

Abstract
Intracellular deposition of aggregated and ubiquitinated proteins is a prominent cytopathological feature of most neurodegenerative disorders frequently correlated with neural cell death. To elucidate mechanisms in neural cell death and degeneration, we characterized the Drosophila ortholog of Sec61α (DSec61α), a component of the translocon that is involved in both protein import and endoplasmic reticulum-associated degradation. Loss-of-function experiments for DSec61α revealed that the translocon contributes to expanded polyglutamine-mediated neuronal toxicity, likely resulting from proteasome inhibition and leading to accumulation of ubiquitinated proteins. Taken together, proteasome inhibition by expanded polyglutamine tracts may lead to the accumulation of toxic undegraded proteins normally transported by the Sec61α translocon.
In a wide variety of neurodegenerative diseases, unfolded polypeptides accumulate in cells, largely as insoluble inclusions, and appear to play a critical role in disease pathogenesis such as amyotrophic lateral sclerosis, Alzheimer's disease, and Parkinson's disease, and several hereditary diseases caused by the expansion of polyglutamine tracts (e.g., Huntington's disease or the spinocerebeller ataxias). In all of these neurodegenerative diseases, the pathology and eventual death of specific neuronal populations occurs as a result of the accumulation of distinctly abnormal polypeptides (1). Many observations suggest that these various types of inclusions arise through common mechanisms that elicit similar host responses. For example, they all contain components of the ubiquitin-proteasome degradative pathway as well as molecular chaperones, representing the two main systems that protect eukaryotic cells against the buildup of unfolded polypeptides (1). In fact, the transient expression of an expanded polyglutamine tract causes nearly partial inhibition of the ubiquitin-proteasome system in cell culture system (2, 3). Because of the central role of ubiquitin-dependent proteolysis in regulating fundamental cellular events such as cell division and apoptosis, many recent studies suggest a potential mechanism linking protein aggregation to cellular disregulation and cell death (2, 3). However, the execution mechanisms underlying the neuronal cell death mediated by pathogenic inclusions and proteasome inhibition remain poorly understood, especially regarding the genetic evidence involving unfolded proteins in neurotoxicity in vivo.
We used a technique for genetic screening in Drosophila to identify gene products that can cause neural death or degeneration (4, 5) and revealed that one of these gene products, a homologue of Sec61α translocon, plays a critical role in neural cell death and degeneration (H.K. and M.M., unpublished data). Protein-conducting channels of eukaryotic endoplasmic reticulum (ER) are composed of three transmembrane subunits: Sec61α, Sec61β, and Sec61γ (6–8), collectively termed a translocon. Translocons are known to be required for both protein import (for the translocation of membrane or secretory preproteins) and export [for ER-associated protein degradation (ERAD)] at the ER lumen (9–11). Eukaryotic cells have complex degradation machinery enabling elimination of misfolded or unassembled secretory proteins from the ER. These proteins are retained in the ER/pre-Golgi compartment until retrograde translocation (dislocation) from the ER to the cytoplasm delivers them to the ubiquitin-proteasome system in ERAD. This dislocation process is mediated by Sec61α together with Sec61β and Sec61γ. Many recent observations suggest that increased protein dislocation from the ER or inhibition of the ubiquitinproteasome system may lead to the accumulation of ER-derived misfolded proteins in the cytosol, thereby contributing to the pathogenesis of several severe human diseases (9). Recently, Drosophila has been used to elucidate mechanisms of human neurodegenerative disease, including Alzheimer's, Parkinson's, and Huntington's diseases (12–14). Therefore, we examined the function of Drosophila Sec61α in the induction of neuronal cell death and degeneration, particularly during expanded polyglutamine-mediated neurotoxicity. Sec61α was found to be necessary for polyglutamine-induced neurodegeneration, and loss of Sec61α function in Drosophila reduced amounts of ubiquitinated protein accumulation induced by expanded polyglutamine. Our results suggest that the Sec61α translocon mediates protein deposition from the ER to the cytoplasm and that toxicity results from the proteasome's inability to degrade these proteins in the presence of expanded polyglutamine proteins.
Experimental Procedures
Fly Stocks. All general fly stocks and GAL4 lines were obtained from Drosophila stock centers. Fly culture and crosses were performed at 25°C. The UAS-MJDtr-Q78 and GMR-huntingtin120Q flies were a gift from Nancy Bonini (University of Pennsylvania, Philadelphia) and Lary Zipursky (University of California, Los Angeles) (13, 14).
Histology. Flies were prepared for semithin sections and toluidine blue staining as described (15). All fluorescent labeling was examined with an Axioskop2plus fluorescence microscope (Zeiss) and LSM510 confocal microscope (Zeiss). For light microscopic images of the adult eye, flies were anesthetized and examined with a Nikon SMZ1000 microscope equipped with an AxioCam digital camera (Zeiss).
Plasmid Construction. A DSec61α fragment from an EST clone (LP02784) was inserted into pUAST and pBSSK to generate pUAST-DSec61α and pBSSK-DSec61α. A fragment for FLAG-tagged DSec61α amplified by PCR was inserted into pUAST (pUAST-FLAG-DSec61α) and pCaspeR-hs (pCaspeR-hs-FLAGDSec61α). The EGFP fragment from pCaspeR-hs-EGFP (15) was inserted into pBSSK (pBSSK-EGFP). The fragments of tNhtt-Q17-EGFP, tNhtt-Q60-EGFP, and tNhtt-Q150-EGFP (16) were inserted into pUAST (pUAST-htt17QEGFP, pUAST-htt60QEGFP, and pUAST-htt150QEGFP, respectively) and pCaspeR-hs (pCaspeR-hs-htt17QEGFP, pCaspeR-hshtt60QEGFP, and pCaspeR-hs-htt150QEGFP, respectively). A driver plasmid that expresses GAL4 under the control of the actin5C promoter (pWAGAL4), pUAST-GFP, and pCaspeR-hslacZ has been described (17, 18).
Cell Culture, Transfections, and Viability Assays. Drosophila S2 cells and neural cell line BG2-c6 (BG2 cells) (19) were cultured and transfected as described (18, 19). For a cell death assay, these cells were cultured in 24-well plates (1 × 105 cells per well) and transfected with various amounts of the indicated plasmids plus 2ngof pWAGAL4, which expresses the GAL4 protein under the control of the actin5C promoter. In the RNA interference experiments, described amounts of double-stranded RNA (dsRNA) were transfected by the same procedure. At 24 h after transfection, cells were heat-shocked at 37°C for total 2 h, cultured at 27°C for 24 h, and then subjected to cell death assay as described (18). For a viability assay in Drosophila embryos, plasmids that can be driven by heat-shock promoter (pCaspeR-hs constructs) were dissolved in injection buffer (5 mM KCl in 0.1 mM phosphate buffer, pH 8.0) at 50 or 100 ng/μl. Embryos were collected within 30 min after egg laying at 25°C, dechorionated, and attached to a coverslip with double-stick tape. Embryos were then covered in halocarbon oil and injected at precellularized stage. Injection location was in the posterior domain extending from 50% to 75% egg length. The DNA solution was injected by a syringe. The injection volume ranged from 50 to 150 pL as determined by measuring the diameter of droplets injected into halocarbon oil. At 12 h after injection, embryos were heat-shocked at 37°C for 1 h and developed for 12 h, and hatched larvae were counted.
Immunoblotting. For detection of ubiquitinated proteins and FLAG-tagged or Myc-tagged proteins, S2 cells were cultured in six-well plates (5 × 105 cells per well) and transfected with various amounts of the indicated plasmids. At 24 h after transfection, the cells were lysed in SDS sample buffer. Fly eyes were carefully dissected from anesthetized flies and lysed in SDS sample buffer to assay for ubiquitinated proteins. All samples were separated by 10% SDS/PAGE and subjected to immunoblotting using an anti-ubiquitin mouse antibody (1:250, Stressgen Biotechnologies, Victoria, Canada), anti-myc mouse antibody (1:2,000, Invitrogen), anti-FLAG M2 antibody (1:1,000, Sigma), anti-β-tubulin E7 (1:500, Developmental Studies Hybridoma Bank, Iowa City), and an anti-mouse IgG-horseradish peroxidase antibody (1:1,000, Promega). The signals were visualized with ECL plus (Amersham Pharmacia).
Proteasome Activity. Proteasome activity was assessed in lysates of S2 cells by using synthetic peptide substrates linked to a fluorometric reporter, aminomethylcoumarin (20). S2 cells were cultured in 6-well plates (5 × 105 cells per well) and transfected with various amounts of the indicated plasmids. At 24 h after transfection, cells were collected, washed in PBS, and lysed in buffer H (20 mM Tris/20 mM NaCl/1 mM EDTA/5 mM β-mercaptoethanol, pH 7.6). Cell lysates were collected by centrifugation, and the supernatants were used for the determination of protein concentration and enzymatic activity by using Suc-LLVY-MCA (Peptide Institute, Osaka) as described (20).
RNA Interference in Drosophila Cells. For the production of synthesized dsRNA, the indicated cDNA fragments flanking both the T7 and T3 promoter sequences were amplified by PCR from each pBSSK plasmid (as described above) with the specific primers. The amplified fragments were purified and served as templates for RNA synthesis using T7 or T3 RNA polymerase (Promega) by standard protocols. The RNAs were ethanol-precipitated, dissolved in water, quantified, and mixed for annealing in equal quantities by heating for 10 min at 68°C and cooling to 37°C for 30 min. The resulting dsRNAs were checked to verify their purity and size on agarose gels. For generating a head-to-head inverted repeat construct, a fragment (nucleotides 101–1000 of the ORF) fused with BglII and XbaI sites was inserted into the BglII/XbaI site of pUAST. Next, another fragment (nucleotides 201–1000 of ORF) fused to BglII and EcoRI sites was inserted into the EcoRI/BglII site of the same plasmid to generate pUAST-DSec61α dsRNA. In RNA interference experiments, each dsRNA material was transfected into BG2 or S2 cells by using CellFectin (GIBCO) as described above.
DSec61α Mutant Fly. As the candidate mutant lines for the DSec61α gene, the P560(l(2)k04917) and EP(2)2567 P element strains were identified from the Berkeley Drosophila Genome Project. The P element insertion sites in these lethal lines were found to reside in the first intron of the DSec61α gene. The lethality reverted when the inserted P element of P560 was excised. P element revertants were generated by standard genetic techniques, and the association of the observed phenotype with the P element insertion was confirmed by excision of the P element: one excision line of P560, DSec61αP560ex25, could not improve the expanded polyglutamine-induced eye degeneration (Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). The P560 and EP(2)2567 strains were putative hypomorphic alleles for DSec61α, because RTPCR analysis revealed that the amount of mRNA for DSec61α was significantly reduced in larvae transheterozygous for these strains (data not shown). Thus, we designated these strains DSec61αP560 and DSec61αEP(2)2567.
Results
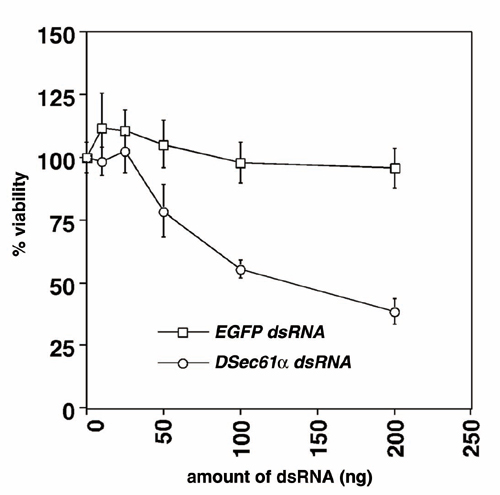
DSec61α Is Involved in Neural Cell Death Induced by Proteasome Inhibition. To elucidate the mechanisms of neural cell death including neurodegeneration, we developed a screen for genes involved in the neuronal cell death associated with neurodegeneration, involving the overexpression of individual genes throughout much of the Drosophila genome. Briefly, to identify neuronal cell death-related genes, we used misexpression fly strains, GS lines, that would misexpress unknown genes driven by GAL4 protein. In our screen we used a rhodopsin 3-GAL4 (rh3-GAL4) line that expresses the GAL4 activator in a specific subset of postmitotic photoreceptor neurons in the adult, expecting that the rh3-GAL4 line combined with GS lines would allow us to identify potential cell death genes of a type that could induce neuronal cell death in a cell-autonomous fashion. As a result of the screen of ≈5,000 fly strains, we identified and characterized the endd2 gene, which encodes the Drosophila ortholog of Sec61α (DSec61α) located in ER, as a component of the neural cell death and degeneration pathways (H.K. and M.M., unpublished data). To understand the mechanisms of this cell death, we examined the role of DSec61α in neural cell death induced by various kinds of stimuli including the proteasome inhibitor lactacystin. In Drosophila, RNA interference has been shown to be an effective method to knock down specific targeted gene products (21–23) and was used in our experiments for the knockdown of DSec61α protein expression (Fig. 1A). Addition of lactacystin, a proteasome inhibitor, caused a severe reduction in cell viability that was partially alleviated through disruption of DSec61α (Fig. 1 B and C). This finding is in contrast to other drugs, including tunicamycin, an inhibitor of N-glycosylation that induces the rapid unfolded protein response (UPR), staurosporine, cycloheximide, and ecdysone, all of which caused cell death independently of DSec61α (Fig. 1B). Thus, these data indicate that DSec61α contributes to neural cell death induced by disruption of proteasome function.
Fig. 1.

DSec61α contributes to cell death induced by proteasome inhibition in Drosophila cells. (A) RNA interference experiments in Drosophila cells. The synthesized dsRNA or the dsRNA expressed as an extended hairpin-loop RNA by the UAS vector were used to knock down the DSec61α protein expression. Briefly, 200 ng each of UAS-FLAG-DSec61α, UAS-Myc-SCAT (used as a control), and pWAGAL4 were cotransfected with 25 ng of synthesized DSec61α dsRNA into Drosophila S2 cells. At 24 h after transfection, the cells were collected and subjected to immunoblotting using an anti-myc and anti-FLAG antibody. (B) The DSec61α gene is partially required for neural cell death induced by lactacystin, which inhibits proteasome activity in cells. dsRNA (25 ng of EGFP and DSec61α) was transfected into Drosophila BG2 neural cells. Twenty-four hours after transfection, 10 μg/ml tunicamycin, 1 μM staurosporine, 1 μg/ml cycloheximide, 10 μM ecdysone, and 20 μM lactacystin were added to the media. The cells were then incubated for 24 h, and the cell viability was determined. (C) Synthesized dsRNA (25 ng of DSec61α dsRNA) or dsRNA expression vector (10 ng of UAS-DSec61α dsRNA with 2 ng of pWAGAL4) was transfected into Drosophila S2 cells. Twenty-four hours after transfection, lactacystin was added to the media (1, 5, 20, and 50 μM). The cells were then incubated for 24 h more, and the cell viability was determined.
Expanded Polyglutamine Provokes Proteasome Inhibition. A number of studies have focused on the potential role of protein aggregation and disruption of the proteasome proteolytic pathway in polyglutamine-mediated neurodegeneration (2, 3). Because our results suggested involvement of DSec61α in a cell death signaling pathway evoked by proteasome inhibition, we analyzed a possible connection between expanded polyglutamine-mediated toxicity and the DSec61α translocon protein. The expression of truncated versions of the pathogenic human MJD/SCA3 gene or the human Huntingtin gene in postmitotic neurons elicits lateonset progressive degeneration and the loss of photoreceptor neurons in a manner largely independent of caspase-dependent cell death pathways (13, 14). We first examined the correlation between polyglutamine-induced neural degeneration and the ubiquitin-proteasome function. In agreement with previous finding (24), reduced gene dosage of Pros26 (described below) clearly enhanced the neurodegeneration in flies expressing Machado–Joseph disease (MJD) protein or Huntingtin (Fig. 2 A–H). Similarly, overexpression of expanded polyglutamine reduced proteasome activity in a tract length-dependent manner in S2 cells and treatment of S2 cells with the proteasome inhibitor, lactacystin, blocked proteasome activity, resulting in the accumulation of ubiquitin conjugates (Fig. 2 I and J). Furthermore, the accumulation of ubiquitin conjugates from whole eyes and S2 cells expressing expanded polyglutamine exhibited a stronger high-molecular-weight smear of ubiquitin immunoreactivity (Fig. 2 J and K). The proteolytic machinery of regulated protein degradation is a large, multisubunit complex known as the 26S proteasome. This complex is made up of two components: a 20S core particle composed of four stacked heptameric rings and 19S regulatory complexes capping each end. Drosophila Pros261 is missense mutations in the 20S proteasome subunits, β6. In a mutant heterozygous for this 26S proteasome subunit gene (Pros261/+), the accumulation of ubiquitin conjugates from whole eyes expressing expanded polyglutamine was significantly enhanced (Fig. 2K). These data indicate that the mechanism underlying the neural degeneration induced by expanded polyglutamine tracts involves inhibition of proteasome function, which can lead to the accumulation of undegraded, ubiquitin-conjugated proteins.
Fig. 2.

The DSec61α translocon is required for the accumulation of undegraded proteins caused by expanded polyglutamine. (A–H) In the fly models for the polyglutamine diseases MJD and Huntington's disease, the partial loss of the 26S proteasome subunit clearly enhanced the phenotypes of neurodegeneration in the eye. Light microscopic (A–D) and semithin section (E–H) images of the compound eyes are shown (day 15 after eclosion). (Magnifications: ×63.) The following genotypes are shown: w;GMR-GAL4/+ (A and E), w;GMR-GAL4/+;UAS-MJD(M)/+ (B), w;GMR-GAL4/+;UAS-MJD(M)/Pros261 (C), w;+/+;Pros261/+ (D and H), w;+/+; GMR-huntingtin120Q/+ (F), and w;+/+;GMR-huntingtin120Q/Pros261 (G). (I) Proteasome activity in Drosophila cells. Plasmids (800 ng) encoding the expanded polyglutamine tracts (httQ17, httQ60, and httQ150) were overexpressed in Drosophila S2 cells with 200 ng of driver plasmid pWAGAL4. Other S2 cells were treated with lactacystin (1, 5, and 20 μM). The proteasome activities (a cleavage activity of Suc-LLVY-MCA; chymotrypsin-like) were measured 24 h after transfection. Enzymatic activity is expressed as the percentage of the activity in control lysates. (J) Immunoblotting of the cell lysates prepared in I with the anti-ubiquitin (αUb) antibody. In some cases, 30 ng of UAS-DSec61α dsRNA and 200 ng of driver plasmid pWAGAL4 were cotransfected. Similar amounts of protein were loaded, as determined by using an anti-β-tubulin (αbeta-tubulin) antibody. (K) The accumulation of ubiquitinated proteins induced by the expanded polyglutamine tracts in the Drosophila eye was remarkably reduced in flies that were transheterozygous for the mutant DSec61α allele. Eighteen fly eyes from each line of the indicated genotype (day 1 after eclosion) were carefully dissected and subjected to immunoblotting with an anti-ubiquitin (αUb) antibody. Similar amounts of protein were loaded, as determined with an anti-β-tubulin (αbeta-tubulin) antibody. The following genotypes are shown: w;GMR-GAL4/+ (lane 1), w;GMR-GAL4/+;UAS-MJD(M)/+ (lane 2), w;GMR-GAL4/+;UAS-MJD(M)/Pros261 (lane 3), w;GMR-GAL4 DSec61αP560/DSec61αEP(2)2567;UAS-MJD(M)/+ (lane 4), w;+/+;Pros261/+ (lane 5), and w;DSec61αP560/DSec61αEP(2)2567;+/+ (lane 6).
DSec61α Is Involved in Polyglutamine-Induced Neural Cell Death and Degeneration. Like MJD/SCA3 or Huntingtin overexpression, DSec61α overexpression induced cell death and degeneration partially through a caspase-independent pathway that is distinct from that used by other known Drosophila killer proteins, Reaper and Hid (data not shown). The finding that DSec61α function is partially involved in a caspase-independent signaling pathway also suggests a possible connection between endogenous DSec61α protein and polyglutamine toxicity in neural tissues. To further explore this connection in vivo, we crossed lines expressing truncated MJD protein with an expanded polyglutamine tract (78 repeats) or Huntingtin with hypomorphic DSec61α mutants (DSec61αP560 and DSec61αEP(2)2567). WT flies expressing expanded polyglutamine protein exhibited severe, progressive eye deterioration (Fig. 3 A and B), and extensive photoreceptor degeneration contributing to an unusual ommatidium morphology (Fig. 3 D and E). Strikingly, a reduced gene dosage of DSec61α significantly suppressed these phenotypes, ameliorating the loss of pigment (Fig. 3 B and C) and photoreceptor degeneration (Fig. 3 E and F). To confirm this suppression depended on the loss of function of DSec61α we generated a revertant and found it could not suppress the polyglutamine-mediated neurodegeneration (Fig. 5). Next, we overexpressed the first exon of human Huntingtin, containing expanded polyglutamine region (17, 60, and 150 repeats), fused to enhanced GFP (EGFP) (16), in the Drosophila neural cell line BG2. The level of polyglutamine aggregation, as indicated by the EGFP fluorescence, depended on the length of the overexpressed polyglutamine repeats (Fig. 3G Top) with a corresponding change in cell viability 72 h after transfection (Fig. 3G Bottom). Again, DSec61α was shown to be indispensable in this toxicity; although knockdown of DSec61α through cotransfection of DSec61α dsRNA did not affect the rate of aggregation (Fig. 3G Middle), neural cell death was significantly reduced (Fig. 3G Bottom). Similar results were seen when expanded polyglutamine-EGFP fusion proteins were overexpressed in Drosophila embryos. Direct injection of cDNA containing 150 polyglutamine repeats, but not shorter repeats, clearly exhibited many aggregate-like structures in embryos (Fig. 3H) and resulted in severe lethality of WT embryos (Fig. 3I and Table 1). This finding is in contrast to DSec61α mutant embryos in which expression of expanded polyglutamine did not significantly exert toxic effects on embryos (Fig. 3I). However, when DSec61α was coexpressed with polyglutamine in DSec61α mutant embryos, lethality was once again seen. This toxicity depended on coexpression of DSec61α and polyglutamine, as expression of DSec61α alone did not cause lethality (Table 1). Thus, DSec61α is involved in expanded polyglutamine-induced neural cell death and degeneration.
Fig. 3.

The DSec61α translocon is involved in expanded polyglutamineinduced neural cell death and degeneration in Drosophila. (A–F) The degenerative eye phenotype of the different Drosophila models for polyglutamine disease was improved in mutants for the DSec61α gene. Light microscopic (A–C) and semithin section (D–F) images of the compound eyes are shown (day 30 after eclosion). (Magnifications: ×100.) The following genotypes are shown: w;GMR-GAL4/+ (A and D), w;GMR-GAL4/+;UAS-MJD(M)/+ (B), w;GMR-GAL4 DSec61αP560/DSec61αEP(2)2567;UAS-MJD(M)/+ (C), w;+/+;GMR-huntingtin120Q/+ (E), and w;DSec61αP560/DSec61αEP(2)2567;GMR-huntingtin120Q/+ (F). (G) The neural cell death induced by expanded polyglutamine tracts was attenuated in the DSec61α gene knockdown cells. (Top) Plasmids (200 ng of each type) encoding the expanded polyglutamine tracts with the first exon of human Huntingtin fused to EGFP (pUAST vectors) were overexpressed in Drosophila BG2 neural cells with 200 ng of driver plasmid pWAGAL4. The fluorescence indicating GFP-positive cells was examined 24 h after transfection. (Middle) Plasmids (200 ng of each type) encoding the expanded polyglutamine tracts were cotransfected with 25 ng of synthesized dsRNA (EGFP or DSec61α) in Drosophila BG2 neural cells with 200 ng of driver plasmid pWAGAL4. The numbers indicate the percentages of the population of GFP-positive cells that contained fluorescent aggregates (EGFP dsRNA, ○; DSec61α dsRNA, •). (Bottom) Plasmids (5 ng of each type) encoding the expanded polyglutamine tracts were cotransfected with 25 ng of synthesized dsRNA (EGFP or DSec61α) in Drosophila BG2 neural cells with 2 ng of driver plasmid pWAGAL4, and cell viability was determined. (H and I) The aggregate-like structure was produced by expanded polyglutamine in Drosophila embryos, which affected viability of embryos. Three types of expanded polyglutamine tracts with the first exon of human Huntingtin fused to EGFP and 100 ng/μl pCaspeR-hs vectors were injected into Drosophila embryos (w1118 and DSec61αEP(2)2567/DSec61αP560). At 12 h after injection, embryos were heat-shocked at 37°C for 1 h and developed at 25°C for 12 h. The fluorescence indicating GFP-positive cells was examined 18 h after injection (H). And then the number of hatched larvae was counted (I).
Table 1. DSec61α function is involved in polyglutamine-mediated toxicity in Drosophila embryos.
| Genotype | Injected gene(s) | Ratio of viable larvae, % (total N) |
|---|---|---|
| w1118 | hs-GFP | 29.0 (178) |
| hs-htt150Q | 9.0 (187) | |
| DSec61αEP(2)2567/DSec61αP560 | hs-GFP + hs-lacZ | 31.3 (213) |
| hs-htt150Q + hs-lacZ | 25.0 (203) | |
| hs-GFP + hs-DSec61α wt | 29.8 (196) | |
| hs-htt150Q + hs-DSec61α wt | 10.1 (208) |
Precellularized embryos from WT (w1118) or DSec61α mutant (DSec61αEP(2)2567/DSec61αP560) flies were injected with 100 ng/μl hs-GFP or hs-htt150Q in addition to 50 ng/μl hs-lacZ or hs-DSec61α wt and allowed to develop at 25°C. At 12 h after injection, embryos were heat-shocked at 37°C for 1 h and developed at 29°C for 12 h, and the numbers of hatched larvae were counted.
Accumulation of Ubiquitinated Proteins by Expanded Polyglutamine Is Mediated by DSec61α. To gain insight into the mechanism by which the Sec61α translocon regulates polyglutamine-mediated neural degeneration, we focused on the function of DSec61α.We hypothesized that for polyglutamine tracts to exert a toxic effect, DSec61α must be able to dislocate potentially toxic proteins to the cytoplasm where the inhibited proteasome would no longer be able to degrade them. Interestingly, when the expression level of DSec61α mRNA was down-regulated by treatment with dsRNA, the accumulation of ubiquitinated proteins induced by lactacystin in Drosophila cells was clearly reduced (compare Fig. 2 J, lanes 2–4 and 8–10). Similar results were observed when expanded polyglutamine was overexpressed in S2 cells (compare Fig. 2 J lanes 5–7 and 11–13). Furthermore, we observed a remarkable reduction of ubiquitinated proteins induced by expanded polyglutamine in the eyes of transheterozygous DSec61α mutants (compare Fig. 2K, lanes 2 and 4). Correlated with this observations, we examined the expression of Drosophila BiP (dBiP) in cells and fly embryos. The UPR directly correlates with the acute up-regulation of the molecular chaperone BiP/GRP78, and inhibition of ERAD in yeast induces the UPR induction (see Supporting Text, which is published as supporting information on the PNAS web site). As a result, the reduction of DSec61α caused the rapid induction of dBiP (Fig. 6, which is published as supporting information on the PNAS web site), indicative of rapid induction of the UPR, suggesting that loss of DSec61α may lead to a reduction of protein dislocation. Because severe reduction of DSec61α expression in DSec61αP560/DSec61αP560 fly or in cells transfected with higher amount of DSec61α dsRNA clearly induced more UPR but also cell death (Fig. 6 and Fig. 7, which is published as supporting information on the PNAS web site), the mild loss of Sec61α translocon may contribute to the improvement of polyglutamine-mediated neural degeneration. Therefore, the increase in ubiquitinated proteins in response to polyglutamine requires a functional DSec61α translocon.
Discussion
The present study provides some important insights into neural cell death and degeneration caused by protein aggregates and unfolded proteins. Many recent studies suggest that proteasome inhibition might be a common link between the different genetic triggers of neurodegenerative diseases, especially amyotrophic lateral sclerosis, Parkinson's disease, and polyglutamine disease (1). For example, one hypothesis for the etiology of Parkinson's disease is that subsets of neurons are vulnerable to a failure in proteasome-mediated protein turnover. Overexpression of mutant α-synuclein increases sensitivity to proteasome inhibitors by decreasing proteasome function, whereas overexpression of Parkin decreases sensitivity to proteasome inhibitors in a manner dependent on Parkin's ubiquitin-protein E3 ligase activity (25). It is obvious that proteasome-inhibition induces accumulation of undegraded proteins that can trigger some kind of signal for cell death and degeneration; however, an important unanswered question remains: from where do these undegraded proteins come? The ubiquitin-proteasome system degrades proteins derived from all cellular components including the ER. Our present observations suggest that one source of undegraded protein might be the ER, thereby providing genetic evidence implicating the ER as an important source of undegraded, toxic proteins.
We characterized the Drosophila ortholog of Sec61α (DSec61α), a component of the translocon that is involved in ERAD in neural cell death and degeneration pathways. Loss-of-function experiments for DSec61α revealed that the translocon contributes to the expanded polyglutamine-mediated neuronal toxicity and accumulation of ubiquitinated proteins caused by inhibiting the proteasome. These observations provide a possible mechanism for polyglutamine toxicity: expanded polyglutamine inhibits the ubiquitin-proteasome function, thereby leading to an accumulation of unfolded proteins in the cytosol. Furthermore, these unfolded proteins are likely dislocated through the Sec61α translocon and other protein dislocation machinery (Fig. 4). Supporting this idea, the AAA ATPase Cdc48/p97/VCP, which is also involved in the ERAD system mediated by Sec61α (26, 27), has recently been identified as a positive regulator of polyglutamine-mediated toxicity in Drosophila; loss of Drosophila Cdc48/p97/VCP can improve the neurodegenerative phenotypes induced by expanded polyglutamine (28). Moreover, reduction of DSec61α may not only inhibit the retrograde translocation of unfolded proteins from the ER, but also suppress the translocation of nascent secretory protein into the ER, leading to a decrease of undegraded proteins in the cytosol (Fig. 4). Additionally, it might be possible that acute UPR induction that is caused by loss of DSec61α regulates the amount of ubiquitinated protein. Because UPR induction is correlated with up-regulation of molecular chaperone proteins (e.g., BiP/GRP78), this will lead to reduction of unfolded proteins in ER. Sequentially, the amount of unfolded proteins that should be dislocated into cytosol by ERAD system is decreased, resulting in significant reduction of ubiquitinated proteins that can contribute to polyglutamine-mediated toxicity. UPR may also reduce the expression of proteins in the cell and eventually reduce the level of ubiquitinated proteins. However, it is still less effective to improve the phenotypes of polyglutamine-expressing flies by loss of DSec61α function, suggesting that the accumulation of ubiquitinated protein, added to the nuclear functions of expanded polyglutamine such as transcriptional repression, may be a partial contributor to the degeneration, and not solely responsible for polyglutamine-mediated degeneration. Thus, such an entire translocation/dislocation system of unfolded proteins through the ER might contribute to accumulation of undesirable proteins in cells when proteasome activity is inhibited in specific neurodegenerative disease conditions.
Fig. 4.

A proposed model for Sec61α function in regulation of cell death. In normal cells (A), the translocation of secretory preproteins into ER is mediated by Sec61α. Unfolded proteins produced in ER are dislocated by Sec61α into the cytosol, which leads to protein degradation by the ubiquitin-proteasome system, a process known as ERAD. In contrast, when expanded polyglutamine is expressed, the proteasome activities are reduced. This inhibition of proteasome activity causes accumulation of ubiquitinated proteins because of an impaired ERAD system and continuous supply of these cytoplasmic undegraded proteins may induce cell death (B). However, a reduction of Sec61α function causes a decrease of translocated unfolded proteins into cytosol, resulting in accumulation of such proteins in ER. These unfolded proteins might be decreased by the UPR system in ER (C).
Many recent observations suggest that there could be specific kinds of toxic proteins in whole amounts of undegraded proteins. The Pael receptor, a substrate of the ubiquitin ligase Parkin, is proposed to be degraded by ERAD and exhibits neural toxicity when the ubiquitin-proteasome system is impaired; this cell death may be partially accompanied by cytosolic Pael receptor aggregates (29). Because accumulation of mutant forms of α-synuclein or loss of Parkin E3 ligase activity causes inhibition of ubiquitin-proteasome system (25), it might be possible that unfolded Pael receptor is dislocated by the Sec61α translocon from the ER in dopaminergic neurons in Parkinson's disease. Another example stems from changes in prion protein (PrP) folding that are associated with fatal neurodegenerative disorders such as bovine spongiform encephalopathy, scrapie, and Creutzfeldt–Jakob disease. However, the neurotoxic species is unknown. Like other proteins that traffic through the ER, misfolded PrP is retrograde transported to the cytosol for degradation by proteasomes. The transmembrane form of the prion protein (CtmPrP) is rapidly degraded by the ubiquitinproteasome system under normal conditions (30, 31); however, under certain circumstances, the specific isoform of PrP is dislocated from the ER, and accumulation of even small amounts of cytosolic PrP leads to neurotoxicity in cultured cells and transgenic mice (32, 33). Similarly the dislocation of these neurotoxic PrPs might be carried through the Sec61α translocon. These lines of evidence show that the ERAD machinery must be tightly regulated to avoid cellular disorders, while maintaining efficient proteasome function, otherwise misfolded or unassembled potentially toxic secretory proteins may accumulate in the cytosol. Thus, considering the importance of the DSec61α translocon in these processes, specific regulators may prove to be valuable therapeutic tools for the treatment of several severe human neurodegenerative diseases.
Supplementary Material
Acknowledgments
We are grateful to R. Akai for technical support, N. Nukina, Y. Hiromi, K. Ui-Tei, N. Bonini, and L. Zipursky, for materials and flies, the Developmental Studies Hybridoma Bank for antibody, the Berkeley Drosophila Genome Project for providing various reagents and information, and the Bloomington Stock Center for fly stocks. We are also grateful to N. Bence, N. Nukina, R. Takahashi, and R. Schekman for valuable discussions. This work was supported in part by grants from the Japanese Ministry of Education, Science, Sports, Culture, and Technology (to H.O. and M.M.), Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Corporation (to H.O.), and a RIKEN Bioarchitect Research Grant (to M.M.). T.I. is a research fellow of the Japan Society for the Promotion of Science. E.K. is a research fellow of the Junior Research Associate Program, RIKEN. H.K. is a research fellow of the Special Postdoctoral Researchers Program, RIKEN.
Abbreviations: ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; dsRNA, double-stranded RNA; UPR, unfolded protein response; MJD, Machado–Joseph disease; EGFP, enhanced GFP; PrP, prion protein.
References
- 1.Sherman, M. Y. & Goldberg, A. L. (2001) Neuron 29, 15–32. [DOI] [PubMed] [Google Scholar]
- 2.Bence, N. F., Sampat, R. M. & Kopito, R. R. (2001) Science 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- 3.Jana, N. R., Zemskov, E. A., Wang, G. H. & Nukina, N. (2001) Hum. Mol. Genet. 10, 1049–1059. [DOI] [PubMed] [Google Scholar]
- 4.Kanuka, H. & Miura, M. (2002) Cell Death Differ. 9, 231–233. [DOI] [PubMed] [Google Scholar]
- 5.Toba, G., Ohsako, T., Miyata, N., Ohtsuka, T., Seong, K. H. & Aigaki, T. (1999) Genetics 151, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorlich, D. & Rapoport, T. A. (1993) Cell 75, 615–630. [DOI] [PubMed] [Google Scholar]
- 7.Hanein, D., Matlack, K. E., Jungnickel, B., Plath, K., Kalies, K. U., Miller, K. R., Rapoport, T. A. & Akey, C. W. (1996) Cell 87, 721–732. [DOI] [PubMed] [Google Scholar]
- 8.Schekman, R. (1996) Cell 87, 593–595. [DOI] [PubMed] [Google Scholar]
- 9.Plemper, R. K. & Wolf, D. H. (1999) Trends Biochem. Sci. 24, 266–270. [DOI] [PubMed] [Google Scholar]
- 10.Pilon, M., Schekman, R. & Romisch, K. (1997) EMBO J. 16, 4540–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou, M. & Schekman, R. (1999) Mol. Cell 4, 925–934. [DOI] [PubMed] [Google Scholar]
- 12.Fortini, M. E. & Bonini, N. M. (2000) Trends Genet. 16, 161–167. [DOI] [PubMed] [Google Scholar]
- 13.Warrick, J. M., Paulson, H. L., Gray, B. G., Bui, Q. T., Fischbeck, K. H., Pittman, R. N. & Bonini, N. M. (1998) Cell 93, 939–949. [DOI] [PubMed] [Google Scholar]
- 14.Jackson, G. R., Salecker, I., Dong, X., Yao, X., Arnheim, N., Faber, P. W., MacDonald, M. E. & Zipursky, S. L. (1998) Neuron 21, 633–642. [DOI] [PubMed] [Google Scholar]
- 15.Kanuka, H., Sawamoto, K., Inohara, N., Matsuno, K., Okano, H. & Miura, M. (1999) Mol. Cell 4, 757–769. [DOI] [PubMed] [Google Scholar]
- 16.Wang, G. H., Mitsui, K., Kotliarova, S., Yamashita, A., Nagao, Y., Tokuhiro, S., Iwatsubo, T., Kanazawa, I. & Nukina, N. (1999) NeuroReport 10, 2435–2438. [DOI] [PubMed] [Google Scholar]
- 17.Usui, T., Shima, Y., Shimada, Y., Hirano, S., Burgess, R. W., Schwarz, T. L., Takeichi, M. & Uemura, T. (1999) Cell 98, 585–595. [DOI] [PubMed] [Google Scholar]
- 18.Igaki, T., Kanuka, H., Inohara, N., Sawamoto, K., Nunez, G., Okano, H. & Miura, M. (2000) Proc. Natl. Acad. Sci. USA 97, 662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ui, T. K., Sato, S., Miyake, T. & Miyata, Y. (1996) Neurosci. Lett. 203, 191–194. [DOI] [PubMed] [Google Scholar]
- 20.Wojcik, C. & DeMartino, G. N. (2002) J. Biol. Chem. 277, 6188–6197. [DOI] [PubMed] [Google Scholar]
- 21.Clemens, J. C., Worby, C. A., Simonson, L. N., Muda, M., Maehama, T., Hemmings, B. A. & Dixon, J. E. (2000) Proc. Natl. Acad. Sci. USA 97, 6499–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammond, S. M., Bernstein, E., Beach, D. & Hannon, G. J. (2000) Nature 404, 293–296. [DOI] [PubMed] [Google Scholar]
- 23.Kennerdell, J. R. & Carthew, R. W. (2000) Nat. Biotechnol. 18, 896–898. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez, F. P., Nino, R. M., de Gouyon, B., She, W. C., Luchak, J. M., Martinez, P., Turiegano, E., Benito, J., Capovilla, M., Skinner, P. J., et al. (2000) Nature 408, 101–106. [DOI] [PubMed] [Google Scholar]
- 25.Petrucelli, L., O'Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., Choi, P., Wolozin, B., Farrer, M., Hardy, J. & Cookson, M. R. (2002) Neuron 36, 1007–1019. [DOI] [PubMed] [Google Scholar]
- 26.Ye, Y., Meyer, H. H. & Rapoport, T. A. (2001) Nature 414, 652–656. [DOI] [PubMed] [Google Scholar]
- 27.Jarosch, E., Taxis, C., Volkwein, C., Bordallo, J., Finley, D., Wolf, D. H. & Sommer, T. (2002) Nat. Cell Biol. 4, 134–139. [DOI] [PubMed] [Google Scholar]
- 28.Higashiyama, H., Hirose, F., Yamaguchi, M., Inoue, Y. H., Fujikake, N., Matsukage, A. & Kakizuka, A. (2002) Cell Death Differ. 9, 264–273. [DOI] [PubMed] [Google Scholar]
- 29.Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y. & Takahashi, R. (2001) Cell 105, 891–902. [DOI] [PubMed] [Google Scholar]
- 30.Hegde, R. S., Mastrianni, J. A., Scott, M. R., DeFea, K. A., Tremblay, P., Torchia, M., DeArmond, S. J., Prusiner, S. B. & Lingappa, V. R. (1998) Science 279, 827–834. [DOI] [PubMed] [Google Scholar]
- 31.Yedidia, Y., Horonchik, L., Tzaban, S., Yanai, A. & Taraboulos, A. (2001) EMBO J. 20, 5383–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma, J., Wollmann, R. & Lindquist, S. (2002) Science 298, 1781–1785. [DOI] [PubMed] [Google Scholar]
- 33.Ma, J. & Lindquist, S. (2002) Science 298, 1785–1788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}