

Abstract
T cell–specific adapter protein (TSAd) is a SRC-homology-2 (SH2) domain–containing intracellular signaling molecule that is required for T cell antigen receptor (TCR)–induced cytokine synthesis in T cells. How TSAd functions in TCR signal transduction is not clear. Previous work has suggested a nuclear role for this adapter. However, other evidence suggests that TSAd also functions in the cytoplasm. Using T cells from TSAd-deficient mice, we now show that the major role of TSAd in the cytoplasm is in activation of the LCK protein tyrosine kinase at the outset of TCR signal transduction. Consequently, TSAd regulates several downstream signaling events, including intracellular calcium mobilization and activation of the Ras–extracellular signal–regulated kinase signaling pathway. TSAd regulates LCK activity directly through physical interaction with LCK SH3 and SH2 domains. These studies reveal TSAd as a positive regulator of proximal TCR signal transduction and provide important new information on the mechanism of TCR-induced LCK activation.
TCR binding to antigen-MHC molecules displayed on APCs triggers the activation of protein tyrosine kinases (PTKs) (1). The first PTK to become activated is the SRC family PTK, LCK, which phosphorylates tyrosine residues in immunoreceptor tyrosine-based activation motifs (ITAMs) present in TCR signaling chains such as TCRζ. Phosphorylated ITAMS are recognized by SRC-homology-2 (SH2) domains of the Syk-family PTK, ZAP-70, which is thus recruited to the complex and activated, in part through transphosphorylation by LCK. In turn, activated ZAP-70 phosphorylates the LAT transmembrane adaptor protein leading to SH2 domain–mediated interaction with different signaling molecules including phospholipase Cγ1 (PLCγ1), and the cytosolic adaptor proteins, Grb-2 and GADS. Recruitment of these signaling molecules to the membrane triggers the activation of distinct signaling pathways that culminate in the activation of AP-1, NFκB, and NFAT transcription factors. Together, these transcription factors induce the expression of numerous genes that drive T cell proliferation and differentiation into effector cells.
T cell–specific adaptor protein (TSAd) is a relatively recently described SH2 domain–containing intracellular adaptor molecule that was initially reported to be restricted in expression to the T cell lineage (2, 3). TSAd appears to play an important role in TCR signal transduction as indicated by the fact that T cells from TSAd-deficient mice secrete reduced quantities of the cytokines IL-2, IL-4, and IFN-γ in response to TCR engagement (4, 5). Exactly how TSAd participates in TCR signal transduction is unknown. In previous studies, a role for TSAd as a direct regulator of cytokine gene transcription in the nucleus has been suggested (6). In addition, TSAd has been shown to interact physically with different cytoplasmic protein kinases, suggesting an important function in this cellular compartment as well (3, 4, 7). One such kinase that TSAd interacts with is LCK (3, 8). Physical interaction with LCK has been demonstrated in yeast–hybrid systems and T cell lines, although the functional significance of this interaction is not clearly understood. Under conditions of strong overexpression in T cell lines, TSAd can inhibit LCK activity (8, 9). However, the notion that TSAd, a positive regulator of T cell cytokine synthesis, acts as a physiological inhibitor of LCK is at odds with the fact that LCK is required for cytokine synthesis and other T cell responses (10).
In the present studies, we have used TSAd-deficient mice to further address the role of TSAd in the T cell cytoplasm. We report that TSAd controls multiple TCR-initiated cytoplasmic signaling pathways that can be explained on the basis that TSAd is in fact essential for the activation of LCK.
RESULTS AND DISCUSSION
We examined if TSAd is required for the activation of extracellular signal–regulated kinase (ERK), c-Jun NH2-terminal protein kinase (JNK), and p38 kinases that regulate cytokine secretion through phosphorylation of AP-1 and other transcription factors (11). LN T cells from TSAd (+/+) and (−/−) mice were stimulated with mAb against CD3ε (a component of the TCR complex) and the CD28 costimulatory receptor and kinase activation was assessed by Western blotting using phospho-specific antibodies (Fig. 1 A). ERK1, ERK2, and p38 were activated weakly and with delayed kinetics in TSAd (−/−) compared with (+/+) T cells. Activation of JNK was also impaired and delayed although to a lesser extent than ERKs and p38 (Fig. 1 A). As determined in Raf-1–Ras-binding domain (RBD) pull-down assays, activation of the Ras small G-protein was delayed in TSAd (−/−) T cells, which explains the blocked ERK response (Fig. 1 B).
Figure 1.

Defective activation of ERK, JNK, and p38 kinases and impaired intracellular calcium mobilization in TSAd-deficient T cells. (A and B) LN T cells from TSAd (+/+) and TSAd (−/−) mice were stimulated with optimal concentrations of CD3 and CD28 mAb for the indicated times (in min). White lines indicate that intervening lanes have been spliced out. (A) ERK, JNK, and p38 activation was assessed by Western blotting of whole cell lysates using phospho-specific antibodies. Relative densitometry scanning readings for phospho-JNK1 are indicated below the image. (B) Active GTP-bound Ras was pulled down from lysates of LN T cells with a Raf-1–RBD fusion protein and quantified by Western blotting using a Ras antibody. (C) Fura-2–loaded LN T cells were stimulated with optimal or suboptimal concentrations of CD3 plus CD28 mAb at the indicated time point. Shown is the ratio (R) of light emission at a 510-nm wavelength at excitation wavelengths of 340 and 380 nm over time. Ionomycin (Iono) was added to cells at the indicated point as a positive control.
We also examined if TSAd was required for CD3/CD28-induced intracellular calcium mobilization that is necessary for nuclear translocation of NFAT and cytokine gene transcription (12). Calcium mobilization was monitored with the use of the Fura-2 calcium-sensitive fluorescent dye (Fig. 1 C). Using relatively high concentrations of stimulating CD3 and CD28 mAb (optimal), only a small difference in the calcium response between TSAd (−/−) and (+/+) T cells was noted. However, using 20-fold lower concentrations of mAb (suboptimal), TSAd (−/−) T cells showed a clear defective calcium response.
Through its ability to generate InsP3 and DAG, which promotes recruitment of RasGRP1 to the plasma membrane, PLCγ1 lies upstream of calcium and Ras–ERK signaling pathways (12–14). We determined that PLCγ1 was not activated properly in TSAd (−/−) T cells (Fig. 2 A). This finding, therefore, provides an explanation for the defective calcium and Ras–ERK responses. Activation of PLCγ1 in T cells is dependent on SH2 domain–mediated recruitment of PLCγ1 to phosphorylated Y136 of LAT (15, 16). In addition, Grb-2 binds to phosphorylated Y175, Y195, and Y235 of LAT via its SH2 domain and in so doing contributes to the activation of Ras (15, 16). Therefore, we examined if LAT interacted with PLCγ1 and Grb-2 in TSAd (−/−) T cells (Fig. 2 B). As shown, reduced quantities of PLCγ1 and Grb-2 were coimmunoprecipitated with LAT after CD3/CD28 engagement and this correlated with reduced phosphorylation of LAT tyrosine residues Y136 and Y195, respectively. Because ZAP-70 is the principal PTK responsible for phosphorylation of LAT, we examined ZAP-70 activation in TSAd (−/−) T cells by Western blotting (Fig. 2 C). ZAP-70 was indeed only weakly activated in response to CD3/CD28 engagement.
Figure 2.

Impaired LAT phosphorylation and interaction with PLCγ1 and Grb-2 in TSAd-deficient T cells. LN T cells from TSAd (+/+) and (−/−) mice were stimulated with optimal concentrations of CD3 and CD28 mAb for the indicated times (in min). (A) PLCγ1 activation was assessed by Western blotting of whole cell lysates using a phospho-specific antibody. (B) LAT was immunoprecipitated from lysates. Phosphorylation of immunoprecipitated LAT on Y136 and Y195 and coimmunoprecipitation of PLCγ1 and Grb-2 was detected by Western blotting using specific antibodies. (C) Activation of ZAP-70 was assessed by Western blotting of whole cell lysates using a phospho-specific antibody.
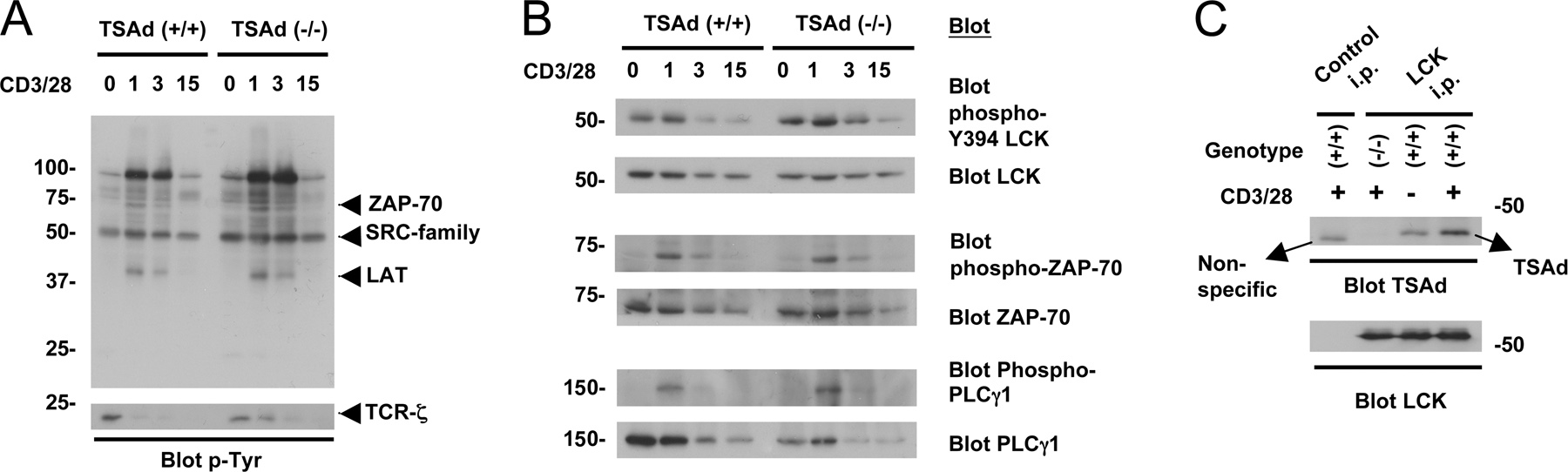
ZAP-70 activation during TCR signal transduction is dependent on kinases of the SRC family, particularly LCK (1). Consistent with the possibility that TSAd is required for the activation of LCK, bands corresponding to the molecular weight of SRC family kinases and the TCRζ chain were found to be hypophosphorylated in whole phosphotyrosine Western blots of CD3/CD28-stimulated TSAd (−/−) T cells (Fig. 3 A). After confirming that TSAd and LCK interact physically with another in wild-type LN T cells in response to CD3/CD28 engagement (Fig. 3 B), we examined LCK activation directly. As shown in both Western blotting experiments and in vitro autokinase assays, LCK was activated poorly in TSAd (−/−) T cells in response to CD3/CD28 (Fig. 3, C and D). Thus, in primary T cells, TSAd functions as a positive and not a negative regulator of LCK, consistent with the finding that TSAd is required for normal induction of cytokines (4, 5). Most likely, previous reports of an inhibitory effect of TSAd upon LCK activation are an artifact of strong overexpression of TSAd and or/the use of the Jurkat leukemia cell line in experiments (8, 9). Indeed, under conditions of mild overexpression, TSAd can augment IL-2 gene transcription in Jurkat (6).
Figure 3.

LCK activation in TSAd-deficient T cells. LN T cells from TSAd (+/+) and (−/−) mice were stimulated with optimal concentrations of CD3 and CD28 mAb for the indicated times in minutes (1 min in B). (A) Whole cell protein phosphorylation was determined by Western blotting using a phosphotyrosine antibody. Also shown is a longer exposure of the low molecular weight range of the phosphotyrosine blot to reveal the TCRζ chain. (B) LCK was immunoprecipitated from lysates and coimmunoprecipitated TSAd was detected by Western blotting. (C) Activation of LCK was assessed by Western blotting of whole cell lysates using a phospho-Y394 LCK antibody. (D) Kinase activity of immunoprecipitated LCK was assessed by an ability to self-incorporate 32P in autokinase assays. Separate aliquots from autokinase assays were analyzed by Western blotting using an LCK antibody to check for equivalent immunoprecipitation of LCK.
We examined if TSAd has the capacity to activate LCK kinase activity directly (Fig. 4 A). In an in vitro SRC peptide transphosphorylation assay, recombinant tyrosine-phosphorylated TSAd was able to activate LCK kinase activity in a dose-dependent fashion from concentrations as meager as 0.01 μM up to 1.0 μM (although, at 10 μM, the effect of TSAd was diminished). However, even at 1.0 μM, the degree of LCK activation was less than that observed after stimulation of wild-type LN T cells with CD3/CD28 antibodies (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051637/DC1). This suggests that, although TSAd is required for LCK activation in vivo (Fig. 3, C and D), TSAd is not sufficient for full LCK activation and that full LCK activation in vivo may depend on TSAd cooperation with other factors (e.g., CD45).
Figure 4.

TSAd activation of LCK kinase activity. (A) LCK was immunoprecipitated from TSAd-deficient LN T cells and tested for its ability to transfer 32P to a SRC peptide substrate in the presence of a tyrosine-phosphorylated His6-Smt3–tagged TSAd fusion protein. (B) Yeast were transformed with the indicated TetR-fused LCK SH3-SH2 bait proteins and TA-fused TSAd COOH region prey proteins. WT, wild-type bait or prey proteins; 3Y-F and 3P-A, Y275F/Y292F/Y317F and P262A/P263A/P265A triple mutants of the TSAd COOH region, respectively; W-R, inactivating W97R mutant of the LCK SH3 domain; R-K, inactivating R154K mutant of the LCK SH2 domain. Yeast were also transformed with the kinase domain of LCK fused to LexA to phosphorylate the TSAd COOH region. The LexA moiety is irrelevant to the system and the kinase domain fusion protein is predicted to diffuse freely within the cell. Growth of yeast on Uracil dropout plates was assessed after 48 h of incubation and taken as an indicator of bait–prey interaction. Note that mutation of tyrosine residues and the proline-rich stretch of the TSAd COOH region are necessary to prevent interaction with LCK. Note also that mutation of the SH2 and SH3 domains of LCK is necessary to prevent interaction with TSAd when the latter is phosphorylated (by LCK). (C) Experiments were conducted as in A using the indicated TSAd fusion proteins. SRC-peptide phosphorylation (left) and autokinase activity (right) was determined. (D) Splenic T cells from TSAd (−/−) mice were transfected with wild-type, 3Y-F/3P-A TSAd, or vector alone (None) and stimulated with optimal concentrations of CD3 and CD28 mAb for 2 min. Phosphorylation of LCK on Y394 was assessed by Western blotting of whole cell lysates using a phospho-specific antibody. Blots were reprobed with LCK and TSAd antibodies to show equivalent loading of LCK and expression of TSAd, respectively. TSAd is observed as a triplet of three proteins of very similar molecular weights in these experiments. The species of intermediate molecular weight comigrates with a nonspecific band that can be identified in T cells transfected with vector alone.
Using a yeast multihybrid system, we established that physical interaction of LCK and TSAd was mediated through both LCK SH2 domain recognition of LCK-phosphorylated tyrosines in the TSAd COOH region (Y275, Y292, and Y317) and SH3 domain recognition of a TSAd COOH region proline-rich stretch (259RPRPPIPAK267) (Fig. 4 B) (17). This is significant since LCK SH2 domain binding to phosphorylated LCK Y505 (located in a short peptide COOH to the kinase domain) and LCK SH3 domain binding to an LCK proline-rich stretch (located in a linker region between the LCK SH2 and kinase domains) are both thought to restrain the kinase in an inactive conformation (10). Therefore, by interacting with LCK SH2 and SH3 domains, TSAd might relieve LCK from these autoinhibitory mechanisms and activate the kinase. To confirm the importance of the different TSAd COOH region residues, the effect of their mutation upon an ability of TSAd to activate LCK in vitro was examined (Fig. 4 C). As shown, mutation of all of the three TSAd tyrosine residues to phenylalanine (3Y-F) profoundly reduced the ability of TSAd to activate LCK kinase activity. In addition, mutation of three of the proline residues (3P-A) in the proline-rich stretch (P262, P263, and P265) to alanine reduced the ability of TSAd to activate LCK kinase activity, although to a lesser degree than the 3Y-F mutation. It is predicted that the SH2 domain–Y505 interaction would not directly inhibit kinase activity but indirectly by promoting SH3 domain interaction with the LCK linker region. In this regard, one possible explanation for the lesser influence of the TSAd 3P-A mutation upon LCK activity could be that the 3P-A mutant would still be capable of disrupting the SH2–Y505 interaction and hence, to a degree, the SH3–linker interaction also. Conversely, even though the TSAd 3Y-F mutant would still contain the proline stretch, this might not have access to the LCK SH3 domain so long as the LCK SH2 domain–Y505 interaction remained intact. As expected, a TSAd protein containing both types of mutation (3Y-F/3P-A) was unable to activate LCK kinase activity.
We examined if TSAd COOH region tyrosine residues and the proline-rich stretch were required for CD3/CD28-induced activation of LCK in vivo (Fig. 4 D). For this purpose, resting splenic T cells from TSAd (−/−) mice were reconstituted with wild-type or 3Y-F/3P-A TSAd. In wild-type TSAd-reconstituted T cells, activation of LCK in response to CD3/CD28 was restored. In contrast, in mutant TSAd-reconstituted T cells, activation of LCK was not apparent. These results are consistent with the idea that physical interaction of TSAd with the SH2 and SH3 domains of LCK is necessary for activation of LCK kinase activity induced through CD3/CD28 ligation.
The mechanism by which the TCR induces LCK activation is only partially understood. A prevailing view is that TCR engagement triggers LCK into open forms and induces their local aggregation, leading to transphosphorylation of Y394 in the kinase domain and resultant full activation of enzymatic activity (10). How the TCR promotes the open conformation is unclear, although the CD45 protein tyrosine phosphatase, by dephosphorylating Y505, is considered to play an important role (18). Based upon the findings here, we propose an updated model of TCR-induced LCK activation. In this model, TCR engagement would be envisaged to stimulate an initial increase in kinase activity that would be independent of TSAd. This would be possible either through the action of CD45 and/or the local aggregation of LCK molecules that preexist in open conformations. The initial increase in kinase activity would then lead to phosphorylation of TSAd COOH tyrosine residues that may expose the COOH region proline-rich stretch. As a result, TSAd would have the capability to interact with the LCK SH2 and SH3 domains and, in so doing, would have the effect of triggering additional LCK molecules (perhaps in concert with CD45) into open and, subsequently, fully activated conformations.
Apart from LCK, TSAd has also been described to interact with the Tec family PTK, Rlk and Itk, and the mitogen-activated protein 3-kinase, MEKK2 (4, 7). Because Tec family PTK reside downstream of SRC family PTK in T cells, it is probable that their activation is also impaired in TSAd-deficient T cells and that this contributes to deficient induction of IL-2 (19). However, whether TSAd directly modulates Tec family kinases in T cells is uncertain because physical interaction has hitherto been demonstrated only in yeast hybrid systems or under conditions of strong overexpression of both molecules. With regards to MEKK2, physical interaction has been shown to be necessary for MEKK2 activation in an epithelial cell line (7). However, physical interaction between endogenous molecules in T cells has also yet to be demonstrated. Moreover, the notion that TSAd is required for MEKK2 activation in T cells is difficult to reconcile with the finding that T cells from MEKK2-deficient mice produce increased quantities of IL-2 in response to TCR stimulation (20).
In contrast with peripheral T cells, TSAd is not required for the activation of LCK in thymocytes despite the fact that TSAd is well expressed in thymocytes from the CD4+CD8+ double-positive stage (4) (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20051637/DC1). This is consistent with the finding that TSAd is not required for either positive or negative selection in the thymus as determined using non-TCR transgenic and TCR transgenic mice (4, 21) (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20051637/DC1). The molecular mechanisms responsible for an uncoupling of TSAd from LCK in thymocytes are unclear at present, although this is unlikely to be explained by lack of association of the two molecules that can be readily detected in thymocytes (Fig. S2). It is possible that other regulatory proteins can compensate for the loss of TSAd in thymocytes. Candidates include Unc119, which is expressed at low levels in all hematopoietic cells and the recently described TSAd-related molecule, ALX, whose expression in thymus has been documented previously (22, 23).
In summary, we reveal a novel aspect of the regulation of LCK in T cells that involves TSAd. By promoting LCK activation during TCR triggering, TSAd controls different downstream cytoplasmic signaling events that regulate cytokine synthesis. Together with a proposed nuclear role of TSAd, these findings provide a molecular explanation for the role of TSAd in T cell cytokine induction. How cytoplasmic and nuclear signaling functions of TSAd are integrated during T cell activation remains to be determined.
MATERIALS AND METHODS
Mice.
TSAd (+/+) and (−/−) C57BL/6 mice were obtained from Dr. Jeff Bluestone (University of California, San Francisco, San Francisco, CA) and were bred in our laboratory (4). All mice used in this study were <6 wk of age at the time of sacrifice (i.e., before the onset of autoimmune disease in TSAd-deficient mice). Extensive phenotypic analysis of lymphoid organs revealed that T cells from TSAd (−/−) mice were normal (compared with wild-type mice) at this age and did not show any signs of increased past or recent activation (Fig. S3). All experiments were performed in compliance with University of Michigan guidelines and were approved by the University Committee on the Use and Care of Animals.
Antibodies.
Primary mAb against the following antigens were used: CD3ε (145-2C11), CD28 (37.51) (BD Biosciences); Ras (RAS10) (Upstate Biotechnology); LAT (a gift from W. Zhang, Duke University, Durham, NC); LCK (3A5) (Santa Cruz Biotechnology, Inc.); and protein phosphotyrosine (PY99) (Santa Cruz Biotechnology, Inc.). Primary rabbit polyclonal antibodies were as follows: ERK, phospho-ERK, JNK, phospho-JNK, PLCγ1, phospho-Y783 PLCγ1, phospho-Y195 LAT, ZAP-70 and phospho-Y319 ZAP-70 (Cell Signaling Technology); p-38α and Grb-2 (Santa Cruz Biotechnology, Inc.); phospho-Y136 LAT (BD Biosciences); LCK (Upstate Biotechnology); and phospho-Y394 LCK (a gift from A. Shaw, Washington University, St. Louis, MO). A rabbit polyclonal TSAd antibody was produced by immunization of rabbits with a His6-Smt3–murine TSAd fusion protein.
Protein phosphorylation, coimmunoprecipitation, Ras activation, and kinase assays.
LN T cells (purified by negative selection) were coated with CD3ε and CD28 mAb (1 μg each/5 × 106 cells) for 20 min at 4°C. Secondary cross-linking antibody was added and cells were transferred to 37°C for varying times before resuspension in lysis buffer containing 1% NP-40 and 0.5% n-Dodecyl-β-D-maltoside. Western blot analyses of protein phosphorylation in whole cell lysates and coimmunoprecipitation studies were performed as described using 3 and 50 × 106 T cells per condition, respectively (6, 17). To assay Ras activation, lysates of 50 × 106 T cells were rotated with GST-Raf-1-RBD–coated agarose beads (Upstate Biotechnology) for 40 min at 4°C. Beads were washed extensively and any bound Ras-GTP was detected by Western blotting. LCK autokinase activity and SRC peptide phosphorylation was assessed as described using LCK immunoprecipitated from 7 × 106 LN T cells (24). Tyrosine-phosphorylated His6-Smt3–murine TSAd fusion protein, produced in TKB1 bacteria (BL21 that express a tyrosine kinase) (Stratagene), was incorporated into kinase reactions from the outset.
Calcium mobilization.
Intracellular calcium flux experiments were performed as described previously (25). Fura-2–labeled (Invitrogen) LN T cells (107/2 ml) were stimulated by the addition of CD3ε plus CD28 mAb (optimal stimulation: 1 μg/ml; suboptimal stimulation: 0.05 μg/ml) and a secondary cross-linking antibody. Light emission at 510 nM wavelength in response to excitation with light of wavelengths of 340 and 380 nM was recorded over time in a fluorimeter (PerkinElmer).
Yeast-hybrid experiments.
The modified yeast two-bait interaction trap system was used to dissect the mechanism of TSAd–LCK interaction (17). CXWY2 yeast were transformed with TetR DNA-binding domain-LCK SH3-SH2 (residues 56–232) bait proteins, transcription activator (TA)-TSAd COOH region (residues 192–366), prey proteins, and a LexA DNA-binding domain-LCK kinase domain (residues 241– 509) fusion protein to phosphorylate the TSAd prey. After selection of transformants on LHW dropout plates, yeast were plated onto LHWUra dropout plates and growth was assessed after 48 h.
TSAd reconstitution.
T cells were purified from spleens of C57BL/6 TSAd (−/−) mice by negative selection and were transfected with murine TSAd or TSAd 3Y-F/3P-A contained in pcDNA3.1 (Invitrogen) or with pcDNA3.1 alone using an AMAXA nucleofection device. After 20 h of culture, cells were stimulated with CD3ε/CD28 mAb and lysed. Activation of LCK was determined by Western blotting of whole cell lysates.
Online supplemental material.
Fig. S1 compares LCK activation induced by CD3/28 in vivo and recombinant TSAd in vitro. Fig. S2 shows normal activation of LCK in TSAd (−/−) thymocytes and LCK association with TSAd in TSAd (+/+) thymocytes. Fig. S3 is a flow cytometric and histological analysis of lymphoid organs from 6-wk-old TSAd (+/+) and TSAd (−/−) mice. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051637/DC1.
Supplemental Material
Acknowledgments
We thank M. Moliner for assistance with graphics.
This work was supported by Public Health Service grant nos. AI044926 and AI050699 (to P.D. King).
The authors have no conflicting financial interests.
References
- 1.Mustelin, T., and K. Tasken. 2003. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem. J. 371: 15– 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spurkland, A., J.E. Brinchmann, G. Markussen, F. Pedeutour, E. Munthe, T. Lea, F. Vartdal, and H.C. Aasheim. 1998. Molecular cloning of a T cell-specific adapter protein (TSAd) containing an Src homology (SH) 2 domain and putative SH3 and phosphotyrosine binding sites. J. Biol. Chem. 273:4539–4546. [DOI] [PubMed] [Google Scholar]
- 3.Choi, Y.B., C.K. Kim, and Y. Yun. 1999. Lad, an adapter protein interacting with the SH2 domain of p56lck, is required for T cell activation. J. Immunol. 163:5242–5249. [PubMed] [Google Scholar]
- 4.Rajagopal, K., C.L. Sommers, D.C. Decker, E.O. Mitchell, U. Korthauer, A.I. Sperling, C.A. Kozak, P.E. Love, and J.A. Bluestone. 1999. RIBP, a novel Rlk/Txk- and itk-binding adaptor protein that regulates T cell activation. J. Exp. Med. 190:1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drappa, J., L.A. Kamen, E. Chan, M. Georgiev, D. Ashany, F. Marti, and P.D. King. 2003. Impaired T cell death and lupus-like autoimmunity in T cell–specific adapter protein-deficient mice. J. Exp. Med. 198:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marti, F., N.H. Post, E. Chan, and P.D. King. 2001. A transcription function for the T cell–specific adapter (TSAd) protein in T cells: critical role of the TSAd Src homology 2 domain. J. Exp. Med. 193:1425–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun, W., X. Wei, K. Kesavan, T.P. Garrington, R. Fan, J. Mei, S.M. Anderson, E.W. Gelfand, and G.L. Johnson. 2003. MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol. Cell. Biol. 23:2298–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sundvold-Gjerstad, V., S. Granum, T. Mustelin, T.C. Andersen, T. Berge, M.J. Shapiro, V.S. Shapiro, A. Spurkland, and T. Lea. 2005. The C terminus of T cell-specific adapter protein (TSAd) is necessary for TSAd-mediated inhibition of Lck activity. Eur. J. Immunol. 35:1612–1620. [DOI] [PubMed] [Google Scholar]
- 9.Sundvold, V., K.M. Torgersen, N.H. Post, F. Marti, P.D. King, J.A. Rottingen, A. Spurkland, and T. Lea. 2000. T cell-specific adapter protein inhibits T cell activation by modulating Lck activity. J. Immunol. 165:2927–2931. [DOI] [PubMed] [Google Scholar]
- 10.Palacios, E.H., and A. Weiss. 2004. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 23:7990–8000. [DOI] [PubMed] [Google Scholar]
- 11.Dong, C., R.J. Davis, and R.A. Flavell. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55–72. [DOI] [PubMed] [Google Scholar]
- 12.Hogan, P.G., L. Chen, J. Nardone, and A. Rao. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17:2205– 2232. [DOI] [PubMed] [Google Scholar]
- 13.Roose, J.P., M. Mollenauer, V.A. Gupta, J. Stone, and A. Weiss. 2005. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol. Cell. Biol. 25:4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katan, M. 1998. Families of phosphoinositide-specific phospholipase C: structure and function. Biochim. Biophys. Acta. 1436:5–17. [DOI] [PubMed] [Google Scholar]
- 15.Finco, T.S., T. Kadlecek, W. Zhang, L.E. Samelson, and A. Weiss. 1998. LAT is required for TCR-mediated activation of PLCγ1 and the Ras pathway. Immunity. 9:617–626. [DOI] [PubMed] [Google Scholar]
- 16.Zhang, W., R.P. Trible, M. Zhu, S.K. Liu, C.J. McGlade, and L.E. Samelson. 2000. Association of Grb2, Gads, and phospholipase C-γ 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J. Biol. Chem. 275:23355–23361. [DOI] [PubMed] [Google Scholar]
- 17.Marti, F., C.W. Xu, A. Selvakumar, R. Brent, B. Dupont, and P.D. King. 1998. LCK-phosphorylated human killer cell-inhibitory receptors recruit and activate phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 95:11810–11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hermiston, M.L., Z. Xu, and A. Weiss. 2003. CD45: a critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 21:107–137. [DOI] [PubMed] [Google Scholar]
- 19.Berg, L.J., L.D. Finkelstein, J.A. Lucas, and P.L. Schwartzberg. 2005. Tec family kinases in T lymphocyte development and function. Annu. Rev. Immunol. 23:549–600. [DOI] [PubMed] [Google Scholar]
- 20.Guo, Z., G. Clydesdale, J. Cheng, K. Kim, L. Gan, D.J. McConkey, S.E. Ullrich, Y. Zhuang, and B. Su. 2002. Disruption of Mekk2 in mice reveals an unexpected role for MEKK2 in modulating T-cell receptor signal transduction. Mol. Cell. Biol. 22:5761–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lapinski, P.E. M.J.N., Marti F., and King P.D. 2006. The T cell-specific adapter protein functions as regulator of peripheral but not central immunological tolerance. Adv. Exp. Med. Biol. In press. [DOI] [PubMed]
- 22.Greene, T.A., P. Powell, C. Nzerem, M.J. Shapiro, and V.S. Shapiro. 2003. Cloning and characterization of ALX, an adaptor downstream of CD28. J. Biol. Chem. 278:45128–45134. [DOI] [PubMed] [Google Scholar]
- 23.Gorska, M.M., S.J. Stafford, O. Cen, S. Sur, and R. Alam. 2004. Unc119, a novel activator of Lck/Fyn, is essential for T cell activation. J. Exp. Med. 199:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King, P.D., A. Sadra, J.M. Teng, L. Xiao-Rong, A. Han, A. Selvakumar, A. August, and B. Dupont. 1997. Analysis of CD28 cytoplasmic tail tyrosine residues as regulators and substrates for the protein tyrosine kinases, EMT and LCK. J. Immunol. 158:580–590. [PubMed] [Google Scholar]
- 25.Pfeifhofer, C., K. Kofler, T. Gruber, N.G. Tabrizi, C. Lutz, K. Maly, M. Leitges, and G. Baier. 2003. Protein kinase C θ affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J. Exp. Med. 197:1525–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}