

Abstract
Transcription of the highly pathogenic Ebola virus depends on VP30, a nucleocapsid-associated Ebola virus-specific transcription factor. The transcription activator VP30 was shown to play an essential role in Ebola virus replication, most likely by stabilizing nascent mRNA. Here we present the crystal structure of the C-terminal domain (CTD) of VP30 (VP30CTD) at 2.0-Å resolution. VP30CTD folds independently into a dimeric helical assembly. The VP30CTD dimers assemble into hexamers that are present in virions, by an oligomerization domain located in the N terminus of VP30. Mutagenesis of conserved charged amino acids on VP30CTD revealed that two regions, namely a basic cluster around Lys-180 and Glu-197, are required for nucleocapsid interaction. However, only mutagenesis of the basic cluster was shown to impair transcription activation, suggesting that both processes are regulated independently. The structure and the mutagenesis results reveal a potential pocket for small-molecule inhibitors that might prevent VP30 activity and thus virus propagation as it has been shown previously by peptides, which interfere with VP30 homooligomerization.
Keywords: nucleocapsid interaction
Ebola virus (EBOV), a filovirus in the order Mononegavirales, causes unpredictable sporadic outbreaks of a fatal hemorrhagic fever in Central Africa (1–4). To date, neither a vaccine nor a treatment of the EBOV infection is available for human use.
Most viruses in the order Mononegavirales contain three proteins, the nucleoprotein N (NP), the phosphoprotein P, and the polymerase L, that drive the processes of replication (synthesis of genomic RNA) and transcription (synthesis of viral mRNAs). NP is the major factor that encapsidates the viral genome into the nucleocapsid that serves as a template for the viral polymerase complex composed of the catalytic subunit L and the cofactor P (5).
EBOV follows a unique transcription strategy using the activity of the viral NP protein VP30 (6), which is essential for the activation of the synthesis of viral mRNAs (6). Recent investigations showed that EBOV transcription could take place in the absence of VP30 if the secondary structure of the RNA that includes the transcription start site of the first gene (NP) is changed (7). It has been thus hypothesized that VP30 may help to overcome this barrier for transcriptional activation, consistent with its proposed role at an early stage of transcription (6). Although VP30 is present in nucleocapsids, its incorporation is not mandatory for the nucleocapsid to function as a template for transcription, because it can be provided in trans (8). Transcription activation by VP30 strongly depends on the phosphorylation state of VP30. Although none or weakly phosphorylated VP30 activates transcription, fully phosphorylated VP30 acts as an efficient inhibitor (9). VP30 was also shown to constitute a potential target for antiviral treatment, because its essential homooligomerization could be inhibited by synthetic peptides, which in turn inhibited EBOV propagation in cell culture (10). To gain further insight into the function of VP30, we performed structural studies.
Here we present the crystal structure of the C-terminal dimeric domain (CTD) of VP30, which is sufficient for VP30–nucleocapsid interaction. We show further that full length VP30 forms hexamers in solution and in infectious EBOV particles. We identified a basic cluster (residues 179, 180, and 183) and Glu-197 as being critical for NP–nucleocapsid interaction. Mutation of the basic cluster also disabled VP30's transcription activation function, whereas exchange of Glu-197 left transcription activation function intact. Although the presence of VP30 in nucleocapsids does not seem to be essential for transcription activation, its association with the nucleocapsid is essential to activate initial transcription in newly infected target cells (8).
Results
VP30 Forms Hexamers in Vitro and in Vivo.
VP30 activates EBOV-specific transcription and associates with the nucleocapsid complex by interaction with NP (9). Transcription activation is exerted by a homooligomer of VP30, which requires a hydrophobic stretch of leucine residues in the N terminus (10). To elucidate the stoichiometry of the VP30 homooligomer and obtain structural information on VP30, we expressed and purified full length VP30 from insect cells and found that the protein associated tightly with cellular nucleic acids and formed soluble aggregates that were further analyzed by protease digestion. Based on the protease digestion results and analyses of secondary structure elements, we designed N-terminal truncated forms of VP30 for Escherichia coli expression. This expression of VP30 mutants produced two soluble protein fragments containing residues 142–272 (VP30CTD, 15 kDa) and residues 89–272 (VP3089–272 25 kDa). In gel-filtration analysis, VP3089–272 eluted at 13.8 ml [supporting information (SI) Fig. 5A] from a Superdex200 column. The marker protein aldolase (150 kDa) eluted at 13.7 ml, suggesting that VP3089–272 is present in a hexameric form in solution. Chemical cross-linking of VP3089–272 produced a new band migrating at ≈50 kDa (corresponding to a VP3089–272 dimer), some potential intermediate forms, and a final cross-linked product migrating at ≈150 kDa that might thus correspond to a hexameric form of VP30 (Fig. 1B). This result indicates that VP30 forms hexamers in solution by an N-terminal oligomerization domain contained within residues 94–112, as determined previously (10). In density-gradient analysis, VP30 peaked in fractions 7 and 8 between the protein markers aldolase (SI Fig. 5B; 150 kDa, fractions 6 and 7) and β-amylase (200 kDa, fractions 8 and 9). In line with this finding, VP3089–272, which contained the N-terminal oligomerization site, peaked in fractions 6 and 7, corresponding to a molecular weight of ≈150 kDa (SI Fig. 5C). These experiments thus suggest a hexameric stoichiometry of VP30. In contrast, chemical cross-linking of VP30CTD, which lacks the N-terminal oligomerization domain, revealed dimerization, because increasing amounts of chemical cross-linker produced a new band on SDS/PAGE migrating at ≈30 kDa (Fig. 1A). Thus a second oligomerization domain is present in the CTD. To test whether the oligomerization observed in vitro corresponds to the oligomeric state of VP30 in vivo, we analyzed VP30 present in EBOV particles using a ProSieve native gel system. This Western blot analysis showed the appearance of a band corresponding to VP30 wild-type monomers migrating slightly larger than the 30-kDa marker protein, a second band migrating at ≈66 kDa that corresponds to the dimeric form of VP30, and a third band migrating close to the 220-kDa marker protein that might thus correspond to a hexamer of VP30 (Fig. 1C), thus suggesting that VP30 functions as a hexameric unit.
Fig. 1.

Stoichiometry of homooligomers of VP30 and VP30 fragments. Chemical cross-linking of VP30 fragments. Fragments VP30CTD (A) and VP3089–272 (B) were purified and cross-linked with 0.5–5 mM ethyleneglycol bis (succinimidyl-succinate) (EGS), separated on 10% ProSieve-acrylamide SDS/PAGE, and stained with Coomassie blue. (C) An aliquot of a γ-irradiated preparation of EBOV structural proteins was mixed with 0.1% SDS sample buffer and separated on a 10% ProSieve-acrylamide SDS/PAGE. Proteins were blotted onto PVDF membranes and stained with a polyclonal α-VP30 antibody (dilution 1:500). Bound antibodies were detected by using a secondary horseradish peroxidase-coupled goat anti-rabbit antibody (Dianova; dilution 1:30,000).
Crystal Structure of a CTD of VP30.
VP30CTD was crystallized in space group P212121 containing two monomers per asymmetric unit, consistent with the dimerization observed in solution. The structure was solved from a selenomethionine-containing crystal using the single-wavelength anomalous dispersion method and diffraction data to 2.2-Å resolution combined with phase extension to a resolution of 2.0 Å with data collected from the same crystal (SI Table 1). This analysis resulted in a readily interpretable electron-density map (Fig. 2A). Each monomer folds into a compact core helical arrangement consisting of six helices (Fig. 2B). A seventh helix reaches over to the neighboring monomer by a long linker, allowing both monomers to pack head to head (Fig. 2C). C-terminal residues 267–272 show no electron density and are presumably disordered. The dimerization interface is extensive and covers ≈3,727 Å2. The majority of the contacts are polar interactions between side-chain atoms and C-α backbone amides and carbonyls predominantly between the loop region connecting helices 6 and 7 that nestles into a groove on the neighboring monomer (residues marked by *) made up by helices 1 and 2 (Fig. 2D). The dimerization interface includes a series of conserved hydrogen bonds (L247 O–L144* N, L247 O–T143* OG1, C251 O–K180* NZ, C251 N–I142*O, E252 O–I142* N, and L264 O–W154* NE1) and a salt bridge between E152 (OE2) and K149 (NZ) (Fig. 2D and SI Fig. 6). Only one direct contact is observed between the core structure (K180 O and Q185* NE2), which, however, is not conserved (SI Fig. 6). This observation indicates that dimerization is mainly achieved by domain swapping of helix 7. It is therefore conceivable that the movement of helix 7 would be sufficient to disrupt dimerization. The conservation of the interactions indicates that both EBOV and Marburg virus VP30 can form such VP30 dimers (SI Fig. 6).
Fig. 2.
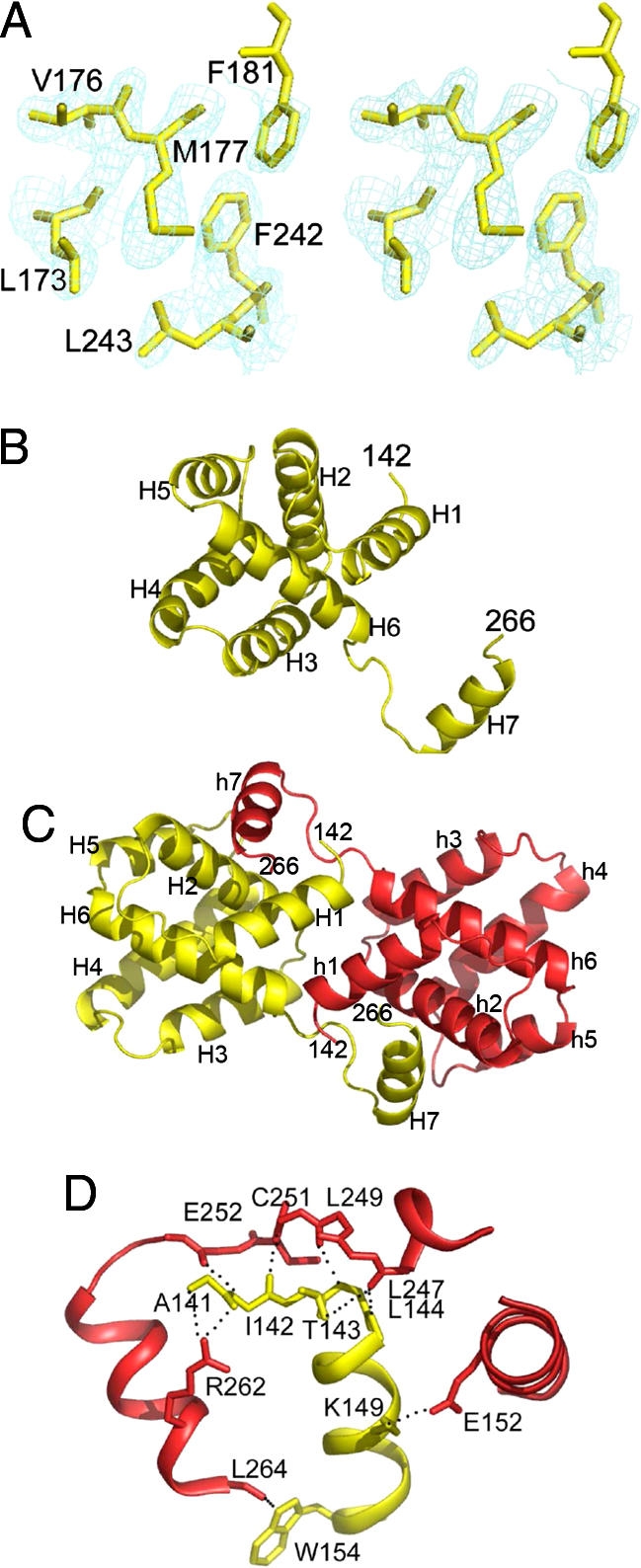
Crystal structure of dimeric VP30CTD. (A) Stereo image of the experimental electron-density map obtained from single-wavelength anomalous dispersion phases and noncrystallographic symmetry averaging showing the hydrophobic core of VP30. (B) Ribbon representation of the VP30CTD monomer that is dominated by a compact core composed of six helices and a seventh helix pointing away from the core. Secondary-structure elements are indicated. (C) Ribbon representation of the VP30CTD dimer in yellow and red. Head-to-head dimerization is supported by helix 7 interactions with the neighboring core of VP30CTD. Both N termini point away from the same side of the dimer and could potentially link up to form parallel hexamers as formed by VP3089–272 in vitro and VP30 present in EBOV particles. (D) Closeup of the VP30CTD dimerization interface. Residues mediating polar interactions are shown as all-atom model. Note that most of the interactions are between helix 7 and the loop region connecting helices 6 and 7 and the opposing helix 1.
VP30CTD Is Sufficient for Nucleocapsid Interaction.
Expression of full length VP30, VP30NTD (NTD, N-terminal domain; amino acids 1–141), and VP30CTD in HEK 293 cells showed that all three forms of VP30 colocalize with inclusions that are induced by NP, which were shown to contain nucleocapsid-like structures (SI Fig. 7A; ref. 11). However, only wild-type VP30 and VP30CTD coimmunopreciptated NP (SI Fig. 7B) and were incorporated into nucleocapsid-containing virus-like particles (VLPs) released from HEK 293 cells (SI Fig. 7C; refs. 8 and 12), whereas VP30NTD was found only in the cell lysate and was not present in VLPs (SI Fig. 7D). These results suggested that dimeric VP30CTD is sufficient for nucleocapsid interaction. When coimmunoprecipitation was performed in the presence of RNase, interaction between VP30 and NP was not impaired, suggesting that binding was not mediated by RNA (data not shown). To map the potential interaction site on VP30, we mutated charged amino acids that lie within highly conserved surface patches of dimeric VP30CTD, which is characterized by one conserved face of the molecule, whereas the opposite site is much less conserved (SI Fig. 8A). We replaced a basic cluster (residues R179, K180, and K183) by neutral or negatively charged residues (VP30mut1, SI Fig. 8B, first image), D158 and E197 by alanine (VP30mut3), and D158 alone by alanine (VP30mut2) (SI Fig. 8B, third image) within full length VP30. Expression of the VP30 mutants in HUH7 cells revealed an intracellular distribution of mutant VP30 indistinguishable from VP30 wild-type expression (data not shown). Upon coexpression with NP, VP30mut2 was efficiently recruited into the NP-induced inclusions like VP30 wild type, indicating that the conserved D158 is not essential for interaction (Fig. 3A). In contrast, VP30mut1 as well as VP30mut3 showed only partial colocalization with NP (Fig. 3A). This pattern of interaction was further confirmed by coimmunoprecipitation of VP30 and NP. Although VP30mut2 coprecipitated NP similar to wild-type VP30 (Fig. 3B, lanes 2 and 4), both VP30mut1 and VP30mut3 were largely reduced in their capacity to immunoprecipitate NP (Fig. 3B, lanes 3 and 5).
Fig. 3.
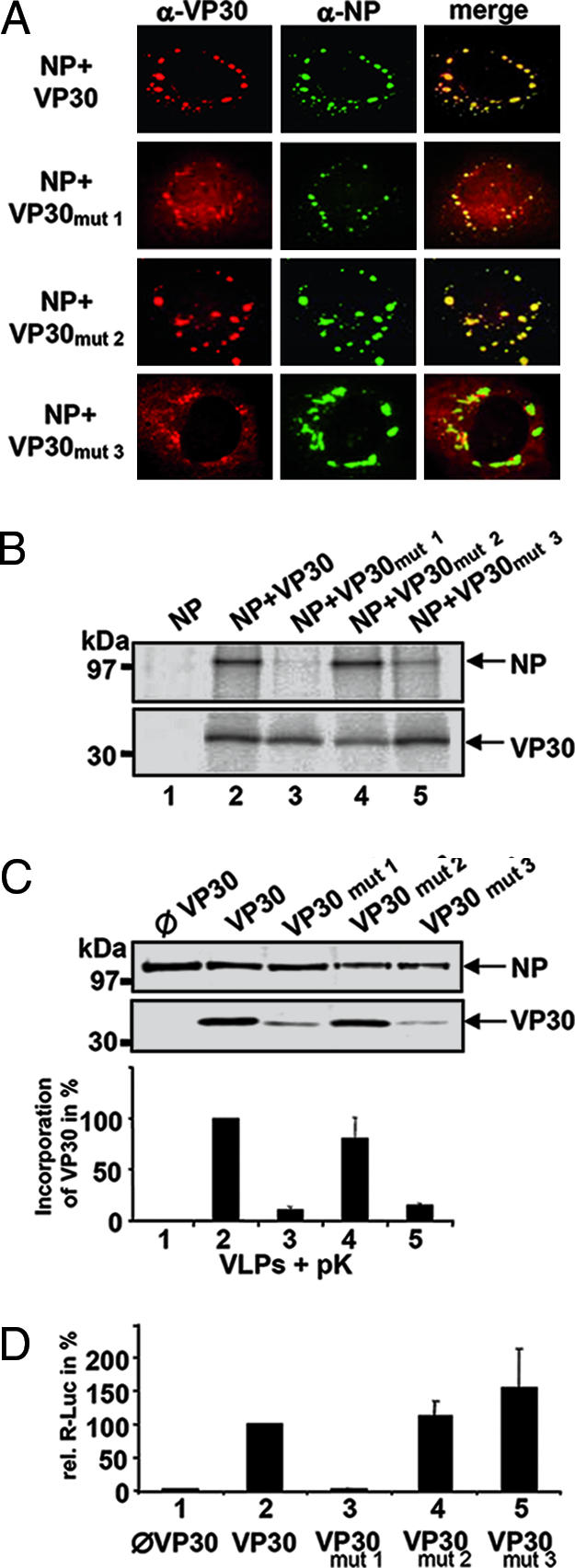
Nucleocapsid association of and transcription activation by substitution mutants of VP30. (A) Colocalization studies of NP with VP30 substitution mutants. Plasmids encoding NP and VP30 substitution mutants were transfected into HUH7 cells. At 16 h after transfection, cells were fixed, and immunofluorescence analysis was performed by using a mouse monoclonal anti-VP30 IgM (dilution 1:10) and a mouse monoclonal IgG anti-NP antibody (dilution 1:20). Bound antibodies were detected with FITC-labeled goat anti-mouse IgG serum (Dianova; dilution 1:200) and rhodamine-labeled goat anti-mouse IgM antibody (Dianova; dilution 1:100). (B) Coimmunoprecipitation analysis of VP30 and VP30 mutants with NP. HUH7 cells were cotransfected with plasmids encoding wild-type VP30 or VP30 mutants and NP. Cells were metabolically labeled and immunoprecipitated as described in Experimental Procedures. Immune complexes were separated by SDS/PAGE and exposed to a Bio-Imager (Fuji, Düsseldorf, Germany) plate. The positions of VP30 and VP30 mutants and cosedimented NP are shown by arrows. (C) Incorporation of VP30 substitution mutants into VLPs. HEK 293 cells were transfected with plasmids encoding all structural proteins of EBOV, a minigenome with the Renilla luciferase reporter gene, the T7 DNA-dependent RNA polymerase, and the firefly luciferase. If indicated, plasmid-encoding VP30 was replaced by VP30 mutants. Released VLPs were purified through a 20% sucrose cushion and treated with proteinase K for 1 h. Digestion was stopped with PMSF and samples were separated by 12% SDS/PAGE and blotted onto a nitrocellulose membrane. Immunostaining was performed with a mouse anti-flag antibody (dilution 1:15,000) and a mouse anti-NP antibody (dilution 1:200). Bound antibodies were detected with an IRDye700DX goat anti-mouse antibody (Rockland, Gilbertsville, PA; dilution 1:5,000) and analyzed with LiCor Odyssey Infrared Imaging System (Li-Cor Biosciences, Bad Homburg, Germany). (Upper) Proteinase K digested VLPs. (Lower) Quantification of wild-type VP30 or VP30 mutants incorporated into VLPs. Incorporation of VP30 and VP30 mutants was normalized against the incorporated amount of NP. Incorporation of wild-type VP30 was set to 100%. (D) Analysis of EBOV-specific transcription activation by VP30 and VP30 substitution mutants. HEK 293 cells were transfected with plasmids expressing the nucleocapsid proteins of EBOV, an EBOV-specific minigenome containing the Renilla luciferase reporter gene, and the T7 DNA-dependent RNA polymerase. As control for cellular transcription, the vector pGL2 was transfected, which contained the gene for the firefly luciferase under a simian virus 40 promoter. At 3 days after transfection, cells were lysed, and activities of Renilla luciferase and firefly luciferase were measured. Renilla luciferase activity was normalized by firefly luciferase activity, and the activity gained with wild-type VP30 was set to 100%.
Next, using a VLP assay (12), it was investigated whether VP30 mutants were incorporated into nucleocapsids and released from the cell. Incorporation efficiencies of VP30mut1 and VP30mut3 were significantly lower than that of VP30wt or VP30mut2 (Fig. 3C; VP30mut1, 11%; VP30mut3, 16%; compare lanes 3 and 5 with lanes 2 and 4). This result indicated that the basic cluster and E197 both play a role in VP30–nucleocapsid association, whereas the conserved residue D158 seems to be dispensable.
We then investigated whether the decreased incorporation of VP30 mutants into the nucleocapsid correlated with the ability to activate EBOV transcription (6) by using an EBOV-specific transcription system. As expected, the presence of VP30 activated transcription of an EBOV-specific minigenome (Fig. 3D; lanes 1 and 2). However, VP30mut1 was unable to activate transcription, whereas VP30mut2 and VP30mut3 both activated EBOV transcription similar to or better than VP30wt (Fig. 3D; lanes 4 and 5). When the oligomerization pattern of the mutants was compared with wild-type VP30 by using density-gradient centrifugation analysis, VP30mut3 revealed a migration pattern similar to that of wild-type VP30 (Fig. 4A and C). In contrast, the migration pattern of VP30mut1 suggested monomers and high-molecular-weight aggregates (Fig. 4B, fractions 3–5, 13, and 14) but no VP30 hexamers. Although the mutations in VP30mut1 should not affect CTD dimerization, they might favor VP30mut1 polymerization, as observed in the gradient. Together, our data suggest that a cluster of basic amino acids around position 180 was necessary for both nucleocapsid association and transcription activation, whereas Glu-197 was found to be essential for incorporation of VP30 into nucleocapsids. Furthermore, incorporation of VP30 into released nucleocapsids was necessary for activation of viral transcription in early steps of EBOV infection.
Fig. 4.
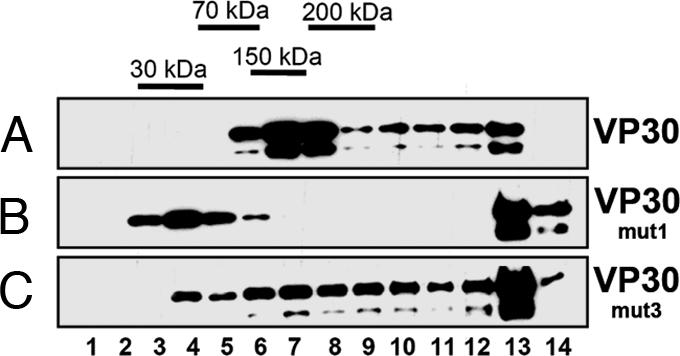
Nycodenz gradient sedimentation of VP30 and VP30 mutants. Flag-tagged VP30, VP30mut1, or VP30mut3 were expressed in HEK 293 cells. At 24 h after transfection, cells were lysed. Lysates were loaded on 10–45% Nycodenz gradients and gradient fractions were separated by SDS/PAGE and blotted onto PVDF membrane. Immunostaining was performed with a mouse anti-flag antibody (dilution 1:5,000). Bound antibodies were detected with a peroxidase-coupled goat anti-mouse antibody (Dianova; dilution 1:30,000). (A) Wild-type VP30. (B) VP30mut1. (C) VP30mut3.
Discussion
Oligomerization of VP30.
We have shown that transcription activation function of VP30 depends on oligomerization of VP30, which is mediated by a stretch of hydrophobic amino acids at position 94–112 (10). Here we show that VP30 homooligomerization leads to hexamerization. We also provide evidence that VP30 contains a second homooligomerization domain, composed of the C-terminal half of VP30 (VP30CTD). Structural analyses show that VP30CTD folds independently into a helical arrangement of a VP30CTD head-to-head dimer, with no significant structural similarity to known protein structures (13). Although the dimerization interface is extensive, most of the interactions are between the loop region connecting helices 6 and 7 and helix 1*. It is thus conceivable that the displacement of helix 7 could be sufficient to disrupt the dimerization observed in VP30CTD and allow other VP30 conformers. This idea would be consistent with the observation that VP30mut1 is also found in a monomeric conformation in solution. Nevertheless, our structural data suggest that the dimer might be the building block for a hexameric form of VP30 detected in vitro (VP3089–272) and in vivo in infectious EBOV particles. This suggestion is also consistent with the position of the N termini in the crystallized VP30 fragment that could connect to the N-terminal oligomerization domain (10), because they both point in the same orientation. However, we cannot exclude the possibility that the dimer as seen in VP30CTD might dissociate depending on its function. Conformational plasticity of VP30 might be induced by phosphorylation of the N-terminal oligomerization domain, which has been shown to regulate transcriptional activation by VP30 (9). When we mutated the N-terminal oligomerization domain, hexamerization was impeded (VP305LA; ref. 10). However, this mutant still forms dimers, as detected by chemical cross-linking and density-gradient analyses (not shown). Although the role of the CTD dimerization within oligomeric VP30 is still somewhat unclear, mutagenesis of the dimer interface suggests that dimer formation is important. Dimer interface mutants were either aggregated in vitro or could not be efficiently detected upon expression in eukaryotic cells (data not shown), suggesting misfolding and immediate degradation.
A previous study revealed that VP30's ability to activate transcription depends on its intracellular concentration. At catalytic concentrations, VP30 readily activates EBOV transcription. In contrast, transcription activity is suppressed with increasing concentrations of VP30 (6). The activity of VP30 may be regulated similar to the function of transcription factors such as bacterial TyrR that forms dimers and hexamers in a concentration-dependent way (14, 15). Although dimers of TyrR activate, bacterial transcription hexamers tend to be inhibitory. It is thus conceivable that an equilibrium between activating (hexamers) and nonactivating (dimers) forms of VP30 might regulate EBOV transcription activity and may thus control the balance of transcription and replication. This presumption is also consistent with the analysis of VP30 contained in EBOV particles, which showed monomers and dimers and a fraction of potential hexamers that are essential for transcription activation.
Interaction with Nucleocapsids.
We show that VP30CTD interacts with NP and is incorporated into VLPs released from the cells (8, 12). In contrast, VP30NTD, although recruited into NP-induced inclusions, is not released in VLPs containing nucleocapsids. This result suggests that VP30 may interact with NP by its NTD and its CTD, although the latter interaction is tighter, because it can be detected by immunoprecipitation. Therefore, VP30 recognition of NP may be split into two functions: (i) VP30 interacts loosely by its NTD with NP-RNA helical-coil structures that are transcription-competent, and (ii) VP30's tight interaction with packaged NP-RNA nucleocapsid complexes requires an intact VP30CTD to be transported to the site of assembly and incorporated into VLPs. VP30CTD nucleocapsid interaction is supported by our mutagenesis studies, which show that an extensive conserved basic region of VP30CTD and E197, which is part of an acid patch on each end of the dimer, are required for efficient VP30 incorporation into VLPs. The basic region plays also a pivotal role in the transcriptional control activity exerted by VP30, suggesting that both processes of nucleocapsid or NP interaction and transcriptional control are tightly linked. This idea is in accordance with the fact that the viral L-P polymerase complex acts only on the NP-RNA complex during transcription and replication (ref. 5, p. 145). Alternatively, mutagenesis of the basic patch, which impaired oligomerization, induced an inactive conformation of VP30 that directly prevents efficient nucleocapsid interaction and/or might hinder a conformational change of VP30 required for transcriptional activation control. The second hypothesis is also in agreement with the result that E197 is essential for nucleocapsid incorporation but not for transcription activation, thus indicating that both processes are independently controlled, which is in agreement with previous data showing that the ability of VP30 to activate transcription is independent of its ability to accumulate inside NP inclusions (9)
Because of its essential function in viral transcription, VP30 is a potential candidate for antiviral therapy. This result has already shown for the N-terminal oligomerization of VP30, which can be inhibited by synthetic peptides leading to inactive VP30, which does not support viral propagation. Dimerization of VP30CTD generates a 7 × 11-Å-wide cavity whose interior is lined by hydrophobic residues (Leu-147, Ile-148, and Phe-181) and its opening by conserved hydrophilic residues Lys-180, Arg-179, Gln-185, Glu-252, and Ser-184. Our mutagenesis data concerning the requirement of the basic cluster for transcription activation predict that targeting this site by a small molecule might inhibit transcription activation and thus productive infection of target cells. In summary, our structural and functional studies of VP30 produced further insight into the versatile role of VP30 in EBOV replication and may serve as a platform to decipher the process of EBOV transcription.
Experimental Procedures
Bacterial Expression of VP30.
VP30 DNA corresponding to residues 89–272 and 142–272 was amplified by PCR and cloned into the bacterial expression vector pProEx HTb (Invitrogen, Carlsbad, CA) using standard procedures. The sequence was verified by DNA sequencing.
Protein expression was performed in E. coli BL21 codon plus cells (Invitrogen) at 37°C. Cell lysis was performed by sonication in a lysis buffer (containing 50 mM Tris·HCl, pH 8.0; 100 mM NaCl; 3 mM 2-mercaptoethanol) after addition of 5 μl of Benzonase (Merck, Whitehouse Station, NJ) and complete EDTA-free protease inhibitors (Pierce, Rockford, IL). The cell lysate was cleared by centrifugation and loaded onto a Ni2+ chelating Sepharose (Amersham Pharmacia Biotech, Uppsala, Sweden) column. The column was sequentially washed with lysis buffer alone, lysis buffer plus 1 M NaCl, and lysis buffer plus 1 M KCl. VP30 was then eluted by applying an imidazole gradient in lysis buffer and was further purified on a Q-Sepharose column (Amersham Pharmacia Biotech) using the lysis buffer. A final purification step included gel filtration on a Superdex75 column for VP30CTD and on a Superdex200 column for VP3089–272 in a buffer containing 25 mM Tris·HCl, pH 8.0; 100 mM NaCl; 3 mM 2-mercaptoethanol. Selenomethionine-labeled VP30CTD was produced as described (16) and purified as described above using 5 mM 2-mercaptoethanol in all buffers.
Chemical Cross-Linking.
Purified VP30 fragments were dialyzed against a buffer containing 50 mM Hepes, pH 8.0; 100 mM NaCl; 3 mM 2-mercaptoethanol before cross-linking with ethyleneglycol bis(-succinimidylsuccinate) (Pierce) at the indicated concentrations. Cross-linking was performed for 30 min at room temperature, and reactions were quenched with 50 mM Tris, pH 8.0. The cross-linked samples were separated on SDS/PAGE, and bands were visualized by Coomassie brilliant blue staining.
Nycodenz Gradient Sedimentation Analysis.
Gradients were generated by a gradient maker. Cells were lysed in lysis buffer (50 mM Tris, pH 8.0/50 mM NaCl/1% Nonidet P-40), and lysates were cleared by centrifugation (10 min at 10,000 × g), loaded on top of a 10–45% linear Nycodenz gradient in TNE buffer (10 mM Tris·HCl, pH 7.5/1 mM EDTA/100 mM NaCl), and centrifuged at 350,000 × g in a swing-out Beckman Coulter (Fullerton, CA) SW60 rotor for 22 h at 4°C. Gradients were fractionated from top to bottom and aliquots of the fractions separated by SDS/PAGE and blotted onto PVDF membrane. Immunostaining was performed with a mouse anti-flag antibody (dilution 1:5,000). Bound antibodies were detected with a peroxidase-coupled goat anti-mouse antibody (Dianova, Hamburg, Germany; dilution 1:30,000). Protein markers were sedimented under the same conditions (30 kDa carbonic anhydrase, 70 kDa bovine serum albumine, 150 kDa aldolase, and 200 kDa β-amylase).
Crystallization and Data Collection.
Crystals were obtained at 293 K from hanging drops containing a 1:1 mixture of VP30CTD at 16 mg/ml and well buffer (0.1 M ammonium acetate/15 mM magnesium acetate tetrahydrate/0.05 M sodium cacodylate, pH 6.5/10% vol/vol isopropanol). For cryogenic data collection, crystals were equilibrated in the reservoir buffer supplemented with 25% glycerol and flash-cooled in a gaseous nitrogen stream at 100 K. Data were collected at beamline BM14CRG at the European Synchrotron Light Source (ESRF, Grenoble, France). Two data sets from one selenomethionine-labeled crystal were collected at wavelengths 0.9763 (remote) and 0.9795 Å (peak) (SI Table 1). The diffraction data were processed by using the programs XDS and SCALA (17, 18) (SI Table 1). The crystal belonged to space group P212121 and contained two molecules in the asymmetric unit.
Structure Determination and Refinement.
The program BnP (19) was used to find four selenium positions in the peak wavelength data set by using data to 2.2-Å resolution. The programs Solve and Resolve were then used to validate the sites, and the correct hand and phases were extended to 2.0-Å resolution (20, 21). The density-modification procedure as implemented in Resolve produced an excellent electron-density map. The program Arp/Warp (22) was used to build 244 of 302 amino acids as polyalanine and for the sequence assignment of 201 residues. The model was completed by manual model building by using the program COOT (23) and refined with the programs CNS (24) and REFMAC (25). The model has been refined to an R factor of 19.5 and an Rfree of 24.1, exhibiting good stereochemistry, and, according to the program PROCHECK (17), 97.9% and 2.1% of the residues are in the most-favored and additionally allowed regions of a Ramachandran plot (SI Table 1). The model consists of 259 amino acids (chain A, residues 142–266 plus two N-terminal residues from the expression vector; chain B, 142–266 plus eight N-terminal extra residues from the expression vector) and 176 water molecules. The coordinates have been deposited in the RCSB Protein Data Bank (PDB) (PDB ID code 2I8B).
Structure Analysis.
Figs. 1–4 and SI Figs. 5–8 were generated by using ESPript (26), GRASP (27), and PYMOL (28). Sequences were aligned by using Clustalx (29). Secondary-structure elements were assigned by using the program DSSP (30).
Construction of Mammalian Expression Plasmids.
Plasmids encoding the EBOV structural proteins NP, VP35, VP40, VP24, and L were subcloned from the expression vector pTM1 into vector pCAGGS by using unique restriction sites. pCAGGS-GP was subcloned from vector pCDNA3 by using EcoRI and XhoI. Renilla luciferase minigenome was a generous gift of Julie Dyall (Apath, St. Louis, MO) and was cloned by replacing the CAT reporter gene from the EBOV-specific minigenome 3E5E (6) with the Renilla luciferase reporter gene. The resulting plasmid was named 3E-5E-RLuc. Plasmids of VP30 mutants and VP30 destined for mammalian expression were all provided with a C-terminal Flag-epitope (f) by PCR. The Flag-epitope has no influence on distribution and function of VP30 (9). pCAGGS-VP30f was generated by subcloning from pTM1-VP30f with EcoRI and StuI. The fragments VP30NTD (amino acids 1–141) and VP30CTD (amino acids 142–272) were amplified by PCR by using reverse primers with flag sequence and pTM1-VP30f as template and cloned into pCAGGS with EcoRI and SacI. Cloning of VP30 substitution mutants was performed with the QuikChange site-directed mutagenesis kit (Stratagene) by using pTM1-VP30f as template. Verified mutants were subcloned into pCAGGS with EcoRI and SmaI. The following substitution mutants were generated: pCAGGS-VP30mut1 (R179E, K180A, and K183E), pCAGGS-VP30mut2 (D158), and pCAGGS-VP30mut3 (D158A and E197A). All constructs were verified by sequencing. Detailed cloning strategies are available on request.
Purification and Analysis of Native ZEBOV Structural Proteins.
ZEBOV strain Mayinga was grown and passaged under biosafety level 4 conditions, as described (31). The virus was purified through a 20% sucrose cushion and inactivated by γ-irradiation (60 kGy). An aliquot of the virus preparation was mixed with 0.1% SDS loading buffer [100 mM Tris·HCl (pH 6.8)/0.2% bromophenol blue/20% glycerol/10% 2-mercaptoethanol/0.1% SDS] and separated on a native 10% ProSieve50-Acrylamide (Cambrex Bioscience, Walkersville, MD) gel with SDS running buffer. Afterward, a Western blot was performed and stained with indicated antibodies.
Cell Lines.
HEK 293 cells and human hepatoma HUH-7 cells were maintained in DMEM supplemented with 10% FCS/l-glutamine/penicillin–streptomycin solution. The cells were grown in an incubator at 37°C under 5% CO2.
Transfection, Metabolic Labeling, and Coimmunoprecipitation.
HUH-7 cells were transfected with pCAGGS-plasmids encoding for NP (500 ng), VP30 (500 ng), or VP30 mutants (500 ng) by using Fugene (Roche Applied Science, Indianapolis, IN) and OptiMem (Invitrogen) according to the manufacturer's instructions. At 16 h after transfection, cells were starved for 1 h with methionine- and cysteine-deficient DMEM. Medium was removed, and cells were labeled for 2 h with 25 μCi (1 Ci = 37 GBq) of [35S] Promix (Amersham Pharmacia Biosciences) in 1 ml of methionine- and cysteine-deficient DMEM. After labeling, cells were washed twice with ice-cold PBS (140 mM NaCl/3 mM KCl/8 mM Na2HPO4/1 mM KH2PO4) and scraped into 1 ml of ice-cold coimmunoprecipitation buffer (20 mM Tris·HCl, pH 7.5/100 mM NaCl/1% Nonidet P-40/17.5 mM EDTA/1 mM PMSF/1 mM DTT). Subsequently, cells were sonicated, and cell debris was removed by centrifugation (10,000 × g, 10-min speed duration). Supernatants were preincubated with 25 μl of protein A Sepharose (Sigma, St. Louis, MO). Cleared supernatant was divided, one part was incubated with a monoclonal α-flag antibody (M2; Sigma; dilution 1:500), and one part was incubated with M2 antibody and a mouse monoclonal ZEBOV α-NP IgG (B6G5) antibody (dilution 1:200). Immune complexes were bound to protein A Sepharose, precipitated, and washed three times with coimmunoprecipitation buffer. Final pellets were resuspended in loading buffer [100 mM Tris·HCl (pH 6.8)/0.2% bromophenol blue/20% glycerol/10% 2-mercaptoethanol/4% SDS] and separated on SDS/PAGE. The radioactive signals were detected with a BAS-1000 Bio-Imaging Analyzer (Fujifilm, Valhalla, NY) and TINA 2.0 software (Raytest, Wilmington, NC).
Indirect Immunofluorescence Analysis.
HUH-7 cells were transfected with pCAGGS-NP (400 ng) and pCAGGS-VP30 or pCAGGS-VP30 mutants (600 ng) by using Fugene (Roche Applied Science). Immunofluorescence analysis was performed as described (32). The antibodies used and their respective dilutions are given in legends to Figs. 1–4 and SI Figs. 5–8. Cross-reactivity of the used antibodies was not observed.
VLP Assay and Infectious VLP Assay.
VLP assay with proteinase K digestion was performed as described (8). Afterward, samples were separated on SDS/PAGE and analyzed by Western blot as described (32). For detection of transcription activity, cells were scraped off in PBS and spun down, and the pellet was lysed in 250 μl of passive lysis buffer (Promega, Madison, WI) The samples were stored at −80°C until determination of Renilla and firefly luciferase activity by using the dual luciferase reporter assay (Promega) and the luminometer Centro LD 960 (Berthold, Bad Wildbad, Germany).
Supplementary Material
Acknowledgments
We thank all members of the European Molecular Biology Laboratory (EMBL)/European Synchrotron Light Source (ESRF) Joint Structural Biology Group for access to the ESRF beam lines and Dr. J. Marquez and his team for crystallization screening. We also thank Angelika Lander for expert technical assistance. This work was supported by the EMBL (W.W.) and Deutsche Forschungsgemeinschaft (Grant SFB 593, to W.W. and S.B.).
Abbreviations
- EBOV
Ebola virus
- NP
nucleoprotein
- CTD
C-terminal domain
- VLP
virus-like particle
- NTD
N-terminal domain.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0606730104/DC1.
References
- 1.Bowen ET, Lloyd G, Harris WJ, Platt GS, Baskerville A, Vella EE. Lancet. 1977;1:571–573. doi: 10.1016/s0140-6736(77)92001-3. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Epidemiol Bull. 1995;16:16. [Google Scholar]
- 3.World Health Organization. Weekly Epidemiol Rec. 1999;74:145. [Google Scholar]
- 4.World Health Organization. Weekly Epidemiol Rec. 2001;76:41–48. [Google Scholar]
- 5.Conzelmann KK. Annu Rev Genet. 1998;32:123–162. doi: 10.1146/annurev.genet.32.1.123. [DOI] [PubMed] [Google Scholar]
- 6.Mühlberger E, Weik M, Volchkov VE, Klenk H-D, Becker S. J Virol. 1999;73:2333–2342. doi: 10.1128/jvi.73.3.2333-2342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weik M, Modrof J, Klenk HD, Becker S, Mühlberger E. J Virol. 2002;76:8532–8539. doi: 10.1128/JVI.76.17.8532-8539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoenen T, Groseth A, Kolesnikova L, Theriault S, Ebihara H, Hartlieb B, Bamberg S, Feldmann H, Stroher U, Becker S. J Virol. 2006;80:7260–7264. doi: 10.1128/JVI.00051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Modrof J, Mühlberger E, Klenk HD, Becker S. J Biol Chem. 2002;277:33099–33104. doi: 10.1074/jbc.M203775200. [DOI] [PubMed] [Google Scholar]
- 10.Hartlieb B, Modrof J, Mühlberger E, Klenk HD, Becker S. J Biol Chem. 2003;278:41830–41836. doi: 10.1074/jbc.M307036200. [DOI] [PubMed] [Google Scholar]
- 11.Kolesnikova L, Mühlberger E, Ryabchikova E, Becker S. J Virol. 2000;74:3899–3904. doi: 10.1128/jvi.74.8.3899-3904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe S, Watanabe T, Noda T, Takada A, Feldmann H, Jasenosky LD, Kawaoka Y. J Virol. 2004;78:999–1005. doi: 10.1128/JVI.78.2.999-1005.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm L, Sander C. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 14.Dixon MP, Pau RN, Howlett GJ, Dunstan DE, Sawyer WH, Davidson BE. J Biol Chem. 2002;277:23186–23192. doi: 10.1074/jbc.M112184200. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Hwang JS, Camakaris H, Irawaty W, Ishihama A, Pittard J. Mol Microbiol. 2004;52:243–256. doi: 10.1111/j.1365-2958.2003.03965.x. [DOI] [PubMed] [Google Scholar]
- 16.Doublie SC, CW . In: Crystallization of Nucleic Acids and Proteins. A Practical Approach. Ducruix A, Giege R, editors. Oxford: Oxford Univ Press; 1992. [Google Scholar]
- 17.CCP4. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 18.Kabsch W. J Appl Crystallogr. 1988;21:916–924. [Google Scholar]
- 19.Weeks CM, Miller R. Acta Crystallogr D. 1999;55:492–500. doi: 10.1107/s0907444998012633. [DOI] [PubMed] [Google Scholar]
- 20.Terwilliger TC. Acta Crystallogr D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terwilliger TC, Berendzen J. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris RJ, Perrakis A, Lamzin VS. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 26.Gouet P, Robert X, Courcelle E. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholls A, Sharp KA, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 28.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 29.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 31.Becker S, Feldmann H, Will C, Slenczka W. Med Microbiol Immunol (Berlin) 1992;181:43–55. doi: 10.1007/BF00193395. [DOI] [PubMed] [Google Scholar]
- 32.Kolesnikova L, Bamberg S, Berghofer B, Becker S. J Virol. 2004;78:2382–2393. doi: 10.1128/JVI.78.5.2382-2393.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.