

Abstract
Toll-like receptors (TLRs) function as primary sensors that elicit coordinated innate immune defenses through recognition of microbial products and induction of immune and proinflammatory genes. Here we report the identification and biological characterization of a lipopolysaccharide (LPS)-like molecule extracted from the cyanobacterium Oscillatoria Planktothrix FP1 (cyanobacterial product [CyP]) that is not stimulatory per se but acts as a potent and selective antagonist of bacterial LPS. CyP binds to MD-2 and efficiently competes with LPS for binding to the TLR4–MD-2 receptor complex. The addition of CyP together with LPS completely inhibited both MyD88- and TRIF-dependent pathways and suppressed the whole LPS-induced gene transcription program in human dendritic cells (DCs). CyP protected mice from endotoxin shock in spite of a lower capacity to inhibit LPS stimulation of mouse DCs. Interestingly, the delayed addition of CyP to DCs responding to LPS strongly inhibited signaling and cytokine production by immediate down-regulation of inflammatory cytokine mRNAs while not affecting other aspects of DC maturation, such as expression of major histocompatibility complex molecules, costimulatory molecules, and CCR7. Collectively, these results indicate that CyP is a potent competitive inhibitor of LPS in vitro and in vivo and reveal the requirement of sustained TLR4 stimulation for induction of cytokine genes in human DCs.
DCs control the adaptive immune response by providing a quantitative and qualitative framework for T cell antigen recognition (1–3). DCs are present in a resting immature state and rapidly respond to microbial and inflammatory stimuli by undergoing a maturation process that leads to their migration to secondary lymphoid organs, up-regulation of MHC and costimulatory molecules, and production of T cell–polarizing cytokines. Abundant data from the literature provide convincing evidence of the existence of multiple pathways of DC activation elicited by engagement of a variety of receptors, including members of the IL-1/Toll-like receptor (TLR) family and the TNF-R family (TNF-R and CD40; reference 4). It has been proposed that DCs are flexible and adapt immune responses to invading pathogens according to the specific receptor engaged and the presence of certain cytokines in the environment at the time of activation (5, 6).
Human monocyte-derived immature DCs (7) have been extensively used to identify maturation stimuli and to study the interplay between synergizing and inhibitory stimuli. Based on their sensitivity and high grade of flexibility, we thought of exploiting this cell system for the identification and isolation of novel bioactive compounds through bioassay-guided fractionation of natural extracts.
Cyanobacteria, also termed blue-green algae, are a large group of photosynthetic oxygenic prokariots with a high degree of biological adaptation. They represent one of the oldest forms of life on earth (8) and are a rich source of natural products with unique pharmacological activities. Bioactive metabolites isolated from cyanobacteria have been found to display neuroprotective, cytotoxic, antibacterial, antifungal, antiviral, and antiinflammatory properties (9–14). LPS derived from cyanobacteria and from Gram-negative bacteria differs in both chemical and biological characteristics. In general, LPS from cyanobacteria lacks l-glycero-d-mannoheptose and phosphate groups, has long-chain saturated and unsaturated fatty acids, and very low content of 2-keto-3-deoxyoctonate (15–19). Furthermore, cyanobacterial LPS shows minimal or no toxicity in mice, whereas no data are available concerning its activity on human cells (16, 17).
In this study, we describe the biological activity of an LPS-like molecule extracted from the freshwater cyanobacterium Oscillatoria Planktothrix FP1, which we named CyP. We show that CyP is a potent antagonist of bacterial LPS, which, depending on the time of addition, can either completely block LPS-induced activation of DCs or prevent secretion of cytokines without affecting phenotypic maturation. Notably, when tested in vivo, CyP is able to protect mice against lethal endotoxin shock. These results open promising perspectives for the use of CyP as a therapeutic agent able to modulate innate and adaptive immune responses and reveal the requirement of sustained TLR4 stimulation for induction of cytokine gene expression in human DCs.
RESULTS
Extracts from Oscillatoria Planktothrix FP1 potently inhibit the response of human DCs to LPS
Oscillatoria Planktothrix FP1 is a filamentous freshwater cyanobacterium isolated from an algal bloom in Lake Varese, Italy (20). To study the LPS of this cyanobacterium, extracts were prepared by a phenol and guanidinium thiocyanate–based procedure as described previously (21), taking care of removing free lipids, phospholipids, and protein contaminants. The extracts represented 2% of cell dry weight and contained mainly an LPS-like product as detected in silver-stained deoxycholate PAGE (DOC-PAGE; Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20060136/DC1) and to which we refer to as CyP hereafter. The 2-keto- 3-deoxyoctonate content of CyP was 0.15% (wt/wt), which is in the low range previously reported for LPS isolated from other cyanobacteria, and the endotoxin activity, measured by the limulus amebocyte assay, was very low at 4 EU/μg as compared with 8,000 EU/μg of Salmonella abortus equi LPS and 15,000 EU/μg of Escherichia coli 055:B5 LPS.
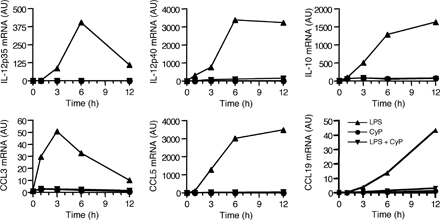
CyP was compared with bacterial LPS for its capacity to stimulate human monocyte-derived DCs. Although E. coli LPS induced up-regulation of CD80, CD86, and CD83, CyP failed to do so (Fig. 1 A). Remarkably, however, when added with LPS, CyP was able to completely inhibit the LPS-induced up-regulation of these molecules. In addition, CyP did not induce cytokine (TNF, IL-6, IL-10, IL-12p35, and p40) and chemokine (CCL3, CCL5, and CCL19) gene transcripts but strongly inhibited their induction by LPS (Fig. 1 B and Fig. S2, which is available at http://www.jem.org/cgi/content/full/jem.20060136/DC1). Secretion of TNF and IL-6 proteins in LPS-stimulated DCs was inhibited in a dose-dependent fashion by the addition of CyP (Fig. 1 C). Furthermore, IL-10 was below the detection limit in supernatants of CyP-treated cells as well as in supernatants of cells stimulated with LPS in the presence of CyP, whereas the inhibitory activity of CyP was still observed in the presence of anti–IL-10 blocking antibodies, excluding CyP functions through IL-10 induction (unpublished data).
Figure 1.

CyP inhibits LPS-induced activation of human DCs in a dose-dependent manner. (A) Human monocyte-derived DCs were treated with 1 μg/ml LPS and 20 μg/ml CyP alone or in combinations. After 16 h, expression of CD80, CD86, and CD83 was measured. Untreated DCs are shown in each panel as a gray profile. One representative experiment of six is shown. (B) Kinetics of TNF and IL-6 mRNA expression in DCs treated with LPS (▴), CyP (•), or LPS and CyP (▾) as measured by quantitative real-time RT-PCR. One representative experiment of five is shown. Fig. S2 shows additional transcript analysis. (C) DCs were stimulated with 10 (white bars) or 1 (black bars) μg/ml LPS in the absence or presence of graded amounts of CyP. After 20 h, TNF and IL-6 were measured in the culture supernatants by ELISA. One representative experiment of three is shown. (D) Nascent mRNA was isolated from the nuclei of DCs before (unst) and 1 or 3 h after stimulation by LPS in the absence (−) or presence (+) of CyP, and PCR was performed using specific primers.
On a weight basis, CyP behaved as a very potent inhibitor because 50% inhibition of cytokine production could be elicited by concentrations of CyP 10-fold lower than LPS. Inhibition of cytokine and chemokine production was always >90%, regardless of the source of stimulatory LPS, which included several different serotypes of E. coli, S. abortus equi, and Salmonella minnesota Re 595 (unpublished data).
To understand whether CyP affects cytokine mRNA transcription and/or stability, we measured TNF and IL-6 nascent mRNAs in the nuclei of DCs stimulated by E. coli LPS in the absence or presence of CyP. IL-6 gene transcription was completely inhibited in the presence of CyP, whereas TNF gene transcription was not because the amount of nascent mRNA in untreated and CyP-treated DCs was comparable (Fig. 1 D). These results suggest that CyP inhibits cytokine production by affecting both gene transcription and mRNA stability and are consistent with previous findings showing that posttranscriptional mechanisms play a major role in the regulation of TNF expression in LPS-stimulated DCs (22).
We conclude that CyP behaves as a potent inhibitor of LPS-induced DC-phenotypic maturation and cytokine production.
CyP is a selective inhibitor of LPS-induced activation of DCs
LPS induces DC maturation by triggering TLR4. Because DCs express several other receptors that can mediate maturation and cytokine production, including TLRs, IL-1R, and CD40, we investigated whether CyP might interfere with DC activation induced by other stimuli. Although CyP completely inhibited TNF and IL-6 production by DCs stimulated with LPS, it did not interfere with that elicited by peptidoglican (PGN), poly(I:C), R848 (which trigger TLR2, TLR3, and TLR8, respectively) IL-1β, or CD40L (Fig. 2 A). CyP also failed to inhibit CpG-induced IFN-α release by plasmacytoid DCs (unpublished data), indicating that it does not interfere with TLR9 stimulation. These results indicate that CyP behaves as a selective inhibitor of the LPS–TLR4 axis.
Figure 2.

CyP specifically inhibits LPS stimulation of DCs. (A) DCs were stimulated for 16 h with different TLR agonists (1 μg/ml LPS, 10 μg/ml PGN, 20 μg/ml poly(I:C), and 2.5 μg/ml R848), 20 ng/ml IL-1β, or 1 μg/ml soluble CD40L in the absence or presence of 20 μg/ml CyP. Data are expressed as the percentage of the response (TNF production, black bars; IL-6 production, white bars) obtained with the specific agonists in the absence of CyP and represent the mean ± SD of four independent experiments. Inhibition of LPS was found to be statistically significant (P < 0.0001), whereas inhibition of all other stimuli was found to be nonsignificant (P > 0.05). (B) DCs were challenged with graded numbers of DH5α bacteria in the absence (white bars) or presence (black bars) of 20 μg/ml CyP. TNF was measured in the 20-h culture supernatants by ELISA. CyP did not affect bacterial growth. One representative experiment of three is shown. (C) DCs were stimulated with LPS, 10 ng/ml IFN-γ, soluble CD40L, or R848 alone or in the indicated combinations in the absence (white bars) or presence (black bars) of CyP. IL-12p70 was measured in the 24-h culture supernatants. One representative experiment of four is shown.
Given the potent inhibitory activity of CyP on LPS stimulation, we wondered whether CyP might be able to suppress activation of DCs by LPS-containing Gram-negative bacteria or by LPS combined with stimuli that have been shown to synergize in induction of IL-12p70 (23–25). When E. coli DH5α bacteria were titrated into DC cultures in the presence of CyP, a 700-fold increase in the number of bacteria was required to elicit a comparable amount of TNF release (Fig. 2 B). In addition, when DCs were triggered by LPS in combination with IFN-γ, CD40L, or R848 in the presence of CyP, a marked inhibition of IL-12p70 production was observed (Fig. 2 C), consistent with a complete ablation of the LPS-dependent component.
Collectively, these experiments show that CyP specifically suppresses LPS-induced activation of DCs and by doing so it hampers mechanisms of amplification of immune responses by agonists acting in synergy with LPS.
CyP inhibits LPS binding to the TLR4–MD-2 receptor complex
Detection of LPS by immune cells depends upon the proper function of the TLR4–MD-2 receptor complex (26–28). Although TLR4 is the signal transduction component of the LPS receptor, MD-2 has been shown to be the endotoxin binding unit (29–31). We therefore asked whether the inhibitory activity of CyP is mediated through inhibition of TLR4 intracellular signaling or through competition for binding to the receptor complex. First, we transfected Jurkat cells with expression vectors encoding either TLR4, TLR9, or a chimera of extracellular TLR9 and intracellular TLR4 (TLR9N4C; reference 32) and measured the activity of a luciferase reporter gene in response to LPS or CpG. CyP was able to inhibit the LPS response in TLR4-transfected cells but was ineffective on the CpG-induced response of cells expressing the chimeric receptor (Fig. 3 A). Interestingly, the constitutive signaling of TLR4 transfectants that can be measured in the absence of LPS (33) was also inhibited by CyP (Fig. 3 B). Together with the finding that CyP did not affect limulus amebocyte lysate activity of E. coli LPS (unpublished data), this result indicates that CyP acts at the extracellular level and does not function by complexing with LPS and neutralizing its activity.
Figure 3.

CyP inhibits LPS at the level of MD-2–TLR4 extracellular domain. (A) Luciferase activity of Jurkat cells transfected with a 3×NF-κB–driven luciferase reporter together with empty vector (Mock) or expression vectors encoding either TLR4, TLR9, or a chimera of extracellular TLR9 and intracellular TLR4 (TLR9N4C) and stimulated with 1 μg/ml LPS or 3 μM CpG. Reporter activity was measured after 16 h of LPS stimulation in the absence (white bars) or presence (black bars) of 20 μg/ml CyP. Similar results were obtained at 6 h of stimulation. Data represent the mean ± SD of duplicates of one experiment of two performed with identical results. (B) Spontaneous luciferase activity in Jurkat cells transfected with a 3×NF-κB–driven luciferase reporter together with an empty vector (Mock) or an expression vector encoding TLR4. Reporter activity in the absence (white bars) or presence (black bars) of CyP was measured 40 h after transfection in a 6-h assay. (C) Monocytes were stained with LPS conjugated to Alexa Fluor 488 (0.25 μg/ml LPS-AF488) in the absence (thick line) or presence (thin lines) of increasing concentrations of CyP (0.25, 12.5, 125, and 250 μg/ml) and analyzed by FACS. Background fluorescence of monocytes is shown as a gray profile. One representative experiment of four is shown. Fig. S3 shows the EC50 of CyP inhibiting LPS binding. (D) HEK293T cells mock transfected or transfected with MD-2–FLAG were either lysed and probed for MD-2 expression with anti-FLAG antibodies or treated with 20 μg/ml biotinylated CyP. Biotinylated CyP was then captured with immobilized streptavidin, and MD-2 coprecipitates were detected with anti-FLAG antibodies. Stripped blots were subsequently probed with anti–MD-2 antibodies. Arrows indicate specific bands of differentially glycosylated MD-2 (reference 63). (E) Recombinant human MD-2 (1 μg/ml, fixed concentration) was incubated in wells coated with CyP (left) or LPS (right) in the presence of increasing concentrations of soluble LPS or CyP (0.24, 0.74, 2.22, 6.66, 20, and 60 μg/ml). MD-2 bound to the coated plate was then detected by a specific anti–MD-2 antibody followed by horseradish peroxidase–conjugated secondary antibody. EC50 values were calculated on sigmoidal dose–response curves (variable slope; R squared, 0.9953 or 0.9939 and 0.9692 or 0.9996 for CyP and LPS on CyP or LPS coat, respectively). Data shown are from one experiment of two performed with identical results.
Collectively, the data described above suggest that CyP directly interacts with the TLR4–MD-2 complex and hence interferes with LPS binding. To directly test this possibility, we used three different experimental approaches. First, we incubated human monocytes that express high levels of surface TLR4 and MD-2 with fluorescent LPS in the presence of increasing concentrations of CyP. CyP inhibited in a dose-dependent fashion LPS binding with a 50% value being reached at a concentration 3.5-fold higher than that of LPS (Fig. 3 C and Fig. S3, which is available at http://www.jem.org/cgi/content/full/jem.20060136/DC1). Second, CyP was biotinylated and incubated with HEK293T cells transfected with MD-2-FLAG. Precipitation of cell lysates using streptavidin beads revealed the association of CyP with MD-2, detected in Western blot by anti-FLAG or anti–MD-2 antibodies (Fig. 3 D). Finally, we set up ELISA assays using coated LPS or CyP and anti–MD-2 antibodies and found that soluble recombinant MD-2 binds to LPS and CyP and that these bindings can be inhibited in a concentration-dependent fashion by both LPS and CyP (Fig. 3 E). Collectively, these experiments are consistent with the notion that CyP functions as an LPS antagonist by competitively binding to MD-2.
CyP inhibits both MyD88-dependent and -independent signal transduction and gene expression
To ask whether CyP might behave as a partial antagonist, we analyzed the TLR4 signaling pathways and performed a global gene transcription analysis in DCs stimulated by LPS and CyP alone or in combination. At least two distinct intracellular signaling pathways are activated by TLR4. The “MyD88-dependent” pathway requires the adaptor protein Mal (also known as TIRAP) and MyD88 to signal through IRAK and TRAF6 and lead to activation of NF-κB and mitogen-activated protein kinases, such as p38, ERK, and JNK. In contrast, the adaptor molecules TRAM and TRIF transduce “MyD88-independent” signaling through activation of IRF3, which is required for the induction of IFN-β synthesis (34). CyP inhibited LPS-induced activation of both MyD88-dependent and -independent pathways, as assessed by the almost complete inhibition of p38, ERK, and c-Jun phosphorylation (Fig. 4 A) as well as nuclear translocation of IRF3 (Fig. 4 B). Moreover, as expected for an inhibitor that prevents LPS binding and signaling through TLR4–MD-2, LPS-induced degradation of IRAK (an upstream event in signal transduction) did not occur in the presence of CyP (Fig. 4 A). Similarly, CyP prevented LPS-induced IκBα degradation and subsequent induction, both indicative of NF-κB activation (Fig. 4 A). Of note, when DCs were cultured in the presence of CyP alone, MyD88, TRAF6, and IRAK expression were not affected (Fig. 4 C), indicating that CyP does not induce degradation of signaling components.
Figure 4.

CyP inhibits MyD88-dependent and -independent signaling. Immunoblot of cell lysates of DCs stimulated for different times with 0.4 μg/ml LPS in the absence or presence of 20 μg/ml CyP (A and B) or with CyP alone (C). IRF3 in B was detected in the nuclear fraction. One representative experiment of four is shown.
An Affymetrix microarray analysis showed that the addition of CyP to DCs had a very limited effect on gene transcription (Fig. 5, A and B). After a 1- or 3-h treatment with CyP, only 7 and 8 genes, respectively, out of 12,656 expressed were significantly (P < 0.05) up-regulated or down-regulated (greater than twofold change) compared with unstimulated control (Table S1, available at http://www.jem.org/cgi/content/full/jem.20060136/DC1). In contrast, LPS stimulation resulted in a significant greater than twofold induction or suppression of 274 genes. Remarkably, cells challenged with LPS in the presence of CyP showed an almost complete suppression of all LPS–up-regulated or –down-regulated genes, including the most responsive ones (Fig. 5 and Table S1). These results indicate that CyP is a full TLR4 antagonist and inhibits the entire LPS-induced activation program impeding all intracellular responses.
Figure 5.

CyP inhibits LPS-induced gene expression. Affymetrix methodology was used for global analysis of gene transcription. DCs from two different donors were left untreated (control), treated with 20 μg/ml CyP for 1 or 3 h, or treated with 0.5 μg/ml ultrapure LPS in the absence or presence of CyP for 3 h. (A) Genes showing a statistically significant change (P < 0.05) in at least one experimental condition versus control (321 genes) are ordered on the x axis according to decreasing “fold change” expression (relative to control) induced by LPS treatment. For each gene, the fold changes subsequent to different treatments are shown on the y axis. (B) Volcano plots of the fold changes between treated (1 h CyP, 3 h CyP, and 3 h LPS in the presence or absence of CyP) and untreated DCs for the 12,656 expressed genes. Green, transcripts with at least twofold up-regulation with P < 0.05; blue, transcripts with at least twofold down-regulation with P < 0.05; red, transcripts with fold change less than 2 or P > 0.05. See Table S1 for the list of all expressed gene transcripts.
CyP inhibits cytokine gene expression when added several hours after LPS
The results described above indicate that CyP behaves as a potent and selective LPS receptor antagonist, implying that it exerts its activity when given before or together with LPS. Consistent with this notion, we found that CyP added 15 min or 1 h after LPS failed to inhibit the up-regulation of B7 costimulatory and MHC molecules (Fig. 6 A). Moreover, 15-min stimulation by LPS was sufficient to induce high expression of CCR7. In line with this result, the T cell stimulatory capacity of LPS-matured DCs was inhibited when CyP was given together with LPS but was not affected by the delayed addition of CyP (Fig. 6 B).
Figure 6.

CyP interferes with LPS-induced cytokine production even when added several hours after LPS. DCs were stimulated with 0.4 μg/ml LPS, and 20 μg/ml CyP was added at various time intervals. (A) Cells were analyzed for CD80, CD86, and HLA-DR 20 h after stimulation and for CCR7 44 h after stimulation. Shadowed profiles correspond to unstimulated cells. One representative experiment out of five is shown. (B) Graded numbers of DCs stimulated as in A were cultured with allogeneic naive CD4+ T cells, and proliferation was measured on day 5 by [3H]thymidine incorporation and expressed as cpm. Data represent the mean ± SD of duplicates of one experiment of four performed. (C) DCs were stimulated with LPS, and CyP was subsequently added at the indicated time points. Cytokines released in the culture supernatants at 20 h were measured by ELISA and expressed as percentage of production in absence of CyP addition (−). Data represent the mean ± SD of three independent experiments. (D) DCs were stimulated with LPS in replicate cultures. The cells were left untreated (•) or CyP was added after 1 (▿), 3 (⋄), or 6 (○) h. The kinetics of cytokine-specific transcripts were determined by quantitative real-time RT-PCR. One representative experiment of three is shown. Fig. S4 reports the kinetics of CCL5 transcripts. (E) After 4 h of LPS stimulation, DCs were left untreated or CyP was added to the culture. Cell lysates were prepared 20 min, 1 h, or 2 h after the addition of CyP and analyzed by Western blot with antibodies specific for phosphorilated c-Jun, phosphorilated p38, or IκBα.
In sharp contrast, LPS-induced cytokine production was dramatically altered even when CyP was added several hours after LPS (Fig. 6 C). In particular, a significant (>60%) inhibition of IL-12p70 and IL-6 was still observed when CyP was added 6 h after LPS. This effect was further investigated by performing a detailed kinetics of cytokine mRNA levels. As shown in Fig. 6 D, at any time after LPS stimulation, the addition of CyP resulted in an abrupt decrease in the amount of TNF, IL-6, IL-12p40, and IL-12p35 mRNAs. It should be noted that for other induced genes, kinetics of inhibition of mRNA transcripts varied considerably, and in some cases (like CCL5) a 1-h LPS stimulation was sufficient to induce maximal expression (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20060136/DC1, and unpublished data). In addition, CyP given 4 h after LPS led to a decrease of late phosphorilation of c-Jun and p38 as well as an increase in IκBα protein levels (Fig. 6 E).
Collectively, the results described above indicate distinct temporal requirements of TLR4 signaling in the induction of different DC maturation programs. At variance with B7, MHC, and CCR7 induction, which can be triggered by a very brief TLR4 stimulation, prolonged TLR4 engagement is required to sustain expression of some cytokine genes. Importantly, this sustained TLR triggering can be interrupted by the addition of a specific receptor antagonist, thus limiting cytokine production.
CyP protects mice from lethal endotoxin shock
LPS plays a key role in Gram-negative sepsis by inducing production of cytokines, such as TNF, IL-6, and IL-1, which mediate hyperactivation of the inflammatory system, ultimately leading to death by endotoxin shock (35). The potent suppression of LPS-induced proinflammatory cytokine production observed in vitro on human cells, the interference with LPS synergy with other stimuli in DC activation, and the effectiveness at late time points after stimulation prompted us to investigate whether CyP might inhibit LPS-induced endotoxin shock in mice.
Because some LPS antagonists such as lipid IVa are species specific, in that they are antagonist on human cells but agonist on murine cells (36), we first assessed the activity of CyP in vitro on the mouse macrophage line RAW 264.7 and on mouse bone marrow–derived DCs. In these cells, CyP did not show any agonistic activity and antagonized LPS-induced cytokine release, although the potency was 50–200-fold lower than that measured in human cells. Indeed, a 20-fold excess of CyP, which completely inhibited cytokine production in human DCs, was able to reduce the production of TNF and IL-6 by mouse DCs from 35 to 75% in separate experiments (unpublished data).
We then investigated whether CyP might protect mice from LPS-induced endotoxin shock. A group of mice was sensitized with d-galactosamine and injected i.p. with 25 ng LPS (Fig. 7 A). Within the first 4 h, mice developed signs of endotoxemia and died within 8 h due to liver injury induced by TNF-dependent apoptotic death of sensitized hepatocytes (37). Coinjection of 750 μg CyP conferred significant protection: mice showed milder signs of distress, death was delayed, and 58% of the mice survived (Fig. 7 A). In a second group of mice, endotoxin shock was induced in the absence of d-galactosamine sensitization by i.p. injection of 1.5 mg LPS (Fig. 7 B). In this setting, which better reflects the complexity of endotoxin shock, mice developed ruffled fur, diarrhea, abnormal secretion of eyes, ataxia, twitching, hunched posture, and lethargy, and death occurred between 24 and 40 h after injection due to multi organ failure. Coinjection of 850 μg CyP conferred significant protection from death in 80% of the mice that generally showed only mild signs of endotoxemia (Fig. 7 B). No late death occurred over more than 1 wk, indicating that CyP did not merely delay the onset of LPS lethality. CyP also protected from i.p. injection of bacteria and when given intravenously (unpublished data). Collectively, these results demonstrate that CyP exerts anti-endotoxin effects in vivo.
Figure 7.

CyP inhibits LPS-induced endotoxin shock in mice. (A) C57BL/6 mice were sensitized with d-galactosamine and injected i.p. with 25 ng S. abortus equi LPS alone (13 mice, ⋄) or in combination with 750 μg CyP (12 mice, •). (B) C57BL/6 mice were injected i.p. with 1.5 mg E. coli 055:B5 LPS alone (6 mice, ⋄) or in combination with 850 μg CyP (10 mice, •). p-values calculated by the Kaplan-Meyer log-rank test are indicated.
DISCUSSION
In this study, we functionally characterize a cyanobacterial LPS-like product (CyP) that acts as a potent and selective TLR4–MD-2 receptor antagonist. CyP is nontoxic at high concentrations on both human and mouse cells, and, when added to human DCs, it does not elicit any response detectable by global transcriptional analysis. Furthermore, when given together with LPS, CyP completely inhibits DC activation in vitro as well as endotoxic shock in vivo.
Several reports have described natural and synthetic inhibitors of LPS-induced inflammatory responses in human cells, and some compounds are currently being tested in vivo (38, 39). These fall in two broad categories: receptor antagonists and LPS-neutralizing molecules. Among the first are LPS-like molecules isolated from bacteria (Rhodobacter capsulatus and sphaeroides, Porphyromonas gingivalis, and Helicobacter pylori), deacylated LPS, lipid IVa, synthetic Lipid A analogues (including E5531, E5564, DT-5461, and GLA-47), and certain cationic peptides (40–49). LPS-neutralizing molecules include polymixin B and synthetic peptides (50–52). Some of the compounds listed above behave as agonists when given at high concentrations or in mice. In contrast, CyP is devoid of any agonistic activity even at high concentrations and acts as a pure LPS antagonist on human and mouse cells. Indeed, no lethality was observed in mice injected with up to 1 mg CyP even upon sensitization with d-galactosamine (unpublished data). It is worth noting that CyP behaves as a specific TLR4–MD-2 antagonist and does not interfere with stimulation of other TLRs. However, by selectively inhibiting LPS, CyP removes a potent stimulus that synergizes with CD40L, IFN-γ, or R848 in DC activation (23–25). Remarkably, the inhibitory effect of CyP is observed even when DCs are challenged with live Gram-negative bacteria.
It has recently been reported that the synthetic LPS antagonist E5564 inhibits the binding of LPS to the TLR4–MD-2 receptor complex (31). We found that CyP can directly bind to MD-2, and by doing so, it competitively inhibits binding of LPS. In addition, CyP directly binds to the LPS binding protein (unpublished data), indicating that it may subtract serum components that strongly enhance LPS activity. Thus, by targeting both essential and enhancing mediators of LPS responses, CyP may represent a promising candidate for the development of drugs to treat endotoxin-dependent diseases.
The biological activity of CyP is likely to be ascribed to the LPS-like glycolipid that is visualized by silver staining of purified extracts separated by DOC-PAGE. Indeed, CyP is obtained following a classical extraction procedure for LPS. Its activity is maintained after boiling, it behaves as a glycolipid in chromatographic purification procedures we are currently using, and finally, it binds to MD-2, LPS binding protein, and polymixin B (unpublished data). To date, only few LPSs from cyanobacteria have been isolated and chemically characterized. Compared with LPS from Enterobacteriaceae, cyanobacterial LPS from different genera revealed peculiar sugar and fatty acid composition and was generally shown to be nontoxic when injected into mice (15–19). Extracts from filamentous cyanobateria other than Oscillatoria Planktothrix FP1 (Oscillatoria Neglecta, environmentally isolated Oscillatoria Sp. CCAP1459-26, and Leptolyngbya Sp.) were not stimulatory on human DCs but were not able to suppress LPS-induced activation (unpublished data), indicating that the LPS antagonistic activity is a distinct property of CyP.
A striking finding made in our study is that CyP exerts its inhibitory activity on cytokine production even when given to maturing DCs several hours after LPS. Biochemical studies showed that CyP added 4 h after LPS led to a sharp decrease in c-Jun and p38 phosphorylation, providing a mechanistic basis for the inhibition of cytokine production (53–55). We also found that a very short TLR4 stimulation (CyP added 15 min after LPS) was sufficient to induce full phenotypic maturation of DCs with acquisition of T cell stimulatory capacity, but did not lead to significant production of TNF, IL-6, and IL-12p70. In contrast, these cytokines were induced only in response to sustained TLR stimulation. These results reveal the existence of distinct thresholds for the induction of surface markers versus cytokines and are consistent with the existence of a temporal window during which signals emanated from TLR4 are kinetically integrated before they convey into a specific response.
Several recent studies examined the impact of the kinetics of receptor stimulation on expression of immune response genes. It has been shown that accessibility of NF-κB to the promoter of early- and late-induced genes depends on the state of histone acetylation (56). In that study, a short stimulation was sufficient to recruit NF-κB to the promoter of IκBα and other constitutively and immediately accessible genes, but not to the promoter of late genes that need to be acetylated to become transcriptionally competent. Another study showed that a transient stimulus was not sufficient to ensure NF-κB activity at late time points due to the rapid induction of IκBα that provides a rapid negative feedback on NF-κB activity (57). Further evidence that specificity in gene expression could be accomplished by using signal transduction pathways in temporally distinct ways came from computational modeling of signaling events downstream of TLR4 (58). Based on these and our results, we speculate that sustained TLR stimulation is required to maintain NF-κB activity in spite of IκBα resynthesis at late time points when some genes, such as IL-12, become transcriptionally accessible. Consistent with this model, we found that if CyP is added at late time points during a response to LPS, IκBα protein levels increase, suggesting that CyP inhibits ongoing IκB degradation. It is also possible that the expression of late genes might also require synthesis of transcription factors that synergize with NF-κB.
It was recently shown that TLR stimulation is absolutely required for the generation of fully activated “effector” DCs capable of directing T cell differentiation, whereas inflammatory cytokines are not sufficient (59, 60). Our results reveal that besides nature and concentration of microbial stimulus, duration of TLR stimulation is a previously unappreciated parameter that critically contributes to the effector function of DCs by regulating the expression of selected cytokine genes, such as IL-12. It will be interesting to assess the functional consequences of the split of DC maturation induced by transient TLR triggering, especially for the magnitude and quality of T cell response and generation of memory (61).
In conclusion, our findings indicate that a TLR receptor antagonist can be used to block endotoxin shock in vivo and to modulate DC maturation in vitro, revealing for the first time a requirement of sustained TLR stimulation for the expression of a selected set of immunoregulatory genes in maturing DCs. These results suggest that CyP might be a candidate for the development of a therapeutic agent able to control endotoxin shock and modulate immune responses.
MATERIALS AND METHODS
Extracts from cyanobacteria.
Cultures from cyanobacterium Oscillatoria Planktothrix FP1 were grown in 250-ml flasks containing 150 ml of BG11 medium (Sigma-Aldrich) at 27°C under constant irradiance of cool white light at an intensity of 20 μmol.m−2.s−1 until stationary phase was reached. Microorganisms were then collected by centrifugation and frozen before processing with TRI reagent (Sigma-Aldrich) according to a method described previously (21). Extracts in the acqueous phase were precipitated with 10 mM sodium acetate and 2 volumes of acetone and recovered by centrifugation at 2,000 g for 10 min. Pellets were washed twice with 70% ethanol to remove phospholipids, digested overnight at 37°C with 100 μg/ml proteinase K in 25 mM Tris, pH 8.5, to remove protein contaminants, and subjected to Folch partition to remove free lipids. Purified extracts (named CyP) were then dissolved in PBS at 2 mg/ml. The biological activity was not affected by boiling the solution for 15 min or by repeated freezing and thawing.
Isolation and stimulation of DCs.
Buffy coats were obtained from the Swiss Blood Center, Basel. Permission to do experiments on human primary cells was obtained from the Federal Office of Public Health. Monocytes were purified from peripheral blood mononuclear cells by positive sorting using anti-CD14–conjugated magnetic microbeads (Miltenyi Biotec). DCs were generated as described previously by culturing monocytes in RPMI 10% FBS supplemented with granulocyte/macrophage colony-stimulating factor and IL-4 (7). Cells were stimulated with LPS extracted from E. coli (serotypes 026:B6, 055:B5, and 0111:B4; Sigma-Aldrich) or S. abortus equi (S-form, pure TLR4 agonist; Qbiogene) at concentrations ranging from 0.4 to 10 μg/ml as specified in the figure legends. Alternatively, cells were stimulated with 10 μg/ml PGN (Sigma-Aldrich), 20 μg/ml poly(I:C) (GE Healthcare), 2.5 μg/ml CpG 2006 (5′-TCGTCGTTTTGTCGTTTTGTCGTT-3′ phosphorotioate; Microsynth), 2.5 μg/ml R848 (GLSyhthesis), 1 μg/ml CD40L plus 1 μg/ml Enhancer (Qbiogene), 20 ng/ml IL-1β (R&D Systems), or 10 ng/ml IFN-γ (provided by Roche).
FACS analysis.
The following monoclonal antibodies were used: anti-CD80 (MAB104), anti-CD83 (HB15a; Immunotech), anti-CD86 (IT2.2: BD Biosciences), and anti–HLA-DR (clone HB55; American Type Culture Collection [ATCC]) followed by appropriate rat anti–mouse IgG1 (BD Biosciences) or goat anti–mouse IgG2a or IgG2b conjugated to FITC (SouthernBiotech) secondary antibodies; and anti-CCR7 (3D12; provided by M. Lipp, Max Delbroeck Center, Berlin, Germany) followed by biotin-conjugated mouse anti–rat IgG2a (BD Biosciences) and APC-conjugated streptavidin (Invitrogen). Samples were analyzed on a FACSCalibur cytometer (Becton Dickinson) using propidium iodide (Sigma-Aldrich) to exclude dead cells.
Cytokine detection.
Cytokine production was measured in the supernatants of DCs (0.5 × 106 cells/ml) stimulated for 16–24 h using matched paired antibodies specific for human TNF, IL-6, and IL-12p70 (DuoSet ELISA development kits; R&D Systems). Data shown are from measurements with standard deviations <5%.
Real-time fluorogenic RT-PCR.
Total RNA was isolated with the RNeasy kit (QIAGEN). cDNA was synthesized with random hexamers (Invitrogen) and MMLV (Invitrogen Life Technologies). Gene expression was determined by real-time fluorogenic PCR with the ABI PRISM 7700 Sequence Detection System (Applied Biosystems) by using predesigned TaqMan Gene expression assays and reagents according to the manufacturer's instructions (Applied Biosystems). For each sample, mRNA expression levels for specific transcripts were normalized to the amount of 18S rRNA and expressed as arbitrary units.
Analysis of nascent transcripts.
Nascent transcripts were isolated as described previously (62) starting from 2.5 × 106 cells, lysing nuclei in 850 μl and extracting chromatin in 1 ml TRIzol (Invitrogen Life Technologies). Samples were treated with 20 U DNase I, RNase-free (Roche), in the presence of 80 U RNase OUT (Invitrogen) for 1 h at 37°C, extracted with TRIzol, and dissolved in water. PCR for specific transcripts was performed using the following primers: TNF, 5′-GATGGTAGGCAGAACTTGGAGAC-3′ (located in intron 3–4) and 5′-CAGCTGGTTATCTCTCAGCTCCA-3′; and IL-6, 5′-AAATTCGGTACATCCTCGACGGC-3′ and 5′-TGGTGGGCTCTGAGGTATGAATC-3′ (located in intron 2–3). β-actin was amplified as internal control (5′-TCACCCACACTGTGCCCATCTACGA-3′ and 5′-CAGCGGAACCGCTCATTGCCAATG G-3′). Contamination of genomic DNA was always tested and found to be below the detection limit.
Transfection assays.
Jurkat cells (expressing TLR9 and MD-2) were transfected with 2 μg expression vectors encoding TLR9, a fusion of extracellular TLR9, and intracellular TLR4 (TLR9N4C; provided by R. Medzhitov, Yale University, New Haven, CT), or TLR4 together with 2 μg of 3×NF-κB Luc reporter vector (provided by G. Natoli, IFOM, Milano, Italy). Transfection was performed with FuGENE6 according to the manufacturer's instructions (Roche). After 16 h, cells were washed, resuspended in 12 ml, and plated in a 12-well plate with 1 ml of cells for each stimulatory condition (1 μg/ml LPS or 3 μM CpG 1826 5′-TCCATGACGTTCCTGACGTT-3′ in the absence or presence of 20 μg/ml CyP). After a 6- or 16-h stimulation, cells were harvested and lysed, and reporter gene activity was measured using the Luciferase Assay System (Promega) and Veritas luminometer (Turner BioSystems).
LPS binding assay.
LPS binding assay was performed on freshly isolated monocytes as described previously (41). In brief, cells were preincubated for 30 min at 37°C in 180 μl SEBDAF buffer (20 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM EDTA, 300 μg/ml BSA, 10 mM NaN3, 2 mM NaF, 5 mM deoxyglucose) to prevent ligand internalization. Appropriate dilutions of CyP were prepared in 20 μl FBS containing 2.5 μg/ml E. coli 055:B5 LPS conjugated to Alexa Fluor 488 (Invitrogen) and added to the samples to produce the final concentrations indicated in Fig. 3 C. After 30 min of incubation at 37°C, cells were centrifuged and washed once in ice-cold buffer before FACS analysis.
MD-2 binding assays.
Detection of binding of CyP to surface MD-2 was performed in HEK293T cells (ATCC) transfected with pEFBOS expression vector encoding human MD-2-FLAG-His6 (provided by K. Miyake, University of Tokyo, Japan), according to previously described procedures (31). In brief, CyP was labeled using biotin hydrazide according to the manufacturer's instructions (Pierce Chemical Co.) and added to cells at 20 μg/ml for 1h at 37°C. Immobilized streptavidin (Pierce Chemical Co.) was then added to cell lysates (20 mM Tris, pH 7.4, 137 mM NaCl, 1% Triton X-100, 2 mM EDTA, 10% glycerol) and captured complexes separated on SDS-PAGE under reducing conditions, blotted, and probed with anti-FLAG (M2; Sigma-Aldrich) or anti–MD-2 (R&D Systems) antibodies. Alternatively, binding of MD-2 to CyP and competitive inhibition of the binding of MD-2 to LPS was studied in ELISA assays. LPS from E. coli 055:B5 or CyP (30 μg/ml in PBS) was coated overnight at 37°C on MaxiSorp 96-well plates (Nunc). After blocking with 0.05% BSA in wash buffer (50 mM Hepes, 150 mM NaCl), dilutions of LPS or CyP (as indicated in Fig. 3 E) in 1 μg/ml recombinant human MD-2 (R&D Systems) were added to the plates for 2 h 30 min at 37°C. MD-2 bound to coated LPS or CyP was then detected by incubation with 2 μg/ml mouse anti–human MD-2 first, followed by horseradish peroxidase–conjugated sheep anti–mouse IgG secondary antibody (GE Healthcare). Plates were developed by the addition of 300 μg/ml 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt (Sigma-Aldrich) in 0.1 M citric acid, pH 4.35, 0.03% hydrogen peroxide.
Immunoblot analysis.
Equal amounts of protein lysates were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Filters were blocked with 5% dry skim milk and probed with the following specific primary antibodies: anti-MyD88 and anti-IκBα (Imgenex); anti-TRAF6, anti-IRF3, anti-IRAK-1, and anti-total p38 (Santa Cruz Biotechnology, Inc.); and anti-phosphoJun (Ser73; Upstate Biotechnology), anti-diphosphorilated ERK-1 and -2, and anti-diphosphorilated p38 (Sigma-Aldrich). Blots were then incubated with appropriate horseradish peroxidase–conjugated donkey anti–rabbit IgG or sheep anti–mouse IgG secondary antibody (GE Healthcare) and developed with SuperSignal WestPico chemiluminescence Substrate (Pierce Chemical Co.).
Microarray and data analysis.
DCs from two different donors were left untreated or treated with either 20 μg/ml CyP for 1 or 3 h or 0.5 μg/ml LPS from S. abortus equi S-form in the presence or absence of CyP for 3 h. RNA was extracted with the TRIzol method (Invitrogen Life Technologies). 5 μg total RNA was labeled and hybridized with GeneChip Expression 3′ Amplification One-Cycle Target Labeling kit on Affymetrix U133 2.0 Plus microarrays (Affymetrix). Washes and scanning were performed according to Affymetrix protocols with Fluidics Station 400 and GeneChip Scanner 3000. Raw signals and the “present” or “absent” calls were obtained using Affymetrix GCOS 1.2 software. Additional data analysis was performed with GeneSpring 7.2 (Silicon Genetics). Signal values <0.01 were set to 0.01. The percentile of all the measurements in each sample was calculated using all genes not marked absent, and each gene measurement was divided by the 50th percentile. Each gene was then divided by the mean of its normalized measurements in the two untreated samples. Genes that did not present a signal intensity >50 and that were not marked as present in at least one condition of each replicate were discarded: 12,656 probes out of 54,675 passed this filter. “Fold changes” between treated and untreated cells at 1 and 3 h were calculated on the 321 genes showing a statistically differential expression in at least one of the conditions using a parametric test with variance assumed equal (ANOVA) and a p-value cut-off of 0.05, followed by the Benjamini and Hochberg false discovery rate multiple test correction, and a Tukey Post Hoc test. Accession code GEO: GSE4748.
Sorting and priming of naive T cells.
Naive CD4+ T cells were sorted from peripheral blood mononuclear cells with anti-CD4 magnetic beads (Miltenyi Biotec), followed by positive selection of cells stained with PE-conjugated CD45RA antibody (Immunotech). DCs were stimulated for 12 h with LPS in the absence or presence of CyP, washed, and irradiated (40 Gy), and graded numbers were plated with 37,500 allogeneic naive T cells. After 4 d, 4 μCi/ml of [methyl-3H]thymidine (GE Healthcare) was added for the last 12 h.
LPS-induced endotoxin shock in mice.
All animal experiments were performed in accordance with approved protocols specified in the permission obtained from the Swiss Federal Veterinary Office. Mice used in all survival studies were 8–10-wk-old C57BL/6 females (Charles River Laboratories). In the d-galactosamine–sensitized model, mice were injected i.p. with 20 mg d-galactosamine (Sigma-Aldrich) with 25 ng LPS from S. abortus equi (S-form) alone or in combination with 750 μg CyP diluted in PBS. Alternatively, mice were injected i.p. with 1.5 mg LPS from E. coli serotype 055:B5 diluted in PBS with or without 850 μg CyP.
Statistical analysis.
Statistical significance was assessed by Student's paired t test. Differences with p-values of <0.05 were considered statistically significant. The concentration of competitor that competes for half of the specific binding (EC50) was calculated on sigmoidal dose–response curves (variable slope). Datasets of survival curves were analyzed by the Kaplan-Meyer log-rank test. Statistical analysis was conducted using Prism 4 GraphPad software.
Online supplemental material.
Purity of phenolic extracts from cyanobacterium Oscillatoria Planktothrix FP1 (CyP) was evaluated by separation on DOC-PAGE and silver staining as shown in Fig. S1. Fig. S2 shows kinetics of cytokine and chemokine gene expression induced by LPS in the presence of CyP and complements data from Fig. 1 B. The EC50 of CyP-inhibiting binding of LPS-AF488 to monocytes shown if Fig. 3 C is calculated in Fig. S3. Kinetics of CCL5 mRNA induction upon LPS stimulation in the presence of CyP complements the data of Fig. 6 D and is shown in Fig. S4. A list of all 12,656 probe sets that in microarray analysis were marked as present in at least one condition can be found in Table S1. The supplemental material is available at http://www.jem.org/cgi/content/full/jem.20060136/DC1.
Supplemental Material
Acknowledgments
We are grateful to Ruslan Medzhitov, Gioacchino Natoli, and Kensuke Miyake for providing plasmids used in transfection experiments. We also thank Alfonso Martín-Fontecha and Grata Guarda for help with the in vivo mouse experiments, Jeremy Luban for help with the transfection experiments, all members of Lanzavecchia and Sallusto's laboratory for discussion, and Mariagrazia Uguccioni for critical reading of the manuscript.
This work was partially supported by the European Commission FP6 “Network of Excellence” initiative under contract numbers LSHG-CT-2003-502935 MAIN and LSHB-CT-2004-512074 DC-THERA.
The authors have no conflicting financial interests.
Abbreviations used: CyP, cyanobacterial product; PGN, peptidoglican; TLR, Toll-like receptor.
References
- 1.Iwasaki, A., and R. Medzhitov. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5:987–995. [DOI] [PubMed] [Google Scholar]
- 2.Lanzavecchia, A., and F. Sallusto. 2001. Regulation of T cell immunity by dendritic cells. Cell. 106:263–266. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 4.Reis e Sousa, C. 2001. Dendritic cells as sensors of infection. Immunity. 14:495–498. [DOI] [PubMed] [Google Scholar]
- 5.Kapsenberg, M.L. 2003. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 3:984–993. [DOI] [PubMed] [Google Scholar]
- 6.Reis e Sousa, C. 2004. Activation of dendritic cells: translating innate into adaptive immunity. Curr. Opin. Immunol. 16:21–25. [DOI] [PubMed] [Google Scholar]
- 7.Sallusto, F., and A. Lanzavecchia. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor α. J. Exp. Med. 179:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanier, R.Y., and G. Cohen-Bazire. 1977. Phototrophic prokaryotes: the cyanobacteria. Annu. Rev. Microbiol. 31:225–274. [DOI] [PubMed] [Google Scholar]
- 9.Romay, C., N. Ledon, and R. Gonzalez. 1998. Further studies on anti-inflammatory activity of phycocyanin in some animal models of inflammation. Inflamm. Res. 47:334–338. [DOI] [PubMed] [Google Scholar]
- 10.Oufdou, K., N. Mezrioui, B. Oudra, M. Loudiki, M. Barakate, and B. Sbiyyaa. 2001. Bioactive compounds from Pseudanabaena species (Cyanobacteria). Microbios. 106:21–29. [PubMed] [Google Scholar]
- 11.Shi, Q., J. Cui, J. Zhang, F.X. Kong, Z.C. Hua, and P.P. Shen. 2004. Expression modulation of multiple cytokines in vivo by cyanobacteria blooms extract from Taihu Lake, China. Toxicon. 44:871–879. [DOI] [PubMed] [Google Scholar]
- 12.Zainuddin, E.N., S. Mundt, U. Wegner, and R. Mentel. 2002. Cyanobacteria a potential source of antiviral substances against influenza virus. Med. Microbiol. Immunol. (Berl.). 191:181–182. [DOI] [PubMed] [Google Scholar]
- 13.Garbacki, N., V. Gloaguen, J. Damas, L. Hoffmann, M. Tits, and L. Angenot. 2000. Inhibition of croton oil-induced oedema in mice ear skin by capsular polysaccharides from cyanobacteria. Naunyn Schmiedebergs Arch. Pharmacol. 361:460–464. [DOI] [PubMed] [Google Scholar]
- 14.Prinsep, M.R., R.A. Thomson, M.L. West, and B.L. Wylie. 1996. Tolypodiol, an antiinflammatory diterpenoid from the cyanobacterium Tolypothrix nodosa. J. Nat. Prod. 59:786–788. [DOI] [PubMed] [Google Scholar]
- 15.Buttke, T.M., and L.O. Ingram. 1975. Comparison of lipopolysaccharides from Agmenellum quadruplicatum to Escherichia coli and Salmonella typhimurium by using thin-layer chromatography. J. Bacteriol. 124:1566–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keleti, G., and J.L. Sykora. 1982. Production and properties of cyanobacterial endotoxins. Appl. Environ. Microbiol. 43:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keleti, G., J.L. Sykora, E.C. Lippy, and M.A. Shapiro. 1979. Composition and biological properties of lipopolysaccharides isolated from Schizothrix calcicola (Ag.) Gomont (Cyanobacteria). Appl. Environ. Microbiol. 38:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikheyskaya, L.V., R.G. Ovodova, and Y.S. Ovodov. 1977. Isolation and characterization of lipopolysaccharides from cell walls of blue-green algae of the genus Phormidium. J. Bacteriol. 130:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weckesser, J., A. Katz, G. Drews, H. Mayer, and I. Fromme. 1974. Lipopolysaccharide containing l-acofriose in the filamentous blue-green alga Anabaena variabilis. J. Bacteriol. 120:672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pomati, F., S. Sacchi, C. Rossetti, S. Giovannardi, H. Onodera, Y. Oshima, and B.A. Neilan. 2000. The freshwater cyanobacterium Planktothrix sp. FP1: molecular identification and detection of paralytic shellfish poisoning toxins. J. Phycol. 36:553–563. [DOI] [PubMed] [Google Scholar]
- 21.Yi, E.C., and M. Hackett. 2000. Rapid isolation method for lipopolysaccharide and lipid A from gram-negative bacteria. Analyst. 125:651–656. [DOI] [PubMed] [Google Scholar]
- 22.Kontoyiannis, D., M. Pasparakis, T.T. Pizarro, F. Cominelli, and G. Kollias. 1999. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 10:387–398. [DOI] [PubMed] [Google Scholar]
- 23.Hilkens, C.M., P. Kalinski, M. de Boer, and M.L. Kapsenberg. 1997. Human dendritic cells require exogenous interleukin-12-inducing factors to direct the development of naive T-helper cells toward the Th1 phenotype. Blood. 90:1920–1926. [PubMed] [Google Scholar]
- 24.Snijders, A., P. Kalinski, C.M. Hilkens, and M.L. Kapsenberg. 1998. High-level IL-12 production by human dendritic cells requires two signals. Int. Immunol. 10:1593–1598. [DOI] [PubMed] [Google Scholar]
- 25.Napolitani, G., A. Rinaldi, F. Bertoni, F. Sallusto, and A. Lanzavecchia. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagai, Y., S. Akashi, M. Nagafuku, M. Ogata, Y. Iwakura, S. Akira, T. Kitamura, A. Kosugi, M. Kimoto, and K. Miyake. 2002. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat. Immunol. 3:667–672. [DOI] [PubMed] [Google Scholar]
- 27.Poltorak, A., X. He, I. Smirnova, M.Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 28.Shimazu, R., S. Akashi, H. Ogata, Y. Nagai, K. Fukudome, K. Miyake, and M. Kimoto. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189:1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medzhitov, R., P. Preston-Hurlburt, and C.A. Janeway Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388:394–397. [DOI] [PubMed] [Google Scholar]
- 30.Viriyakosol, S., P.S. Tobias, R.L. Kitchens, and T.N. Kirkland. 2001. MD-2 binds to bacterial lipopolysaccharide. J. Biol. Chem. 276:38044–38051. [DOI] [PubMed] [Google Scholar]
- 31.Visintin, A., K.A. Halmen, E. Latz, B.G. Monks, and D.T. Golenbock. 2005. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J. Immunol. 175:6465–6472. [DOI] [PubMed] [Google Scholar]
- 32.Barton, G.M., J.C. Kagan, and R. Medzhitov. 2006. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 7:49–56. [DOI] [PubMed] [Google Scholar]
- 33.Muzio, M., G. Natoli, S. Saccani, M. Levrero, and A. Mantovani. 1998. The human Toll signaling pathway: divergence of nuclear factor κB and JNK/SAPK activation upstream of tumor necrosis factor receptor–associated factor 6 (TRAF6). J. Exp. Med. 187:2097–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511. [DOI] [PubMed] [Google Scholar]
- 35.Cohen, J. 2002. The immunopathogenesis of sepsis. Nature. 420:885–891. [DOI] [PubMed] [Google Scholar]
- 36.Golenbock, D.T., R.Y. Hampton, N. Qureshi, K. Takayama, and C.R. Raetz. 1991. Lipid A-like molecules that antagonize the effects of endotoxins on human monocytes. J. Biol. Chem. 266:19490–19498. [PubMed] [Google Scholar]
- 37.Mignon, A., N. Rouquet, M. Fabre, S. Martin, J.C. Pages, J.F. Dhainaut, A. Kahn, P. Briand, and V. Joulin. 1999. LPS challenge in d-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am. J. Respir. Crit. Care Med. 159:1308–1315. [DOI] [PubMed] [Google Scholar]
- 38.Lynn, M., D.P. Rossignol, J.L. Wheeler, R.J. Kao, C.A. Perdomo, R. Noveck, R. Vargas, T. D'Angelo, S. Gotzkowsky, and F.G. McMahon. 2003. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J. Infect. Dis. 187:631–639. [DOI] [PubMed] [Google Scholar]
- 39.Ondiveeran, H.K., and A. Fox-Robichaud. 2004. Drug evaluation: E-5564. IDrugs. 7:582–590. [PubMed] [Google Scholar]
- 40.Christ, W.J., O. Asano, A.L. Robidoux, M. Perez, Y. Wang, G.R. Dubuc, W.E. Gavin, L.D. Hawkins, P.D. McGuinness, M.A. Mullarkey, et al. 1995. E5531, a pure endotoxin antagonist of high potency. Science. 268:80–83. [DOI] [PubMed] [Google Scholar]
- 41.Kitchens, R.L., and R.S. Munford. 1995. Enzymatically deacylated lipopolysaccharide (LPS) can antagonize LPS at multiple sites in the LPS recognition pathway. J. Biol. Chem. 270:9904–9910. [DOI] [PubMed] [Google Scholar]
- 42.Lepper, P.M., M. Triantafilou, C. Schumann, E.M. Schneider, and K. Triantafilou. 2005. Lipopolysaccharides from Helicobacter pylori can act as antagonists for Toll-like receptor 4. Cell. Microbiol. 7:519–528. [DOI] [PubMed] [Google Scholar]
- 43.Loppnow, H., P. Libby, M. Freudenberg, J.H. Krauss, J. Weckesser, and H. Mayer. 1990. Cytokine induction by lipopolysaccharide (LPS) corresponds to lethal toxicity and is inhibited by nontoxic Rhodobacter capsulatus LPS. Infect. Immun. 58:3743–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuura, M., M. Kiso, and A. Hasegawa. 1999. Activity of monosaccharide lipid A analogues in human monocytic cells as agonists or antagonists of bacterial lipopolysaccharide. Infect. Immun. 67:6286–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato, K., Y.C. Yoo, A. Fukushima, I. Saiki, T.A. Takahashi, M. Fujihara, S. Tono-Oka, and I. Azuma. 1995. A novel synthetic lipid A analog with low endotoxicity, DT-5461, prevents lethal endotoxemia. Infect. Immun. 63:2859–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scott, M.G., D.J. Davidson, M.R. Gold, D. Bowdish, and R.E. Hancock. 2002. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 169:3883–3891. [DOI] [PubMed] [Google Scholar]
- 47.Yang, Q.W., L. Mou, F.L. Lv, P.F. Zhu, Z.G. Wang, J.X. Jiang, and J.Z. Wang. 2005. Novel TLR4-antagonizing peptides inhibit LPS-induced release of inflammatory mediators by monocytes. Biochem. Biophys. Res. Commun. 329:846–854. [DOI] [PubMed] [Google Scholar]
- 48.Yoshimura, A., T. Kaneko, Y. Kato, D.T. Golenbock, and Y. Hara. 2002. Lipopolysaccharides from periodontopathic bacteria Porphyromonas gingivalis and Capnocytophaga ochracea are antagonists for human toll-like receptor 4. Infect. Immun. 70:218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rossignol, D.P., and M. Lynn. 2002. Antagonism of in vivo and ex vivo response to endotoxin by E5564, a synthetic lipid A analogue. J. Endotoxin Res. 8:483–488. [DOI] [PubMed] [Google Scholar]
- 50.Morrison, D.C., and D.M. Jacobs. 1976. Binding of polymyxin B to the lipid A portion of bacterial lipopolysaccharides. Immunochemistry. 13:813–818. [DOI] [PubMed] [Google Scholar]
- 51.Rustici, A., M. Velucchi, R. Faggioni, M. Sironi, P. Ghezzi, S. Quataert, B. Green, and M. Porro. 1993. Molecular mapping and detoxification of the lipid A binding site by synthetic peptides. Science. 259:361–365. [DOI] [PubMed] [Google Scholar]
- 52.Taylor, A.H., G. Heavner, M. Nedelman, D. Sherris, E. Brunt, D. Knight, and J. Ghrayeb. 1995. Lipopolysaccharide (LPS) neutralizing peptides reveal a lipid A binding site of LPS binding protein. J. Biol. Chem. 270:17934–17938. [DOI] [PubMed] [Google Scholar]
- 53.Arrighi, J.F., M. Rebsamen, F. Rousset, V. Kindler, and C. Hauser. 2001. A critical role for p38 mitogen-activated protein kinase in the maturation of human blood-derived dendritic cells induced by lipopolysaccharide, TNF-alpha, and contact sensitizers. J. Immunol. 166:3837–3845. [DOI] [PubMed] [Google Scholar]
- 54.Puig-Kroger, A., M. Relloso, O. Fernandez-Capetillo, A. Zubiaga, A. Silva, C. Bernabeu, and A.L. Corbi. 2001. Extracellular signal-regulated protein kinase signaling pathway negatively regulates the phenotypic and functional maturation of monocyte-derived human dendritic cells. Blood. 98:2175–2182. [DOI] [PubMed] [Google Scholar]
- 55.Nakahara, T., H. Uchi, K. Urabe, Q. Chen, M. Furue, and Y. Moroi. 2004. Role of c-Jun N-terminal kinase on lipopolysaccharide induced maturation of human monocyte-derived dendritic cells. Int. Immunol. 16:1701–1709. [DOI] [PubMed] [Google Scholar]
- 56.Saccani, S., S. Pantano, and G. Natoli. 2001. Two waves of nuclear factor kappaB recruitment to target promoters. J. Exp. Med. 193:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann, A., A. Levchenko, M.L. Scott, and D. Baltimore. 2002. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 298:1241–1245. [DOI] [PubMed] [Google Scholar]
- 58.Covert, M.W., T.H. Leung, J.E. Gaston, and D. Baltimore. 2005. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 309:1854–1857. [DOI] [PubMed] [Google Scholar]
- 59.Sporri, R., and C. Reis e Sousa. 2005. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat. Immunol. 6:163–170. [DOI] [PubMed] [Google Scholar]
- 60.Pasare, C., and R. Medzhitov. 2004. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 21:733–741. [DOI] [PubMed] [Google Scholar]
- 61.Lutz, M.B., and G. Schuler. 2002. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 23:445–449. [DOI] [PubMed] [Google Scholar]
- 62.Masternak, K., N. Peyraud, M. Krawczyk, E. Barras, and W. Reith. 2003. Chromatin remodeling and extragenic transcription at the MHC class II locus control region. Nat. Immunol. 4:132–137. [DOI] [PubMed] [Google Scholar]
- 63.da Silva Correia, J., and R.J. Ulevitch. 2002. MD-2 and TLR4 N-linked glycosylations are important for a functional lipopolysaccharide receptor. J. Biol. Chem. 277:1845–1854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}