

Abstract
The live attenuated yellow fever vaccine 17D (YF-17D) is one of the most effective vaccines available, with a 65-yr history of use in >400 million people globally. Despite this efficacy, there is presently no information about the immunological mechanisms by which YF-17D acts. Here, we present data that suggest that YF-17D activates multiple Toll-like receptors (TLRs) on dendritic cells (DCs) to elicit a broad spectrum of innate and adaptive immune responses. Specifically, YF-17D activates multiple DC subsets via TLRs 2, 7, 8, and 9 to elicit the proinflammatory cytokines interleukin (IL)-12p40, IL-6, and interferon-α. Interestingly, the resulting adaptive immune responses are characterized by a mixed T helper cell (Th)1/Th2 cytokine profile and antigen-specific CD8+ T cells. Furthermore, distinct TLRs appear to differentially control the Th1/Th2 balance; thus, whilst MyD88-deficient mice show a profound impairment of Th1 cytokines, TLR2-deficient mice show greatly enhanced Th1 and Tc1 responses to YF-17D. Together, these data enhance our understanding of the molecular mechanism of action of YF-17D, and highlight the potential of vaccination strategies that use combinations of different TLR ligands to stimulate polyvalent immune responses.
Yellow fever vaccine 17D (YF-17D) is considered to be one of the most effective vaccines available. During the 65 yr since its development, the vaccine has been administered to >400 million people world wide with minimal incident of severe side effects. YF-17D was developed by Max Theiler and associates, who in the 1930's experimentally attenuated the Asibi strain of yellow fever virus by >200 serial passages through monkeys, mouse embryonic tissue, and chicken embryonic tissue cultures (1, 2). Protection is achieved in >98% of its recipients for at least 10 yr, and in many individuals, neutralizing antibody titers have been found as many as 35 yr after a single vaccination (3, 4). Though neutralizing antibody titers are thought to be important in conferring protection against infections with yellow fever, YF-17D also has been demonstrated as a potent inducer of cytotoxic T cell responses (5).
The efficacy of this vaccine has brought it to the attention of several researchers who have used YF-17D as a viral vector to develop novel vaccines against other infectious diseases. For example, clinical trials are currently under way with chimeric viruses generated by inserting genes from other flaviviruses such as Japanese encephalitis, dengue, and West Nile virus into YF-17D. Recombinant forms of YF-17D even have been demonstrated as effective vaccines against malaria and cancer (6–15).
Despite the vaccine's efficacy in controlling yellow fever over the past several decades and its promise as a vaccine vector, the cellular and molecular mechanisms by which it elicits such broad based immunity are unclear. In this regard, recent evidence suggests that the innate immune system is a critical determinant of the strength and quality of the adaptive immune response (16–20). Within the innate immune system, DCs occupy a preeminent position, as they play critical roles in sensing microbial stimuli and in initiating and modulating adaptive immune responses. Moreover, DCs are equipped to sense microbial signatures through pathogen recognition receptors (PRRs), such as the Toll-like receptors (TLRs) (17, 21, 22). TLRs constitute an evolutionarily conserved family of receptors involved in microbe recognition, of which 11 have been described in mammals. Different TLRs appear to recognize distinct microbial components. For example, lipopolysaccharides from Escherichia coli are recognized by TLR4 (23), whereas certain lipopeptides, LPS from leptospira (24, 25) and Porphyromonas gingivalis (26, 27), and the yeast cell wall zymosan (28) are recognized by TLR2. Furthermore, unmethylated DNA from bacteria and viruses are recognized by TLR9 (29–32), single-stranded RNA is recognized by TLR7/8 (33–35), and double-stranded RNA is recognized by TLR3 (36–38).
Upon recognition of a pathogen via TLRs, the immature DCs at the site of pathogen entry undergo a maturation process during which they exit the site and migrate to the T cell–rich areas of the neighboring lymph nodes, where the mature DCs present their acquired antigens and stimulate antigen-specific T cells, thus initiating adaptive immunity and immune memory.
Interestingly, emerging evidence suggests that the quality of the adaptive immune response is partly determined by the particular TLR triggered, as well as by the particular subset of DC activated (17, 21, 22). Thus, signaling through TLR9 or 7/8 is known to induce robust IL-12(p70) from myeloid DCs (TLR 7/8) (30, 39) and IFN-α production from plasmacytoid DCs (40, 41), and cross-presentation of exogenous antigens and stimulation of cytotoxic T cells (42, 43). In contrast, stimulation of TLR2 by several ligands has been shown to stimulate IL-10 and induce Th2 or T regulatory responses (44–49).
Given the pivotal role of TLRs and DCs in initiating and tuning the adaptive immune response, there is at present much interest in exploiting these in the development of novel vaccines. However, the converse question of whether some of our best, empirically derived vaccines actually work by stimulating TLRs is largely unexplored. In this context, we sought to determine whether YF-17D triggered the activation of innate immunity and DCs via TLRs and, if so, what effect this had on the quality of the innate and adaptive immune response in the present study. Our data suggest that YF-17D activates multiple subsets of DCs by signaling through multiple TLRs, including TLR2, 7, 8, and 9, resulting in diverse types of adaptive immune responses.
RESULTS
YF-17D activates multiple subsets of human and mouse DCs in vitro
To determine whether YF-17D activates DCs, we first cultured human monocyte-derived DCs [mDCs] with YF-17D (Fig. 1). UV irradiated or heated YF-17D was used as a negative control. These mDCs had a dose-dependent response to YF-17D with increasing multiplicities of infection (MOIs) of virus leading to greater induction of proinflammatory cytokines, including IL-6, TNF-α, MCP-1, and IP-10 (Fig. 1 A). YF-17D also induced both IL-12p40 and IL-12p70 from mDCs, although the amounts induced were lower than with LPS, and induction of IL-12p70 was only apparent when an additional CD40 ligand signal was provided (in the form of a fibroblast cell line, expressing the CD40 ligand signal), consistent with numerous previous studies (e.g., reference 45 and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051720/DC1). Stimulation of DCs with CD40 ligand–expressing fibroblasts alone resulted in little or no IL-12p70. Furthermore, YF-17D was also an efficient inducer of the costimulatory molecules CD80 and CD86 (Fig. 1 B). Both UV irradiation and heat treatment for 1 h at 56°C of YF-17D impaired its ability to stimulate cytokines or induce costimulatory molecules on mDCs, although the effects of UV irradiation varied with the cytokine being examined. Thus, there was only a modest effect of UV irradiation on production of IL-10 (Fig. 1) and IL-12p40 (Fig. 2). Therefore, it is possible that induction of IL-10 (Fig. 1) and IL-12p40 (Fig. 2) by YF-17D is not critically dependent on viral replication, and that nonreplicating virions, or non-RNA components such as viral proteins, may mediate this.
Figure 1.

YF-17D activates human monocyte–derived DCs and plasmacytoid DCs. Human mDCs were cultured with LPS; YF-17D at an MOI of 1, 0.1, or 0.01; or they were YF-17D UV or heat inactivated. Cytokines (A) and costimulatory molecule expression (B) were measured after 48 h. (A) The mean fluorescent intensities, are indicated at the top left of each histogram. (B, top) In the histograms, the numbers in parentheses represent the mean fluorescent intensities of the DCs cultured in media alone. (C) PDCs were isolated from human blood using BDCA-2 microbeads and culture for 48 h before measuring IFN-α. Error bars in A and C represent standard deviation. These data are representative of at least four independent experiments.
Figure 2.

YF-17D activates DCs through multiple TLRs and multiple T LR adaptor proteins. CD11c+ DCs were isolated from the spleens of Flt-3L–injected knockout and age-matched, sex-matched, wild-type mice and cultured for 48 h in the presence of TLR agonists and YF. Cell culture supernatants were assayed for IL-12p40 and IL-6. (A) MyD88−/− mice versus control mice; (B) TIRAP−/− mice versus control mice; (C) TLR4−/− mice versus control mice; (D) TLR2−/− mice versus control mice; (E) TLR9−/− mice versus control mice; and (F) TLR7−/− mice versus control mice. Error bars represent standard deviation. These data are representative of at least four independent experiments. *, indicates statistically significant difference (P < 0.005 by paired Student's t test) between wild type (black) and knockout (gray) DCs.
To evaluate whether YF-17D infects DCs, human mDCs were exposed to YF-17D and the intracellular expression of nonstructural proteins NS4A and NS4B and evaluated 48 h after infection. As shown by intracellular FACS staining in Fig. S2 (available at http://www.jem.org/cgi/content/full/jem.20051720/DC1) and consistent with similar, recently published observations (50), a significant proportion of DCs contain intracellular NS4A and NS4B. Interestingly, enhanced induction of the DC activation marker CD86, relative to mock-infected DCs, appears to occur in all DCs (Fig. S2). Therefore, although productive viral infection induces robust activation of DCs, DCs that are not productively infected also appear to be activated, perhaps as a result of viral entry or surface binding to a PRR or bystander activation.
Plasmacytoid DCs (pDCs) play a key role in many viral infections by producing IFNα, which in turn activates other immune cells and induces an antiviral program in cells. To determine if YF-17D can activate pDCs, pDCs were isolated from human blood and cultured with the virus. Cell culture supernatants were assayed for IFNα. pDCs produced high levels of IFNα in response to an MOI of 1 of the virus and this cytokine production was greatly inhibited by heating the virus to 56°C for 1 h (Fig. 1 C). UV irradiation did not have as much of an effect on reducing the activation of the pDCs.
YF-17D activates DCs through multiple TLRs (TLR2, 7, 8, and 9) and multiple TLR adaptors (MyD88 and TIRAP)
Given that YF-17D induced potent activation of DCs, we sought to determine the innate immune receptors through which YF-17D mediated its effects. For these studies, we used mouse DCs based on the inherent advantages in using animals deficient in specific genes that might provide insights into mechanism. Splenic CD11chigh DCs from Flt3 ligand–treated mice were cultured in vitro with YF-17D, and 6 h later, the induction costimulatory molecules were evaluated by FACS. As with human DCs, YF-17D induced CD86, CD80, and CD40 on splenic DCs (unpublished data). As previous studies suggest that several viruses can activate DCs via TLRs, we assessed the capacity of YF-17D to activate DCs from various TLR-deficient mice strains.
MyD88 is an adaptor protein that is associated with all TLRs, except TLR3 (36, 51, 52) and that mediates the signal transduction from TLRs. Thus, as a first measure of whether any TLRs are involved in the response to YF-17D, CD11c+ DCs from MyD88−/− mice or wild-type mice were cultured in the presence of live or inactivated virus. As shown in Fig. 2 A, there was a profound impairment in the induction of IL-12p40 and IL-6 by YF-17D in MyD88−/− DCs, relative to control DCs from wild-type mice. Furthermore, inactivation of the virus by UV irradiation or heating to 56°C for 1 h largely abrogates its ability to induce the cytokine response from the wild-type cells. In the case of murine DCs, we could not detect significant numbers of infected cells, either by FACS analysis of the intracellular expression of nonstructural proteins NS4A and NS4B, or by a PCR-based assay (unpublished data). The reasons for the differences in infection between human (Fig. S2) and murine DCs are not clear. Despite the apparent lack of productive infection of murine DCs, clearly YF-17D does induce robust cytokine production from such cells. The likely explanation for this might be viral entry or surface binding to a PRR.
In addition to MyD88, at least three other adaptor proteins (TIRAP, TRIF, and TRAM) have been described previously (51–57). TIRAP appears only to be associated with TLR2 and 4 (52, 55), whereas TRIF is only associated with TLR3 and 4 (53–56). We cultured splenic CD11chigh DCs from TIRAP−/− mice with YF-17D. As shown in Fig. 2 B, there was a very significant diminution of both IL-12p40 and IL-6 in response to YF-17D in TIRAP−/−, relative to wild-type controls. Thus, YF-17D activates DCs through at least two different TLR adaptor proteins, MyD88 and TIRAP.
Next, we determined precisely which TLRs were activated by YF-17D. DCs from wild-type mice, TLR2−/−, TLR4−/−, TLR7−/−, and TLR9−/− mice were cultured in vitro with YF-17D (Fig. 2, C–F). No difference in IL-12p40 and IL-6 production was observed between the TLR4−/− and wild-type cultures. This indicates that TLR4 does not play a role in the response to YF-17D. In contrast, TLR2 does play a role, as TLR2−/− DCs had a greatly impaired production of IL-12p40 and IL-6. This is consistent with recent reports that certain viruses such as measles (58), HCV (59, 60), and LCMV (61) also signal through TLR2. Furthermore, TLR7 also appears to be important as DCs from TLR7−/− mice display impaired production of IL-12p40 (Fig. 2 F), consistent with the fact that YF-17D is a single-stranded RNA virus and previous observations that TLR7 is a receptor for single-stranded RNA (33–35, 40). However, we could not detect a consistent effect on IL-6 production in TLR 7−/− mice (unpublished data). The reason for this is at present unclear.
Until recently, TLR9 has been shown to respond only to particular unmethylated CpG DNA motifs found in certain bacteria and viruses (29). Given that YF-17D is an RNA virus, we expected to exclude a role for TLR9 in the vaccine response. Surprisingly, TLR9−/− DCs had a clear diminution in both IL-12p40 and IL-6, although the induction of these cytokines by YF-17D in this strain and its littermate was weaker than with the other strains, likely due their mixed C57BL/6 × 129 genetic backgrounds. A possible explanation for TLR9 signaling by YF-17D was that there was contaminating DNA from the SW480 cells used to grow the virus. To address this issue, YF-17D or control CpG DNA were pretreated with DNase buffer and with preincubations for 30 min in ice, or at 37°C, or at 37°C in the presence of DNase. pDCs were cultured with these various stimuli and supernatants were assayed for IFN-α (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20051720/DC1). Incubation at 37°C had no effect on YF-17D and CpG DNA relative to the stimuli on ice (unpublished data). DNase treatment substantially reduced cytokine output from cultures treated with DNase digested CpG DNA, but had no effect on YF-17D, suggesting that the contaminant cellular DNA likely did not play a role in the effects of YF-17D on DCs. This observation is consistent with the recent finding that Newcastle Disease virus, another single-stranded RNA virus, also activates DCs through both TLR7 and TLR9 (62). In addition, as demonstrated recently, certain metabolites such as the hemozoin of malaria parasites may signal through TLR9 (63). Thus, TLR9 may sense not only unmethylated, CpG-rich DNA, but also other microbial or viral components.
In addition to TLR7, TLR8 has also been shown to respond to certain single-stranded RNA viruses such as HIV, human parechovirus 1, and Cocksackie virus B (34, 64, 65). To determine whether YF-17D also signals through TLR8, HEK293 cells stably transfected with human TLR8 and an NFκB-luciferase reporter were cultured with YF-17D or the synthetic TLR7/8 agonist 3M-003. Cell lysates were measured for luciferase activity as a measure of NFκB activation downstream of TLR8 (Fig. 3). After 1 d of culture, an 11-fold increase in luciferase activity was measured from the cells treated with 3M-003, but there was only a modest increase in activity from YF. However, on the second day of culture (after the virus had a chance to begin replication), YF produced an 18-fold increase in luciferase activity. The very slight decrease in the response induced by 3M-003 is likely a reflection of the earlier effect of the compound through the NFκB promoter and decay of the NFκB response, and not decay of the compound itself. The comparatively delayed response by the virus is more likely to be explained by the kinetics of cellular infection and subsequent synthesis of viral components required to stimulate the TLR8–NFκB pathway. Thus, YF-17D appears to activate DCs via multiple TLRs.
Figure 3.

YF-17D signals through TLR 8. Stably transfected HEK293 cells expressing human TLR8 and an NFκB-inducible luciferase reporter were stimulated with the TLR7/8 agonist 3M-003 or an MOI of 0.1 of YF-17D. After 1 and 2 d, the cells were lysed an analyzed for luciferase activity.
YF-17D induces DC activation and a mixed Th1/Th2 T cell response in vivo
Given that YF-17D activates multiple DC subsets via distinct TLRs, we sought to determine the quality of the adaptive immune responses stimulated by this vaccine. We first wanted to ascertain whether DCs were activated at all in vivo. YF-17D or control TLR ligands were injected subcutaneously and, 1 d later, the draining lymph nodes were isolated and the phenotype and activation status of DCs was evaluated by flow cytometry. The costimulatory molecules CD86 and CD40 in addition to the MHC class I molecule I-Ab were all clearly up-regulated when compared with the PBS injection at equivalent levels to that induced by LPS (Fig. 4), although we were unable to detect any substantial up-regulation of CD80.
Figure 4.

YF-17D induces DC activation in vivo. Mice were injected with YF-17D, LPS, Pam3Cys, or PBS subcutaneously and the draining lymph nodes were collected 24 h later. Costimulatory molecule expression on CD11c+ DCs was analyzed by flow cytometry.
It has been shown previously that most TLR ligands induce robust Th1 responses (66, 67), but several studies suggest that signaling via TLR 2 results in very weak induction of IL-12p70 in DCs and a bias toward the Th2 or T regulatory pathway (44–49, 68–71). Because YF-17D signaled through TLR2, 7, 8 and 9, it was important to characterize the quality of the T cell response after the DC activation in vivo. For this purpose, we obtained YF-Ova8, which expresses a cytotoxic T lymphocyte epitope derived from chicken ovalbumin (SIINFEKL) (15). These recombinant viruses replicated comparably to the YF-17D vaccine strain in cell culture (unpublished data) and stably expressed the SIINFEKL peptide, and infected cells presented the antigen in the context of MHC class I. YF-Ova8 was injected subcutaneously into Thy1.1+ mice that had received 2.5 × 106 OT-I transgenic T cells (Thy 1.2+) 1 d earlier; 5 d after vaccination, the cells in the draining lymph nodes were either analyzed directly by flow cytometry or restimulated in vitro with SIINFEKL for T cell proliferation assays and cytokine analysis of cell culture supernatants. YF-Ova8 induced clonal expansion of antigen-specific CD8+ T cells, as indicated by the increased frequency of CD8+ Thy 1.2+ cells (Fig. 5 A). 5 d after vaccination, the draining lymph nodes were isolated, and single cell suspensions were prepared and cultured in vitro with SIINFEKL peptide or in media. 72 h later, the induction of Th1 and Th2 cytokines, IFN-γ (Fig. 5 B) and IL-4, IL-5, IL-13, and IL-10 (Fig. 5 C), in culture was evaluated by ELISA and by intracellular flow cytometry. As indicated in Fig. 5 B, YF-Ova8 induced the production of IFN-γ by CD8+ T cells. This cytokine production was largely specific to the SIINFEKL peptide as indicated by much higher frequency of CD8+IFN-γ+ cells in response to YF-Ova8 relative to immunization with nonrecombinant YF-17D (Fig. 5 B). However, there were still nanograms/ml amounts produced (Fig. 5 B) by cells incubated with media alone (presumably as a result of viral carry over) and the ensuing T cell responses to endogenous YF-17D proteins. In contrast with IFN-γ, the production of the Th2 cytokines IL-4, IL-5, IL-13, and IL-10 did not appear to be specific to OVA, as restimulation with SIINFEKL did not increase their production. This is likely a result of the fact that the OT-1 CD8+ transgenic T cells may have an intrinsic bias to secrete IFN-γ and their relatively high frequency in this adoptive transfer system would result in much higher levels of IFN-γ (but not IL-4, IL-5, or IL-13) being secreted. In contrast, the endogenous YF-17D proteins may stimulate specific T cells that produce a mixed Th1/Th2 profile; thus, the bulk of the Th2 cytokines detected in this system may well be produced by such T cells and, thus, be produced independently of SIINFEKL stimulation (Fig. 5 C).
Figure 5.

YF-17D induces efficient induction of antigen-specific CD8+ T cells, and a mixed Th1/Th2 cytokine profile in vivo. Mice were injected with 2.5 × 106 OT-I TCR transgenic T cells and allowed to rest overnight. YF-Ova8 vaccinations were administered subcutaneously and the draining lymph nodes were collected 5 d later. A portion of cells was stained directly ex vivo for CD8 and Thy 1.2 to evaluate clonal expansion of SIINFEKL-specific CD8+ T cells by FACS (A). The remaining cells were placed into culture and restimulated with SIINFEKL or cultured in media alone. After 3 d, the induction of IFN-γ, IL-4, IL-5, and IL-13 cytokines was measured by intracellular FACS staining (B, IFN-γ) or by ELISA (B and C). Error bars represent standard deviation. These data are representative of three independent experiments.
Distinct TLRs differentially regulate the Th1/Th2 bias in response to YF-17D
We next sought to determine the contribution that individual TLRs made to the quality of the T cell response induced by YF-17D. Based on previous findings that MyD88−/− mice show a selective defect in Th1 responses (66), one possibility was that YF-17D signaling through TLRs only resulted in a Th1 response and that the Th2 responses were mediated by some other innate immune receptors. Alternatively, signaling via TLR2 might mediate Th2 or T regulatory responses (44–49, 68–71). To distinguish between these possibilities, wild-type or MyD88−/− mice were injected subcutaneously with either YF-17D or with supernatant from mock-infected SW-480 cells. 5 or 10 d later, the draining lymph nodes were removed and single cell suspensions from these lymph nodes were cultured in vitro either in media alone or with a mixture of CD4+ T cell epitopes and CD8+ T cell epitopes that had been previously identified (72). The CD4+ T cell epitope is an I-Ab–restricted epitope from the viral envelope protein (residues 231–247 from the envelope protein, LVEFEPPHAATIRVL), whereas the CD8+ T cell epitopes are comprised of one subdominant H-2Db–restricted epitope (residues 1–15 from the E1 envelope protein, AHCIGITDRDFIEGV), and another H-2Kb–restricted dominant epitope mapped to the NS3 protein (residues 261–275 from the NS3 protein, VIDAMCHATLTYRML) (72). After 3 d, the supernatants were assayed for cytokines (IFN-γ, IL-4, IL-5) and intracellular staining of CD8+, CD4+, and IFN-γ on the cells was performed. As shown in Fig. 6 A, in wild-type mice, YF-17D induced high levels of IFN-γ. This induction occurred even when the cells were cultured in media alone, likely reflecting carry over of viral components that served as antigen for in vitro restimulation. In MyD88−/− mice, however, there was a profound reduction in IFN-γ levels (Fig. 6 A). These trends were reflected in the intracellular staining assays for IFN-γ+ cells shown in Fig. 6 B. Similar trends were also observed with CD4+IFN-γ+ cells (Fig. 6 C). Thus, as described previously (66), MyD88 appears to be critical for the induction of Th1 and Tc1 responses. Also, in agreement with previous findings, MyD88−/− mice displayed enhanced induction of IL-4 and IL-5 (unpublished data).
Figure 6.

Induction of Th1 and Tc1 responses by YF-17D is dependent on MyD88. Wild-type or MyD88−/− mice were vaccinated subcutaneously with YF-17D. Control mice were injected with supernatants from mock transfected SW-480 cells. 10 d later, the draining lymph nodes were harvested and single cell suspensions were cultured in vitro in media alone or with a combination of CD8+ T and CD4+ T cell epitopes from YF-17D as described in Materials and methods. 3 d later, the induction of IFN-γ was measured by ELISA (A) and by intracellular cytokine staining (B and C). These data are representative of three independent experiments with three mice per group in each experiment.
Next, we evaluated immune responses in TLR2−/− mice. The experiments were performed exactly as described in the previous paragraph with MyD88−/− mice. In brief, we immunized wild-type or TLR2−/− mice subcutaneously with either YF-17D or with supernatant from mock-infected SW-480 cells. 5 or 10 d later, the draining lymph nodes were removed, and single cell suspensions from these lymph nodes were cultured in vitro, either in media alone or with a mixture of CD4+ T cell epitopes and CD8+ T cell epitopes, which had been previously identified as described in the previous paragraph (72). After 3 d, the supernatants were assayed for cytokines (IFN-γ, IL-4, IL-5) and intracellular staining of CD8+, CD4+, and IFN-γ on the cells was performed. As shown in Fig. 7 A, the frequency of CD8+IFN-γ+ T cells in wild-type mice at day 10 is 1.05%. However, at this time point, the TLR2−/− mice contained 3.63%. A similar trend was observed at day 5. Furthermore, in the case of CD4+IFN-γ+ T cells (Fig. 7 B), at day 5 after immunization, the response in TLR2−/− mice appeared to be markedly higher than in wild-type mice. However, by day 10, the CD4+IFN-γ+ response appeared to have subsided, and no differences were apparent. This suggests that signaling through TLR 2 may exert an inhibitory effect on the induction of CD4+IFN-γ+ or CD8+ IFN-γ+ cells in response to YF-17D. These results are consistent with the enhanced secretion of IFN-γ in TLR2 knockout splenocytes, relative to wild-type splenocytes as evaluated by ELISA (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20051720/DC1). These results are consistent with previous findings that TLR2 signaling induces Th2 or T regulatory responses (26, 44–49, 68–71). Interestingly, in the case of the Th2 cytokines IL-4 and IL-5, there appeared to be a modest decrease in TLR 2−/− mice (unpublished data). Collectively, these data suggest that TLR2 might regulate the induction of Th1/Th2 balance in response to YF-17D.
Figure 7.

TLR2 exerts an inhibitory effect on Th1 and Tc1 responses against YF-17D. Wild-type or TLR2−/− mice were vaccinated subcutaneously with YF-17D. Control mice were injected with supernatants from mock transfected SW-480 cells. 5 or 10 d later, the draining lymph nodes were harvested and single cell suspensions cultured in vitro in media alone, or with a combination of CD8+ T and CD4+ T cell epitopes from YF-17D. 3 d later, the induction of IFN-γ was measured by intracellular cytokine staining (A and B). These data are representative of four independent experiments with three mice per group in each experiment.
DISCUSSION
The data presented here, together with that reported recently by Barba-Spaeth et al. (50), offer some insights into the mechanism of action of YF-17D, one of the most effective vaccines available. In particular, our data suggest that YF-17D activates DCs via multiple TLRs, including TLR2, 7, 8 and 9, and that signaling via distinct TLRs exerts differential effects on the quality of the antigen-specific T cell response induced by YF-17D. Whether other TLRs, particularly TLR3, which is a receptor for double-stranded RNA (36), are also involved in YF-17D recognition is at present unclear.
The biological significance of activation of several TLRs is highlighted by two intriguing aspects of the data. First, the absence of a single TLR appears to result in a significant impairment of IL-12p40 and IL-6 (Fig. 2). This is surprising because it might have been expected that other TLRs would compensate for this deficiency. One explanation for this observation is that the TLRs signal in a synergistic manner, and that YF-17D contains suboptimal amounts of individual TLR ligands, which by themselves stimulate suboptimal responses, but which can signal in concert, to produce a synergistic effect. This hypothesis is consistent with two recent reports that suggest that selected TLR combinations can indeed synergistically trigger a Th1 polarizing program in human DCs (73, 74). In the case of YF-17D, whether such synergy might occur by triggering multiple TLRs on a single DC subset, by triggering multiple TLRs on distinct DC subsets, or by both mechanisms is yet to be determined. A second intriguing aspect to the data is that signaling via distinct TLRs appear to differentially bias the Th1/Th2 balance. Thus, eliminating signaling via MyD88 impairs Th1 responses, whereas eliminating TLR2 signaling enhances Th1 responses (Figs. 5 and 6). This is consistent with emerging data that distinct TLRs may differentially affect the Th1/Th2 balance (44–49, 68–71). Therefore, vaccines that target multiple TLRs might facilitate both immune synergy as well as polyvalent immune responses to generate broad-based protective immunity. In this context, a recent study suggests that conjugating HIV Gag protein to a TLR7/8 agonist results in activation of both myeloid and pDCs, and enhances the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates (75).
The question of which types of immune responses offer protection against challenge with the pathogenic strain of yellow fever virus is of considerable interest. This was not addressed in the present study, primarily because yellow fever appears to be rather host (primate) restricted and mice are not very susceptible. Even immunosuppressed mice appear to be relatively poor hosts. In fact, several investigators appear to have used nonhuman primates for their experiments with yellow fever and chimeric yellow fever viruses (6–15). However, it is known that YF-17D does kill mice after intracerebral challenge (9), but the relevance of such a model to yellow fever infection in humans is questionable. These issues notwithstanding, one might speculate on which types of responses confer protection against yellow fever. Though neutralizing antibody titers are thought to be important, a role for cytotoxic T cell responses has also been suggested (5). However, what influence the Th1/Th2 balance might have on protection is unknown. Because Th1 versus Th2 cytokines are thought to favor the development of cytotoxic T cells versus antibodies, respectively, it is conceivable that both Th1 and Th2 responses are important for protection. Thus, intracerebral infection of MyD88 knockout mice or TLR2 knockout mice might have both resulted in impaired survival of the hosts as a result of perturbed Th1/Th2 balances.
Finally, it is also important to address the question of precisely which viral components act through which innate immune receptors. In this context, it is important to note that live viral vaccines contain not only viral proteins and nucleic acids, but also cellular contaminants derived from the cell lines in which the vaccines were grown. Such cellular contaminants (Janeway's “dirty little secrets” [reference 76] or Matzinger's “danger signals” [reference 77]), may well play important roles in the induction and modulation of vaccine induced immune responses. Thus, the chicken embryos and cell lines in which YF-17D are grown are potential sources of these dirty little secrets or danger signals. In this context, avian retroviral proteins, ovalbumin, and chicken DNA are all found in YF-17D preparations for human use (78). It is thus likely that these factors and other nonviral components of the YF-17D vaccine, such as heat shock proteins and uric acid, contribute to the induction of an immune response. Both heat shock proteins and uric acid are released from stressed cells and are potent activators of DCs and monocytes (for review see reference 79). Thus, future vaccinologists should not only concern themselves with using the right combination of adjuvants but also having vaccine preparations that are “clean,” but which recapitulate the immunogenicity of the “dirty” vaccines. Furthermore, whatever the source of the molecules that stimulate a given TLR or PRR, clearly further work is required to determine the specific combinations of PRRs (the correct “code” of innate immune buttons) that promote a particular facet of the immune response, such Th1/Th2 balance, neutralizing antibody responses, long-lived antibody-producing cells that home to mucosal tissues and continuously secrete high-affinity neutralizing antibody, and long-lived memory T cells.
In conclusion, the present data enhance our understanding of the molecular mechanisms of action of YF-17D and highlight the potential of vaccination strategies that use combinations of different TLR ligands to stimulate polyvalent and synergistic immune responses. The present approach of determining the TLRs that YF-17D signals through could be used to deconstruct the immunological basis for the efficacy of other successful vaccines. It is likely that many of these vaccines, which consist of live attenuated viruses or bacteria, may well signal through multiple TLRs, as well as other non-TLRs such as RIG-1 (62). Conversely, it conceivable that “poor” vaccines, which require repetitive boosts to generate robust protective responses, do not trigger TLRs at all or even inhibit TLR signaling. A common method used in the construction of many developing vaccines is to use a single adjuvant, such as CpG DNA, in combination with an antigen. However, such vaccines maybe limited in the quantity and quality of the immune response generated and, therefore, may not provide the most effective prevention or treatment possible. Thus, future vaccine strategies aimed at incorporating multiple TLR ligands and antigens into delivery systems such as nanoparticles, which can target specific DC subsets in vivo, is likely to be of great benefit in conferring the degree of immunogenicity and protection mediated by our best vaccines such as the yellow fever vaccine YF-17D.
MATERIALS AND METHODS
Mice.
Mutant mice (MyD88−/−, TIRAP−/−, TLR2−/−, TLR4−/−, TLR7−/−) were provided by S. Akira (Osaka University, Osaka, Japan) and were either in the C57BL/6 background or in a mixed C57BL/6 × 129 background (backcrossed at least five times to C57BL/6). C3H/HeJ and C3H/HeN were purchased from The Jackson Laboratory and Taconic Farms, respectively. Spleens from TLR 9−/− mice on the mixed C57BL/6 × 129 background were provided by R. Medzhitov (Yale University School of Medicine, New Haven, CT). In each case, the appropriate littermate control was used. All mutant mice and controls were sex- and age-matched and between 6 and 16 wk old. For adoptive transfer studies, OT-1 TCR transgenic mice and B6.PL mice were purchased from The Jackson Laboratory and bred at the Yerkes Vivarium. B6.PL recipients were injected i.v. with 2.5 × 106 OT-1 TCR transgenic T cells. All animal studies were approved by the Institutional Animal Care and Use Committee of Emory University.
Virus.
YF-17D subpassaged from YF-VAX (Aventis Pasteur) in SW-480 cells was a gift from R. Ahmed (Emory University, Atlanta, GA), and YF-Ova8 (encoding the SIINFEKL peptide) and YF-FL were gifts from R. Andino (University of California, San Francisco, CA) (15). Viruses were passaged in SW-480 cells in DMEM + 10% FBS + penicillin/streptomycin. Cell culture supernatants were collected ∼6 d after infection when ∼20% cytopathic effect was noted. Supernatants were clarified by centrifugation twice at 750 g for a total of 20 min. This crude virus stock was purified further, as follows: sucrose purified virus was generated by centrifuging the virus through a PBS + 35% sucrose + 10 μM Tris cushion at 100,000 g for 2 h. The viral pellet was resuspended in PBS and frozen at −80°C in aliquots. Viral titers were calculated by plaque assay using Vero cells. Virus was inactivated by either incubating at 56°C for 1 h or UV irradiated with 120 mJ/cm2 for 5 min.
Stimuli.
Purified E. coli was a gift from T. van Dyke (Boston University, Boston, MA). CpG A (ODN 2336) and CpG B (ODN 2006) DNA was purchased from Coley Pharmaceutical. Pam-3-cys was obtained from EMC Microcollections. 3M-003, a proprietary TLR7/8 agonist was a gift from K. Gorden (3M Corporation, St. Paul, MN).
Purification of mouse splenic CD11c+ DCs.
CD11c+ DCs were purified from the spleens of Flt-3L–injected mice. Mice were injected with 20 μg Flt-3L per day for nine consecutive days. On the 10th day, spleens were digested with Collagenase, Type IV (1 mg/ml; Worthington Biochemical) in complete DMEM + 2% FBS for 30 min at 37°C. Cells were washed 2× with PBS + EDTA + FBS and cell cultures of >95% CD11c+ DC purity were established after enrichment of splenocytes with CD11c+ microbeads (Miltenyi Biotec).
Human DCs.
Human mDCs were generated from apheresis samples of healthy donors. PBMCs were plated at 10 × 106 cells/well in six-well plates. After 2 h, nonadherent cells were removed and the adherent monocytes were cultured in RPMI 1640 + 10% FBS + 50 μg/ml GM-CSF + 10 μg/ml IL-4 (PeproTech) for 6 d. After this time, the nonadherent DCs were collected, washed, and used for experiments. Human pDCs were enriched from PBMCs using BDCA-2 microbeads (Miltenyi Biotec).
In vitro cultures.
The mDCs, pDCs, and mouse CD11c+ DCs were plated at 106 cells/ml in 48-well culture dishes and incubated with the stimuli under humid conditions at 5% CO2 and 37°C. The human DCs were cultured in RPMI 1640 + 10% FBS + pen/strep. Human IL-3 (PeproTech) was added for the pDCs. Mouse DCs were cultured in complete DMEM + 10% FBS. Costimulatory molecule expression was analyzed by flow cytometry after 48 h for human DCs. Alternatively, the supernatants were analyzed after 48 h for cytokines.
Staining for YF-17D in DCs.
Cells were washed in staining medium and stained for surface CD86 for 30 min, 0°C. Subsequently cells were washed twice then fixed and permeabilized using a BD Biosciences cytofix/perm reagent kit following the manufacturer's instructions. A rabbit polyclonal antisera that recognizes the nonstructural proteins NS4A and NS4B (C. Rice, The Rockefeller University, New York, NY) was used to detect intracellular virus (30 min, 0°C). Next, the cells were washed twice to remove unbound antibody and stained with allophycocyanin-labeled anti–rabbit secondary antibody for an additional 30 min at 0°C followed by washing. Data was acquired on FACSCalibur and analyzed using Flowjo analysis software.
Injections.
C57BL/6 mice were injected subcutaneous with either PBS alone, 50 μg Pam-3-cys, 25 μg E. coli LPS, 50 μg CpG B, 2 μg 3M-003, or 106 PFU YF-17D. After 1 d, the draining popliteal and inguinal lymph nodes were dissected and the DCs were isolated with collagenase, type IV digestion. The cells were stained and analyzed by flow cytometry for costimulatory molecule expression. B6.PL (Thy 1.1+) mice reconstituted with OT-1 TCR transgenic T cells (Thy 1.2+) were injected subcutaneously with 50 μg of MHC class I–restricted OVA peptide (SIINFEKL; OVA257-264; BioSynthesis) with 25 μg E. coli LPS or 106 PFU of YF-FL or YF-Ova8. After 4 d, the cells from the draining popliteal and inguinal lymph nodes were subjected to assays as described in the T cell assays section.
Evaluation of infection of DCs.
Infection assays were performed as described previously by Barba-Spaeth et al. (50). In brief, DCs were washed in RPMI 1640 and cultured with YF-17D for 1 h at 37°C. The infection was quenched with 5% pooled human serum and the cells were washed extensively to remove excess virus. After 1 h of infection, DCs were resuspended in conditioned media and incubated for 24 h before monitoring for infection. Surface staining was done in serum containing media at 4°C. Antibodies were obtained from BD Biosciences. Cytoperm/CytoFix Kit (BD Biosciences) was used for fixation and permeabilization. C12 is a rabbit polyclonal antisera that recognizes the nonstructural proteins NS4A and NS4B, and was a gift from C. Rice (50). Secondary antibodies used were PE (Jackson ImmunoResearch Laboratories) or allophycocyanin (Invitrogen).
T cell assays.
Isolated lymph node cells from injected mice were either stained directly ex vivo and analyzed by flow cytometry for T cell clonal expansion, or restimulated in vitro. Cells were plated at 5 × 105 cells/well in 96-well plates and cultured in complete DMEM + 10% FBS alone or with various concentrations of MHC class I–restricted OVA SIINFEKL peptide, or an I-Ab–restricted epitope from the viral envelope protein (residues 231–247 from the envelope protein, LVEFEPPHAATIRVL), or an H-2Db–restricted epitope (residues 1–15 from the E1 envelope protein, AHCIGITDRDFIEGV), or an H-2Kb–restricted dominant epitope mapped to the NS3 protein (residues 261–275 from the NS3 protein, VIDAMCHATLTYRML) (72), or a combination of these epitopes. On the third day, the wells for in vitro proliferation assays had 1 μCi of [3H]thymidine added to them. On the fourth day, supernatants were collected for cytokine measurement, or the cultures were frozen at −20°C to lyse the cells for the measurement of thymidine incorporation.
Cytokine measurement.
For the mouse cytokines, the following kits were used: IL-4 and IFN-γ were obtained from eBioscience; IL-5, IL-6, IL-10, and IL-12p40 were obtained from BD Biosciences; and IL-13 was obtained from R&D Systems. Human IL-6, TNF-α, IL-10, IP-10, and MCP-1 were all measured using kits obtained from BD Bioscience. IFN-α was measured with the kit obtained from Biosource.
Transfection experiments.
The HEK293 cells stably transfected with human TLR8 and an NFκB driven luciferase reporter were a gift from 3M Pharmaceuticals. Cells were plated at 4 × 104 cells per well in 96-well plates with DMEM + 10% FBS + pen/strep in the evening. 18 h later, the cells were stimulated with 10 μM of 3M-003, 4 × 103 PFU of YF-17D, or left in media alone. After 24 or 48 h of stimulation, the luciferase activity of the cells was assayed using the Luciferase Assay System (Promega).
Online supplemental material.
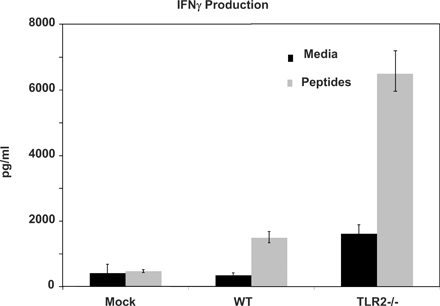
YF-17D induces IL-12p40 and IL-12p70 in both human mDCs (Fig. S1, A and B) and murine splenic CD11c+ DCs (Fig. S1 C). In Fig. S2, YF-17D productively infects human mDCs. Immature human mDCs were exposed to YF-17D, and 48 h later the DCs were harvested stained for intracellular flow cytometric analysis of NS4A and NS4B. Fig. S3 shows that cellular DNA does not contribute to the immune stimulatory effects of YF-17D. Fig. S4 depicts TLR2 exerting an inhibitory effect on Th1 responses induced by IFN-γ. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051720/DC1.
Supplemental Material
Acknowledgments
We would like to thank A. Krieg and members of our lab for discussion and comments on the manuscript.
This work was supported by grants from the National Institutes of Health, nos. U19 AI057266, R01 AI048638, R01 AI056499, R01 DK057665, R21 AI056957, and U54 AI057157.
The authors have no conflicting financial interests.
Abbreviations used: mDC, monocyte-derived DC; MOI, multiplicity of infection; pDC, plasmacytoid DC; PRR, pathogen recognition receptor; TLR, Toll-like receptor; YF-17D, yellow fever vaccine 17D.
References
- 1.Theiler, M., and H.H. Smith. 1937. The use of yellow fever virus modified by in vitro cultivation for human immunization. J. Exp. Med. 65:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theiler, M., and H.H. Smith. 1937. The effect of prolonged cultivation in vitro upon the pathogenicity of yellow fever virus. J. Exp. Med. 65:767–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niedrig, M., M. Lademann, P. Emmerich, and M. Lafrenz. 1999. Assessment of IgG antibodies against yellow fever virus after vaccination with 17D by different assays: neutralization test, haemagglutination inhibition test, immunofluorescence assay and ELISA. Trop. Med. Int. Health. 4:867–871. [DOI] [PubMed] [Google Scholar]
- 4.Poland, J.D., C.H. Calisher, T.P. Monath, W.G. Downs, and K. Murphy. 1981. Persistence of neutralizing antibody 30-35 years after immunization with 17D yellow fever vaccine. Bull. World Health Organ. 59:895–900. [PMC free article] [PubMed] [Google Scholar]
- 5.Co, M.D., M. Terajima, J. Cruz, F.A. Ennis, and A.L. Rothman. 2002. Human cytotoxic T lymphocyte responses to live attenuated 17D yellow fever vaccine: identification of HLA-B35-restricted CTL epitopes on nonstructural proteins NS1, NS2b, NS3, and the structural protein E. Virology. 293:151–163. [DOI] [PubMed] [Google Scholar]
- 6.Arroyo, J., C. Miller, J. Catalan, G.A. Myers, M.S. Ratterree, D.W. Trent, and T.P. Monath. 2004. ChimeriVax-West Nile virus live-attenuated vaccine: preclinical evaluation of safety, immunogenicity, and efficacy. J. Virol. 78:12497–12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhatt, T.R., M.B. Crabtree, F. Guirakhoo, T.P. Monath, and B.R. Miller. 2000. Growth characteristics of the chimeric Japanese encephalitis virus vaccine candidate, ChimeriVax-JE (YF/JE SA14–14–2), in Culex tritaeniorhynchus, Aedes albopictus, and Aedes aegypti mosquitoes. Am. J. Trop. Med. Hyg. 62:480–484. [DOI] [PubMed] [Google Scholar]
- 8.Guirakhoo, F., Z.X. Zhang, T.J. Chambers, S. Delagrave, J. Arroyo, A.D. Barrett, and T.P. Monath. 1999. Immunogenicity, genetic stability, and protective efficacy of a recombinant, chimeric yellow fever-Japanese encephalitis virus (ChimeriVax-JE) as a live, attenuated vaccine candidate against Japanese encephalitis. Virology. 257:363–372. [DOI] [PubMed] [Google Scholar]
- 9.Guirakhoo, F., J. Arroyo, K.V. Pugachev, C. Miller, Z.X. Zhang, R. Weltzin, K. Georgakopoulos, J. Catalan, S. Ocran, K. Soike, M. Ratterree, and T.P. Monath. 2001. Construction, safety, and immunogenicity in nonhuman primates of a chimeric yellow fever-dengue virus tetravalent vaccine. J. Virol. 75:7290–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pugachev, K.V., F. Guirakhoo, F. Mitchell, S.W. Ocran, M. Parsons, B.W. Johnson, O.L. Kosoy, R.S. Lanciotti, J.T. Roehrig, D.W. Trent, and T.P. Monath. 2004. Construction of yellow fever/St. Louis encephalitis chimeric virus and the use of chimeras as a diagnostic tool. Am. J. Trop. Med. Hyg. 71:639–645. [PubMed] [Google Scholar]
- 11.Guirakhoo, F., K. Pugachev, Z. Zhang, G. Myers, I. Levenbook, K. Draper, J. Lang, S. Ocran, F. Mitchell, M. Parsons, et al. 2004. Safety and efficacy of chimeric yellow Fever-dengue virus tetravalent vaccine formulations in nonhuman primates. J. Virol. 78:4761–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molenkamp, R., E.A. Kooi, M.A. Lucassen, S. Greve, J.C. Thijssen, W.J. Spaan, and P.J. Bredenbeek. 2003. Yellow fever virus replicons as an expression system for hepatitis C virus structural proteins. J. Virol. 77:1644–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monath, T.P., F. Guirakhoo, R. Nichols, S. Yoksan, R. Schrader, C. Murphy, P. Blum, S. Woodward, K. McCarthy, D. Mathis, C. Johnson, and P. Bedford. 2003. Chimeric live, attenuated vaccine against Japanese encephalitis (ChimeriVax-JE): phase 2 clinical trials for safety and immunogenicity, effect of vaccine dose and schedule, and memory response to challenge with inactivated Japanese encephalitis antigen. J. Infect. Dis. 188:1213–1230. [DOI] [PubMed] [Google Scholar]
- 14.Tao, D., G. Barba-Spaeth, U. Rai, V. Nussenzweig, C.M. Rice, and R.S. Nussenzweig. 2005. Yellow fever 17D as a vaccine vector for microbial CTL epitopes: protection in a rodent malaria model. J. Exp. Med. 201:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAllister, A., A.E. Arbetman, S. Mandl, C. Pena-Rossi, and R. Andino. 2000. Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine experimental solid tumors and pulmonary metastases. J. Virol. 74:9197–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beutler, B. 2004. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 430:257–263. [DOI] [PubMed] [Google Scholar]
- 17.Iwasaki, A., and R. Medzhitov. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5:987–995. [DOI] [PubMed] [Google Scholar]
- 18.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 19.Kawai, T., and S. Akira. 2005. Pathogen recognition with Toll-like receptors. Curr. Opin. Immunol. 17:338–344. [DOI] [PubMed] [Google Scholar]
- 20.Germain, R.N. 2004. An innately interesting decade of research in immunology. Nat. Med. 10:1307–1320. [DOI] [PubMed] [Google Scholar]
- 21.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 22.Pulendran, B. 2005. Variegation of the immune response with dendritic cells and pathogen recognition receptors. J. Immunol. 174:2457–2465. [DOI] [PubMed] [Google Scholar]
- 23.Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 24.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 11:443–451. [DOI] [PubMed] [Google Scholar]
- 25.Werts, C., R.I. Tapping, J.C. Mathison, T.H. Chuang, V. Kravchenko, I. Saint Girons, D.A. Haake, P.J. Godowski, F. Hayashi, A. Ozinsky, et al. 2001. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat. Immunol. 2:346–352. [DOI] [PubMed] [Google Scholar]
- 26.Hirschfeld, M., J.J. Weis, V. Toshchakov, C.A. Salkowski, M.J. Cody, D.C. Ward, N. Qureshi, S.M. Michalek, and S.N. Vogel. 2001. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect. Immun. 69:1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiramine, H., K. Watanabe, N. Hamada, and T. Umemoto. 2003. Porphyromonas gingivalis 67-kDa fimbriae induced cytokine production and osteoclast differentiation utilizing TLR 2. FEMS Microbiol. Lett. 229:49–55. [DOI] [PubMed] [Google Scholar]
- 28.Underhill, D.M., A. Ozinsky, A.M. Hajjar, A. Stevens, C.B. Wilson, M. Bassetti, and A. Aderem. 1999. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 401:811–815. [DOI] [PubMed] [Google Scholar]
- 29.Krieg, A.M. 2002. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 20:709–760. [DOI] [PubMed] [Google Scholar]
- 30.Krug, A., A.R. French, W. Barchet, J.A. Fischer, A. Dzionek, J.T. Pingel, M.M. Orihuela, S. Akira, W.M. Yokoyama, and M. Colonna. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 21:107–119. [DOI] [PubMed] [Google Scholar]
- 31.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeshita, F., C.A. Leifer, I. Gursel, K.J. Ishii, S. Takeshita, M. Gursel, and D.M. Klinman. 2001. Cutting edge: Role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J. Immunol. 167:3555–3558. [DOI] [PubMed] [Google Scholar]
- 33.Diebold, S.S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 34.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 35.Lund, J.M., L. Alexopoulou, A. Sato, M. Karow, N.C. Adams, N.W. Gale, A. Iwasaki, and R.A. Flavell. 2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA. 101:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 37.Guillot, L., R. Le Goffic, S. Bloch, N. Escriou, S. Akira, M. Chignard, and M. Si-Tahar. 2005. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 280:5571–5580. [DOI] [PubMed] [Google Scholar]
- 38.Tabeta, K., P. Georgel, E. Janssen, X. Du, K. Hoebe, K. Crozat, S. Mudd, L. Shamel, S. Sovath, J. Goode, et al. 2004. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA. 101:3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorden, K.B., K.S. Gorski, S.J. Gibson, R.M. Kedl, W.C. Kieper, X. Qiu, M.A. Tomai, S.S. Alkan, and J.P. Vasilakos. 2005. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR 8. J. Immunol. 174:1259–1268. [DOI] [PubMed] [Google Scholar]
- 40.Ito, T., Y.H. Wang, and Y.J. Liu. 2005. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR 9. Springer Semin. Immunopathol. 26:221–229. [DOI] [PubMed] [Google Scholar]
- 41.Coccia, E.M., M. Severa, E. Giacomini, D. Monneron, M.E. Remoli, I. Julkunen, M. Cella, R. Lande, and G. Uze. 2004. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 34:796–805. [DOI] [PubMed] [Google Scholar]
- 42.Heit, A., T. Maurer, H. Hochrein, S. Bauer, K.M. Huster, D.H. Busch, and H. Wagner. 2003. Cutting edge: Toll-like receptor 9 expression is not required for CpG DNA-aided cross-presentation of DNA-conjugated antigens but essential for cross-priming of CD8 T cells. J. Immunol. 170:2802–2805. [DOI] [PubMed] [Google Scholar]
- 43.Datta, S.K., V. Redecke, K.R. Prilliman, K. Takabayashi, M. Corr, T. Tallant, J. DiDonato, R. Dziarski, S. Akira, S.P. Schoenberger, and E. Raz. 2003. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J. Immunol. 170:4102–4110. [DOI] [PubMed] [Google Scholar]
- 44.Pulendran, B., P. Kumar, C.W. Cutler, M. Mohamadzadeh, T. Van Dyke, and J. Banchereau. 2001. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J. Immunol. 167:5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dillon, S., A. Agrawal, T. Van Dyke, G. Landreth, L. McCauley, A. Koh, C. Maliszewski, S. Akira, and B. Pulendran. 2004. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J. Immunol. 172:4733–4743. [DOI] [PubMed] [Google Scholar]
- 46.Netea, M.G., R. Sutmuller, C. Hermann, C.A. Van der Graaf, J.W. Van der Meer, J.H. van Krieken, T. Hartung, G. Adema, and B.J. Kullberg. 2004. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J. Immunol. 172:3712–3718. [DOI] [PubMed] [Google Scholar]
- 47.Sing, A., D. Rost, N. Tvardovskaia, A. Roggenkamp, A. Wiedemann, C.J. Kirschning, M. Aepfelbacher, and J. Heesemann. 2002. Yersinia V-antigen exploits toll-like receptor 2 and CD14 for interleukin 10-mediated immunosuppression. J. Exp. Med. 196:1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agrawal, S., A. Agrawal, B. Doughty, A. Gerwitz, J. Blenis, T. Van Dyke, and B. Pulendran. 2003. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J. Immunol. 171:4984–4989. [DOI] [PubMed] [Google Scholar]
- 49.Redecke, V., H. Hacker, S.K. Datta, A. Fermin, P.M. Pitha, D.H. Broide, and E. Raz. 2004. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. J. Immunol. 172:2739–2743. [DOI] [PubMed] [Google Scholar]
- 50.Barba-Spaeth, G., R.S. Longman, M.L. Albert, and C.M. Rice. 2005. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J. Exp. Med. 202:1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 301:640–643. [DOI] [PubMed] [Google Scholar]
- 52.Hoebe, K., X. Du, P. Georgel, E. Janssen, K. Tabeta, S.O. Kim, J. Goode, P. Lin, N. Mann, S. Mudd, et al. 2003. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 424:743–748. [DOI] [PubMed] [Google Scholar]
- 53.Horng, T., G.M. Barton, and R. Medzhitov. 2001. TIRAP: an adapter molecule in the Toll signaling pathway. Nat. Immunol. 2:835–841. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, and S. Akira. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 169:6668–6672. [DOI] [PubMed] [Google Scholar]
- 55.Fitzgerald, K.A., D.C. Rowe, B.J. Barnes, D.R. Caffrey, A. Visintin, E. Latz, B. Monks, P.M. Pitha, and D.T. Golenbock. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto, M., S. Sato, H. Hemmi, H. Sanjo, S. Uematsu, T. Kaisho, K. Hoshino, O. Takeuchi, M. Kobayashi, T. Fujita, K. Takeda, and S. Akira. 2002. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR 4. Nature. 420:324–329. [DOI] [PubMed] [Google Scholar]
- 57.Oshiumi, H., M. Matsumoto, K. Funami, T. Akazawa, and T. Seya. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4:161–167. [DOI] [PubMed] [Google Scholar]
- 58.Bieback, K., E. Lien, I.M. Klagge, E. Avota, J. Schneider-Schaulies, W.P. Duprex, H. Wagner, C.J. Kirschning, V. Ter Meulen, and S. Schneider-Schaulies. 2002. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 76:8729–8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duesberg, U., A. von dem Bussche, C. Kirschning, K. Miyake, T. Sauerbruch, and U. Spengler. 2002. Cell activation by synthetic lipopeptides of the hepatitis C virus (HCV)–core protein is mediated by toll like receptors (TLRs) 2 and 4. Immunol. Lett. 84:89–95. [DOI] [PubMed] [Google Scholar]
- 60.Dolganiuc, A., S. Oak, K. Kodys, D.T. Golenbock, R.W. Finberg, E. Kurt-Jones, and G. Szabo. 2004. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 127:1513–1524. [DOI] [PubMed] [Google Scholar]
- 61.Zhou, S., E.A. Kurt-Jones, L. Mandell, A. Cerny, M. Chan, D.T. Golenbock, and R.W. Finberg. 2005. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 35:822–830. [DOI] [PubMed] [Google Scholar]
- 62.Kato, H., S. Sato, M. Yoneyama, M. Yamamoto, S. Uematsu, K. Matsui, T. Tsujimura, K. Takeda, T. Fujita, O. Takeuchi, and S. Akira. 2005. Cell Type-Specific Involvement of RIG-I in Antiviral Response. Immunity. 23:19–28. [DOI] [PubMed] [Google Scholar]
- 63.Coban, C., K.J. Ishii, T. Kawai, H. Hemmi, S. Sato, S. Uematsu, M. Yamamoto, O. Takeuchi, S. Itagaki, N. Kumar, T. Horii, and S. Akira. 2005. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Triantafilou, K., E. Vakakis, G. Orthopoulos, M.A. Ahmed, C. Schumann, P.M. Lepper, and M. Triantafilou. 2005. TLR8 and TLR7 are involved in the host's immune response to human parechovirus 1. Eur. J. Immunol. 35:2416–2423. [DOI] [PubMed] [Google Scholar]
- 65.Triantafilou, K., G. Orthopoulos, E. Vakakis, M.A. Ahmed, D.T. Golenbock, P.M. Lepper, and M. Triantafilou. 2005. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 7:1117–1126. [DOI] [PubMed] [Google Scholar]
- 66.Schnare, M., G.M. Barton, A.C. Holt, K. Takeda, S. Akira, and R. Medzhitov. 2001. Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2:947–950. [DOI] [PubMed] [Google Scholar]
- 67.Brightbill, H.D., D.H. Libraty, S.R. Krutzik, R.B. Yang, J.T. Belisle, J.R. Bleharski, M. Maitland, M.V. Norgard, S.E. Plevy, S.T. Smale, et al. 1999. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 285:732–736. [DOI] [PubMed] [Google Scholar]
- 68.Re, F., and J.L. Strominger. 2001. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J. Biol. Chem. 276:37692–37699. [DOI] [PubMed] [Google Scholar]
- 69.Re, F., and J.L. Strominger. 2004. IL-10 released by concomitant TLR2 stimulation blocks the induction of a subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in human dendritic cells. J. Immunol. 173:7548–7555. [DOI] [PubMed] [Google Scholar]
- 70.Schaub, B., A. Bellou, F.K. Gibbons, G. Velasco, M. Campo, H. He, Y. Liang, M.W. Gillman, D. Gold, S.T. Weiss, D.L. Perkins, and P.W. Finn. 2004. TLR2 and TLR4 stimulation differentially induce cytokine secretion in human neonatal, adult, and murine mononuclear cells. J. Interferon Cytokine Res. 24:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ropert, C., and R.T. Gazzinelli. 2004. Regulatory role of Toll-like receptor 2 during infection with Trypanosoma cruzi. J. Endotoxin Res. 10:425–430. [DOI] [PubMed] [Google Scholar]
- 72.van der Most, R.G., L.E. Harrington, V. Giuggio, P.L. Mahar, and R. Ahmed. 2002. Yellow fever virus 17D envelope and NS3 proteins are major targets of the antiviral T cell response in mice. Virology. 296:117–124. [DOI] [PubMed] [Google Scholar]
- 73.Napolitani, G., A. Rinaldi, F. Bertoni, F. Sallusto, and A. Lanzavecchia. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roelofs, M.F., L.A. Joosten, S. Abdollahi-Roodsaz, A.W. Van Lieshout, T. Sprong, F.H. van den Hoogen, W.B. Van Den Berg, and T.R. Radstake. 2005. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 52:2313–2322. [DOI] [PubMed] [Google Scholar]
- 75.Wille-Reece, U., B.J. Flynn, K. Lore, R.A. Koup, R.M. Kedl, J.J. Mattapallil, W.R. Weiss, M. Roederer, and R.A. Seder. 2005. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc. Natl. Acad. Sci. USA. 102:15190–15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janeway, C.A., Jr. 1989. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54:1–13. [DOI] [PubMed] [Google Scholar]
- 77.Matzinger, P. 1994. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12:991–1045. [DOI] [PubMed] [Google Scholar]
- 78.Hussain, A.I., J.A. Johnson, M. Da Silva Freire, and W. Heneine. 2003. Identification and characterization of avian retroviruses in chicken embryo-derived yellow fever vaccines: investigation of transmission to vaccine recipients. J. Virol. 77:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pulendran, B. 2004. Immune activation: death, danger and dendritic cells. Curr. Biol. 14:R30–R32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}