on fluorophore separation. The fluorophores can therefore be placed at any convenient position on the molecule. This contrasts with fluorescence resonance energy transfer experiments wherein the transfer efficiency is highly dependent on the fluorophore separation. The absence of this constraint is a significant advantage of the TCCD method.
Single-Molecule Level Analysis.
Our first objective was to obtain single-molecule level fluorescence detection of T cell surface receptors. Three different murine T cell hybridomas were used in the present study: the Vβ8+ DO11.10 T cell hybridoma (24); a second Vβ8+ hybridoma, YAe5B3K (25); and the Vβ8− hybridoma, KMAC92.6 (26). The two Vβ8+ hybridomas were used to minimize hybridoma-specific artifacts, whereas the third was used as a labeling control. Where necessary, the DO11.10 cells were transduced with expression constructs encoding cytoplasmic domain-lacking forms of the known monomeric and dimeric receptors, human CD86 (27, 28) and CD28 (28, 29), respectively. All experiments were performed at 37°C. To label particular receptors, anti-murine Vβ8 TCR (F23.1), anti-murine CD3ε (KT3), anti-human CD86 (BU63), and anti-human CD28 (7.3B6) antibodies were used to generate fluorescent Fab fragments. These were prepared and labeled with either Alexa488 or Alexa647 dyes. On average, each Fab molecule was labeled with a single fluorophore (see supplementary information (SI) Materials and Methods) and saturating concentrations were used in all experiments. Since the Fabs had off-rates of ≈4 × 10−4 s−1 (measured in the presence of sodium azide; see SI Materials and Methods and SI Fig. 7) and cell surface proteins are likely to be gradually internalized [at rates of 1–3.8 × 10−4 s−1 (30)], all data were recorded in the first 20 min of incubation of the cells with the Fabs to ensure >50% occupancy of the receptor. Data were collected at the apical rather than the proximal membrane where contact with the glass slide could have impeded diffusion of the labeled proteins. This approach avoided the detection of fluorescence from glass-adsorbed Fab fragments.
Control experiments with anti-TCRβ and anti-CD3ε Fab fragments confirmed that the fluorescence bursts we could detect by using the apparatus resulted from the diffusion over the cell surface of receptors bound by single, fluorophore-labeled Fabs (Fig. 2). (i) In experiments in which the anti-TCRβ Fab nonbinding, anti-CD3ε Fab-binding KMAC92.6 cells were labeled with a stoichiometric mix of both Fabs, fluorescence bursts were only observed in the CD3ε channel (Fig. 2A). This indicates that fluorescence bursts are associated only with the target antigen of the Fab fragments. (ii) Unlabeled cells excited with both the red and the blue laser gave no fluorescence bursts in either channel. The addition of fluorescent Fab to the medium at a solution concentration of 50 pM also failed to give detectable bursts of fluorescence at the cell surface, indicating that solution-phase Fabs are not the source of the fluorescence bursts (data not shown). (iii) Using DO11.10 cells labeled with anti-TCRβ Fab fragments, the cell was scanned systematically from 2 μm below the glass slide to 21 μm above it. Two peaks of fluorescence at distances corresponding to the perimeter of the cell were detected, indicating that it is only proteins at the cell surface that are being observed (Fig. 2B). In addition, when a membrane dye (DiO) was added, bursts of fluorescence were detected at the same focus height at the top of the cell as Fab fluorescence (Fig. 2C). (iv) When the Fab-labeled T cell surface was “fixed” with paraformaldehyde, constant fluorescence was detected. This indicates that the bursts are due to labeled proteins diffusing over the surface rather than movements of the cell or cell membrane. (v) Identical results were obtained for two different T cell hybridomas (DO11.10 and Yae5B3K). (vi) A very low laser power (1–2 μW) was used to avoid the optical trapping effects seen in refs. 31–33. To confirm this, the excitation beam power was halved and no difference in the bursts was observed, except that the intensity was reduced by 50% (Fig. 2D). (vii) Autocorrelation analysis of the fluorescence time trajectory gave a TCR diffusion constant of 0.06 ± 0.01 μm2 s−1, in good agreement with previous work (34; see SI Materials and Methods for details). (viii) Identical results were obtained with and without sodium azide (10 mM), a chemical that inhibits receptor internalization and thus allowed analysis of the cells for a longer period.
Fig. 2.

Control experiments confirming single-molecule level fluorescence detection. (A) Vβ8-KMAC92.6 cells incubated with Alexa488-labeled anti-TCRβ Fab fragments (nonbinding, blue trace) and anti-CD3ε Fab fragments (binding, red trace). Fluorescence bursts are only observed for the anti-CD3ε Fab, indicating that the bursts result from the binding of the fluorescent Fabs to their target antigens at the cell surface. (B) A DO11.10 cell incubated with Alexa488-labeled anti-TCRβ Fab fragments was scanned across the diameter of the cell by using a scan rate of 1 μm/25 s. Fluorescence bursts are only observed at the perimeter of the cell. (C) A DO11.10 cell stained with the DiO membrane probe; single-molecule fluorescence bursts are observed at the same focus height (15 μm) as the bursts detected in Fig. 2B. (D) Fluorescence from Yae5B3K cells incubated with Alexa647-labeled anti-CD3ε Fab fragments and excited at different laser powers. No differences in the bursts was observed, beyond a 50% reduction in overall fluorescence, for cells illuminated with the laser power routinely used (1 μW; dark-blue trace) versus half that typically used (0.5 μW; blue trace), suggesting that optical trapping effects are absent.
TCCD Analysis of the T Cell Surface.
Having established that the experimental setup allowed single-molecule level fluorescence detection, we undertook two-color TCCD experiments. Fig. 3 shows typical data obtained in a TCCD experiment, in this case a human CD28-expressing DO11.10 T cell incubated with Alexa488- and Alexa647-labeled anti-CD28 Fabs. Under the conditions of these experiments, individual fluorescence bursts are clearly visible on both channels, indicating single-molecule detection. The raw data were analyzed with the only variable being the threshold used on each channel to count events. Selection of the correct threshold is necessary to determine the fraction of all events that are coincident above the statistical coincident background, which we define as the association quotient, Q. We have found that Q reaches a maximum for particular threshold values on both channels, allowing these thresholds to be optimized (22). For the present experiments, because the autofluorescence background level varied with time and from cell to cell, we optimized the threshold for the red and blue channels for individual files containing 200 s of data (see Materials and Methods for details). The optimized threshold that is applied increases with molecule density, reducing the effective probe area and, as a result, the average rate of red and blue events. In this way the numbers of detected molecules in the effective probe area remain comparable despite differences in molecule density, as shown in Table 1 and discussed in detail in SI Materials and Methods. The value of Q measured in the following experiments corresponds to the fraction of all detected proteins that are associated (21). Because all of the Fab-labeled proteins are detectable (see SI Materials and Methods), and the event rates are similar over separate experiments, the values of Q can be directly compared. Before examining the subunit composition of the TCR, we first confirmed that Q could be used to distinguish between dual-labeled receptors (e.g., dimers) and receptors carrying single labels (e.g., monomers) in two types of control experiments and established the maximum value of Q for oligomers.
Fig. 3.

Typical fluorescence versus time traces obtained after dual-color excitation of anti-human CD28 Fabs labeled with Alexa488 (blue trace) or Alexa647 (red trace), bound to the CD28-expressing DO11.10 T cell hybridoma. Raw data were collected at 25-ms resolution with 2 μW at 488 nm excitation and 1 μW at 633 nm excitation. The dotted line indicates the threshold for the individual channel above which bursts are detected.
Table 1.
Association quotients for T cell surface proteins labeled at 37°C with singly or dually fluorescently labeled WGA, or with pairs of fluorescently labeled Fab fragments, together with the significant event rate (the rate of coincident events above the rate due to random diffusion), the rate of red and blue events, and the numbers of cells and events analyzed
| Association quotient × 103, Q | Coincident event rates, s−1 | Red event rates, s−1 | Blue event rates, s−1 | No. of cells analyzed | No. of events analyzed | No. of detectable molecules in probe area | |
|---|---|---|---|---|---|---|---|
| Single WGA | 3.3 ± 1.5 | 0.88 | 25.0 | 19.5 | 34 | 690,000 | 0.45 |
| Dual WGA | 16.4 ± 2.8 | 1.27 | 19.8 | 20.7 | 18 | 800,000 | 0.41 |
| CD86/CD86 | 8.0 ± 2.6 | 0.46 | 10.6 | 7.1 | 23 | 280,000 | 0.44 |
| CD28/CD28 | 18.3 ± 3.3 | 0.47 | 8.2 | 6.2 | 32 | 250,000 | 0.36 |
| TCRβ/CD3ε | 18.0 ± 2.3 | 0.62 | 10.7 | 10.3 | 170 | 1,900,000 | 0.53 |
| CD3ε/CD3ε | 17.3 ± 2.4 | 0.64 | 11.3 | 9.4 | 59 | 450,000 | 0.52 |
| TCRβ/TCRβ | 7.1 ± 1.1 | 0.47 | 10.8 | 9.8 | 150 | 1,400,000 | 0.52 |
The last column shows the average number of red and blue molecules in the probe area in each experiment.
Cells labeled with wheat germ agglutinin.
In the first set of experiments, we showed that Q was significantly higher for cells incubated with dual-labeled wheat germ agglutinin (WGA) molecules than singly labeled WGA. Cells were incubated with 100 pM Alexa647-labeled WGA and Atto488-labeled WGA, washed, and then analyzed. This low labeling level, which is well below saturation, allowed single-molecule analysis of WGA binding and gave the expected low value of Q of (3.3 ± 1.5) × 10−3, close to zero (Fig. 4A; Table 1). A sample of Alexa488- and Alexa647-labeled WGA was then prepared and analyzed in free solution, showing that 22% of the molecules were dual-labeled (data not shown). This sample was then used to label the T cell hybridoma, giving a value of Q of (16.4 ± 2.8) × 10−3, significantly higher than that obtained with the singly labeled forms of WGA (Fig. 4A; Table 1). This established that TCCD can be used to detect oligomers on the cell surface and confirmed that monomers give a value for Q close to zero. The experiment also allowed us to determine the “coincidence detection efficiency” at the apical surface of the cell: because 22% of the WGA was dual-labeled, the coincidence detection efficiency is estimated to be ≈7%. This value is reduced compared with focusing in free solution (≈20% efficiency), and presumably reflects the contribution of additional losses due to scattering, reflection, and refraction in the cell and the background autofluorescence. Thus, the estimated maximum value of Q in the present TCCD experiments on live cells would be ≈70 × 10−3 (i.e., ≈7%).
Fig. 4.

Association quotients (Q) for cells expressing T cell surface proteins incubated at 37°C with Alexa488- and Alexa647-labeled lectin and Fab fragments. (A) The DO11.10 T cell hybridoma incubated with dually and singly labeled WGA. (B) Truncated CD86 monomer- and CD28 homodimer-expressing DO11.10 T cell hybridomas incubated with Alexa488- and Alexa647-labeled anti-CD86 and anti-CD28 Fab fragments. (C) The DO11.10 T cell hybridoma incubated with Alexa488- and Alexa647-labeled anti-TCRβ and anti-CD3ε Fab fragments in the combinations indicated. The first Fab fragment of each pair shown was labeled with Alexa488, and the second was labeled with Alexa647. The maximum coincidence detection efficiency in these experiments is estimated to be 7%; see text and SI Materials and Methods for details.
Analysis of membrane receptors of known stoichiometry.
In a second set of experiments we showed that Q could be used to distinguish between proteins with known, distinct stoichiometries. To do this, DO11.10 (i.e., murine) T cells were stably transduced with the human genes encoding either the monomer, CD86 (27, 28), or the homodimer, CD28 (28, 29). The cytoplasmic regions of both molecules were deleted to avoid complications in the stoichiometric analysis arising from potential cytoskeletal interactions. The transduced DO11.10 cells expressed CD28 and CD86 at ≈4,000 and ≈8,000 copies per cell, respectively; DO11.10 cells express 26,000 copies of TCRβ at the cell surface (see SI Materials and Methods). We analyzed 23 cells giving a total of 280,000 events for CD86-expressing DO11.10 T cells incubated with Alexa488- and Alexa647-labeled anti-CD86 Fabs. The value of Q thus obtained was (8.0 ± 2.6) × 10−3 (Fig. 4B; Table 1). This value was marginally higher than that obtained in the experiment in which singly labeled WGA was analyzed, but is more likely to reflect the actual level of coincidence characteristic of randomly diffusing, nonspecifically interacting cell surface proteins. Human CD28-expressing DO11.10 T cells incubated with anti-CD28 Fab fragments labeled with Alexa488 and Alexa647, on the other hand, gave a significantly higher value for Q of (18.3 ± 3.3) × 10−3 (Fig. 4B; Table 1). This established the maximum value of Q obtainable using fluorophore-labeled Fabs under these experimental conditions and showed that the coincidence signal from oligomeric proteins cannot only be detected for live T cells, but is also easily resolved from both the signal associated with the random interactions of receptors and the statistical background.
TCCD-Based Analysis of the Subunit Composition of the TCR Complex.
Having established that coincident bursts could be detected from fluorescent Fabs colocalized at the cell surface by associations with oligomeric cell surface proteins, we turned our attention to the TCR complex of DO11.10 T cells. The anti-TCRβ Fab bound stoichiometrically to the TCR without cross-linking it or interfering with anti-CD3 antibody-induced TCR triggering (see SI Materials and Methods and SI Figs. 5 and 6), implying that the Fab fragment is very unlikely to alter the subunit composition of the TCR or otherwise affect the behavior of the hybridoma. Initially, the cells were incubated with the anti-TCRβ Fab and an anti-CD3ε Fab fragment, which were labeled with Alexa488 and Alexa647, respectively (i.e., the TCRβ/CD3ε experiment). In a second experiment, we incubated the cells with anti-CD3ε Fab fragments labeled with Alexa488 and Alexa647 (i.e., the CD3ε/CD3ε experiment). Finally, the cells were incubated with anti-TCRβ Fab fragments labeled with Alexa488 and Alexa647 (i.e., the TCRβ/TCRβ experiment), to determine whether there is more than one αβ heterodimer per TCR complex.
The CD3ε/CD3ε and TCRβ/CD3ε experiments yielded values for Q of (17.3 ± 2.4) × 10−3 and (18.0 ± 2.3) × 10−3, respectively, which were not significantly different from the values obtained for cell-attached, dual-labeled WGA or for cells expressing the homodimer, CD28, in the presence of anti-CD28 Fab fragments labeled with Alexa488 and Alexa647 (Fig. 4C; Table 1). These results accord well with the notion that each TCR complex comprises two CD3ε subunits, in addition to the TCRβ subunit. The TCRβ/TCRβ experiment, on the other hand, yielded a value for Q of (7.1 ± 1.1) × 10−3, which was statistically indistinguishable from that obtained for cells expressing the monomer, CD86, in the presence of anti-CD86 Fab fragments labeled with Alexa488 and Alexa647 (Fig. 4C; Table 1). TCR complexes consisting of more than one αβ heterodimer were therefore not detected, which indicates that the dominant form of the complex is composed of a single αβ heterodimer.
Discussion
We present a general method for studying the organizational properties of mobile proteins in living cells based on a molecule-by-molecule analysis using TCCD. Our method is based on the analysis of the raw data with the only adjustable parameter being the threshold used to count events (23, 35). The method is general and applicable to any mobile protein that can be labeled with fluorescent antibody Fab fragments or tagged with autofluorescent proteins, provided that single-molecule analysis is possible. The subunit composition of protein complexes present in different structures within or on the surface of cells can be studied by focusing the lasers on the relevant cellular substructure. In the present work, we probed receptor organization at the cell surface by using Fab fragments to label endogenously expressed protein, thereby avoiding artifacts associated with over- or underexpression.
The new method appears to have the potential to provide quantitative information about the effect of membrane structure on the organization of its components, because nonzero values of Q were measured for proteins that are known to be monomeric at the cell surface, such as CD86. This observation suggests some low-level, time-dependent correlation of the movement of such proteins within the cell membrane. Whether this is the result of transient confinement or the diffuse clustering of molecules within the membrane on a length scale comparable to the diameter of our laser (600 nm) requires further investigation. This finding is, however, in good agreement with other studies of cell membrane structure. Transient confinement and nonrandom diffusion have been observed (16, 36–38), and the distribution of T cell plasma membrane-associated proteins within fixed membrane preparations was recently proposed to be nonrandom and clustered (39). Since the form of CD86 used in these experiments lacked a cytoplasmic domain, it would seem that cytoskeletal associations are unlikely to be responsible for this type of membrane heterogeneity. In investigating the source of this effect, it might be particularly useful if lipids are also labeled.
Having established the method, our goal was to use it to study the organizational properties of important T cell surface molecules in situ. The notion that the TCR consists of pairs of αβ heterodimers emerged when an apparent imbalance between the charges of the transmembrane domains of TCR components was noted (40). Immunoprecipitation analyses (3, 4) that suggested that the TCR instead consists of individual αβ heterodimers could not be considered definitive, because these experiments employed detergents that could, in principle, disrupt weak higher-order assemblies. Fluorescence resonance energy transfer experiments using whole, labeled anti-TCR antibodies were used to make the case that the TCR forms obligate dimers (5). However, on that occasion, in addition to the likelihood that whole antibodies might lead to artifactual TCR dimerization, the study did not adequately control for the possibility that changes in the donor environment rather than the presence of the acceptor were responsible for the observed donor quenching (taken as a measure of fluorescence resonance energy transfer). Similarly, recent electron microscopic analysis of TCR organization suggesting that the TCR consists of a mixture of monomers and oligomers relied on multivalent antibody-coupled beads, which would themselves be expected to artifactually cluster the TCR (6). Our data have been obtained on live cells by using Fabs that cannot cross-link the TCR. The T cells also trigger normally with saturating binding of the Fabs, indicating that the monomer detected on the cell surface is fully functional. Although we cannot formally exclude the possibility that the Fabs disrupt a weak dimer on the cell surface, such a dimer could have no role in TCR triggering.
A functionally monovalent TCR has important implications for T cell function. The participation of single TCR heterodimers in antigen recognition is likely to ensure that, at the level of individual triggering events, TCRs depend only on the intrinsic antigenicity of individual, fully formed MHC-peptide ligands. The “competitive advantage” of bivalent over monovalent recognition, in terms of dissociation rates, is likely to be of the order of 100-fold (40). Had it been true that T cell responsiveness depended on whether the efficiency with which peptides are processed was high enough to generate sufficient TCR ligands for bivalent recognition, the breadth of the T cell response to a given pathogen would in all probability have been markedly reduced. In conjunction with data indicating that TCRs can respond to single MHC-peptide agonists (41), our data also places constraints on possible triggering mechanisms. It seems much more likely that triggering relies, in the very first instance at least, on the passive association of individual, monovalent TCR complexes with MHC molecules, rather than on the reorganization of existing bi- or multivalent TCR complexes (42).
Materials and Methods
Details of labeled Fab production, expression of human genes in DO11.10 cells, determination of receptor numbers, and labeling of WGA are given in SI Materials and Methods.
T Cell Hybridoma Labeling.
DO11.10 (F23.1+) (24), YAe5B3K (F23.1+) (25), and KMAC92.6 (F23.1−) (26) T cell hybridomas were cultured in MEM (without phenol red) supplemented with 10% FCS, glutamine and antibiotics, at 37°C and 5% CO2. 5 × 105 cells were centrifuged at 600 × g for 2 min at room temperature, resuspended with either 1 ml of 0.1% BSA, PBS, or additionally with 10 mM sodium azide to prevent internalization of the TCRs, and incubated at 0°C for 30 min (experiments with and without azide gave comparable data). The pelleted cells were then incubated with ≈50 pmol of each Fab at 0°C for at least 30 min with regular agitation. A 2-μl aliquot of stained cells was then added to 1.5 ml of ice-cold buffer and centrifuged as before. Supernatant was removed with a syringe, the pellet resuspended in 37°C buffer, and 100 μl of the suspension immediately placed on a preheated glass slide on the microscope to allow the cells to settle. An analogous protocol was used to label the T cells with WGA (see SI Materials and Methods for details).
Fluorescence Measurements.
A schematic of the experimental setup is shown in Fig. 1 and was described in ref. 23. Maximum overlap between the two-laser focal volumes was found to be ≈30% (23). The cells were placed on a coverslip maintained at a constant temperature of 37°C by using a temperature stage (PE60, Linkam Scientific Instruments, Surrey, U.K.). The cells were allowed to settle onto the coverglass before data acquisition. The cells were changed every 15–20 min and replaced with “freshly” labeled cells to limit the effects of the Fab off-rate and internalization of Fab-labeled proteins. Throughout the experiment, 25-ms integration (bin) times on both multichannel scalar cards were used.
Data Analysis.
Each experimental data set consisted of matched file pairs of fluorescence data collected from the Alexa488 and Alexa647 channels simultaneously. The data sets were then analyzed to identify coincident events by using a method that has been validated in solution studies of model samples of DNA (21). The resulting association quotient (Q) gives the fraction of total events, that is, fluorescence bursts, that are coincident above the statistical background:
where A and B are the rates of events in the two channels, C is the rate of coincident events, and E = A·B·τ is the rate of coincident events expected to occur by chance, with τ being the integration (bin) time. In brief, the fluorescence thresholds were varied to maximize the value of Q for each pair of files (22). This provides a systematic way to select thresholds to apply to two-color data and can be used in situations where it is not possible to perform adequate control experiments for this aspect of the method, for example, when the background varies. The value of Q from each optimized file pair was then averaged over the entire experimental data set. In contrast to our previous work in solution (22), for cells this process had to be done on a file-by-file basis as the background varied between file pairs, as well as between data sets. This generates a statistical offset, because the method consistently selects for positive statistical fluctuations in Q. File-by-file analysis was therefore also performed on nonpaired red and blue data files for each experiment to measure the size of this offset (22) since, in this case, all observed coincident events must be random, that is, Q should equal zero. The offset was then subtracted from the initial estimate of Q, giving the final values referred to in the text.
We also analyzed a large fraction of the data by using a Bayesian approach to identify events (see SI Materials and Methods and SI Figs. 9 and 10). This gave essentially the same results, albeit with larger errors, since the method does not identify all coincident events. Thus two very different analysis methods gave the same results.
Supplementary Material
Acknowledgments
We thank D. Zhou for preparation of the labeled wheat germ agglutinin, T. Hünig for provision of the 7.3B6 antibody and A. Bruckbauer for assistance with the experiments. We also thank L. Ying, E. Evans, and P. Klenerman for constructive comments during the course of this work and P. Marrack for providing the T cell hybridomas. This work was funded by the Biotechnology and Biological Sciences Research Council, and by the Wellcome Trust.
Abbreviations
- CD
cluster of differentiation
- TCCD
two-color coincidence detection
- TCR
T cell receptor
- WGA
wheat germ agglutinin.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.L.D. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700411104/DC1.
References
- 1.Evans EJ, Hene L, Sparks LM, Dong T, Retiere C, Fennelly JA, Manso-Sancho R, Powell J, Braud VM, Rowland-Jones SL, et al. Immunity. 2003;19:213–223. doi: 10.1016/s1074-7613(03)00198-5. [DOI] [PubMed] [Google Scholar]
- 2.Rudolph MG, Luz JG, Wilson IA. Annu Rev Biophys Biomol Struct. 2002;31:121–149. doi: 10.1146/annurev.biophys.31.082901.134423. [DOI] [PubMed] [Google Scholar]
- 3.Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. Cell. 2002;111:967–979. doi: 10.1016/s0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Punt JA, Roberts JL, Kearse KP, Singer A. J Exp Med. 1994;180:587–593. doi: 10.1084/jem.180.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez-Miguel G, Alarcon B, Iglesias A, Bluethmann H, Alvarez-Mon M, Sanz E, de la Hera A. Proc Natl Acad Sci USA. 1999;96:1547–1552. doi: 10.1073/pnas.96.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schamel WWA, Arechaga I, Risueno RM, van Santen HM, Cabezas P, Risco C, Valpuesta JM, Alarcon B. J Exp Med. 2005;202:493–503. doi: 10.1084/jem.20042155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin A. J Biol Chem. 2003;278:28274–28283. doi: 10.1074/jbc.M210652200. [DOI] [PubMed] [Google Scholar]
- 8.Zal T, Gascoigne NR. Curr Opin Immunol. 2004;16:418–427. doi: 10.1016/j.coi.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 9.de la Hera A, Muller U, Olsson C, Isaaz S, Tunnacliffe A. J Exp Med. 1991;173:7–17. doi: 10.1084/jem.173.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.San Jose E, Sahuquillo AG, Bragado R, Alarcon B. Eur J Immunol. 1998;28:12–21. doi: 10.1002/(SICI)1521-4141(199801)28:01<12::AID-IMMU12>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Blumberg RS, Ley S, Sancho J, Lonberg N, Lacy E, McDermott F, Schad V, Greenstein JL, Terhorst C. Proc Natl Acad Sci USA. 1990;87:7220–7224. doi: 10.1073/pnas.87.18.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sako Y, Minoghchi S, Yanagida T. Nat Cell Biol. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 13.Schutz GJ, Kada G, Pastushenko VP, Schindler H. EMBO J. 2000;19:892–901. doi: 10.1093/emboj/19.5.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harms GS, Cognet L, Lommerse PH, Blab GA, Kahr H, Gamsjager R, Spaink HP, Soldatov NM, Romanin C, Schmidt T. Biophys J. 2001;81:2639–2646. doi: 10.1016/S0006-3495(01)75907-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mörtelmaier M, Kögler EJ, Hesse J, Sonnleitner M, Huber LA, Schütz GJ. Single Mol. 2002;3:225–231. [Google Scholar]
- 16.Douglass AD, Vale RD. Cell. 2005;121:937–950. doi: 10.1016/j.cell.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigler R, Pramanik A, Jonasson P, Kratz G, Jansson OT, Nygren PA, Stahl S, Ekberg K, Johansson BL, Uhlen S, et al. Proc Natl Acad Sci USA. 1999;96:13318–13323. doi: 10.1073/pnas.96.23.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwille P, Haupts U, Maiti S, Webb WW. Biophys J. 1999;77:2251–2265. doi: 10.1016/S0006-3495(99)77065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maier C, Runzler D, Wagner L, Grabner G, Kohler G, Luger A. Single Molecules. 2002;3:211–216. [Google Scholar]
- 20.Sengupta P, Balaji J, Maiti S. Methods. 2002;27:374–387. doi: 10.1016/s1046-2023(02)00096-8. [DOI] [PubMed] [Google Scholar]
- 21.Orte A, Clarke R, Balasubramanian S, Klenerman D. Anal Chem. 2006;78:7707–7715. doi: 10.1021/ac061122y. [DOI] [PubMed] [Google Scholar]
- 22.Clarke RW, Orte A, Klenerman D. Anal Chem. 2007;79:2771–2777. doi: 10.1021/ac062188w. [DOI] [PubMed] [Google Scholar]
- 23.Li HT, Ying LM, Green JJ, Balasubramanian S, Klenerman D. Anal Chem. 2003;75:1664–1670. doi: 10.1021/ac026367z. [DOI] [PubMed] [Google Scholar]
- 24.Yague J, White J, Coleclough C, Kappler J, Palmer E, Marrack P. Cell. 1985;42:81–87. doi: 10.1016/s0092-8674(85)80103-3. [DOI] [PubMed] [Google Scholar]
- 25.Marrack P, Bender J, Jordan M, Rees W, Robertson J, Schaefer BC, Kappler J. J Immunol. 2001;167:617–621. doi: 10.4049/jimmunol.167.2.617. [DOI] [PubMed] [Google Scholar]
- 26.Liu CP, Parker D, Kappler J, Marrack P. J Exp Med. 1997;186:1441–1450. doi: 10.1084/jem.186.9.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA, Davis SJ. Immunity. 2002;17:201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 28.James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. Nat Methods. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- 29.Evans EJ, Esnouf RM, Manso-Sancho R, Gilbert RJ, James JR, Yu C, Fennelly JA, Vowles C, Hanke T, Walse B, et al. Nat Immunol. 2005;6:271–279. doi: 10.1038/ni1170. [DOI] [PubMed] [Google Scholar]
- 30.Liu HY, Rhodes M, Wiest DL, Vignali DAA. Immunity. 2000;13:665–675. doi: 10.1016/s1074-7613(00)00066-2. [DOI] [PubMed] [Google Scholar]
- 31.Bar-Ziv R, Menes R, Moses E, Safran SA. Phys Rev Lett. 1995;75:3356–3359. doi: 10.1103/PhysRevLett.75.3356. [DOI] [PubMed] [Google Scholar]
- 32.Chiu DT, Zare RN. J Am Chem Soc. 1996;118:6512–6513. [Google Scholar]
- 33.Osborne MA, Balasubramanian S, Furey WS, Klenerman D. J Phys Chem B. 1998;102:3160–3167. [Google Scholar]
- 34.Favier B, Burroughs NJ, Wedderburn L, Valitutti S. Int Immunol. 2001;13:1525–1532. doi: 10.1093/intimm/13.12.1525. [DOI] [PubMed] [Google Scholar]
- 35.Ren X, Li H, Clarke RW, Alves DA, Ying L, Klenerman D, Balasubramanian S. J Am Chem Soc. 2006;128:4992–5000. doi: 10.1021/ja056613z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kusumi A, Nakada C, Ritchie K, Murase K, Suzuki K, Murakoshi H, Kasai RS, Kondo J, Fujiwara T. Annu Rev Biophys Biomol Struct. 2005;34:351–378. doi: 10.1146/annurev.biophys.34.040204.144637. [DOI] [PubMed] [Google Scholar]
- 37.Saxton MJ, Jacobson K. Annu Rev Biophys Biomol Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- 38.Vereb G, Szollosi J, Matko J, Nagy P, Farkas T, Vigh L, Matyus L, Waldmann TA, Damjanovich S. Proc Natl Acad Sci USA. 2003;100:8053–8058. doi: 10.1073/pnas.1332550100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lillemeier BF, Pfeiffer JR, Surviladze Z, Wilson BS, Davis MM. Proc Natl Acad Sci USA. 2006;103:18992–18997. doi: 10.1073/pnas.0609009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobs H. Immunol Today. 1997;18:565–569. [PubMed] [Google Scholar]
- 41.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- 42.Schamel WW, Risueno RM, Minguet S, Ortiz AR, Alarcon B. Trends Immunol. 2006;27:176–182. doi: 10.1016/j.it.2006.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
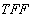{kind=link}
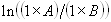{kind=link}
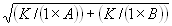{kind=link}
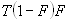{kind=link}
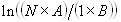{kind=link}
{kind=link}
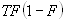{kind=link}
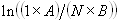{kind=link}
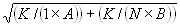{kind=link}
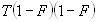{kind=link}
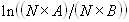{kind=link}
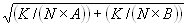{kind=link}
{kind=link}
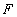{kind=link}
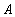{kind=link}
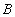{kind=link}
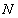{kind=link}
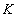{kind=link}
{kind=link}
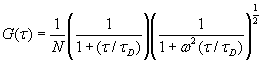{kind=link}
{kind=link}
{kind=link}
{kind=link}
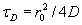{kind=link}
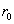{kind=link}
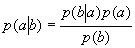{kind=link}
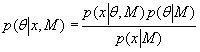{kind=link}
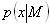{kind=link}
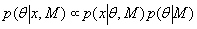{kind=link}