

Abstract
The control of cellular physiology and gene expression in response to extracellular signals is a basic property of living systems. We have constructed a synthetic bacterial signal transduction pathway in which gene expression is controlled by extracellular Zn2+. In this system a computationally designed Zn2+-binding periplasmic receptor senses the extracellular solute and triggers a two-component signal transduction pathway via a chimeric transmembrane protein, resulting in transcriptional up-regulation of a β-galactosidase reporter gene. The Zn2+-binding site in the designed receptor is based on a four-coordinate, tetrahedral primary coordination sphere consisting of histidines and glutamates. In addition, mutations were introduced in a secondary coordination sphere to satisfy the residual hydrogen-bonding potential of the histidines coordinated to the metal. The importance of the secondary shell interactions is demonstrated by their effect on metal affinity and selectivity, as well as protein stability. Three designed protein sequences, comprising two distinct metal-binding positions, were all shown to bind Zn2+ and to function in the cell-based assay, indicating the generality of the design methodology. These experiments demonstrate that biological systems can be manipulated with computationally designed proteins that have drastically altered ligand-binding specificities, thereby extending the repertoire of genetic control by extracellular signals.
There is considerable interest in constructing synthetic signal transduction pathways to control cellular physiology and gene transcription (1, 2). In addition to the potential benefit in the study of complex networks that are not readily amenable to genetic analysis (3, 4), such engineered systems have numerous potential biotechnological applications (5, 6). Here we report the rational design of a synthetic bacterial transcriptional control system that is activated by extracellular Zn2+. Using structure-based computational design methods (7), we have converted the periplasmic Escherichia coli ribose-binding protein (RBP) into a specific receptor for Zn2+ and demonstrated its potential utility as a biosensor. This designed protein has also been introduced into a strain containing a chimeric two-component signal transduction pathway linking binding of ligand by RBP to transcriptional activation of a reporter gene (8). The construction of such a system demonstrates the opportunities for developing metal-mediated synthetic signal transduction pathways for which there are no naturally evolved examples known. Synthetic pathways may be used for cell-based detection of metals in the environment (9), or for the control of gene expression with inexpensive zinc salts in bioprocessing (6).
The bacterial periplasmic binding proteins (PBPs), including RBP, are involved in chemotaxis and solute transport (10). PBPs are well suited for the design of proteins with radically altered ligand-binding specificities by computational design techniques (11–13). Metal-binding sites can be designed in proteins by using an automated computational design procedure that predicts positions in the three-dimensional protein structure at which the residues interacting directly with the metal according to a predefined geometry can be introduced by mutagenesis to form the desired primary coordination sphere (PCS) (14). Using this algorithm, a variety of metal centers have been constructed in several proteins (15–21), including maltose-binding protein (12), another member of the PBP family. Most of these earlier metalloprotein designs considered only mutations necessary to construct the PCS, without taking into account additional changes to optimize the interactions of the PCS with the surrounding protein matrix. It has been shown that residues in this secondary coordination sphere (SCS) make important contributions to metal affinity (11, 22–27), selectivity (28), and control of activity (12, 19). Here we design both the PCS and SCS simultaneously, rather than by optimizing the SCS in design iterations subsequent to design, construction, and characterization of the PCS. The mutations required to construct the SCS were predicted by using a deterministic algorithm that designs sequences to optimize packing interactions in proteins (29, 30).
The periplasmic binding protein family has also proved useful for the construction of reagentless biosensors (31–33). PBP family members representing a wide array of ligand-binding specificity (sugars, amino acids, metals, anions, and cations) have been converted into reagentless, protein-based biosensors through the use of systematic cysteine-scanning mutagenesis, coupled with conjugation of environmentally sensitive fluorophores (31, 34, 35) or redox centers (33). The resulting optical and electrochemical biosensors have been shown to be useful for ligand concentration determination, even in complex mixtures (33). The signal change for the optical and electrochemical biosensors is coupled to the large conformational change that occurs on ligand binding, resulting in a substantially altered chemical environment around the fluorophore or redox center. The structures of both the open, ligand-free conformation and the closed, ligand-bound conformation have been determined for RBP (36, 37). The equilibrium between the two conformations, as well as a structural model of the response of an environmentally sensitive fluorophore molecule, is depicted in Fig. 1.
Fig. 1.

Ligand-mediated conformational changes in RBP. On binding of ribose the protein switches from an open conformation (Left; Protein Data Bank ID code 1URP; ref. 36) to a closed conformation (Right; Protein Data Bank ID code 2DRI; ref. 37). The conformational interchange can be monitored by covalent coupling of a thiol-reactive fluorophore to Cys-236 (31). A model of the conjugated styryl dye JPW4042 in each conformation was generated by identification of the minimum energy dye conformation (based on an in vacuo calculation using a semiempirical force field). The differences in the two conformations of the dye are primarily due to changes in the interactions between the dye and protein near the point of attachment. Also indicated are the positions of the Zn2+ ions in the three designed sites relative to the ribose in the closed conformation. Colors indicate the domain structure, fluorescent conjugate, and ligands of the receptor: green, N-terminal domain; blue, C-terminal domain; purple, hinge; yellow, ribose; red, fluorophore JPW4042; orange, zinc sites A (Right) and B (designs B1 and B2; Left).
Two-component signal transduction pathways represent a major mechanism by which prokaryotes and a number of eukaryotes, including plants, respond to their extracellular milieu (38). In E. coli alone, there are at least 32 two-component systems (39) that connect physical and chemical signals to physiological and gene expression responses. Systematic manipulation of such pathways by using engineered receptors responding to nonnatural signals provides a potentially fruitful mechanism by which synthetic organisms can be constructed that respond in a controlled manner to their environment (13).
Materials and Methods
Molecular Modeling. The mutations to construct the PCS were calculated as described (12, 15–20). SCS mutations were constructed around a PCS by using a modified dead-end elimination algorithm (30). SCS calculations used a pairwise, semiempirical potential function to describe the atomic interactions, containing steric and hydrogen-bonding terms to describe short-range interactions, and a semicontinuum model to describe solvation effects (30). Steric interactions were described by a modified Lennard-Jones potential:
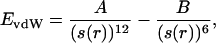 |
[1] |
where A and B are atom pair-dependent parameters, and s(r) is a function that widens the repulsive potential well linearly relative to the standard form of this potential without changing r0, the equilibrium interatomic distance, according to the equation s(r) = min(r, r/α + r0(1–1/α), with a stretching parameter, α (set to 1.1). This effectively constructs a “fuzzy” atom that relaxes steric constraints without altering the preferred (equilibrium) interatomic distances. Solvation energy was described by using an area-dependent term calculated by using an approximation to estimate these areas in pairwise calculations (40). The exposed and buried areas were parameterized as follows: 26 cal·mol–1·Å–2 favoring nonpolar burial, 100 cal·mol–1·Å–2 penalizing polar burial, 26 cal·mol–1·Å–2 penalizing aliphatic exposure, and 200 cal·mol–1·Å–2 penalizing nonpolar aromatic atom exposure. Polar atoms were not considered buried if they are involved in hydrogen bonding interactions. An explicit, geometry- and hybridization-dependent hydrogen-bonding term was used (30). Additionally, a mechanism was used to enforce formation of hydrogen bonds to the noncoordinating imidazole nitrogens of histidine residues in the PCS. This mechanism is implemented as a new elimination filter in the dead-end elimination algorithm. On identification of a situation in which all noneliminated hydrogen bond partners for a demanded atom arise from a single residue position, all non-hydrogen-bonding side chains at that position are eliminated. This is a first-level approximation to the general non-pairwise-decomposable problem of requiring that a particular hydrogen bond donor or acceptor be satisfied (the “hydrogen bond inventory”) (13, 41). All molecular graphics were produced with the program ribbons (42).
Mutagenesis and Protein Expression. Mutagenesis procedures were as described (11). Protein was expressed cytoplasmically in the pAED4 vector (43) and produced by fermentation in BL21 cells grown in 2× YT broth. Protein expression was induced with isopropyl-β-d-thiogalactoside after growth at 37°C to an optical density at 600 nm of 0.5, followed by overnight growth at 22°C. A cleared lysate was prepared as described (11). RBP was purified by ion exchange chromatography, followed by chelation and gel filtration to remove adventitious metal. Cleared lysate was dialyzed against 10 mM Tris (pH 7.5; 30 ml of dialysate/1 liter of buffer; four exchanges), filtered through a 0.45-μm membrane, and applied to a Hi-Prep Q column (Amersham Biosciences). The column was eluted with a 0–600 mM NaCl gradient in 15 column volumes. RBP was eluted at ≈150 mM NaCl. Fractions were pooled and incubated overnight at 4°C with 2mM o-phenanthroline/50 mM EDTA, concentrated 10-fold by ultrafiltration (Amicon YM-10 membrane), applied to a preequilibrated S-100 gel filtration column (Amersham Biosciences), and eluted with 20 mM 3-N-morpholinepropanesulfonic acid (Mops) (pH 6.9)/50 mM NaCl. RBP in the pooled fractions was >95% pure, as judged by PAGE (Coomassie stain). Protein concentrations were determined spectrophotometrically by using an extinction coefficient, ε280 = 1,280 M–1·cm–1, for each tyrosine residue. In all procedures, metal-free buffers were prepared by repeated dithiazone/chloroform extractions (44).
Ligand-Binding Assays. Purified proteins were coupled with the thiol-reactive styryl dye JPW4042 as described (31). Metal binding was determined by monitoring changes in fluorescence intensity on addition of metal (11). All solutions are 20 mM Mops (pH 6.9)/50 mM NaCl at 25°C. Apparent affinities were calculated by fit of the data to a single-site binding isotherm from the total added Zn2+ concentrations (31). The free Zn2+ concentrations in the presence of Mops are not known; a millimolar stability constant of Mops for Zn2+ would adjust the free Zn2+ concentrations to ≈15% of total Zn2+ (45, 46) with concomitant changes in the metal affinities.
Protein Stability. Thermal unfolding transitions were determined by measuring the molar ellipticity at 222 nm [10 μM protein in 20 mM Mops (pH 6.9)/50 mM NaCl] with an Aviv 62DS circular dichroism spectrophotometer (Aviv Associates, Lakewood, NJ). Transition midpoint values (Tm) were determined by fitting to a two-state model (47).
Cellular Signaling. Signal transduction was measured in a variant of RU1012 (8, 13), HH3000, in which the chromosomal copy of RBP, rbsB, was deleted by a gene replacement strategy (48), using a pMAK705 recombinant containing the flanking regions of the rbsB gene [regions 3,933,404–3,933,903 and 3,934,802–3,935,308, numbered according to the complete E. coli genomic sequence (49)], cloned from RU1012 genomic DNA by PCR. Wild-type and designed RBPs were cloned into pACYC under control of the ptac promoter, obtained from the pKK223-2 vector (50) by PCR (pACRBP). The trz HK chimera was constructed by cloning the 663- to 1,353-bp region from envZ and the 1- to 798-bp region of trg, both obtained from genomic DNA [CSH100 strain (51)] by PCR, and recombining these regions into a ptac expression vector, pTRZ, a variant of pMal-2c (11), where the malE region has been replaced with the trz reading frame.
The pACRBP and pTRZ plasmids were cotransformed into HH3000 by electroporation, and maintained by selection with ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml) on 2× YT plates. Signal transduction assays were performed in cultures grown directly from single colonies to mid-log phase (A600 ≈ 0.5) at 37°C (10–12 h incubation) in the presence of 1 mM ribose, or 50 μM Zn2+ added to minimal media [M9 (52), 0.4% glycerol]. Induction of the β-galactosidase reporter gene was monitored enzymatically after lysis of the cultures with chloroform and 0.1% SDS as described (51). Experiments were repeated in triplicate. Induction levels are normalized to β-galactosidase activity in the constructs grown in the absence of ligand.
Results and Discussion
Design. RBP was converted into a Zn2+-binding protein by using computational design techniques (14). The variety of coordination number, geometry, and liganding side-chain identity necessary and sufficient to form a Zn2+-binding site are well understood (22, 53). We elected to design a tetrahedral site, with a PCS composed of carboxylate oxygens and imidazole nitrogens. This geometry is known to form stable Zn2+-binding sites and avoids the experimental difficulties resulting from the use of thiols (17). The calculations were based on a high-resolution x-ray structure of RBP complexed to ribose (37) and were carried out in two phases. First, the program dezymer (14) was used to identify locations in the ribose-binding site of RBP sterically allowing placement of a Zn2+ ion and four liganding groups, taken from the set {histidine (Nδ or Nε), aspartate, glutamate (bidentate carboxylate)}. The resulting PCS designs were rank-ordered according to an energy score that captures the goodness-of-fit of the PCS geometry (14). A subset was selected in which residues span the interdomain interface such that each domain contributes at least one liganding side chain.
In the second phase of the design the SCS was designed, limiting mutations to those residues within a 15-Å sphere of the PCS center (defined by the Zn2+ ion) that make direct van der Waals contact with one or more residues in the PCS. For all designs considered here, the SCS comprises four residues. A dead-end elimination algorithm was then used to optimize the sequence of the residues in this SCS (30). In this procedure, all of the residues in the SCS were replaced with 1017-aa rotamers [based on a published rotamer library (54) that has been augmented by additional sampling of torsional angles around the published minima] representing 17 aa (histidine, cysteine, and proline were omitted from the calculation), keeping the other amino acid side chains and the entire backbone fixed. The optimal sequence was identified by determining the global minimum energy of a potential function that describes the molecular interactions in the system, considering ≈1010 possible sequence and rotamer combinations. The potential function is based on a standard semiempirical force field (30, 55, 56), including a Lennard-Jones potential to represent van der Waals interactions, an explicit, geometry-dependent hydrogen-binding term, and a continuum solvation term to represent the hydrophobic effect. Additionally, a novel term demanding that potential hydrogen bond donors and acceptors in the PCS must be satisfied by the residues in the SCS was found to be critical (13).
For each of the top three PCS candidates, rank-ordered by the geometrical descriptor of metal center coordination geometry, an SCS sequence satisfying the hydrogen-bonding capacity of the PCS histidines was successfully identified (Fig. 2 and Table 1). Two of the sites (B1 and B2) differ by a single change in the PCS, with identical metal location. All sites replace wild-type, ribose-binding residues.
Fig. 2.

Close-up stereoviews of the predicted structures of the three designed Zn2+-binding sites (A, B1, and B2). Gray sphere, Zn2+; red, oxygens; blue, nitrogens. The protein backbone is shown as a ribbon. The sequence numbers indicate the residues in the PCS (underlined) and SCS. Hydrogen bonds between the SCS and PCS are indicated by dashed lines.
Table 1. Mutations constructed in RBP to introduce Zn2+-binding sites.
| Mutations*
|
||
|---|---|---|
| Design | PCS | SCS† |
| A | Phe-15IGlu | Arg-90IAsp(89) |
| Asp-89IHisε | Ser-103HGln(235) | |
| Asp-215IIGlu | Arg-141IIGln | |
| Gln-235HHisδ | Phe-214IISer | |
| B1 and B2 | Asp-89IHisε | Arg-90ISer |
| Thr-135IIGlu | Asn-105IITyr(89) | |
| Ala-137IIGlu/Hisδ‡ | Arg-141IIGln(137§) | |
| Asn-190IIHisε | Phe-164IIThr(190) | |
Subscripts indicate the region in which the residue is located (Fig. 1). I, N-terminal domain; II, C-terminal domain; H, hinge; δ and ε, imidazole nitrogen that coordinates Zn2+.
Numbers in parentheses indicate the PCS histidine to which the SCS residue is hydrogen bonded.
Glu in B1, His in B2.
Gln in both designs, hydrogen bonding in B2 only.
Metal Binding. Binding of zinc was measured for designs with the PCS mutations alone, as well as those with PCS and SCS, using fluorescent conjugates in which the styryl dye JPW4042 was coupled to an engineered cysteine at residue 236 (31) (Fig. 3 and Table 2). The emission intensity of fluorophores conjugated at this position changes in response to ligand-mediated conformations and can therefore be used to monitor ligand binding (31). Wild-type RBP does not bind zinc; conversely, none of the designs bind ribose. In all cases, Zn2+ affinities of designs that contain the PCS alone are one to two orders of magnitude weaker than those of the designs combining the PCS and SCS, clearly indicating the importance of interactions between the SCS and PCS. The combined PCS and SCS designs all have similar affinities for Zn2+ (1–2 μM). Although it is likely that the metal affinity of these designs can be further improved by either removing vestigial interactions with ribose (11) or manipulation of the intrinsic equilibrium between the open and closed states (57), no such optimization was attempted, because the attained affinities approximate that of the original affinity of wild-type RBP for ribose (1 μM) and are sufficiently strong to mediate Zn2+-dependent control of gene expression (see below).
Fig. 3.

Binding of Zn2+ to the redesigned RBPs (design A, circles; design B1, squares; design B2, triangles). In each case, the emission intensity of the styryl dye conjugate increases on binding of Zn2+. [(Inset) Emission spectra of the apoprotein (Apo) and the Zn2+-saturated metalloprotein complex (Sat). Spectra are shown for design A, although all three designs show similar fluorescence response.] Observed intensities are fit to a single-site binding isotherm.
Table 2. Zn-binding affinities and thermal stabilities of the RBP Zn2+-binding designs.
| PCS
|
PCS + SCS
|
|||
|---|---|---|---|---|
| Design | appKd(Zn2+), μM | Tm, °C | appKd(Zn2+), μM | Tm, °C |
| A | 16 | 49.2 | 1.0 | 54.6 |
| B1 | 58 | 50.5 | 1.5 | 55.1 |
| B2 | 430 | 49.7 | 2.5 | 55.2 |
The thermostability of each of the six proteins did not change discernibly on addition of Zn2+. The ribose-binding affinity of wild-type RBP is 1 μM. The thermostability of the wild-type protein is 58°C.
All of the designs are highly selective for Zn2+ (Fig. 4). The first-row transition metals Ni2+, Co2+, and Mn2+ all bind with at least 1,000-fold lower affinities. Similarly, Cd2+ and Hg2+ exhibit very weak affinities. This finding is consistent with formation of a rigid binding site that strongly selects for the intended cognate metal ion by disallowing small structural relaxations to accommodate the preferred coordination geometries of other metals (53). Such a high degree of selectivity is a hallmark of naturally evolved metal centers in proteins (22, 28).
Fig. 4.

Selectivity of the designed sites for Zn2+ over other first-row transition (Ni2+, Co2+, and Mn2+) and group IIB (Cd2+ and Hg2+) metals. Results are reported as the free-energy difference, ΔΔGb, relative to the Zn2+ complex [ΔΔGb = RT ln(appKd(M2+)/appKd(Zn2+])); ΔΔGb > 0 indicates preference for Zn2+].
Thermostability. Thermal denaturation was used to assess the effect of the mutations on protein stability, monitor the presence of native structure by circular dichroism, and compare the midpoints of the thermal denaturation transitions between the apoproteins (Table 2). The stability of wild-type apo-RBP is 58°C. In all cases, the designs that contained the PCS alone are destabilized <10°C, whereas the designs combining the PCS and SCS have stabilities within 3°C of the wild-type protein. The SCS is therefore clearly important for formation of a stable PCS design, consistent both with the metal-binding observations and the design strategy.
Addition of 50 μM Zn2+ to any of the six designs (three full designs and three with PCS alone) has no discernible effect on protein stability (data not shown), suggesting that Zn2+ binds equally well to both the native and denatured states in these experiments, a phenomenon that has been observed in other designs also (17).
Control of Gene Expression. Binding of ribose to wild-type RBP triggers chemotaxis toward the sugar via a two-component signal transduction cascade (58). Ligand-mediated formation of the closed form of this receptor is coupled to binding to a transmembrane histidine kinase, which in turn triggers the cytoplasmic signal transduction cascade (38). A synthetic signal transduction pathway has been constructed that reconnects the ribose-mediated chemotactic response to transcriptional activation of a β-galactosidase reporter gene (8). This artificial pathway uses a chimeric transmembrane receptor, trz, in which the periplasmic domain of trg that interacts with RBP is fused to the cytoplasmic domain of a histidine kinase, envZ. This system was used to characterize the designed receptors in place of the wild-type RBP, thereby testing whether the Zn2+-mediated conformational change is biologically equivalent to the ribose-dependent response. The genes for A, B1, and B2 designs that combine both the PCS and SCS were cloned into a periplasmic expression vector, cotransformed with a plasmid that expresses trz, and assayed in a strain deleted for wild-type RBP. Wild-type RBP exhibits a 10-fold increase in reporter gene expression in response to ribose, but not Zn2+ addition. Conversely, all of the redesigned, Zn2+-binding RBP receptors show the opposite pattern, responding to Zn2+ but not ribose (Fig. 5). The variation in the magnitude of the Zn2+-mediated response of the redesigned RBPs ranges from 6- to 16-fold increases, similar to the 10-fold increase observed for wild-type RBP with ribose. In the absence of the plasmid expressing an RBP receptor, the trz construct shows no response to either ribose or Zn2+. The redesigned, Zn2+-binding RBP receptors therefore mediate control of gene expression by exogenous Zn2+.
Fig. 5.

Comparison of ligand-dependent expression of the β-galactosidase reporter gene mediated by the wild-type and redesigned RBPs. Error bars indicate the standard deviation of three independent measurements. The reported β-galactosidase activity is normalized relative to cell cultures grown in the absence of ligand. There is no Zn2+ dependence of β-galactosidase expression by the wild-type RBP construct. Shaded hatched bars, ribose; unshaded hatched bars, Zn2+.
Conclusions
Using automated, computational design methods, we have converted RBP into a Zn2+-binding receptor, with a total of three designed protein sequences comprising two spatially separate metal-binding positions. The redesigned receptors control Zn2+-dependent gene expression in E. coli mediated by a chimeric two-component signal transduction pathway, each showing a response comparable to that mediated by ribose and wild-type RBP.
The three sites presented in this study are all based on a four-coordinate, tetrahedral coordination sphere, with the liganding side chains being contributed by histidine or glutamate residues. A critical aspect of the design strategy is the combined construction of PCS and SCS, in which SCS residues satisfy the hydrogen-bonding capacity of the histidine imidazole nitrogen atoms not coordinated to the metal center. Comparison of the designs that contain the PCS mutations alone with those that combine the PCS and SCS show in all cases that the latter significantly outperforms the former. The full designs result in proteins with thermal stabilities comparable to wild-type RBP, metal-binding affinities equivalent to the original ribose affinity, and selectivities for Zn2+ over other divalent cations similar to that of naturally evolved metalloproteins. This finding clearly demonstrates the importance of the interactions between the PCS and SCS.
Two-component signal transduction pathways such as the one redesigned in this study represent an important class of mechanisms by which bacteria respond to their extracellular chemical milieu (38). These pathways are also widespread in higher organisms such as fungi and plants (59, 60). The systematic redesign of receptor specificity therefore provides a general mechanism for transcriptional control in a large collection of biological systems, which obviates the dependence on intracellular regulatory ligands used by many transcriptional control systems in widespread biotechnological use (61). For instance, it is unlikely that the Zn2+-dependent transcriptional regulation could have been obtained by the reengineering of an intracellular repressor in E. coli, because the cytoplasmic concentration of Zn2+ is tightly clamped to very low intracellular levels in E. coli (45).
One important application for transcriptional control of genes in response to extracellular metals is the construction of cell-based biosensors for the detection of metals in the environment (62). To date, cell-based biosensors have taken advantage of naturally occurring receptors or transcription factors that bind a metal of interest, combined with the fusion of a reporter gene to a promoter controlled by the corresponding downstream signaling pathway (9). By engineering periplasmic receptors that can be connected in a modular fashion to downstream pathways, such as was achieved in this study, it is potentially possible to construct “programmable” cell-based sensors for a wide variety of chemical threats and pollutants. Furthermore, the computational techniques used to construct the PCS and SCS can be generalized to the construction of complementary surfaces around ligands of arbitrary shape. Using these techniques, receptors functioning in the same synthetic signal transduction pathway as presented here have been designed for several small molecule xenobiotics and metabolites (13). The repertoire of controllable, synthetic signal transduction pathways has therefore been extended to include both organic and inorganic ligands.
Acknowledgments
We thank M. Inouye for the kind gift of the RU1012 strain, and L. Loew for the gift of the styryl dye. This work was supported by grants to H.W.H. from the National Institutes of Health and the Defense Advanced Research Project Agency.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PCS, primary coordination sphere; RBP, ribose-binding protein; SCS, secondary coordination sphere.
References
- 1.Hasty, J., McMillen, D. & Collins, J. J. (2002) Nature 420, 224–230. [DOI] [PubMed] [Google Scholar]
- 2.Spencer, D. M., Wandless, T. J., Schreiber, S. L. & Crabtree, G. R. (1993) Science 262, 1019–1024. [DOI] [PubMed] [Google Scholar]
- 3.Bishop, A., Buzko, O., Heyeck-Dumas, S., Jung, I., Liu, Y., Shah, K., Ulrich, S., Witucki, L., Yang, F., Zhang, C. & Shokat, K. M. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 577–606. [DOI] [PubMed] [Google Scholar]
- 4.Stockwell, B. R. (2000) Nat. Rev. Genet. 1, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clackson, T. (2000) Gene Ther. 7, 120–125. [DOI] [PubMed] [Google Scholar]
- 6.Swartz, J. R. (2001) Curr. Opin. Biotechnol. 12, 195–201. [DOI] [PubMed] [Google Scholar]
- 7.Hellinga, H. W. (1998) Folding Des. 3, R1–R5. [DOI] [PubMed] [Google Scholar]
- 8.Baumgartner, J. W., Kim, C., Brisette, R. E., Inouye, M., Park, C. & Hazelbauer, G. L. (1994) J. Bacteriol. 176, 1157–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daunert, S., Barrett, G., Feliciano, J. S., Shetty, R. S., Shrestha, S. & Smith-Spencer, W. (2000) Chem. Rev. 100, 2705–2738. [DOI] [PubMed] [Google Scholar]
- 10.Tam, R. & Saier, M. H., Jr. (1993) Microbiol. Rev. 57, 320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marvin, J. S. & Hellinga, H. W. (2001) Proc. Natl. Acad. Sci. USA 98, 4955–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benson, D. E., Haddy, A. E. & Hellinga, H. W. (2002) Biochemistry 41, 3262–3267. [DOI] [PubMed] [Google Scholar]
- 13.Looger, L. L., Dwyer, M. A. & Hellinga, H. W. (2003) Nature 423, 185–190. [DOI] [PubMed] [Google Scholar]
- 14.Hellinga, H. W. & Richards, F. M. (1991) J. Mol. Biol. 222, 763–785. [DOI] [PubMed] [Google Scholar]
- 15.Pinto, A. L., Hellinga, H. W. & Caradonna, J. P. (1997) Proc. Natl. Acad. Sci. USA 94, 5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coldren, C. D., Hellinga, H. W. & Caradonna, J. P. (1997) Proc. Natl. Acad. Sci. USA 94, 6635–6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wisz, M. S., Garrett, C. Z. & Hellinga, H. W. (1998) Biochemistry 37, 8269–8277. [DOI] [PubMed] [Google Scholar]
- 18.Benson, D. E., Wisz, M. S., Liu, W. & Hellinga, H. W. (1998) Biochemistry 37, 7070–7076. [DOI] [PubMed] [Google Scholar]
- 19.Benson, D. E., Wisz, M. S. & Hellinga, H. W. (2000) Proc. Natl. Acad. Sci. USA 97, 6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, H., Schmidt, J. J., Bachand, G. D., Rizk, S. S., Looger, L. L., Hellinga, H. W. & Montemagno, C. D. (2002) Nat. Mater. 1, 173–177. [DOI] [PubMed] [Google Scholar]
- 21.Yang, W., Jones, L. M., Isley, L., Ye, Y., Lee, H.-W., Wilkins, A., Liu, Z.-R., Hellinga, H. W., Malchow, R., Ghazi, M. & Wang, J. Y. (2003) J. Am. Chem. Soc. 125, 6165–6171. [DOI] [PubMed] [Google Scholar]
- 22.Christianson, D. W. (1991) Adv. Protein Chem. 42, 281–355. [DOI] [PubMed] [Google Scholar]
- 23.Lesburg, C. A. & Christianson, D. W. (1995) J. Am. Chem. Soc. 117, 6838–6844. [Google Scholar]
- 24.Kiefer, L. L., Paterno, S. A. & Fierke, C. A. (1995) J. Am. Chem. Soc. 117, 6831–6837. [Google Scholar]
- 25.Huang, C., Lesburg, C. A., Kiefer, L. L., Fierke, C. A. & Christianson, D. W. (1996) Biochemistry 35, 3439–3446. [DOI] [PubMed] [Google Scholar]
- 26.Christianson, D. W. & Alexander, R. S. (1989) J. Am. Chem. Soc. 111, 6412–6419. [Google Scholar]
- 27.Marino, S. F. & Regan, L. (1999) Chem. Biol. 6, 649–655. [DOI] [PubMed] [Google Scholar]
- 28.Hunt, J. A., Ahmed, M. & Fierke, C. (1999) Biochemistry 38, 9054–9062. [DOI] [PubMed] [Google Scholar]
- 29.Dahiyat, B. I. & Mayo, S. L. (1997) Science 278, 82–87. [DOI] [PubMed] [Google Scholar]
- 30.Looger, L. L. & Hellinga, H. W. (2001) J. Mol. Biol. 307, 429–445. [DOI] [PubMed] [Google Scholar]
- 31.de Lorimier, R. M., Smith, J. J., Dwyer, M. A., Looger, L. L., Sali, K. M., Paavola, C. D., Rizk, S. S., Sadigov, S., Conrad, D. W., Loew, L. & Hellinga, H. W. (2002) Protein Sci. 11, 2655–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hellinga, H. W. & Marvin, J. S. (1998) Trends Biotechnol. 16, 183–189. [DOI] [PubMed] [Google Scholar]
- 33.Benson, D. E., Conrad, D. W., de Lorimier, R. M., Trammell, S. A. & Hellinga, H. W. (2001) Science 293, 1641–1644. [DOI] [PubMed] [Google Scholar]
- 34.Marvin, J. S. & Hellinga, H. W. (1998) J. Am. Chem. Soc. 120, 7–11. [Google Scholar]
- 35.Marvin, J. S., Corcoran, E. E., Hattangadi, N. A., Zhang, J. V., Gere, S. A. & Hellinga, H. W. (1997) Proc. Natl. Acad. Sci. USA 94, 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bjorkman, A. J. & Mowbray, S. L. (1998) J. Mol. Biol. 279, 651–664. [DOI] [PubMed] [Google Scholar]
- 37.Mowbray, S. L. & Cole, L. B. (1992) J. Mol. Biol. 225, 155–175. [DOI] [PubMed] [Google Scholar]
- 38.Stock, A. M., Robinson, V. L. & Goudreau, P. N. (2000) Annu. Rev. Biochem. 69, 183–215. [DOI] [PubMed] [Google Scholar]
- 39.Mizuno, T. (1997) DNA Res. 4, 161–168. [DOI] [PubMed] [Google Scholar]
- 40.Street, A. G. & Mayo, S. L. (1998) Folding Des. 3, 253–258. [DOI] [PubMed] [Google Scholar]
- 41.Fersht, A. R. (1999) Structure and Mechanism in Protein Science (Freeman, New York).
- 42.Carson, M. (1997) Macromol. Crystallogr. B 277, 493–505. [Google Scholar]
- 43.Doering, D. S. & Matsudaira, P. (1996) Biochemistry 35, 12677–12685. [DOI] [PubMed] [Google Scholar]
- 44.Holmquist, B. (1988) Methods Enzymol. 158, 6–12. [DOI] [PubMed] [Google Scholar]
- 45.Outten, C. E. & O'Halloran, T. V. (2001) Science 292, 2488–2492. [DOI] [PubMed] [Google Scholar]
- 46.Fahrni, C. J. & O'Halloran, T. V. (1999) J. Am. Chem. Soc. 121, 11448–11458. [Google Scholar]
- 47.Schellman, J. A. (1955) C. R. Trav. Lab. Carlsberg Ser. Chim. 29, 230–259. [PubMed] [Google Scholar]
- 48.Hamilton, C. M., Aldea, M., Washburn, P. B. & Kushner, S. (1989) J. Bacteriol. 171, 4617–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blattner, F. R., Plunkett, G., III, Bloch, C. A., Perna, N. T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J. D., Rode, C. K., Mayhew, G. F., et al. (1997) Science 277, 1453–1474. [DOI] [PubMed] [Google Scholar]
- 50.Horazdovsky, B. F. & Hogg, R. W. (1987) J. Mol. Biol. 197, 27–35. [DOI] [PubMed] [Google Scholar]
- 51.Miller, J. H. (1992) A Short Course in Bacterial Genetics (Cold Spring Harbor Lab. Press, Plainview, NY).
- 52.Maniatis, T., Fritsch, E. F., Sambrook, J. & Cold Spring Harbor Laboratory (1982) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab., Cold Spring Harbor, NY).
- 53.Glusker, J. P. (1991) Adv. Protein Chem. 42, 1–76. [DOI] [PubMed] [Google Scholar]
- 54.Lovell, S. C., Word, J. M., Richardson, J. S. & Richardson, D. C. (2000) Proteins Struct. Funct. Genet. 40, 389–408. [PubMed] [Google Scholar]
- 55.Gordon, D. B., Marshall, S. A. & Mayo, S. L. (1999) Curr. Opin. Struct. Biol. 9, 509–513. [DOI] [PubMed] [Google Scholar]
- 56.Wang, W., Donini, O., Reyes, C. M. & Kollman, P. A. (2001) Annu. Rev. Biophys. Biomol. Struct. 30, 211–243. [DOI] [PubMed] [Google Scholar]
- 57.Marvin, J. S. & Hellinga, H. W. (2001) Nat. Struct. Biol. 8, 795–798. [DOI] [PubMed] [Google Scholar]
- 58.Bourret, R. B. & Stock, A. M. (2002) J. Biol. Chem. 277, 9625–9628. [DOI] [PubMed] [Google Scholar]
- 59.Oka, A., Sakai, H. & Iwakoshi, S. (2002) Genes Genet. Syst. 77, 383–391. [DOI] [PubMed] [Google Scholar]
- 60.Santos, J. L. & Shiozaki, K. (2001) Sci. STKE 2001, RE1. [DOI] [PubMed] [Google Scholar]
- 61.Donovan, R. S., Robinson, C. W. & Glick, B. R. (1996) J. Ind. Microbiol. 16, 145–154. [DOI] [PubMed] [Google Scholar]
- 62.Kong, I. C., Bitton, G., Koopman, B. & Jung, K. H. (1995) Rev. Environ. Contam. Toxicol. 142, 119–147. [DOI] [PubMed] [Google Scholar]