

Abstract
We recently reported that junctional adhesion molecule (JAM)-C plays a role in leukocyte transendothelial migration. Here, the role of JAM-C in vascular permeability was investigated in vitro and in vivo. As opposed to macrovascular endothelial cells that constitutively expressed JAM-C in cell–cell contacts, in quiescent microvascular endothelial cells, JAM-C localized mainly intracellularly, and was recruited to junctions upon short-term stimulation with vascular endothelial growth factor (VEGF) or histamine. Strikingly, disruption of JAM-C function decreased basal permeability and prevented the VEGF- and histamine-induced increases in human dermal microvascular endothelial cell permeability in vitro and skin permeability in mice. Permeability increases are essential in angiogenesis, and JAM-C blockade reduced hyperpermeability and neovascularization in hypoxia-induced retinal angiogenesis in mice. The underlying mechanisms of the JAM-C–mediated increase in endothelial permeability were studied. JAM-C was essential for the regulation of endothelial actomyosin, as revealed by decreased F-actin, reduced myosin light chain phosphorylation, and actin stress fiber formation due to JAM-C knockdown. Moreover, the loss of JAM-C expression resulted in stabilization of VE-cadherin–mediated interendothelial adhesion in a manner dependent on the small GTPase Rap1. Together, through modulation of endothelial contractility and VE-cadherin–mediated adhesion, JAM-C helps to regulate vascular permeability and pathologic angiogenesis.
The endothelium lining the vasculature constitutes a barrier maintaining the integrity between blood and interstitium and regulating extravasation of fluids and plasma proteins (1, 2). Changes in endothelial barrier function and increases in permeability are essential for neovascularization and tissue repair (1). Tissue hypoxia is associated with the up-regulation of vascular endothelial growth factor (VEGF) that increases vascular permeability as a critical prerequisite step in new vessel formation (3). Contrastingly, during inflammatory responses, dysregulation of vascular permeability contributes to pathological vascular leakage, often seen in conditions associated with edema such as in septic shock (1, 2). In this case, vasoactive substances, including histamine, bradykinin, or TNF-α, are crucial in the regulation of endothelial permeability (2).
Endothelial permeability depends on actomyosin-based cell contractility, as intracellular stress fibers exert centripetal tension to induce permeability, and on the integrity of intercellular junctions (2, 4, 5). In response to vasoactive agents, myosin light chain (MLC) is phosphorylated and can thereby interact with actin filaments resulting in the formation of stress fibers (2). MLC phosphorylation is regulated by MLC kinases and small GTPases (2).
Intercellular junctions (6) important for the endothelial barrier are as follows. (a) Adherens junctions are formed by cadherins that are linked to intracellular catenins. VE-cadherin–mediated cell–cell contacts are stabilized by the small GTPase Rap1 and its effector, the cyclic adenosine monophosphate (cAMP)-activated guanidine exchange factor Epac, thereby decreasing paracellular permeability (7, 8). (b) Tight junctions that are located at the apical-most portion of the lateral interendothelial membrane consist of three major families of transmembrane proteins, claudins, occludin, and junctional adhesion molecules (JAMs) (9).
JAMs, expressed in endothelial and epithelial cells, and on platelets and some leukocytes, consist of two Ig-like domains, and at their final COOH-terminus they have a class II PDZ domain–binding motif, predisposing them to interact with PDZ domain–containing molecules, such as ZO-1, ASIP/PAR-3, or AF-6 (10, 11). JAM-A, JAM-B, and JAM-C regulate leukocyte–endothelial cell interactions by virtue of their ability to undergo heterophilic binding with the leukocyte integrins LFA-1, VLA-4, and Mac-1, respectively (10, 11). In addition, their junctional localization and their propensity to interact homophilically suggests that JAMs may participate in the regulation of tight junction formation in epithelial and endothelial cells and, consequently, in the regulation of paracellular permeability (10–12). A key role of JAM-A in epithelial and endothelial barrier function has been shown (13–15). In contrast, ectopic JAM-C expression in MDCK or CHO cells revealed controversial data as to the function of JAM-C in permeability, whereas recent studies by us and others demonstrated that JAM-C may localize in endothelial junctions associated with ZO-1 and/or ASIP/PAR-3 (16–19). However, no study to date has addressed the role of JAM-C in the process of endothelial permeability.
These diverse observations prompted us to investigate the role of JAM-C in endothelial barrier function. We used microvascular endothelial cells as an appropriate in vitro model for permeability studies. Our findings clearly demonstrate that JAM-C mediates an increase in paracellular permeability, through regulating actomyosin-dependent contractility and VE-cadherin–mediated cell–cell contacts in a Rap1-dependent manner. Furthermore, disruption of JAM-C function thereby blocked both inflammation- and angiogenesis-associated increases in permeability in vitro and in vivo, and consequently JAM-C blockade in vivo, prevented neovascularization.
RESULTS
Regulation of JAM-C localization in microvascular endothelial cells
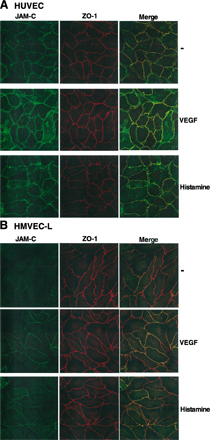
We first investigated the localization of JAM-C in endothelial cells and whether it is affected by vasoactive factors such as VEGF or histamine. In quiescent primary human dermal microvascular endothelial cells (HDMECs) JAM-C was mainly intracellular (Fig. 1 A) and absent from interendothelial junctions. Interestingly, both VEGF and histamine induced a rapid accumulation of JAM-C into the cell–cell borders, where it colocalized with ZO-1 (Fig. 1 A). To analyze the effect of VEGF and histamine on JAM-C localization and expression, we compared total versus surface-exposed JAM-C in resting and stimulated endothelial cells. As shown in Fig. 1 B, under nonstimulated conditions ∼20% of JAM-C is cell surface associated (i.e., the remaining 80% is intracellular). This surface-associated JAM-C is apparently diffusely distributed on the cell surface, as it does not localize in cell–cell junctions (Fig. 1 A). The amount of surface-associated JAM-C increased to 60 and 70% upon stimulation with VEGF and histamine, respectively. Under these conditions most surface-exposed JAM-C is localized to interendothelial contacts (Fig. 1 A). In contrast, the expression of total JAM-C was not affected by VEGF or histamine (Fig. 1 B), which was also verified by flow cytometry and Western blot (unpublished data). Thus, although a small part of JAM-C that is surface associated may also concentrate to junctions upon stimulation, the major mechanism of JAM-C redistribution to the junctions involves exocytosis from intracellular compartments. Interestingly, the same pattern of JAM-C localization was also observed in another microvascular endothelial cell type, the human lung microvascular endothelial cells (HMVEC-L). In contrast, in two macrovascular endothelial cell types, human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAECs), JAM-C was constitutively located mainly at the interendothelial contacts and its localization was not affected by stimulation with VEGF or histamine (Fig. 1 C and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051730/DC1). Thus, JAM-C localization in microvascular endothelial cells is intracellular and differs from the junctional localization in macrovascular cells.
Figure 1.

JAM-C is recruited to the junctions of HDMECs after stimulation by VEGF or histamine. (A) Immunofluorescence analysis followed by confocal microscopy of the interendothelial contacts of HDMECs. Representative stainings of HDMECs that were incubated in the absence (−) or presence of VEGF (1 h, 50 ng/ml) or histamine (1 h, 50 μM), showing the distribution of JAM-C and ZO-1. Double stained images were merged to analyze colocalization. (B) The surface versus total expression of JAM-C in nonpermeabilized versus permeabilized HDMECs, respectively, was analyzed by cell ELISA. HDMECs were pretreated without (−; open bars) or with VEGF (1 h, 50 ng/ml; filled bars) or histamine (1 h, 50 μM; gray bars) as indicated. JAM-C expression is shown as the percentage of control, defined as the total JAM-C expression in the absence of any stimulus. Data are mean ± SD (n = 3) of one experiment typical of three separate experiments. (C) Analysis of the interendothelial contacts of HAECs was performed as in A. Representative immunofluorescence of HAECs that were incubated in the absence (−) or presence of VEGF (1 h, 50 ng/ml) or histamine (1 h, 50 μM). The distribution of JAM-C and ZO-1 is shown. Double stained images were merged to analyze colocalization.
We then studied the kinetics of JAM-C redistribution to the cell–cell contacts in HDMECs. JAM-C appeared partially in junctions already 30 min after VEGF stimulation. The amount of junctional JAM-C peaked at 1 h after stimulation, whereas 2 h after VEGF stimulation junctional JAM-C staining was less and mostly disappeared thereafter. In the later time points and up to 24 h, JAM-C was mostly absent from the junctions. At this latter time point HDMEC junctions were largely disrupted (Fig. 2). The VEGF-mediated increase in HDMEC permeability showed a biphasic response: an early transient increase that peaked at 1 h and a delayed (>12 h) increase that peaked at 24 h (unpublished data and reference 20). Together, JAM-C redistribution to the cell–cell contacts is a rapid and transient process.
Figure 2.

Kinetics of JAM-C redistribution to the interendothelial junctions of HDMECs. Immunofluorescence analysis followed by confocal microscopy of the interendothelial contacts of HDMECs. Representative stainings of HDMECs in the absence (0 h) or presence of VEGF (50 ng/ml) for different time points, showing the distribution of JAM-C and ZO-1. Note the disruption of the junctions at 24 h, as indicated by interendothelial gaps (arrows).
Disruption of JAM-C function results in decreased endothelial permeability
We then investigated a potential role of JAM-C in microvascular endothelial permeability by engaging two different approaches to disrupt JAM-C function. First, we used the recombinant extracellular portion of JAM-C expressed as an Fc fusion protein, and second, knockdown of JAM-C expression was performed with siRNA targeted against human JAM-C. As shown in Fig. 3, A and B, siRNA against JAM-C results in an inhibition of JAM-C expression by 70% in HDMECs. In contrast, no alteration of ZO-1 or actin expression (Fig. 3 A) was observed by JAM-C knockdown. By assessing the influx of FITC-dextran into the lower well in a Transwell filter system, Fc–JAM-C but not control Fc protein significantly reduced the VEGF- or histamine-induced increase in HDMEC permeability (Fig. 3 C). In addition, knockdown of JAM-C expression resulted in a significant decrease (P < 0.05) in basal HDMEC permeability and almost completely abolished the VEGF- and histamine-mediated HDMEC hyperpermeability (Fig. 3 D), whereas nontargeting control siRNA did not affect HDMEC permeability (Fig. 3 D).
Figure 3.

JAM-C mediates an increase in paracellular permeability. (A) The expression of JAM-C, ZO-1, and actin was analyzed by Western blot in HDMECs transfected with siRNA against JAM-C or with control siRNA as indicated. (B) The expression of JAM-C in HDMECs transfected with siRNA against JAM-C was 32 ± 7% of the expression of JAM-C in HDMECs transfected with control siRNA. Data are mean ± SD of six separate experiments. (C) HDMECs were incubated for 1 h without (100% control) or with VEGF (50 ng/ml) or histamine (50 μM) in the absence or presence of Fc (open bars) or Fc–JAM-C (gray bars) (each at 20 μg/ml). Permeability for FITC-conjugated dextran is expressed as the percentage of control, defined as permeability in the absence of any stimulus or competitor. (D) HDMECs transfected with control siRNA (open bars) or with siRNA against JAM-C (gray bars) were incubated for 1 h without (−) or with VEGF (50 ng/ml) or histamine (50 μM). Permeability is expressed as the percentage of control, defined as permeability of the control siRNA-transfected HDMECs in the absence of any stimulus. Data in C and D are mean ± SD (n = 3) of one experiment typical of three separate experiments. *, P < 0.05; ns, not significant. (E) Representative immunofluorescence of histamine-treated HDMECs transfected with control siRNA or histamine-treated HDMECs transfected with siRNA against JAM-C, as indicated, showing the distribution of JAM-C and ZO-1. Note the more contiguous lining of interendothelial contacts in HDMECs upon JAM-C knockdown.
To rule out any unforeseen effects of the siRNA approach and to prove the specificity of the effects of JAM-C knockdown, further controls were engaged (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20051730/DC1). (a) A second siRNA targeting another JAM-C sequence was engaged, and a similar down-regulation of JAM-C concomitant with a reduction in endothelial permeability was observed. (b) Two siRNAs were used that had a single mutation in their sequence compared with the two siRNAs targeting JAM-C, thereby preventing silencing of JAM-C expression. In this case, JAM-C expression and endothelial permeability remained unaffected by these mutated siRNAs. (c) We engaged a siRNA down-regulating an irrelevant gene, cyclophilin, which mediated no changes in endothelial permeability. Thus, the observed effect on the rescue of endothelial barrier function mediated by JAM-C knockdown is specific. Together, these results suggest that JAM-C mediates an increase in HDMEC permeability.
We then studied the effect of JAM-C knockdown on tight junction molecule expression and HDMEC junction morphology. No change in the expression of ZO-1 (Fig. 3 A) or occludin (unpublished data) was found upon JAM-C down-regulation. In contrast, JAM-C knockdown prevented the VEGF- and histamine-mediated disintegration of endothelial junctions, as observed by the more contiguous lining of interendothelial contacts in histamine- or VEGF-treated HDMECs that were transfected with the siRNA against JAM-C (Fig. 3 E; VEGF data is not shown). Thus, the function of JAM-C in HDMEC permeability is associated with morphological changes of the interendothelial contacts rather than changes in expression of junction-associated molecules.
JAM-C is important for actomyosin-driven endothelial contractility
We continued to study the underlying mechanisms for the rescue of endothelial barrier function caused by JAM-C knockdown. Endothelial barrier function depends on the ratio of F-actin to G-actin, and disruption of endothelial barrier function is associated with rearrangement of cortical actin and increased formation of stress fibers (2, 4, 21). Upon JAM-C knockdown in HDMECs, a dramatic decrease in F-actin was observed (Fig. 4 A). Consistently, there was a decrease in the F-actin to G-actin ratio in cells transfected with siRNA against JAM-C compared with cells transfected with control siRNA (Fig. 4 B). Histamine or VEGF increased stress fiber formation in endothelial cells (Fig. 4 A) as also indicated by the increase in F-actin to G-actin ratio (Fig. 4 B; VEGF data not shown); however, consistent with previous reports this change in stress fiber intensity in endothelial cells upon stimulation was not dramatic (21–24). This VEGF- and histamine-induced increase in actin stress fiber formation was prevented by JAM-C down-regulation (Fig. 4 A; VEGF data not shown).
Figure 4.

JAM-C is essential for HDMEC contractility. (A) Representative immunofluorescence of Rhodamine-conjugated phalloidin showing the distribution of F-actin in buffer- or histamine-treated HDMECs transfected with control siRNA or with siRNA against JAM-C. Nuclei are shown by DAPI staining. (B) The F-actin to G-actin ratio of HDMECs transfected with control siRNA or with siRNA against JAM-C in the absence (open bars) or presence of histamine (filled bars) is shown. The F-actin to G-actin ratio is expressed as the percentage of control, defined as the F-actin to G-actin ratio of the control siRNA-transfected HDMECs in the absence of histamine. Data are mean ± SD (n = 3) of three separate experiments. *, P < 0.05; #, P < 0.01. (C) The phosphorylation of MLC was analyzed by Western blot with antibody against phospho-MLC or total MLC in HDMECs transfected with control siRNA or with siRNA against JAM-C. A representative immunoblot for phospho-MLC is shown. The insert shows densitometric analysis of phospho-MLC/total MLC in HDMECs transfected with control siRNA or with siRNA against JAM-C. Data are shown relative to control (phospho-MLC/total MLC in HDMECs transfected with control siRNA was set as 1) and are mean ± SD of three separate experiments. (D) MLC phosphorylation was analyzed as in C in HDMECs transfected with control siRNA or with siRNA against JAM-C in the absence (0 min) or presence of histamine for different time points as indicated.
Stress fiber formation depends on the phosphorylation of the regulatory MLC that regulates the interaction of myosin with actin (2,21). Western blotting with an anti–phospho-MLC antibody revealed a threefold decrease in MLC phosphorylation as a result of the down-regulation of JAM-C expression (Fig. 4 C). JAM-C knockdown also prevented the histamine-induced MLC phosphorylation (Fig. 4 D). In contrast, other signaling pathways, such as extracellular signal-regulated kinase 1 phosphorylation were not affected by JAM-C knockdown (unpublished data). Thus, consistent with its role in endothelial barrier function JAM-C regulates processes related to actomyosin-driven cell contractility.
JAM-C knockdown increases Rap1 activity and VE-cadherin–mediated cell–cell adhesion
Small GTPases have been implicated in the regulation of vascular permeability (2, 4, 5), and JAM-C was recently shown to interact with and regulate the activity of Cdc42 during spermatid polarization (25). Therefore, we studied whether JAM-C knockdown alters the activity of small GTPases. JAM-C knockdown left the activity of Cdc42, Rac, and Ras unchanged (Fig. 5 A and unpublished data). In contrast, a three- to fourfold up-regulation in the endogenous Rap1 activity was found upon JAM-C down-regulation in HDMECs (Fig. 5 A). Recent findings linked the Epac/Rap1 pathway with the strength of the adhesive contacts in adherens junctions and particularly with the homotypic VE-cadherin–mediated interactions between adjacent endothelial cells (7, 8). Consistently, the loss of JAM-C expression resulted in a threefold increase of the homophilic adhesive interaction of VE-cadherin (Fig. 5 B). In contrast, adhesion to platelet endothelial cell adhesion molecule (PECAM)-1, a molecule also involved in endothelial cell–cell contacts (26), was not affected by JAM-C knockdown (Fig. 5 B). VE-cadherin protein expression was not changed because of JAM-C knockdown (Fig. 5 C). In addition, β1-integrin expression and localization that may be regulated by Rap1 (13) were not affected by the expression level of JAM-C (unpublished data).
Figure 5.

JAM-C regulates Rap1 activity and VE-cadherin–mediated contacts. (A) Pull-down assays were performed to isolate active GTP-bound Rap1 or Ras. Western blots for Rap1 or Ras revealed increased active Rap1 in HDMECs transfected with siRNA against JAM-C compared with the HDMECs transfected with control siRNA, whereas active Ras was unchanged. In contrast, no change in the total Rap1 or Ras level was observed. The insert shows densitometric analysis of the ratio between active and total Ras or Rap1 in HDMECs transfected with siRNA against JAM-C (gray bars) or with control siRNA (open bars). Data are shown relative to control (active GTPase/total GTPase in HDMECs transfected with control siRNA was set as 1) and are mean ± SD of three separate experiments. (B) The adhesion of HDMECs transfected with control siRNA (open bars) or HDMECs transfected with siRNA against JAM-C (gray bars) to immobilized BSA, control Fc protein, Fc–VE-cadherin, or Fc–PECAM-1 is demonstrated. Adhesion is shown as the percentage of adherent cells. Data are mean ± SD of a typical experiment; similar results were obtained in three separate experiments. (C) The expression of VE-cadherin was analyzed by Western blot in HDMECs transfected with siRNA against JAM-C or with control siRNA as indicated.
Besides the Epac/Rap1 pathway, protein kinase A (PKA) activity is another cAMP-dependent pathway regulating endothelial barrier function (27). We therefore assessed the contribution of either pathway in the rescue of barrier function by JAM-C knockdown. Whereas JAM-C knockdown resulted in an up-regulation of Rap1 activity (Fig. 5 A), the PKA pathway remained unaffected as assessed by determining the phosphorylation of the transcription factor CREB (unpublished data). The following observations further strengthened the role of Rap1 in mediating the effect of JAM-C on endothelial barrier function. (a) The cAMP-elevating agents forskolin/rolipram and pharmacologic up-regulation of Rap1 activity with an Epac activator (O-Me-cAMP) decreased basal permeability of HDMEC monolayers. The effect of JAM-C knockdown to decrease endothelial permeability was comparable to the effect of O-Me-cAMP (Fig. 6 A). (b) VE-cadherin–mediated cell adhesion was not only increased by JAM-C knockdown but also by O-Me-cAMP, but not by a PKA activator (Sp-8-Br-cAMPS) (Fig. 6 B). (c) The decrease in endothelial permeability and the increase in VE-cadherin–mediated adhesiveness caused by JAM-C knockdown were largely prevented by down-regulation of Rap1 expression and Rap1 activity by engaging a siRNA against Rap1 but not by inhibition of PKA activity by a PKA inhibitor (Rp-8-Br-cAMPS) (Fig. 6, C and D, and unpublished data). Therefore, we conclude that JAM-C knockdown stabilizes VE-cadherin–mediated adhesive interendothelial contacts and rescues endothelial barrier function in a Rap1-dependent manner.
Figure 6.

The rescue of endothelial barrier function and the stimulation of VE-cadherin–mediated adhesiveness caused by JAM-C knockdown is dependent on the activity of Rap1. (A) HDMECs were transfected with control siRNA (open bars) or with siRNA against JAM-C (gray bar). Permeability of HDMECs transfected with control siRNA was analyzed in the absence (−) or in the presence of forskolin (10 μM)/rolipram (20 μM), or the Epac activator O-Me-cAMP (200 μM). Permeability is expressed as the percentage of control, defined as permeability of the control siRNA-transfected HDMECs in the absence of any competitor. (B) The adhesion of HDMECs transfected with control siRNA in the absence (open bars), or in the presence of the PKA activator (Sp-8-Br-cAMPS, 200 μM, filled bars), or in the presence of the Epac activator O-Me-cAMP (200 μM, hatched bars), or of HDMECs transfected with siRNA against JAM-C (gray bars) to immobilized Fc–VE-cadherin or Fc–PECAM-1 is demonstrated. Adhesion is shown as the percentage of adherent cells. (C) HDMECs were transfected with control siRNA (open bars), with control siRNA together with siRNA against Rap1 (shaded bars), with siRNA against JAM-C (gray bars), or with siRNA against JAM-C together with siRNA against Rap1 (hatched bars). Permeability of HDMECs was analyzed in the absence (−) or presence of VEGF (50 ng/ml) or histamine (50 μM). Permeability is expressed as the percentage of control, defined as permeability of the control siRNA-transfected HDMECs in the absence of any stimulus. (D) The adhesion of HDMECs transfected with control siRNA or siRNA against JAM-C to immobilized Fc–VE-cadherin or Fc–PECAM-1 is shown in the absence (open bars), or in the presence of the PKA inhibitor (Rp-8-Br-cAMPS, 200 μM, shaded bars), or in the presence of Rap1 knockdown with siRNA against Rap1 (gray bars). Adhesion is shown as the percentage of adherent cells. Data are mean ± SD of a typical experiment; similar results were obtained in three separate experiments.
Consistent with the previous data, the overexpression of mouse JAM-C in microvascular endothelial cells reversed the increase of VE-cadherin–mediated cell–cell adhesion caused by the knockdown of human JAM-C (Fig. 7). These data underline the specificity of the observed effects mediated by JAM-C knockdown and indicate that mouse JAM-C may undertake similar functions in endothelial cells as human JAM-C.
Figure 7.

Overexpression of mouse JAM-C reverses the increase of VE-cadherin–mediated adhesiveness caused by JAM-C knockdown. (A) The expression of mouse JAM-C and human JAM-C was analyzed by Western blot in HDMECs transfected with control siRNA (1), in HDMECs transfected with control siRNA, and with mouse JAM-C (2), in HDMECs transfected with siRNA against JAM-C (3), or in HDMECs transfected with siRNA against JAM-C and with mouse JAM-C (4) as indicated. (B) The adhesion of HDMECs transfected with control siRNA (open bars), of HDMECs transfected with control siRNA and with mouse JAM-C (shaded bars), of HDMECs transfected with siRNA against JAM-C (gray bars), or of HDMECs transfected with siRNA against JAM-C and with mouse JAM-C (hatched bars) to immobilized control Fc protein, Fc–VE-cadherin, or Fc–PECAM-1 is shown. Adhesion is shown as the percentage of adherent cells. Data are mean ± SD of a typical experiment; similar results were obtained in three separate experiments.
JAM-C blockade decreases microvascular permeability in vivo and hypoxia-induced retinal angiogenesis
To assess the function of JAM-C in vascular permeability in vivo, we studied the effect of JAM-C blockade on VEGF- and histamine-mediated vascular leakage in mice. A modified Miles vascular permeability assay was performed, in which soluble mouse JAM (smJAM)-C or the control 6xHis peptide were injected i.p. 1 h before intradermal injection of VEGF or the compound 48/80 that mediates mast cell degranulation (28). Spectrophotometric measurements of the amount of extravasated Evans blue revealed a four- to fivefold and a six- to sevenfold increase in vascular permeability by VEGF or compound 48/80, respectively. Interestingly, smJAM-C inhibited vascular permeability by at least 50% in both cases (Fig. 8 A), whereas the control 6xHis peptide had no effect (unpublished data). As permeability changes are crucial in cutaneous inflammatory reactions, we investigated the role of smJAM-C in delayed-type hypersensitivity responses that were elicited in mice by using oxazolone as a sensitizing agent. Treatment with smJAM-C significantly reduced macroscopic ear swelling and local edema as assessed by the increased Evans blue leakage in the challenged ear above the baseline of the nonchallenged contralateral control ear were significantly reduced by smJAM-C treatment (unpublished data).
Figure 8.

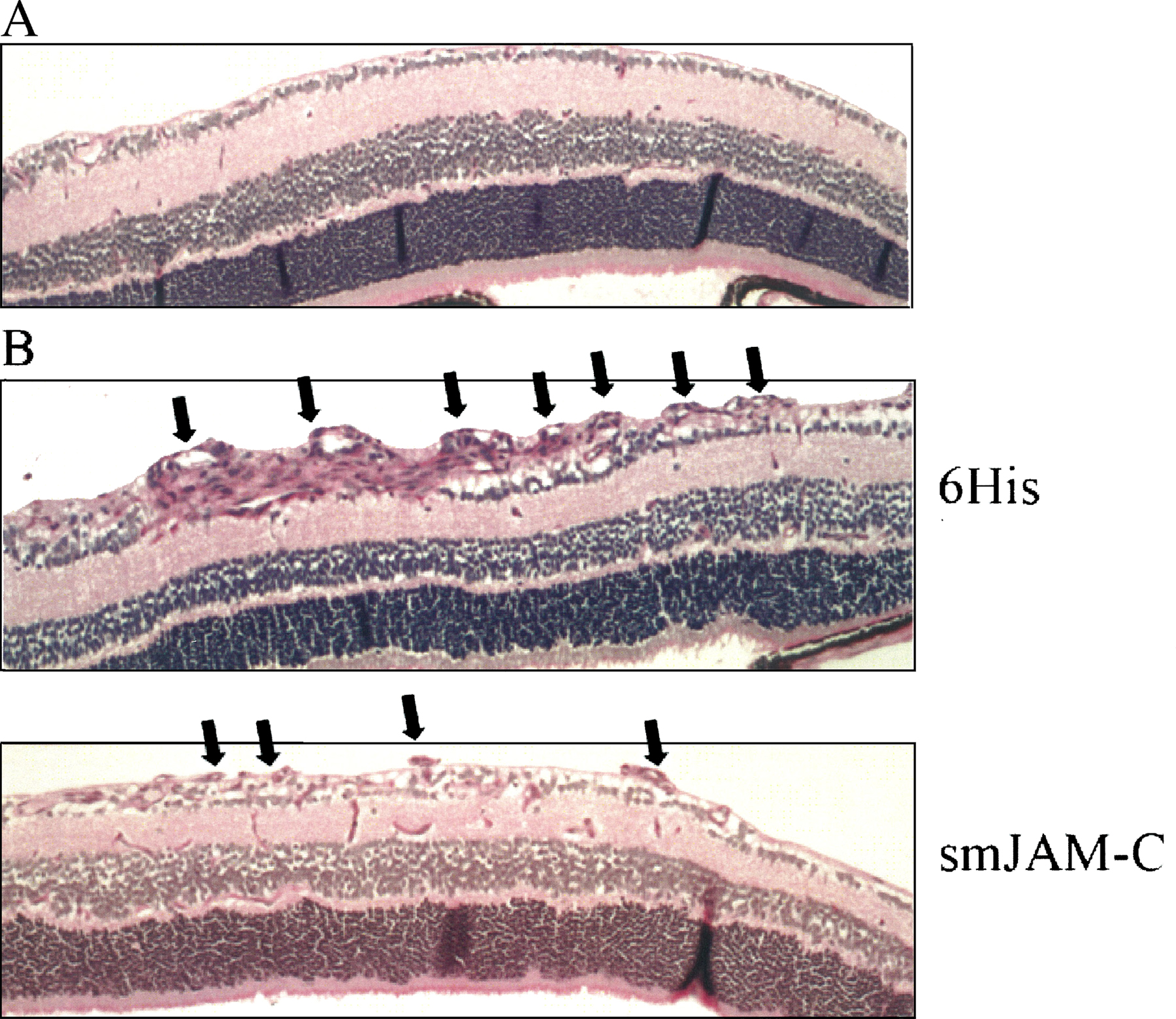
Inhibition of JAM-C decreases microvascular permeability in vivo and hypoxia-induced retina angiogenesis. (A) Skin permeability (as assessed by leakage of Evans blue) induced by intradermal injection of VEGF or compound 48/80 is shown without (open bars) or with pretreatment with smJAM-C (gray bars). The control 6xHis peptide did not affect VEGF- or histamine-induced permeability. Permeability is shown as the percentage of control, defined as the skin permeability after intradermal injection with buffer in the absence of competitors, and data are shown as mean ± SD (n = 5 mice). (B and C) Mice were subjected to the ROP model. (B) The permeability of the retina was assessed by the leakage of Evans blue on day p13. 6 h before assessing the leakage of Evans blue, mice were treated i.p. with buffer, the control 6xHis peptide, or smJAM-C. The leakage of Evans blue was quantified as absorbance/dry weight of the retina, and data are represented as the percentage of control (buffer-treated mice) and are mean ± SD (n = 5 mice). (C) Retinal neovascularization was quantified on day p17 as described in Materials and methods. Mice were treated from day p12 until day p16 once daily i.p. with buffer, the control 6xHis peptide, or smJAM-C. Retinal neovascularization is presented as the percentage of control, defined as neovascularization in the presence of buffer only. Data are mean ± SD (n = 5 mice). *, P < 0.05; ns, not significant.
Besides inflammatory reactions, increases in vascular permeability are important in neovascularization. We therefore examined the role of JAM-C in pathological retinal neovascularization in the mouse model of hypoxia-induced retina angiogenesis (retinopathy of prematurity [ROP]). First, smJAM-C but not the control 6xHis peptide inhibited the increase in retinal vascular permeability that develops during the course of the ROP model. In particular, leakage of Evans blue in retinas of 13-d-old (p13) mice was reduced by 45% in animals treated with smJAM-C (Fig. 8 B). Second, treatment with smJAM-C but not with the control 6xHis peptide from day p12 up to p16 reduced retinal angiogenesis by 50% (Fig. 8 C). Immunohistochemical analysis of axial retinal sections demonstrated a substantial reduction in the extent of neovascularization in mice given smJAM-C compared with mice treated with control 6xHis peptide. In addition, analysis of retina whole mounts indicated that p17 retinas from smJAM-C–treated mice demonstrated a marked reduction in the number of neovascular tufts compared with control-treated animals (Fig. S3 and S4, http://www.jem.org/cgi/content/full/jem.20051730/DC1). Thus, smJAM-C reduced the number and the pathologic morphology of the new vessels in the retina. Together, these data demonstrate that smJAM-C inhibits inflammation- and angiogenesis-related hyperpermeability and pathological retinal neovascularization in vivo.
DISCUSSION
Our present findings clearly demonstrate that JAM-C regulates both inflammation- and angiogenesis-related endothelial permeability. In particular, JAM-C mediates an increase in endothelial permeability by modulating actomyosin-based endothelial contractility and through regulation of VE-cadherin–mediated cell–cell contacts in a Rap1-dependent manner. Consistently, disruption of JAM-C function by soluble JAM-C in vivo decreased vascular permeability in the skin during delayed-type hypersensitivity responses and in the retina during hypoxia-driven retinal neovascularization, and reduced retina angiogenesis.
To date, transmembrane molecules of the tight junctions were thought to function as gatekeepers by interacting in a homotypic fashion (13–15); thus, our findings were somewhat surprising. To our knowledge, JAM-C was defined here as the first junctional molecule that can mediate an increase in permeability. JAM-C knockdown mediated a rescue of endothelial barrier function under basal conditions and prevented VEGF- and histamine-mediated disintegration of endothelial junctions.
Interestingly, the subcellular localization of JAM-C in microvascular endothelial cells was found to be divergent from macrovascular endothelial cells. Consistent with our previous reports and those by others (16, 17), JAM-C in macrovascular endothelial cells was localized in the interendothelial junctions. In contrast, microvascular endothelial JAM-C was mainly intracellular and redistributed rapidly and transiently to the cell–cell contacts upon stimulation with VEGF or histamine. Our findings are in line with the diversity among endothelial cells from different vascular beds especially with respect to the regulation of paracellular permeability or leukocyte transendothelial migration (29, 30). By immunogold electron microscopy we found intracellular JAM-C in HDMECs to be associated with structures some, but not all, of which resembled Weibel-Palade bodies (unpublished data). The complete subcellular localization of JAM-C must be investigated in a future study. The regulated localization of JAM-C and its recruitment to the interendothelial contacts correlate and coincide with the role of JAM-C in regulating VEGF- and histamine-induced permeability. However, although the redistribution of JAM-C to the junctions may be relevant, it is clearly not essential for the effects of JAM-C on endothelial barrier function, as basal nonstimulated endothelial permeability was also decreased by JAM-C knockdown. In fact, intracellular JAM-C clearly regulates signaling, such as the Rap1 pathway, and participates in the regulation of actin stress fiber formation. This may be attributed to the propensity of JAM-C to be associated with AF-6 or other scaffolding molecules that may provide a link between intracellular JAM-C and these signaling pathways. Further studies addressing the detailed molecular mechanism for the intracellular trafficking of JAM-C and the importance of JAM-C localization for endothelial barrier function in microvascular versus macrovascular endothelial cells are required.
JAM-C knockdown in microvascular endothelial cells resulted in a decrease in F-actin and prevented stress fiber formation. In line with these observations JAM-C knockdown decreased the phosphorylation of MLCs. Of note, the changes in actomyosin caused by JAM-C knockdown were considerably higher than the decrease in basal endothelial permeability, indicating that changes in stress fiber formation may only loosely correspond to permeability changes, although these two processes may be functionally linked. By regulating the status of actomyosin, JAM-C may modulate actomyosin-driven contractility, although endothelial contractility was not directly assessed in the present study. Furthermore, knockdown of JAM-C led to the up-regulation of Rap1 activity, whereas the activity of other GTPases remained unaffected. Rap1 is involved in the formation and maintenance of cadherin-mediated cell–cell contacts and adherens junctions in endothelial and epithelial cells (7, 8, 31, 32), thereby improving barrier function. The homophilic ligation of E-cadherin in epithelial cells induces Rap1 activation, and vice versa, the affinity and avidity of cadherin-mediated contacts may be modulated by Rap1-triggered inside-out signaling (31–34). Consistently, JAM-C knockdown strengthened VE-cadherin–mediated cell–cell contacts. This was a Rap1-dependent process, as down-regulation of Rap1 activity largely prevented the rescue of endothelial barrier function and the increase in VE-cadherin–mediated contacts caused by JAM-C knockdown. In contrast, the effects of JAM-C down-regulation were not altered by inhibition of the activity of PKA, another known regulator of endothelial barrier function (27).
Interestingly, JAM-A, which functions to decrease paracellular permeability in endothelial and epithelial cells (13–15), was recently found to regulate Rap1 activity in an opposite way to JAM-C, as JAM-A knockdown decreased Rap1 activity in epithelial cells (14). Although, these authors did not study cadherin-mediated cell–cell contacts, their findings, together with our present data, enable a new mechanism for the regulation of vascular permeability by JAMs to be envisioned: JAM-A acts as a gatekeeper of the endothelial contacts, as its homophilic interaction may participate in the tight junction–mediated adherence, and through stimulating Rap1 it may promote the integrity of adherens junctions. The counter-player, JAM-C, down-regulates Rap1 activity and shifts the equilibrium toward the disruption of VE-cadherin–mediated intercellular contacts. In this intriguing scenario, which needs further experimental proof, it is striking that two structurally very similar molecules, JAM-A and JAM-C, act in an opposite manner to regulate the balance of active versus nonactive Rap1.
The herein reported destabilization of adherens junctions by JAM-C may also be operative in the function of JAM-C to promote transendothelial migration. Besides its interaction with leukocyte Mac-1 during transmigration (16), junctional JAM-C may disrupt adherens junctions and increase the influx of leukocytes. This is consistent with recent findings from transgenic JAM-C–overexpressing mice that revealed increased accumulation of inflammatory cells (35). Moreover, we have previously shown that soluble JAM-C blocks neutrophil transendothelial migration in vitro and in vivo (16). Although soluble JAM-C acts as an inhibitor of Mac-1, it cannot be overlooked that the inhibition of transmigration by soluble JAM-C in vivo may also be attributed to decreased endothelial permeability.
Our findings that JAM-C regulates actin polymerization and actomyosin contractility and VE-cadherin–dependent cell–cell contacts become even more evident in light of two very recent publications deconstructing the cadherin–catenin–actin complex (36, 37). In particular, α-catenin assembled into the cadherin–catenin complex cannot bind actin. Instead, the recruitment of α-catenin to the adherens junctions upon cadherin ligation functions as a molecular switch to ultimately inhibit actin polymerization. This molecular mechanism would explain our present finding that the increase in VE-cadherin adhesiveness caused by JAM-C knockdown is accompanied by a decrease in F-actin; however, direct experimental evidence needs to be generated for this hypothesis. Furthermore, the findings of these two recent publications (36, 37) highlight the emergence of alternate mechanisms that regulate adherens junctions such as the one described in the present study. In epithelial cells, nectins, through their homophilic interaction, regulate the stability of cadherin-based cell–cell contacts both via AF-6 and by modulating the activity of small GTPases, particularly Rap1 (38). JAMs may take over a similar function in regulating the maintenance of adherens junctions in endothelial cells, thereby fine tuning vascular permeability. In addition, the inability of the cadherin–catenin complex to interact with actin (36, 37) highlights the importance of alternate links between these two components. AF-6 may operate as a potential link between the actin cytoskeleton, adherens junctions, and Rap1 (38–40) and may interact with the COOH-terminus of nectins and JAMs (reference 41 and unpublished data). This hypothesis requires further investigation. Nevertheless, our present work uncovers the first functional link between JAMs and VE-cadherin and a novel crosstalk between tight and adherens junctions that may open new directions for studies of endothelial barrier function.
MATERIALS AND METHODS
Production of soluble recombinant JAM-C proteins.
Recombinant smJAM-C was produced as a 6xHis-tagged protein in Schneider Drosophila S2 cells (Invitrogen) as previously described (16). Control 6xHis peptide was purchased from Covance. Fc fusion protein of soluble recombinant human JAM-C (Fc–JAM-C) was produced in High-Five Insect cells as previously described (42). Fc–intercellular adhesion molecule -1 was from R&D Systems, and Fc control protein was purchased from Alexis.
Cell culture and transfection.
HDMECs, HMVEC-L, HAECs, and HUVECs were purchased from Cambrex and cultivated as described by the supplier. All endothelial cells were used in low passages (up to the fourth passage). Cells were grown on culture dishes precoated with 0.2% gelatin (Sigma-Aldrich). For transfections, the following chemically synthesized duplex siRNAs were engaged: two siRNAs directed against human JAM-C, two respective control siRNAs with a single nucleotide mutation, thereby preventing JAM-C silencing, a siRNA against cyclophilin, a nontargeting control siRNA (siGENOME –hJAM3#1 and siGENOME –hJAM3#2, siJAM3#1mut, siJAM3#2mut, si-Cyclophilin-B and siCONTROL nontargeting siRNA#1, respectively) (Dharmacon), and a siRNA against Rap1a (siRap1 Eurogentec). Single siRNAs or combinations of two different siRNA were transfected at a concentration of 100 nM using Oligofectamine reagent (Invitrogen) in OptiMEM medium (Invitrogen). Cells were engaged 48–72 h after transfection in experiments as described below. Transient DNA transfections of microvascular endothelial cells were performed with a nucleofection kit (Amaxa Ink). Mouse full-length JAM-C cloned into pcDNA3.1-Zeo vector was purified with the EndoFree Plasmid Maxi kit (QIAGEN). Cells were nucleofected with 2 μg of DNA according to the manufacturer's protocol and assayed 48 h after nucleofection.
Adhesion assay.
Adhesion of HDMECs to immobilized Fc, Fc–VE-cadherin (R&D Systems), and Fc–PECAM-1 (provided by P.J. Newman, Blood Research Institute, Milwaukee, WI) was tested according to a previously described protocol (43, 44). In brief, wells were coated with Fc, Fc–VE-cadherin, Fc–PECAM-1, or BSA (each 20 μg/ml) in PBS and blocked with 3% BSA. Fluorescence-labeled HDMECs (3 × 104/well) were washed twice and added to the immobilized proteins for 60 min at 37°C in the absence or presence of effectors. After washings, adhesion of cells was quantified as the percentage of total cells added using a fluorescence microplate reader (Bio-Tek).
Cell ELISA for the comparison of total versus surface JAM-C.
Cell ELISA for the comparison of surface versus total JAM-C was performed according to a previously described protocol with modifications (42). In brief, HDMECs were grown to confluence on 24-well plates. Thereafter, cells were washed and incubated in the absence or presence of stimuli in serum-free medium for 1 h at 37°C. After incubation, cells were washed twice with PBS and fixed with 4% PFA. After fixation, a part of the wells was permeabilized with 0.1% Triton X-100. After washing and blocking with PBS containing 3% BSA for 1 h at 22°C, a polyclonal goat anti–human JAM-C (R&D Systems) was incubated, followed by addition of appropriate secondary peroxidase-conjugated antibody and the substrate ABTS. Surface expression (in fixed cells) versus total expression (in fixed and permeabilized cells) was quantitated at 405 nm, and nonspecific binding (binding of the secondary antibody to wells, in which the first antibody was omitted) was used as blank and was subtracted to calculate specific binding.
Endothelial cell permeability.
HDMEC permeability was studied as previously described (20, 44). In brief, cells were grown to confluent monolayers on gelatin-coated membranes in double-chamber tissue culture plates (Transwell membrane, 0.4 μM pore size; Corning Costar). After 2 d, chambers were examined microscopically for integrity and uniformity of endothelial monolayers, medium was switched to serum-free conditions and the stimuli/competitors, VEGF (50 ng/ml; R&D Systems), histamine (50 μM; Calbiochem), the Epac activator 8-pCPT-2′-O-Me-cAMP (O-Me-cAMP; 100–500 μM; Alexis Biochemicals), forskolin (10 μM; Calbiochem), rolipram (20 μM; Calbiochem), the PKA-specific activator Sp-8-Br-cAMPS (200 μM; Calbiochem), and the PKA inhibitor Rp-8-Br-cAMPS (200 μM; Calbiochem), were added in quadruplicate cups at designated times. At the end of the incubation period, FITC-conjugated dextran (1 mg/ml, M r 42,000; Sigma-Aldrich) was added to the upper chambers, and fluorescence in the lower chamber was measured 1 h later with a fluorescence reader (BIOTEK).
Immunofluorescence.
HDMECs were grown to confluence on 0.2% gelatin-coated glass coverslips and then incubated in serum-free medium with stimuli for different time points as indicated in the figure legends. For the staining of F-actin, cells were washed, fixed in 4% PFA for 10 min, and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Cells were blocked with 1% BSA, 5% goat serum in PBS for 1 h at 22°C. Cells were then incubated with AlexaFluor568-conjugated Phalloidin (Molecular Probes) in blocking buffer for 30 min at 22°C. The same protocol was used for the detection of JAM-C and ZO-1 except that cells were fixed in ice-cold methanol for 20 min at −20°C, and the mAbs Gi11 against JAM-C (43) or the rabbit anti–ZO-1 (Zymed) were engaged followed by appropriately conjugated secondary antibodies. Thereafter, cells were washed and mounted in Fluoromount-G (SouthernBiotech). Confocal fluorescence images were captured using a Zeiss laser scanning microscope.
Measurement of endogenous activity of small GTPases.
HDMECs (transfected with siRNA against JAM-C or control siRNA) were grown to confluence. For assaying the activity of Cdc42/Rac, Ras, and Rap1, cells were lysed, and active Cdc42/Rac, Ras, and Rap1 were precipitated with PAK-PBD beads, Raf-PBD beads (Cytoskeleton), or Ral-RBD beads (Upstate Biotechnology), respectively, for 45 min at 4°C. Precipitates were then washed, resuspended in 40 μl of 2× sample buffer, boiled for 5 min at 100°C, and separated by 12% SDS-PAGE. Antibodies against Cdc42 or Rac-1 (BD Biosciences) or antibodies against Ras or Rap1 (Upstate Biotechnology) were used for detection.
Western blot analysis.
HDMECs were washed once with ice-cold PBS and lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.25% Na-deoxycholate, 0.1% SDS, 25 mM NaF, 1 mM sodium orthovanadate, and protease inhibitors), boiled, and separated by SDS-PAGE followed by protein transfer to nitrocellulose membranes. Membranes were blocked for 1 h at 22°C and incubated with antibodies against ZO-1, VE-cadherin (Zymed), MLC, phospho-MLC, p-ERK1/2, phospho-CREB (Cell Signaling), β-actin (Abcam), JAM-C (mAb Gi11), CD29 (Santa Cruz Biotechnology, Inc.), or the goat anti–mouse JAM-C (R&D Systems). After washings, blots were incubated with appropriate peroxidase-coupled secondary antibodies (Abcam) and developed with ECL Plus reagent (GE Healthcare).
Measurement of the F-actin to G-actin ratio.
The F-actin to G-actin ratio was determined as previously described (45). HDMECs were grown to confluence on 0.2% gelatin precoated 24-well plates and stimulated with VEGF or histamine as described for 1 h. Thereafter cells were washed and fixed with 4% PFA, followed by permeabilization with 0.1% Triton X-100. Cells were blocked with 1% BSA, 5% goat serum in PBS for 1 h at 22°C and double stained with AlexaFluor488 DNase I (for monomeric G-actin) and AlexaFluor568 Phalloidin (for polymeric F-actin) (Molecular Probes) for 30 min at 22°C and then rinsed three times with PBS. Quantification of G-actin and F-actin was performed with a spectrofluorometer (BIOTEK).
Hypoxia-induced retinal vascularization (ROP model).
Studies were performed as previously described (44). In brief, 7-d-old (p7) C57BL/6 mice were exposed to 75% oxygen for 5 d in an incubator (BioSpherix) with their nursing mothers. At p12, mice were returned to room air. From p12 until p16, mice were given i.p. injections of smJAM-C, the control 6xHis peptide (each 30 μg/mouse), or buffer alone once daily (five mice/group). Mice were killed at p17, and eyes were processed for quantification of preretinal neovascular nuclei as described (44). To quantify retinal neovascularization, 6-μm paraffin-embedded sections were stained with PAS and hematoxylin, and 10 intact sections of equal length, each 18 μm apart, were evaluated. All retinal vascular cell nuclei anterior to the internal limiting membrane were counted in each section by an observer blinded to the protocol. The mean of the 10 counted sections represents the average neovascular nuclei per section per eye. No neovascular cell nuclei anterior to the internal limiting membrane are observed in normal “room air” mice (44).
Quantification of vascular permeability in the retina of p13 mice subjected to the ROP model was performed by measuring the leakage of Evan's blue, according to a previously described protocol (44, 46). In brief, p13 mice subjected to the ROP model and controls were anesthetized, and 100 μl of Evan's blue (30 mg/kg) was injected into the left ventricle. After removal of the eyes and careful dissection of the retinas, the dye was extracted from each retina by incubation in formamide for 48 h at 55°C. Leakage of Evan's blue was expressed as absorbance at 620 nm/dry weight of the retina. Animal studies were approved by the NCI Animal Care and Use Committee (Bethesda, MD) and the Governmental Office Karlsruhe (Germany).
In vivo vascular permeability assay.
A modified Miles assay was performed as described previously (47). Mice were pretreated by i.p. injection of control 6xHis peptide or smJAM-C (each 100 μg/mouse) 60–90 min before the experiment. Mice were anesthetized and VEGF (100 ng) or compound 48/80 (10 μg; Sigma-Aldrich) in 50 μl PBS was injected intradermally into the shaved back skin. After 30 min, 100 μl Evans blue dye in 0.9% NaCl (30 mg/kg) was injected into the left ventricle. After 10 min mice were killed and the marked region of skin that included the area of the intradermal injection was removed. Evans blue dye was extracted from the skin and the absorbance of extracted dye was measured at 620 nm as described in Hypoxia-induced retinal vascularization (ROP model).
Statistical analysis.
Data were compared using the Student's t test and ANOVA with post-hoc analysis as appropriate; p values <0.05 were regarded as significant.
Online supplemental material.
Fig. S1 shows the localization of JAM-C in HUVEC and HMVEC-L. Fig. S2 shows siRNA control experiments, indicating that the alterations in endothelial permeability caused by JAM-C knockdown are specific. Fig. S3 shows retina whole mounts from mice subjected to the ROP model. Fig. S4 shows axial sections of the retina stained with periodic acid Schiff and hematoxylin, indicating pathologic retinal neovascularization. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051730/DC1.
Supplemental Material
Acknowledgments
The authors would like to acknowledge M.E. Kruhlak (Experimental Immunology Branch, NCI, NIH) for help with confocal microscopy, Robert Silasi-Mansat (Oklahoma Medical Research Foundation) for help with electron microscopy, and Darius Schneider (University Heidelberg) for help with in vivo experiments.
This research was supported by the Intramural Research Program of the NIH, NCI (to T. Chavakis) and by grants from the Deutsche Forschungsgemeinschaft-SFB 547 (to S. Santoso). This work is part of the Ph.D. thesis (Dr. sc. hum.) of V.V. Orlova at the department of Medicine I, University Heidelberg.
The authors have no conflicting financial interests.
Abbreviations used: cAMP, cyclic adenosine monophosphate; HDMEC, human dermal microvascular endothelial cell; HMVEC-L, human lung microvascular endothelial cell; HUVEC, human umbilical vein endothelial cell; HAEC, human aortic endothelial cell; JAM, junctional adhesion molecule; MLC, myosin light chain; PECAM, platelet endothelial cell adhesion molecule; PKA, protein kinase A; ROP, retinopathy of prematurity; smJAM, soluble mouse JAM; VEGF, vascular endothelial growth factor.
References
- 1.van Nieuw Amerongen, G.P., and V.W. van Hinsbergh. 2002. Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul. Pharmacol. 39:257–272. [DOI] [PubMed] [Google Scholar]
- 2.Wojciak-Stothard, B., and A.J. Ridley. 2002. Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 39:187–199. [DOI] [PubMed] [Google Scholar]
- 3.Campochiaro, P.A. 2004. Ocular neovascularisation and excessive vascular permeability. Expert Opin. Biol. Ther. 4:1395–1402. [DOI] [PubMed] [Google Scholar]
- 4.Lee, T.Y., and A.I. Gotlieb. 2003. Microfilaments and microtubules maintain endothelial integrity. Microsc. Res. Tech. 60:115–127. [DOI] [PubMed] [Google Scholar]
- 5.Braga, V.M. 2002. Cell-cell adhesion and signalling. Curr. Opin. Cell Biol. 14:546–556. [DOI] [PubMed] [Google Scholar]
- 6.Bazzoni, G., and E. Dejana. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84:869–901. [DOI] [PubMed] [Google Scholar]
- 7.Cullere, X., S.K. Shaw, L. Andersson, J. Hirahashi, F.W. Luscinskas, and T.N. Mayadas. 2005. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 105:1950–1955. [DOI] [PubMed] [Google Scholar]
- 8.Fukuhara, S., A. Sakurai, H. Sano, A. Yamagishi, S. Somekawa, N. Takakura, Y. Saito, K. Kangawa, and N. Mochizuki. 2005. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol. Cell. Biol. 25:136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matter, K., and M.S. Balda. 2003. Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 4:225–236. [DOI] [PubMed] [Google Scholar]
- 10.Ebnet, K., A. Suzuki, S. Ohno, and D. Vestweber. 2004. Junctional adhesion molecules (JAMs): more molecules with dual functions? J. Cell Sci. 117:19–29. [DOI] [PubMed] [Google Scholar]
- 11.Keiper, T., S. Santoso, P.P. Nawroth, V. Orlova, and T. Chavakis. 2005. The role of junctional adhesion molecules in cell-cell interactions. Histol. Histopathol. 20:197–203. [DOI] [PubMed] [Google Scholar]
- 12.Santoso, S., V. Orlova, K. Song, U.J. Sachs, C.L. Andrei-Selmer, and T. Chavakis. 2005. The homophilic binding of junctional adhesion moplecule-C mediates tumor cell-endothelial cell interactions. J. Biol. Chem. 280:36326–36333. [DOI] [PubMed] [Google Scholar]
- 13.Mandell, K.J., I.C. McCall, and C.A. Parkos. 2004. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J. Biol. Chem. 279:16254–16262. [DOI] [PubMed] [Google Scholar]
- 14.Mandell, K.J., B.A. Babbin, A. Nusrat, and C.A. Parkos. 2005. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J. Biol. Chem. 280:11665–11674. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Padura, I., S. Lostaglio, M. Schneemann, L. Williams, M. Romano, P. Fruscella, C. Panzeri, A. Stoppacciaro, L. Ruco, A. Villa, et al. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavakis, T., T. Keiper, R. Matz-Westphal, K. Hersemeyer, U.J. Sachs, P.P. Nawroth, K.T. Preissner, and S. Santoso. 2004. The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J. Biol. Chem. 279:55602–55608. [DOI] [PubMed] [Google Scholar]
- 17.Ebnet, K., M. Aurrand-Lions, A. Kuhn, F. Kiefer, S. Butz, K. Zander, M.K. Meyer zu Brickwedde, A. Suzuki, B.A. Imhof, and D. Vestweber. 2003. The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: a possible role for JAMs in endothelial cell polarity. J. Cell. Sci. 116:3879–3891. [DOI] [PubMed] [Google Scholar]
- 18.Aurrand-Lions, M., C. Johnson-Leger, C. Wong, L. Du Pasquier, and B.A. Imhof. 2001. Heterogeneity of endothelial junctions is reflected by differential expression and specific cellular localization of the three JAM family members. Blood. 98:3699–3707. [DOI] [PubMed] [Google Scholar]
- 19.Aurrand-Lions, M., L. Duncan, C. Ballestrem, and B.A. Imhof. 2001. JAM-2, a novel immunoglobulin superfamily molecule, expressed by endothelial and lymphatic cells. J. Biol. Chem. 276:2733–2741. [DOI] [PubMed] [Google Scholar]
- 20.Behzadian, M.A., L.J. Windsor, N. Ghaly, G. Liou, N.T. Tsai, and R.B. Caldwell. 2003. VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEB J. 17:752–754. [DOI] [PubMed] [Google Scholar]
- 21.Stockton, R.A., E. Schaefer, and M.A. Schwartz. 2004. p21-activated kinase regulates endothelial permeability through modulation of contractility. J. Biol. Chem. 279:46621–46630. [DOI] [PubMed] [Google Scholar]
- 22.Kiosses, W.B., R.H. Daniels, C. Otey, G.M. Bokoch, and M.A. Schwartz. 1999. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carson, M.R., S.S. Shasby, S.E. Lind, and D.M. Shasby. 1992. Histamine, actin-gelsolin binding, and polyphosphoinositides in human umbilical vein endothelial cells. Am. J. Physiol. 263:L664–L669. [DOI] [PubMed] [Google Scholar]
- 24.Cezar-de-Mello, P.F., V. Nascimento-Silva, C.G. Villela, and I.M. Fierro. 2006. Aspirin-triggered Lipoxin A(4) inhibition of VEGF-induced endothelial cell migration involves actin polymerization and focal adhesion assembly. Oncogene. 25:122–129. [DOI] [PubMed] [Google Scholar]
- 25.Gliki, G., K. Ebnet, M. Aurrand-Lions, B.A. Imhof, and R.H. Adams. 2004. Spermatid differentiation requires the assembly of a cell polarity complex downstream of junctional adhesion molecule-C. Nature. 431:320–324. [DOI] [PubMed] [Google Scholar]
- 26.Mamdouh, Z., X. Chen, L.M. Pierini, F.R. Maxfield, and W.A. Muller. 2003. Targeted recycling of PECAM from endothelial surface-connected compartments during diapedesis. Nature. 421:748–753. [DOI] [PubMed] [Google Scholar]
- 27.Comerford, K.M., D.W. Lawrence, K. Synnestvedt, B.P. Levi, and S.P. Colgan. 2002. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J. 16:583–585. [DOI] [PubMed] [Google Scholar]
- 28.Gatti, S., R. Faggioni, M. Sironi, A. Erroi, and P. Ghezzi. 1993. Mast cells do not contribute to the rapid appearance of TNF in the serum of LPS-treated mice: a study with mast cell-deficient mice. Int. J. Immunopharmacol. 15:551–555. [DOI] [PubMed] [Google Scholar]
- 29.Lim, Y.C., G. Garcia-Cradena, J.R. Allport, M. Zervoglos, A.J. Connolly, M.A. Gimbrone, Jr., and F.W. Luscinskas. 2003. Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am. J. Pathol. 162:1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michel, C.C., and F.E. Curry. 1999. Microvascular permeability. Physiol. Rev. 79:703–761. [DOI] [PubMed] [Google Scholar]
- 31.Price, L.S., A. Hajdo-Milasinovic, J. Zhao, F.J. Zwartkruis, J.G. Collard, and J.L. Bos. 2004. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J. Biol. Chem. 279:35127–35132. [DOI] [PubMed] [Google Scholar]
- 32.Hogan, C., N. Serpente, P. Cogram, C.R. Hosking, C.U. Bialucha, S.M. Feller, V.M. Braga, W. Birchmeier, and Y. Fujita. 2004. Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol. Cell. Biol. 24:6690–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gumbiner, B.M. 2005. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 6:622–634. [DOI] [PubMed] [Google Scholar]
- 34.Bos, J.L., J. de Rooij, and K.A. Reedquist. Rap1 signalling: adhering to new models. 2001. Nat. Rev. Mol. Cell. Biol. 2:369–377. [DOI] [PubMed] [Google Scholar]
- 35.Aurrand-Lions, M., C. Lamagna, J.P. Dangerfield, S. Wang, P. Herrera, S. Nourshargh, and B.A. Imhof. 2005. Junctional adhesion molecule-C regulates the early influx of leukocytes into tissues during inflammation. J. Immunol. 174:6406–6415. [DOI] [PubMed] [Google Scholar]
- 36.Drees, F., S. Pokutta, S. Yamada, W.J. Nelson, and W.I. Weis. 2005. 2005. Alpha-catenin is a molecular switch that binds E-cadherin- beta-catenin and regulates actin-filament assembly. Cell. 123:903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamada, S., S. Pokutta, F. Drees, W.I. Weis, and W.J. Nelson. 2005. Deconstructing the cadherin-catenin-actin complex. Cell. 123:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoshino, T., T. Sakisaka, T. Baba, T. Yamada, T. Kimura, and Y. Takai. 2005. Regulation of E-cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. J. Biol. Chem. 280:24095–24103. [DOI] [PubMed] [Google Scholar]
- 39.Zhang, Z., H. Rehmann, L.S. Price, J. Riedl, and J.L. Bos. 2005. AF6 negatively regulates Rap1-induced cell adhesion. J. Biol. Chem. 280:33200–33205. [DOI] [PubMed] [Google Scholar]
- 40.Boettner, B., E.E. Govek, J. Cross, and L. Van Aelst. 2000. The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc. Natl. Acad. Sci. USA. 97:9064–9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ebnet, K., C.U. Schulz, M.K. Meyer Zu Brickwedde, G.G. Pendl, and D. Vestweber. 2000. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J. Biol. Chem. 275:27979–27988. [DOI] [PubMed] [Google Scholar]
- 42.Keiper, T., N. Al-Fakhri, E. Chavakis, A.N. Athanasopoulos, B. Isermann, S. Herzog, R. Saffrich, K. Hersemeyer, R.M. Bohle, J. Haendeler, K.T. Preissner, S. Santoso, and T. Chavakis, T. 2006. The Role of junctional adhesion molecule-C (JAM-C) in oxidized LDL-mediated leukocyte recruitment. FASEB J. 20:559–561. [DOI] [PubMed] [Google Scholar]
- 43.Santoso, S., U.J. Sachs, H. Kroll, M. Linder, A. Ruf, K.T. Preissner, and T. Chavakis. 2002. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 196:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Economopoulou, M., K. Bdeir, D.B. Cines, F. Fogt, Y. Bdeir, J. Lubkowski, W. Lu, K.T. Preissner, H.P. Hammes, and T. Chavakis. 2005. Inhibition of pathological retinal neovascularization by α-defensins. Blood. 106:3831–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulinilkunnil, T., D. An, S. Ghosh, D. Qi, G. Kewalramani, G. Yuen, N. Virk, A. Abrahani, and B. Rodrigues. 2005. Lysophosphatidic acid-mediated augmentation of cardiomyocyte lipoprotein lipase involves actin cytoskeleton reorganization. Am. J. Physiol. Heart Circ. Physiol. 288:H2802–H2810. [DOI] [PubMed] [Google Scholar]
- 46.Brankin, B., M. Campbell, P. Canning, T.A. Gardiner, and A.W. Stitt. 2005. Endostatin modulates VEGF-mediated barrier dysfunction in the retinal microvascular endothelium. Exp. Eye Res. 81:22–31. [DOI] [PubMed] [Google Scholar]
- 47.Oura, H., J. Bertoncini, P. Velasco, L.F. Brown, P. Carmeliet, and M. Detmar. 2003. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood. 101:560–567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}