

Abstract
Interferon (IFN)-γ, a cytokine critical for resistance to infection and tumors, is produced by CD4+ helper T lymphocytes after stimulation by cultured dendritic cells (DCs) that secrete a cofactor, interleukin (IL)-12. We have identified a major IL-12–independent pathway whereby DCs induce IFN-γ–secreting T helper (Th)1 CD4+ T cells in vivo. This pathway requires the membrane-associated tumor necrosis family member CD70 and was identified by targeting the LACK antigen from Leishmania major within an antibody to CD205 (DEC-205), an uptake receptor on a subset of DCs. Another major DC subset, targeted with 33D1 anti-DCIR2 antibody, also induced IFN-γ in vivo but required IL-12, not CD70. Isolated CD205+ DCs expressed cell surface CD70 when presenting antigen to T cell receptor transgenic T cells, and this distinction was independent of maturation stimuli. CD70 was also essential for CD205+ DC function in vivo. Detection of the IL-12–independent IFN-γ pathway was obscured with nontargeted LACK, which was presented by both DC subsets. This in situ analysis points to CD70 as a decision maker for Th1 differentiation by CD205+ DCs, even in Th2-prone BALB/c animals and potentially in vaccine design. The results indicate that two DC subsets have innate propensities to differentially affect the Th1/Th2 balance in vivo and by distinct mechanisms.
T cell–mediated immunity requires that T lymphocytes undergo both clonal expansion and differentiation to produce bioactive cytokines and cytolysins. The cytokine IFN-γ plays a major role in protection against intracellular pathogens (1–3) and tumors (4) and is produced in large amounts by helper CD4+ T lymphocytes. When T cells differentiate to produce IFN-γ exclusively, they are termed Th1 cells, whereas those that produce IL-4, IL-5, and IL-13, which resist helminth infection and mediate allergy, are termed Th2 (for review see references 5 and 6). Thus, the nature of the cytokines produced by T cells significantly impacts upon host resistance and immunopathology (2, 7).
DCs are specialized antigen-presenting cells that initiate immunity (8, 9). This is illustrated by the fact that the selective targeting of antigens to DCs in immune or lymphoid organs of mice markedly improves the efficiency of clonal expansion (10, 11) and protective immunity (12, 13). DCs support several distinct pathways of T cell differentiation in tissue culture, such as Th1 and Th2 (14). Their capacity to induce IFN-γ in CD4+ T cells is ascribed to secretion of an important cofactor, IL-12 (15–17; for review see references 2, 8, and 18).
The biology of the DC–T cell interaction is also influenced by the existence of different types of DCs, or DC subsets. One expresses the endocytic receptor DEC-205 (here “DEC”; reference 19) as well as a marker of unknown function, CD8αα (20). CD8+ or DEC+ DCs mediate the presentation of antigens on both MHC class I and II products, leading to clonal expansion of killer and Th cells, respectively (21–23). Another DC subset expresses a distinct uptake receptor, DCIR2, recognized by the 33D1 mAb (24, 25) and lacks CD8αα. It primarily expands CD4+ helper lymphocytes (26). When antigen is taken up by isolated DEC+ DCs, they trigger T cells to produce primarily IFN-γ, whereas DEC− DCs induce either IL-4 exclusively or both IFN-γ and IL-4 (27, 28). Consistent with these observations, the IL-12 cofactor for IFN-γ production is produced at higher levels by DEC+ DCs (15, 18, 29, 30).
The function of DC subsets in the induction of cytokine-producing T cells has yet to be analyzed directly in intact lymphoid tissues. A significant enigma is that patients and mice that are genetically deficient in IL-12 or IL-12Rβ1 retain the capacity to produce low levels of IFN-γ and to resist intracellular infections (31–37). These observations indicate the existence of an alternative pathway for IFN-γ induction (for review see reference 38). Here we separately target antigen in vivo to two major subsets of DCs, using the antigen LACK (39) from the parasite Leishmania major. L. major infection of mice is an experimental model that has helped unravel Th1/Th2 CD4+ differentiation and the role of IL-12 in vivo (40–42; for review see reference 43). Surprisingly, we find that the DEC+ subset, which proves to be the more powerful inducer of IFN-γ in vivo, does so in the absence of IL-12. Instead, the membrane-bound cofactor CD70 is vital. These findings outline a new route to the production of a pivotal protective cytokine, IFN-γ, and contrast the mechanisms used by DC subsets to initiate immunity.
RESULTS
Efficient antigen presentation after the targeting of LACK to DC subsets
To examine the role of two major DC subsets in vivo, we selectively delivered the L. major LACK antigen (39) to DEC+ 33D1− and DEC− 33D1+ DCs by producing chimeric anti-DEC and 33D1 LACK chimeric mAbs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070176/DC1). We injected the chimeric LACK mAbs s.c. or i.p., together with the maturation stimuli αCD40 and poly IC, and used LACK-specific TCR transgenic CD4+ T cells as reporters for the successful capture and processing of antigen by DCs in vivo. We found that CD11c+ cells from either lymph nodes (Fig. S2) or spleen (not depicted) were selectively responsible for LACK presentation. When the two subsets of splenic CD11c+ DCs were separated on the basis of DEC expression, the DEC+ DCs exclusively stimulated clonal expansion after injecting DEC-LACK, but not 33D1-LACK, whereas the DEC− DCs were exclusively active after injection of 33D1-LACK (Fig. 1 a), each over a wide range of DC to T cell doses (1:3–1:27; not depicted). In contrast, soluble LACK was presented by both DC subsets, and there was no presentation of control Ig-LACK (Fig. 1 a). To assess antigen presentation in vivo, we adoptively transferred CFSE-labeled, allotype (thy-1.1) -marked T cells into the LACK-targeted mice. Both chimeric mAbs were at least 100-fold more efficient than soluble LACK at initiating clonal expansion in vivo (Fig. 1 b, left); e.g., 0.03 μg of each fusion mAb was more effective in triggering CFSE dilution than 3 μg of nontargeted LACK (Fig. 1 b, right). Thus, the DEC and 33D1 mAbs faithfully and efficiently deliver LACK for presentation by the appropriate DCs, whereas the more standard use of nontargeted antigen leads to presentation by both subsets.
Figure 1.

αDEC and 33D1 mAbs target LACK in vivo to distinct DC subsets. (A) BALB/c mice (five per condition) were immunized i.p. with αDEC-LACK, 33D1-LACK, LACK, and Ig-LACK in the presence of αCD40 and poly IC, or with PBS. 12 h later, splenocytes were enriched for CD11c+ cells by MACS+ selection and separated into DEC+ and DEC− subsets (see Materials and methods). The DCs were added to thy-1.1+ CFSE-labeled, LACK-specific TCR transgenic T cells, and proliferation was assessed by CFSE dilution 3.5 d later. Representative of three experiments. (B) 3 × 106 CFSE-labeled TCR transgenic cells as in A were administered i.v. to thy-1.2+ mice 1 d before immunization i.p. with graded doses of αDEC-LACK, 33D1-LACK, Ig-LACK, or LACK protein. Spleens were harvested at 3 d to enumerate thy-1.1+ T cells that had diluted CFSE. The expansion of transgenic T cells is displayed as total cell numbers on the left, and representative CFSE dilution plots, gated on thy-1.1+ LACK transgenic CD4+ T cells in response to the indicated doses of fusion mAb or LACK protein, are on the right.
Contrasting pathways of T cell differentiation via DC subsets
To examine T cell differentiation in a primary immune response to targeted antigen, we measured IFN-γ production by intracellular cytokine staining. All forms of LACK induced IFN-γ as long as a DC maturation stimulus was also administered (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070176/DC1; references 13 and 44). Consistent with prior research (45), we found that the combination of agonistic anti-CD40 and TLR3 ligand, poly IC, was the most effective stimulus for a primary response to all LACK antigen immunization forms (Fig. S3). Poly IC by itself induced IFN-γ production only when LACK antigen was targeted to DEC+ DCs (Fig. S3). However, poly IC did act synergistically with αCD40 to increase IFN-γ secretion when DEC− DCs were targeted with 33D1-LACK (Fig. S3). The targeting of LACK to maturing DCs enhanced immunization relative to soluble LACK or Ig-LACK control, with DEC-LACK being more effective than 33D1-LACK (Fig. S4). The peptides recognized in the immunized mice (Fig. S5) contained the LACK 158–173 sequence ICFSPSLEHVSGSWD, previously defined to be presented on the I-Ad MHC class II molecule of BALB/c mice (39). These initial data showed that targeting to either DC subset resulted in IFN-γ production in vivo.
We next compared the production of IFN-γ to IL-4 after the targeting of antigens to maturing DC subsets. DEC-LACK immunization exclusively primed for IFN-γ and not IL-4, even though the BALB/c mice we used were prone to produce Th2 responses (Fig. 2 a). In contrast, soluble LACK and 33D1-LACK induced IL-4 and less IFN-γ (Fig. 2 a). Similar results were obtained when we replaced poly IC with LPS (not depicted). These distinct outcomes were corroborated by the isotype of the anti-LACK antibody response. After DEC-LACK immunization, mice primarily produced antibodies of the IgG2a isotype, which is favored by IFN-γ–producing helper CD4+ T cells, whereas 33D1 and soluble LACK induced several isotypes, including particularly high titers of IL-4–dependent IgG1 antibodies (Fig. 2 b). To verify our results with monoclonal T cells, we adoptively transferred specific thy-1.1-marked TCR transgenic T cells into mice that were primed with different forms of LACK. Again, DEC-LACK induced IFN-γ, but not IL-4, whereas 33D1-LACK and soluble LACK induced both IFN-γ and IL-4 (Fig. 2 c). Leishmania organisms produced a strong but exclusive IL-4 response, typical of BALB/c mice (Fig. 2 c). Therefore, in vivo targeting of antigen to different DC subsets leads to different T cell outcomes.
Figure 2.

Contrasting cytokine production by T cells immunized with different forms of LACK. (A) BALB/c mice were injected i.p. with PBS, 10 μg αDEC-LACK, 10 μg 33D1-LACK, 10 μg control Ig-LACK, or 30 μg LACK in the presence of αCD40 and poly IC. 3 wk later, splenocytes were enriched for CD4+ cells (MACS selection) and restimulated with CD11c+ cells plus LACK-dominant peptide. 2 d later, the numbers of IFN-γ– (left) and IL-4– (right) producing cells were revealed by ELISPOT. Three experiments with two mice per group. (B) As in A, but mice were both primed and boosted at 3 wk with different forms of LACK. 6 d later, anti-LACK–specific antibody titers and isotypes were quantified by ELISA. Each symbol is an individual experiment. (C) 3 × 106 CFSE-labeled, thy-1.1+ LACK-specific TCR transgenic cells were transferred i.v. into thy-1.2+ BALB/c mice. 1 d later, the mice were immunized i.p. with the different LACK antigen forms in the presence of αCD40 and poly IC, 106 L. major metacyclic promastigotes, or PBS. 6 d later, the priming of LACK-specific transgenic T cells was assessed for intracellular IFN-γ production by gating on thy-1.1+ splenocytes (left), or for IL-4 production by ELISPOT of positively selected thy-1.1+ splenocytes restimulated for 4 h in the presence of PMA and ionomycin (right). Representative of three experiments.
IL-12–independent IFN-γ production via DEC+ DCs
IL-12 p40/p35 heterodimer is a cofactor for the differentiation of IFN-γ–producing cells (16, 46, 47). To determine the role of IL-12 in T cell differentiation after antigen targeting to DCs, we immunized IL-12 p40 mutant mice (IL-12 p40−/−). Surprisingly, DEC-LACK immunization of IL-12 p40−/− mice induced IFN-γ secretion and no IL-4, and the frequency of IFN-γ–producing cells was comparable to heterozygous littermates (Fig. 3 a) and WT BALB/c mice (not depicted). This was the case over a wide range of doses of DEC-LACK (Fig. 3 b). However, IFN-γ production was ablated in IL-12 p40−/− mice that were immunized with 33D1-LACK, whereas IL-4 was made in increased amounts. Soluble LACK induced less IFN-γ but more IL-4 in IL-12 p40−/− mice (Fig. 3 a), as expected from its targeting to both DC subsets. To determine if these results were restricted to the LACK antigen, we repeated the same experiment using circumsporozoite protein from Plasmodia as described previously (44). DEC-CS, but not 33D1-CS, again induced IFN-γ in an IL-12–independent manner (Fig. 3 c). Thus, in contrast to expectations from prior research with isolated DCs, DEC+ DCs induced IFN-γ in the absence of IL-12 in vivo.
Figure 3.

αDEC-LACK induces IFN-γ secretion by an IL-12–independent pathway. (A) IL-12p40−/− mice and control littermates were immunized i.p. as indicated in Fig. 2 and on the x axis. 3 wk later, the frequency of IFN-γ+ CD4+ CD3+ splenic T cells was determined by intracellular staining (left), or CD4+ splenocytes were restimulated in vitro with CD11c+ cells, medium, or LACK peptide for 36–48 h to measure IL-4– secreting cells by ELISPOT (right). (B) As in A, but each row shows a different dose of the different forms of LACK antigen. (C) As in A, but WT and IL-12 KO mice were immunized with P. yoelli circumsporozoite protein (CSP), either 10 μg DEC-CSP, 10 μg 33D1-CSP, and 10 μg Ig-CSP in the presence of αCD40 and poly IC, or PBS. Representative of three, three, and two individual experiments with two pooled mice per condition, respectively.
DEC− DCs obtain IL-12 from DEC+ DCs to induce IFN-γ
To further analyze the function of isolated DC subsets, we injected mice with different forms of LACK, separated the DEC+ and DEC− DCs, and tested their capacity to differentiate TCR transgenic T cells in vitro. After selective targeting, both DC subsets induced comparable levels of IL-2 (Fig. 4 a, top left), indicating that both types of DCs were presenting antigen effectively. The DEC+ DCs induced the production of IFN-γ (Fig. 4 a, top right), but the isolated DEC− DCs no longer stimulated IFN-γ production and only induced IL-4 and IL-5 (Fig. 4 a, bottom). These data confirm prior work with CD8+ and CD8− DC subsets targeted with antigen in vitro (27, 28).
Figure 4.

DEC− DCs differentiate Th2 type CD4+ T cells but prime IFN-γ secretion with IL-12–bearing DEC+ DCs. (A) BALB/c mice were immunized i.p. as indicated in Fig. 2 and the right inset for 10–12 h. DEC+ and DEC− subsets were sorted from spleens and added for 3.5 d to thy-1.1+ CFSE-labeled, LACK-specific TCR transgenic cells. IL-2, IFN-γ, IL-4, and IL-5 production were measured in the supernatants by ELISA. The symbols represent individual experiments with five mice per condition, and the horizontal bars represent the mean between the different experiments. (B) As in A, but the DC subsets from either WT (shown) or IL-12−/− mice (see Fig. S6) were used to stimulate T cell proliferation (CFSE dilution, x axis) as well as IFN-γ production (y axis; monensin was added overnight in the absence of any restimulation at day 3). (C) DEC− DCs were isolated from mice given 33D1-LACK, LACK in the presence of αCD40 and poly IC, or PBS 12 h earlier (same cells as B), but we then added an equal number of DCs from WT (WT) and IL-12p40−/− mice as indicated on the x axis. The donor mice had been given anti-CD40 and poly IC, but no LACK antigen, 12 h beforehand (mature), or just PBS. As in B, T cell proliferation and IFN-γ production were monitored. One of two similar experiments.
When we compared DCs from WT and IL-12 p40−/− mice, both subsets induced vigorous transgenic T cell proliferation, as shown by CFSE dilution (Fig. 4 b and Fig. S6, x axis, which is available at http://www.jem.org/cgi/content/full/jem.20070176/DC1). However, IFN-γ was only produced when the T cells were cultured with DEC+ DCs, and this was the case with DCs from either WT (Fig. 4 b) or IL-12 p40−/− mice (Fig. S6, y axis). When we injected soluble LACK, both DC subsets induced strong T cell proliferation, but again only DEC+ DCs induced IFN-γ (Fig. 4 b and Fig. S6). Similar results were obtained whether we harvested the DCs from mice that did not receive a maturation stimulus, or when the maturation stimulus was anti-CD40, poly IC, LPS, or both poly IC or LPS and anti-CD40 (not depicted). The fact that only the DEC+ DCs induced IFN-γ secretion after soluble LACK protein presentation strongly suggests that the capacity to induce IFN-γ is a property of the DC subset and not the receptor (DEC205 or DCIR2) that was targeted.
Because isolated DEC− DCs did not induce IFN-γ in vitro (Fig. 4, a and b) but did so in vivo (Fig. 3, a–c), we hypothesized that these DCs obtained the required IL-12 from DEC+ DCs. DEC+ DCs are the major producer of IL-12 (15, 18, 29, 30). We found that 105 DEC+ DCs from mice injected with αCD40 and poly IC secreted 805 pg/ml IL-12 (against a background of 64 pg/ml) when cultured for 2 d, whereas DEC− DCs produced 237 pg/ml (not depicted). We therefore repeated the in vitro experiments with the antigen targeted to WT DEC− DCs, but we added WT or IL-12−/− DEC+ DCs from mice given only anti-CD40 and poly IC (without any LACK antigen). Supplementation of the antigen-targeted DEC− DCs with antigen-free DEC+ DCs restored IFN-γ production, but only if the DCs were mature and from WT mice (Fig. 4 c, left). These data indicate that maturing DEC+ DCs produce sufficient IL-12 to allow DEC− DCs to induce IFN-γ.
CD70 is essential for the function of DEC+ DCs in vitro
To determine how IL-12 p40−/− DEC+ DCs induced IFN-γ, we considered CD70, a member of the TNF family of costimulatory molecules (for review see reference 48). We had found CD70 to be expressed in ongoing studies of DCs maturing in vivo in response to NKT cells (49). In mice injected for 12 h with poly IC and anti-CD40, low levels of CD70 were expressed intracellularly, but not at the cell surface (not depicted), by both DEC+ and DEC− DCs (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20070176/DC1). Surprisingly, 1 d after DC subsets were added to LACK-specific TCR transgenic T cells in culture, CD70 was up-regulated and expressed at the cell surface, but exclusively by antigen-targeted DEC+ DCs (Fig. 5 a). Another TNF family member, 4-1BBL, was not detected on the same DCs (not depicted). We then found that it was not necessary to administer maturation stimuli to mice for antigen-targeted DEC+ DCs to up-regulate CD70 upon co-culture with T cells. These data indicate that surface expression of CD70 is a property of DEC+ DCs presenting antigen to T cells.
Figure 5.

CD70 mediates the stimulatory function of DEC+ DCs, but not DEC− DCs. (A) Mice were immunized i.p. as in Fig. 2 and as shown at the top for 10–12 h. Sorted DEC+ and DEC− subsets were added for 1 d to thy-1.1+ CFSE-labeled, LACK-specific TCR transgenic cells, and the CD11c+ DCs (the only CD70+ cells in the cultures) were monitored by FACS for reactivity to αCD70 (dotted line) or isotype control mAb (gray). (B) As in A, but the cells were cultured for 3.5 d in the presence of αCD70 or control IgG2b (1 μg/ml). Inhibition of T cell growth, assessed by CFSE dilution, is noted with arrowhead and arrows. (C) As in B, but BALB/c mice were injected with αDEC-LACK alone and in the presence of αCD40, poly IC, or both. IFN-γ production by proliferating LACK transgenic cells was determined by intracellular cytokine staining. (D) As in B, but T cell activation was monitored by CD69 staining at days 1.5 and 3.5. (E) As in B, but αCD70 was added 1 d after co-culture. B–E are representative of four, two, five, and two experiments, respectively, with five mice per condition.
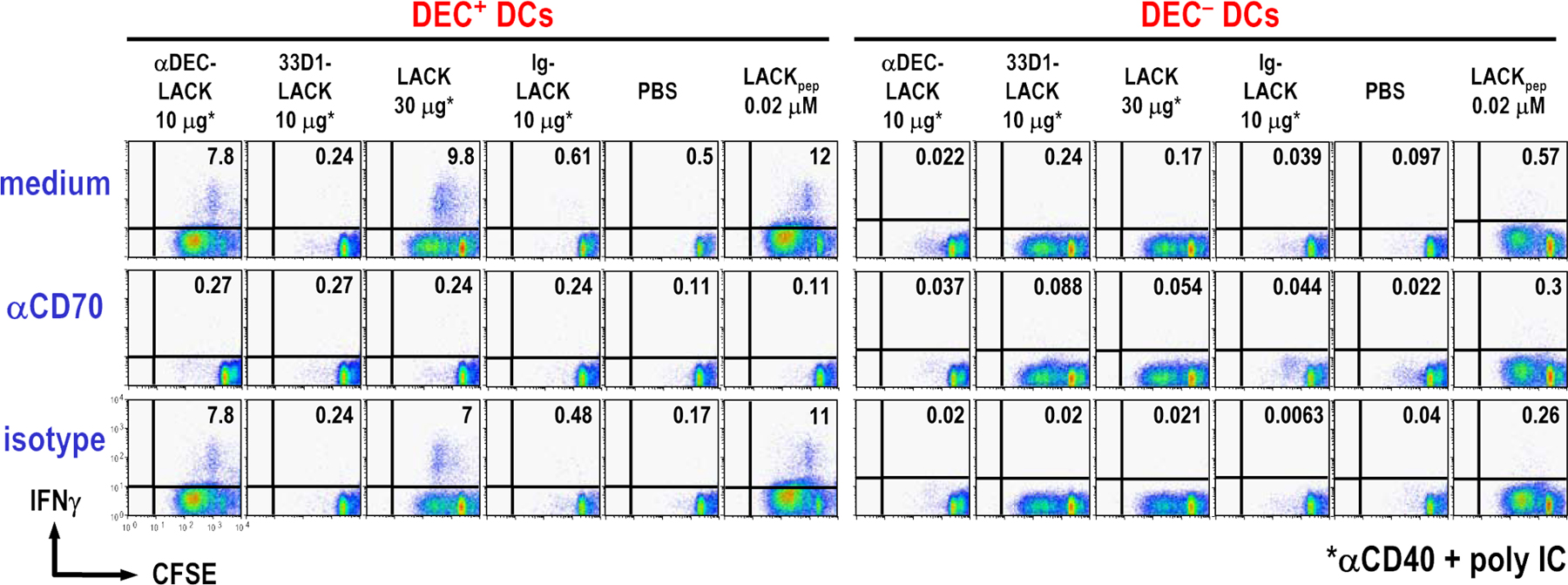
We next tested the effect of a blocking mAb to CD70 (50) on co-cultures of DCs and LACK-specific TCR transgenic T cells. The anti-CD70 mAb, but not an isotype control (or two other isotype-matched mAbs that reacted with DCs, anti-CD8 and anti-FcγR; not depicted), completely blocked T cell proliferation induced by DEC+ DCs (Fig. 5 b, left, arrow) as well as the production of IFN-γ (not depicted). This was the case when DEC+ DCs had captured DEC-LACK or soluble LACK in vivo (Fig. 5 b, arrows), or when DEC+ DCs were presenting LACK peptide in vitro (Fig. 5 b, arrowhead). In contrast, anti-CD70 did not block presentation by DEC− DCs targeted with 33D1-LACK, soluble LACK, or LACK peptide (Fig. 5 b). Identical results were obtained with DCs from IL-12 p40−/− mice; i.e., DEC+ DCs required CD70 to stimulate T cell proliferation and IFN-γ production (Fig. S6). We also found that in vivo–targeted DEC+ DCs did not require TLR or CD40 engagement to efficiently induce IFN-γ production from LACK transgenic T cells in a CD70-dependent manner (Fig. 5 c). Therefore, CD70 is required by DEC+ DCs to stimulate T cell responses, but not by DEC− DCs, even when the two DC subsets are targeted with the same form of antigen, LACK protein, or LACK peptide.
To further address the consequences of CD70 blockade, we first verified that αCD70 did not alter the number of DCs in the culture (not depicted) or inhibit the early steps in antigen presentation because the CD69 activation antigen was up-regulated normally when αCD70 was present for 1.5 or 3.5 d (Fig. 5 d). To test if αCD70 could block IFN-γ production after T cell proliferation had taken place, we added the blocking mAb to co-cultures of DC subsets and LACK transgenic T cells at 1 (Fig. 5 e) or 2 d after the initiation of the DC–T cell co-culture (not depicted). αCD70 at these later time points did not block T cell proliferation (Fig. 5 e). However, it completely inhibited IFN- γ induction by antigen-bearing DEC+ DCs, but not IL-4 production induced by DEC− DCs (Table I), showing that CD70 contributes to both T cell proliferation and IFN-γ production by DEC+ DCs.
Table I.
αCD70 blocks IFN-γ secretion by LACK transgenic cells primed by DEC+ DCs, but not by DEC− DCs
| Form of LACK antigen to load DCs | ||||||||
|---|---|---|---|---|---|---|---|---|
| DC Subset | Cytokine pg/ml | Blocking mAb | αDEC-LACK | 33D1-LACK | Ig-LACK | LACK | None | LACKpep
0.02 μM |
| DEC+ DCs | IFN-γ | IgG2b | 2,048 | 20 | −66 | 463 | 4 | 2,189 |
| αCD70 | 51 | 34 | 16 | −76 | −61 | 23 | ||
| IL-4 | IgG2b | −41 | −89 | −92 | −77 | −90 | −30 | |
| αCD70 | −29 | −71 | −89 | −70 | −93 | −79 | ||
| DEC− DCs | IFN-γ | IgG2b | 13 | −71 | 25 | −76 | −44 | 80 |
| αCD70 | 15 | 19 | 23 | 11 | −60 | 25 | ||
| IL-4 | IgG2b | −54 | 471 | −64 | 406 | −96 | 485 | |
| αCD70 | −43 | 498 | −70 | 646 | −79 | 602 | ||
BALB/c mice (five mice per condition) were immunized for 10–12 h with 10 μg αDEC-LACK, 10 μg 33D1-LACK, 10 μg Ig-LACK, and 30 μg LACK protein in the presence of αCD40 and poly IC, or PBS. Spleens were harvested and teased in the presence of collagenase, and splenocytes were enriched for CD11c+ cells through MACS+ selection, labeled for CD11c, B220, CD3, and DEC205, and sorted. CD11c+ B220− CD3− DEC+/DEC− subsets were collected and cultured with thy-1.1+ CFSE-labeled LACK TCR transgenic cells at a 1:5 ratio. αCD70 or nonreactive isotype mAb (3 μg/ml) was added to the cultures of DEC+ and DEC− DCs with LACK TCR transgenic cells at day 1 of co-culture. After 3.5 d, T cell priming was assessed by IL-4 and IFN-γ secretion. Representative of two individual experiments.
To exclude the possibility that αCD70 could be acting on CD4+ T cells, we determined CD70 expression levels on LACK TCR transgenic cells 3 d after stimulation by DCs. Presentation by DC subsets loaded with either DEC-LACK or 33D1-LACK induced comparable expression of cell surface (not depicted) and intracellular CD70 by LACK transgenic cells in vivo (Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20070176/DC1). However, αCD70 only blocked stimulation by DEC+ DCs (Fig. 5, c and e), which selectively expressed cell surface CD70 (Fig. S7). Collectively, the data in Figs. 4 and 5 reveal that DEC+ DCs have two mechanisms to induce IFN-γ from CD4+ T cells: a direct pathway that requires CD70 but not IL-12, and an indirect pathway where DEC+ DCs provide IL-12 used by DEC− DCs.
Effect of exogenous IL-12 on T cell responses to DC subsets
To determine whether IL-12 might supersede the CD70–CD27 interaction for the induction of IFN-γ, we added 1 ng/ml IL-12 p70 to cultures containing DC subsets that had been targeted with LACK in vivo. 1 ng/ml of exogenous IL-12 increased the production of IFN-γ by T cells stimulated with either DEC− or DEC+ DCs (Table II, bold data). However, the addition of IL-12 did not overcome the block to T cell proliferation and IFN-γ secretion that was imposed by αCD70 (Table II).
Table II.
IL-12 boosts IFN-γ production in DC–T cell cultures but does not overcome the block imposed by anti-CD70 on DEC+ DC function
| Form of LACK antigen to load DCs (IFN-γ production at day 3.5 of DC–T cell co-culture; pg/ml)
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Additions to cultures of WT15 LACK-specific CD4+ transgenic T cells |
αDEC-LACK 10 μg |
33D1-LACK 10 μg |
Ig-LACK 10 μg |
LACK 30 μg |
None | LACKpep
0.02 μM |
||
| DEC+ DCs | no IL-12 | IgG2b | 1,022 | −118 | −174 | 478 | −161 | 824 |
| αCD70 | −150 | 0 | −33 | 5 | −151 | −4 | ||
| + IL-12 | IgG2b | 6,402 | −54 | 0 | 1,115 | 23 | 3,333 | |
| αCD70 | −53 | 0 | 3 | 10 | 15 | 27 | ||
| DEC− DCs | no IL-12 | IgG2b | 0 | −20 | 55 | 45 | −14 | 0 |
| 0 | −0.136 | 14 | 32 | 71 | −121 | 61 | ||
| + IL-12 | IgG2b | 57 | 2,613 | 37 | 3,421 | 6 | 3,251 | |
| αCD70 | 49 | 3,324 | −42 | 3,447 | 3 | 3,315 | ||
| Additions to cultures of WT15 LACK-specific CD4+ TCR transgenic T cells |
Form of LACK antigen to load DCs (IL-4 at day 3.5 in the medium of DC–T cell co-culture; pg/ml)
|
|||||||
| αDEC-LACK 10 μg |
33D1-LACK 10 μg |
Ig-LACK 10 μg |
LACK 30 μg |
None | LACKpep
0.02 μM |
|||
| DEC+ DCs | no IL-12 | IgG2b | 0 | 4 | 15 | −74 | −70 | −27 |
| αCD70 | −36 | 2 | 2 | 11 | −87 | −30 | ||
| + IL-12 | IgG2b | −90 | −95 | −100 | 4 | 0 | 1 | |
| αCD70 | −85 | −72 | −87 | 15 | 0 | −13 | ||
| DEC− DCs | no IL-12 | IgG2b | −60 | 334 | −94 | 223 | −98 | 422 |
| αCD70 | −36 | 284 | −98 | 358 | −70 | 451 | ||
| + IL-12 | IgG2b | −90 | 55 | −85 | 70 | −89 | 111 | |
| αCD70 | −85 | 66 | −99 | 64 | −87.4 | 79 | ||
BALB/c mice (five mice per condition) were immunized for 10–12 h with αDEC-LACK, 33D1-LACK, Ig-LACK, and LACK protein in the presence of αCD40 and poly IC, or PBS. Spleens were harvested and teased in the presence of collagenase, and splenocytes were enriched for CD11c+ cells through MACS+ selection, labeled for CD11c, B220, CD3, and DEC205, and sorted. CD11c+ B220− CD3− DEC+/DEC− subsets were collected and cultured with thy-1.1+ CFSE-labeled LACK TCR transgenic cells at a 1:5 ratio. 1 ng/ml IL-12 and αCD70 or 3 μg/ml of nonreactive isotype mAb was added to the cultures of DEC+ and DEC− DCs with LACK TCR transgenic cells. After 3.5 d, T cell priming was assessed by IFN-γ secretion. Representative of three individual experiments.
αCD70 blocks the function of DEC+ DCs in vivo
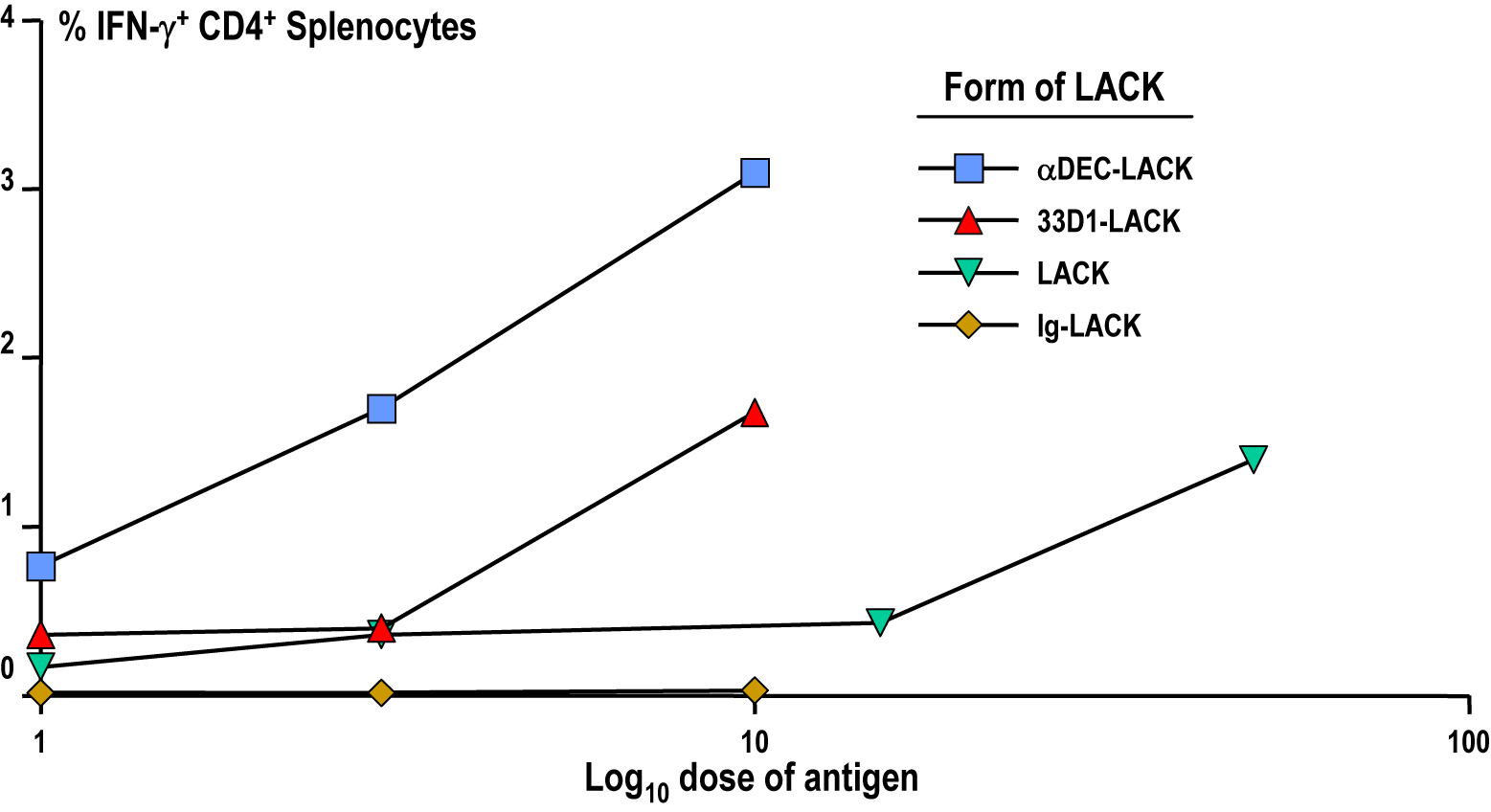
To assess the need for CD70 by DEC+ DCs in vivo, we treated mice with αCD70 or isotype-matched control Ig 1 d before immunization with LACK. CD70 blockade did not alter the frequency of DEC+ or DEC− DCs (not depicted) but did completely abrogate priming via DEC-LACK but not 33D1-LACK (Fig. 6). As expected from its targeting to both DC subsets, LACK priming was only partially reduced by blocking CD70 in vivo. Therefore, CD70 interaction plays a major role in vivo during presentation of antigen to CD4+ T cells by DEC+ DCs.
Figure 6.

Contributions of CD70 to IFN-γ secretion in response to DEC+ DCs in vivo. BALB/c mice were injected i.p. with 50 μg αCD70 mAb 1 d before i.p. immunization with 10 μg DEC-LACK, 10 μg 33D1-LACK, 50 μg LACK protein, and 10 μg Ig-LACK in the presence of αCD40 and poly IC, or PBS. 3 wk later, the frequency of IFN-γ+ CD4+ CD3+ splenic T cells was determined by intracellular staining. One of two similar experiments.
DISCUSSION
Lymphocytes must differentiate to produce effector proteins like IFN-γ to bring about protective and pathologic forms of cell-mediated immunity. By selectively targeting antigen in the presence of αCD40 and poly IC to two major subsets of DEC− and DEC+ DCs in situ, we find that there are two pathways to engage WT CD4+ T cells to make IFN-γ. DEC+ DCs are inherently disposed to induce IFN-γ in an IL-12–independent, CD70-dependent fashion. In contrast, DEC− DCs are disposed to induce IL-4 and IL-5, but will induce IFN-γ in a CD70-independent manner when exogenous IL-12 is provided, e.g., from DEC+ DCs. These distinctions in DC subset function, which were documented both in vivo and in vitro, can be ascribed to the different DC subsets rather than the different DC receptors (DCIR2 and DEC) that we targeted because comparable functional distinctions were made with soluble antigen, which gained access to both DC subsets and even isolated DCs presenting LACK peptide.
We considered the possibility that the observations were dependent on the type of maturation stimulus that was used. However, the distinct Th1- and Th2-polarizing functions of the two DC subsets were apparent when we obtained DCs from unstimulated mice, as documented previously by Pulendran (8) and Moser (9). Likewise, maturation stimuli were not required for the DC subsets to exhibit different requirements for IL-12 and CD70 in culture. In vivo, DEC+ DCs have higher levels of TLR3 message. However, our data showed that DEC− DCs were responsive to poly IC, which served as a suitable stimulus for immune responses when the 33D1 mAb targeted antigen to DEC− DCs (Fig. S3). Also, the distinct outcomes of antigen targeting to DC subsets in vivo were observed when LPS rather than poly IC was used. Therefore, the distinctions in the functions of DC subsets are intrinsic to these cells rather than the use of poly IC as a maturation stimulus.
Our finding that IL-12 is not mandatory for the Th1-inducing function of DEC+ DCs would help to explain why IL-12 p40 KO mice are still able to induce IFN-γ (33–37). The ability of DEC+ DCs to induce IFN-γ in the absence of IL-12 also provides a potential mechanism for the ability of patients with genetic deficiencies in IL-12 or IL-12 receptor to produce IFN-γ and avoid infection with most intracellular pathogens (32).
The function of the DEC+ subset, although independent of IL-12 as a cytokine cofactor, was completely dependent on the membrane cofactor CD70. CD70 was up-regulated intracellularly by both DC subsets in mice given poly IC and anti-CD40 in the absence of LACK. However, CD70 was expressed selectively on the surface of DEC+ DCs when the DCs were presenting antigen to T cells. Although poly IC and anti-CD40 were required for optimal primary immune responses in vivo, these stimuli were not required for the DC subsets to exert their distinct functions in vitro, but again, only the DEC+ DCs up-regulated CD70 expression when presenting antigen to T cells. This up-regulation was consequential because anti-CD70 fully blocked IFN-γ production by DEC+ DCs in vitro and in vivo; in contrast, anti-CD70 had no effect on DEC− DCs. Further studies are needed to determine if the innate propensities of DC subsets to differentially affect the Th1/Th2 balance are independent of the maturation stimulus used. Our initial comparisons using poly IC and LPS as stimuli indicate that DC subset function is similar with these two TLR ligands.
Previous studies have emphasized a contribution of CD70 to CD8+ T cell responses (50–58). In retrospect, the prior emphasis on CD8+ T cells might be attributed to the fact that the CD70-dependent DEC+ DC subset is the main DC that presents antigens on MHC class I products to CD8+ lymphocytes (21, 23, 26, 59). In contrast, although the DEC− subset more efficiently processes antigens onto MHC class II than the DEC+ subset (26), both DEC+ and DEC− DCs present antigen effectively to CD4+ T cells. Previous studies have been unable to show (53, 60, 61) a CD70 requirement for IFN-γ production by CD4+ T cells. The contribution of CD70 to CD4+ T cell responses is probably more difficult to discern when one gives a soluble antigen, which targets both CD70-dependent and -independent DCs. However, CD70 emerges as an essential costimulator when DEC+ DCs are selectively presenting antigen.
When we studied the points at which CD70 was used by DEC+ DCs to stimulate T cells, we found that blockade of this TNF family member did not affect the initial DC–T cell interaction, as indicated by up-regulation of CD69 on T cells, an early activation marker. If anti-CD70 was added early to the culture, T cell proliferation was totally blocked, but if it was added 1 or 2 d later, IFN-γ was shut down but not T cell proliferation. Anti-CD70 blocked the function of DEC+ DCs with all forms of LACK antigen, i.e., DEC-targeted LACK, nontargeted LACK, and even LACK peptide added in vitro. In contrast, anti-CD70 had no effect on T cell stimulation by DEC− DCs that were presenting 33D1-targeted LACK or nontargeted LACK protein or peptide. Importantly, CD70 was selectively expressed by DEC+ DCs. Additional research is required to identify a role for CD70 in response to infections and other challenges, but our data with antigen targeting indicate that a dominant role for CD70 in Th1 responses will only be observed when antigens are being presented predominantly by the DEC+ DC subset in lymphoid tissues. For example, a previous study has reported no role for CD70 during protection against influenza (62), which is to be expected due to influenza presentation by both CD8+ and CD8− DCs (63). We would like to suggest that CD70 is a “decision maker” for DEC+ DCs to induce Th1 T cell differentiation by an IL-12–independent mechanism.
Even in the Th2-prone BALB/c mouse, our findings reveal that the mature DEC+ subset polarizes the T cell response to the Th1 type (IFN-γ+ IL-4−). In contrast, the DEC− subset with the same DC maturation stimuli induces Th2 cytokines (IL-4 and IL-5) as well as IFN-γ. Although DEC+ DCs are a major source of IL-12 (for review see references 2, 8, and 18), IL-12 ironically is not obligatory for the induction of IFN-γ by these DCs in vivo. In sections of human lymph nodes, CD11c+ DCs all express DEC, making this pathway a plausible one in humans (64). The targeting of vaccines to CD70-expressing DEC+ DCs should favor the development of Th1 immunity, a type of response considered to be valuable for protection against several global infectious diseases.
MATERIALS AND METHODS
Mice.
We used BALB/c mice from Taconic, IL-12 p40−/− from The Jackson Laboratory, WT15 BALB/c TCR transgenics specific for the L. major protein LACK (39), and BALB/c thy-1.1 mice from M. and J. Lafaille (New York University, New York, NY). Mice were maintained under specific pathogen-free conditions and used at 7–8 wk of age in accordance with Institutional Animal Care and Use Committee guidelines.
Recombinant L. major LACK protein.
The p24 fragment of LACK protein, whose sequence started at amino acid 136 of our construct, was produced by LACK-transformed Escherichia coli strain BL21CDE3 by isopropylthio-β-D-galactoside induction (Invitrogen). After LACK was purified from lysates on NiATA-agarose columns (GE Healthcare), it was identified on SDS-PAGE and detoxified on LPS columns (Detoxigel Endotosing Removing Gel [Pierce Chemical Co.]; Limulus Amebocyte Lysate assay, QCL-1000 [Bio Whittaker]).
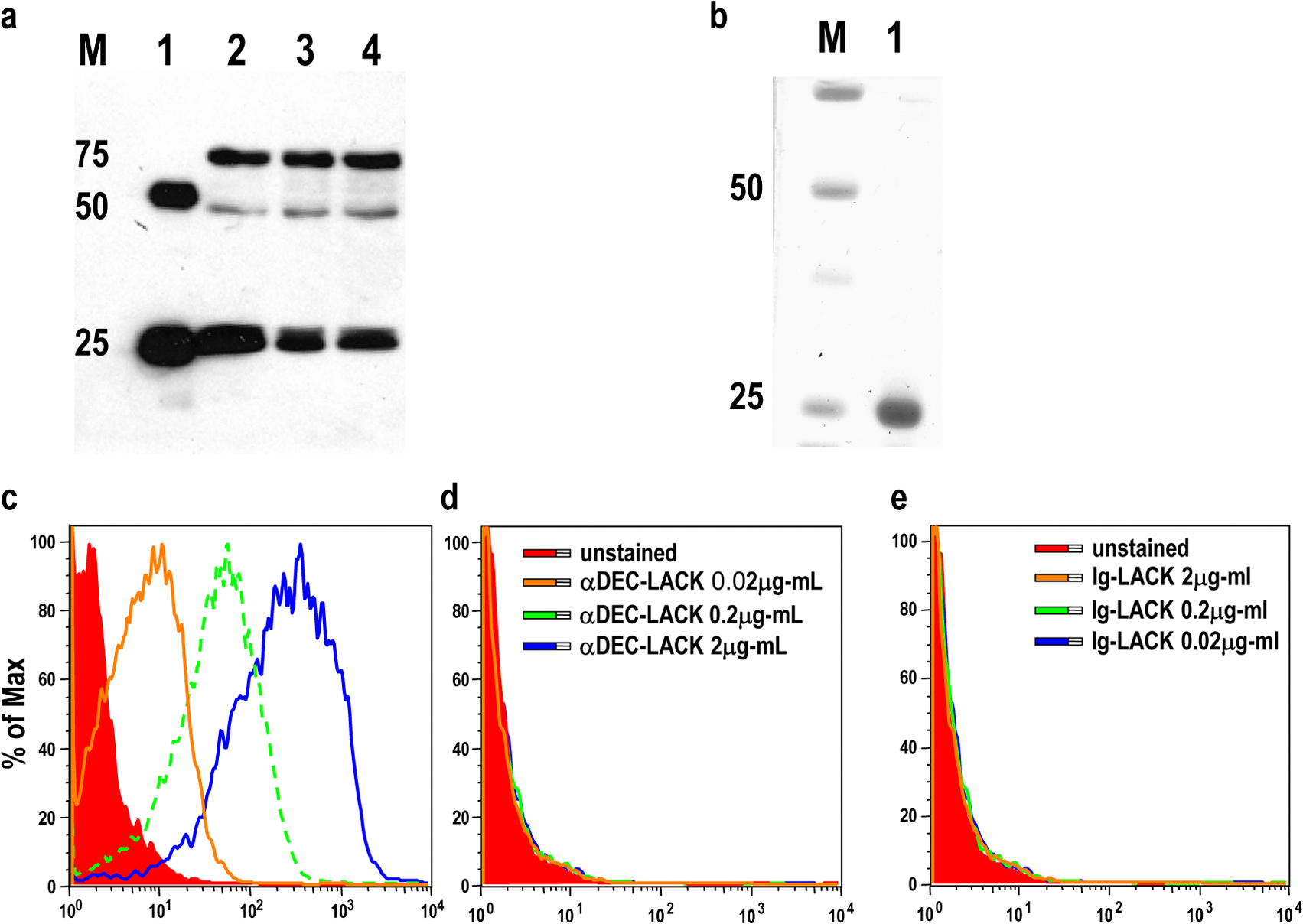
Fusion mAbs containing L. major LACK protein.
DNA from L. major LACK protein (39) was cloned in frame into the C terminus of αDEC-205, 33D1, and control mAb heavy chains (10, 26). The fusion mAbs were expressed by transient transfection (calcium phosphate) in 293 cells in serum-free DMEM supplemented with Nutridoma SP (Roche), purified on protein G columns (GE Healthcare), and characterized by SDS-PAGE and Western blotting (horseradish peroxidase sheep anti–mouse Ig; GE Healthcare). The unmodified heavy and light chains of the α-DEC205 mAb were likewise expressed and purified. All mAbs were endotoxin free.
Antigen targeting and maturation of DCs in vivo.
BALB/c mice were injected i.p. or s.c. (footpads) with LACK fusion mAbs or protein ± stimuli for DC maturation, which were 50 μg poly IC (InVivoGen) and 25 μg 1C10 agonistic αCD40 (65). Alternatively, a combination of αCD40 and 50 μg LPS or 20 μg TLR7/8 agonist was used in some experiments. These stimuli all led to the up-regulation of several costimulatory molecules on splenic DCs in vivo, such as CD40 and CD86.
LACK peptide libraries.
Overlapping (staggered by 4 amino acids) 15-mer peptides spanning the entire LACKp24 sequence were synthesized by the Proteomics Resource Center (The Rockefeller University). The 51-member L. major LACK library was divided into 5 pools of 10 peptides each, except the last pool contained 11 peptides.
Assays for LACK-specific CD4+ T cells.
Spleen cells were restimulated with pools of peptides (2 μg/ml) or medium alone, along with 2 μg/ml of costimulatory αCD28 (clone 37.51) for 6 h. 10 μg/ml brefeldin A (Sigma-Aldrich) was added for the last 4 h. Cells were washed, incubated for 15 min at 4°C with 2.4G2 mAb to block FcγR, and stained with FITC αCD3 (145-2C11) and PE- or PerCP-conjugated αCD4 (RM4-5) for 20 min at 4°C. The cells were permeabilized (Cytofix/Cytoperm Plus; BD Biosciences) and stained with APC-αIFNγ (XMG 1.2) at room temperature (BD Biosciences). We used a FACSCalibur with data analysis in FlowJo (Tree Star). In some experiments, BALB/c mice were injected i.p. with 50 μg anti-CD70 mAb (FR70) or isotype control 1 d before immunization with the various forms of LACK antigen. All plots were gated on CD3+ T cells. For ELISPOT assays, we restimulated positively selected CD4+ T cells with CD11c+ spleen DCs and LACK peptide pools, LACK-immunodominant peptide, or medium alone for 36 h. DCs were enriched from spleens dissociated with collagenase (collagenase D; Roche) using αCD11c-coated magnetic beads (Miltenyi Biotec).
IgG isotype ELISA.
High-binding ELISA plates (Costar) were coated overnight with 5 μg/ml LACK protein in PBS. Plates were washed three times with PBS-Tween 20 0.02% and blocked with PBS-BSA 1% for 1 h at room temperature. Serial dilutions of the sera in PBS-BSA 0.25% were incubated for 2 h at room temperature and visualized with goat anti–mouse subclass-specific antibodies conjugated to horseradish peroxidase (1:500; SouthernBiotech), followed by colorimetric assay using 1-Step ABTS. OD405 was measured using an OPSYS M r microplate reader (ThermoLab Systems). Titers represent highest dilution of serum showing an OD405 of >0.1 and are presented as the log10 antibody titer of each mouse.
Presentation to LACK-specific TCR transgenic CD4+ T cells.
CD11c+ cells from draining lymph nodes and spleens from mice that had been injected 12–16 h previously with different forms of LACK antigen or 106 L. major metacyclic promastigotes were positively selected (Miltenyi Biotec) using αCD11c-coated magnetic beads. Graded doses of CD11c+ DCs or their subsets (see below) were added to 105 WT15 TCR transgenic T cells (negatively selected with anti–MHC class II and anti-CD8) labeled with CFSE (107 cells/ml, 1 μM for 10 min at 37°C; Invitrogen) in round-bottom 96-well plates. For in vivo studies, the T cells were labeled with 5 μM CFSE, and 3 × 106 thy-1.1+ marked cells were i.v. injected into BALB/c thy-1.2+ recipient mice. 24 h later, different forms of LACK or 106 L. major metacyclic promastigotes were injected i.p. or s.c. into the hind footpads. Splenocytes were analyzed for proliferation and cytokine production 6 d later.
In vitro differentiation of LACK-specific, TCR-transgenic CD4+ T cells.
12–16 h after injection of different forms of LACK, CD11c+ cells were selected (Miltenyi Biotec) as described above and sorted on a FACSVantage (BD Biosciences) into B220− CD3− CD11chigh DEC205high and B220− CD3− CD11chigh DEC205low fractions (>99% pure). 6 × 104 DCs were cultured for 3.5 d with 3 × 105 CFSE-labeled WT 15 TCR transgenic cells in round-bottom 96-well plates. Cytokines were detected by APC–anti–IFN-γ (XMG 1.2) intracellular cytokine staining or by ELISAs with mAbs specific for IL-4, IFN-γ, IL-5, and IL-2 (BD OpTEIA; BD Biosciences). 5 ng/ml of mouse rIL-12 (R&D Systems) was added in some experiments.
Fusion mAbs containing Plasmodium yoelli circumsporozoite protein.
These were cloned as described previously (44) and expressed by transient transfection (calcium phosphate) according to the same protocol for fusion mAbs containing LACK protein.
Online supplemental material.
Fig. S1 shows mAb characterization. Fig. S2 shows αDEC-LACK presentation by CD11c+, but not by CD11c−, cells from draining lymph nodes. Fig. S3 shows that a combination of αCD40 and TLR3 agonist, poly IC, confers optimal IFN-γ production by LACK antigen–primed CD4+ T cells. Fig. S4 shows DC targeting as a strategy to efficiently prime CD4+ T cells. Fig. S5 shows LACK protein–unique immunodominant CD4+ epitope. Fig. S6 shows that αCD70 blocks T cell proliferation and IFN-γ secretion by DEC+ DCs, but not DEC− DCs, from IL-12 p40 KO mice in vivo. Fig. S7 shows CD70 expression on DEC+ and DEC− DCs. Fig. S8 shows CD70 expression on LACK transgenic cells. Figs. S1–S8 are available at http://www.jem.org/cgi/content/full/jem.20070176/DC1.
Supplemental Material
Acknowledgments
The authors are grateful to Judy Adams for help with the manuscript.
This work is supported by the Gulbenkian PhD Program, Portugal (to H. Soares), National Institutes of Health grants AI13013 and 40874 (to R.M. Steinman), and a fellowship from the Deutsche Forschungsgemeinschaft DC 548/1-1 (D. Dudziak).
M.C. Nussenzweig and R.M. Steinman have financial interests in Celldex, which is developing anti–DEC-205 antibodies for human use. The other authors have no conflicting financial interests.
References
- 1.Abbas, A.K., K.M. Murphy, and A. Sher. 1996. Functional diversity of helper T lymphocytes. Nature. 383:787–793. [DOI] [PubMed] [Google Scholar]
- 2.O'Garra, A. 1998. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 8:275–283. [DOI] [PubMed] [Google Scholar]
- 3.Murphy, K.M., and S.L. Reiner. 2002. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2:933–944. [DOI] [PubMed] [Google Scholar]
- 4.Shankaran, V., H. Ikeda, A.T. Bruce, J.M. White, P.E. Swanson, L.J. Old, and R.D. Schreiber. 2001. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 410:1107–1111. [DOI] [PubMed] [Google Scholar]
- 5.Mosmann, T.R., and R.L. Coffman. 1989. Th1 and Th2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7:145–173. [DOI] [PubMed] [Google Scholar]
- 6.Cohn, L., J.A. Elias, and G.L. Chupp. 2004. Asthma: mechanisms of disease persistence and progression. Annu. Rev. Immunol. 22:789–815. [DOI] [PubMed] [Google Scholar]
- 7.Powrie, F., and R.L. Coffman. 1993. Cytokine regulation of T-cell function: potential for therapeutic intervention. Immunol. Today. 14:270–274. [DOI] [PubMed] [Google Scholar]
- 8.Pulendran, B. 2004. Modulating vaccine reponses with dendritic cells and toll-like receptors. Immunol. Rev. 199:227–250. [DOI] [PubMed] [Google Scholar]
- 9.Moser, M. 2003. Dendritic cells in immunity and tolerance-do they display opposite functions? Immunity. 19:5–8. [DOI] [PubMed] [Google Scholar]
- 10.Hawiger, D., K. Inaba, Y. Dorsett, K. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonifaz, L., D. Bonnyay, K. Mahnke, M. Rivera, M.C. Nussenzweig, and R.M. Steinman. 2002. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonifaz, L.C., D.P. Bonnyay, A. Charalambous, D.I. Darguste, S. Fujii, H. Soares, M.K. Brimnes, B. Moltedo, T.M. Moran, and R.M. Steinman. 2004. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 199:815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trumpfheller, C., J.S. Finke, C.B. Lopez, T.M. Moran, B. Moltedo, H. Soares, Y. Huang, S.J. Schlesinger, C.G. Park, M.C. Nussenzweig, et al. 2006. Intensified and protective CD4+ T cell immunity at a mucosal surface after a single dose of anti–dendritic cell HIV gag fusion antibody vaccine. J. Exp. Med. 203:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seder, R.A., W.E. Paul, M.M. Davis, and B. Fazekas de St. Groth. 1992. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J. Exp. Med. 176:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pulendran, B., J. Lingappa, M.K. Kennedy, J. Smith, M. Teepe, A. Rudensky, C.R. Maliszewski, and E. Maraskovsky. 1997. Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J. Immunol. 159:2222–2231. [PubMed] [Google Scholar]
- 16.Macatonia, S.E., N.A. Hosken, M. Litton, P. Vieira, C.-S. Hsieh, J.A. Culpepper, M. Wysocka, G. Trinchieri, K.M. Murphy, and A. O'Garra. 1995. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J. Immunol. 154:5071–5079. [PubMed] [Google Scholar]
- 17.Heufler, C., F. Koch, U. Stanzl, G. Topar, M. Wysocka, G. Trinchieri, A. Enk, R.M. Steinman, N. Romani, and G. Schuler. 1996. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur. J. Immunol. 26:659–668. [DOI] [PubMed] [Google Scholar]
- 18.Moser, M., and K.M. Murphy. 2000. Dendritic cell regulation of Th1-Th2 development. Nat. Immunol. 1:199–205. [DOI] [PubMed] [Google Scholar]
- 19.Jiang, W., W.J. Swiggard, C. Heufler, M. Peng, A. Mirza, R.M. Steinman, and M.C. Nussenzweig. 1995. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 375:151–155. [DOI] [PubMed] [Google Scholar]
- 20.Vremec, D., M. Zorbas, R. Scollay, D.J. Saunders, C.F. Ardavin, L. Wu, and K. Shortman. 1992. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J. Exp. Med. 176:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.den Haan, J., S. Lehar, and M. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iyoda, T., S. Shimoyama, K. Liu, Y. Omatsu, Y. Maeda, K. Takahara, Y. Akiyama, R.M. Steinman, and K. Inaba. 2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnorrer, P., G.M. Behrens, N.S. Wilson, J.L. Pooley, C.M. Smith, D. El-Sukkari, G. Davey, F. Kupresanin, M. Li, E. Maraskovsky, et al. 2006. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc. Natl. Acad. Sci. USA. 103:10729–10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nussenzweig, M.C., R.M. Steinman, J.C. Unkeless, M.D. Witmer, B. Gutchinov, and Z.A. Cohn. 1981. Studies of the cell surface of mouse dendritic cells and other leukocytes. J. Exp. Med. 154:168–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley, M., K. Inaba, M. Witmer-Pack, and R.M. Steinman. 1989. The cell surface of mouse dendritic cells: FACS analyses of dendritic cells from different tissues including thymus. Cell. Immunol. 118:108–125. [DOI] [PubMed] [Google Scholar]
- 26.Dudziak, D., A.O. Kamphorst, G.F. Heidkamp, V. Buchholz, C. Trumpfheller, S. Yamazaki, C. Cheong, K. Liu, H.W. Lee, C.G. Park, et al. 2007. Differential antigen processing by dendritic cell subsets in vivo. Science. 315:107–111. [DOI] [PubMed] [Google Scholar]
- 27.Maldonado-Lopez, R., T. De Smedt, P. Michel, J. Godfroid, B. Pajak, C. Heirman, K. Thielemans, O. Leo, J. Urbain, and M. Moser. 1999. CD8α+ and CD8α− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 189:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulendran, B., J.L. Smith, G. Caspary, K. Brasel, D. Pettit, E. Maraskovsky, and C.E. Maliszweski. 1999. Distinct dendritic cell subsets differentially regulate the class of immune responses in vivo. Proc. Natl. Acad. Sci. USA. 96:1036–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reis e Sousa, C., G. Yap, O. Schulz, N. Rogers, M. Schito, J. Aliberti, S. Hieny, and A. Sher. 1999. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 11:637–647. [DOI] [PubMed] [Google Scholar]
- 30.Hochrein, H., K. Shortman, D. Vremec, B. Scott, P. Hertzog, and M. O'Keeffe. 2001. Differential production of IL-12, IFN-α, and IFN-γ by mouse dendritic cell subsets. J. Immunol. 166:5448–5455. [DOI] [PubMed] [Google Scholar]
- 31.Doffinger, R., E. Jouanguy, F. Altare, P. Wood, T. Shirakawa, F. Novelli, D. Lammas, D. Kumararatne, and J.L. Casanova. 1999. Inheritable defects in interleukin-12- and interferon-gamma-mediated immunity and the TH1/TH2 paradigm in man. Allergy. 54:409–412. [DOI] [PubMed] [Google Scholar]
- 32.Fieschi, C., S. Dupuis, E. Catherinot, J. Feinberg, J. Bustamante, A. Breiman, F. Altare, R. Baretto, F. Le Deist, S. Kayal, et al. 2003. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor β1 deficiency: medical and immunological implications. J. Exp. Med. 197:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jankovic, D., M.C. Kullberg, S. Hieny, P. Caspar, C.M. Collazo, and A. Sher. 2002. In the absence of IL-12, CD4+ T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL-10−/− setting. Immunity. 16:429–439. [DOI] [PubMed] [Google Scholar]
- 34.Yang, J., T.L. Murphy, W. Ouyang, and K.M. Murphy. 1999. Induction of interferon-γ production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur. J. Immunol. 29:548–555. [DOI] [PubMed] [Google Scholar]
- 35.Noble, A., M.J. Thomas, and D.M. Kemeny. 2001. Early Th1/Th2 cell polarization in the absence of IL-4 and IL-12: T cell receptor signaling regulates the response to cytokines in CD4 and CD8 T cells. Eur. J. Immunol. 31:2227–2235. [DOI] [PubMed] [Google Scholar]
- 36.Mullen, A.C., F.A. High, A.S. Hutchins, H.W. Lee, A.V. Villarino, D.M. Livingston, A.L. Kung, N. Cereb, T.P. Yao, S.Y. Yang, and S.L. Reiner. 2001. Role of T-bet in commitment of Th1 cells before IL-12-dependent selection. Science. 292:1907–1910. [DOI] [PubMed] [Google Scholar]
- 37.Amsen, D., J.M. Blander, G.R. Lee, K. Tanigaki, T. Honjo, and R.A. Flavell. 2004. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 117:515–526. [DOI] [PubMed] [Google Scholar]
- 38.Trinchieri, G. 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3:133–146. [DOI] [PubMed] [Google Scholar]
- 39.Mougneau, E., F. Altare, A.E. Wakil, S. Zheng, T. Coppola, Z.-E. Wang, R. Waldmann, R.M. Locksley, and N. Glaichenhaus. 1995. Expression cloning of a protective Leishmania antigen. Science. 268:563–565. [DOI] [PubMed] [Google Scholar]
- 40.Heinzel, F.P., R.M. Rerko, F. Ahmed, and E. Pearlman. 1995. Endogenous IL-12 is required for control of Th2 cytokine responses capable of exacerbating leishmaniasis in normally resistant mice. J. Immunol. 155:730–739. [PubMed] [Google Scholar]
- 41.Sypek, J.P., C.L. Chung, S.E.H. Mayor, J.M. Subramanyam, S.J. Goldman, D.S. Sieburth, S.F. Wolf, and R.G. Schaub. 1993. Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med. 177:1797–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park, A.Y., B.D. Hondowicz, and P. Scott. 2000. IL-12 is required to maintain a Th1 response during Leishmania major infection. J. Immunol. 165:896–902. [DOI] [PubMed] [Google Scholar]
- 43.Sacks, D., and N. Noben-Trauth. 2002. The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2:845–858. [DOI] [PubMed] [Google Scholar]
- 44.Boscardin, S.B., J.C. Hafalla, R.F. Masilamani, A.O. Kamphorst, H.A. Zebroski, U. Rai, A. Morrot, F. Zavala, R.M. Steinman, R.S. Nussenzweig, and M.C. Nussenzweig. 2006. Antigen targeting to dendritic cells elicits long-lived T cell help for antibody responses. J. Exp. Med. 203:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahonen, C.L., C.L. Doxsee, S.M. McGurran, T.R. Riter, W.F. Wade, R.J. Barth, J.P. Vasilakos, R.J. Noelle, and R.M. Kedl. 2004. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med. 199:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fearon, D.T., and R.M. Locksley. 1996. The instructive role of innate immunity in the acquired immune response. Science. 272:50–54. [DOI] [PubMed] [Google Scholar]
- 47.Trinchieri, G. 1995. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 13:251–276. [DOI] [PubMed] [Google Scholar]
- 48.Watts, T.H. 2005. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 23:23–68. [DOI] [PubMed] [Google Scholar]
- 49.Fujii, S., K. Liu, C. Smith, A.J. Bonito, and R.M. Steinman. 2004. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J. Exp. Med. 199:1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bullock, T.N., and H. Yagita. 2005. Induction of CD70 on dendritic cells through CD40 or TLR stimulation contributes to the development of CD8+ T cell responses in the absence of CD4+ T cells. J. Immunol. 174:710–717. [DOI] [PubMed] [Google Scholar]
- 51.Hintzen, R.Q., S.M.A. Lens, K. Lammers, H. Kuiper, M.P. Beckmann, and R.A.W. van Lier. 1995. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J. Immunol. 154:2612–2623. [PubMed] [Google Scholar]
- 52.Taraban, V.Y., T.F. Rowley, and A. Al-Shamkhani. 2004. A critical role for CD70 in CD8 T cell priming by CD40-licensed APCs. J. Immunol. 173:6542–6546. [DOI] [PubMed] [Google Scholar]
- 53.Laouar, A., V. Haridas, D. Vargas, X. Zhinan, D. Chaplin, R.A. van Lier, and N. Manjunath. 2005. CD70+ antigen-presenting cells control the proliferation and differentiation of T cells in the intestinal mucosa. Nat. Immunol. 6:698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamada, A., A.D. Salama, M. Sho, N. Najafian, T. Ito, J.P. Forman, R. Kewalramani, S. Sandner, H. Harada, M.R. Clarkson, et al. 2005. CD70 signaling is critical for CD28-independent CD8+ T cell-mediated alloimmune responses in vivo. J. Immunol. 174:1357–1364. [DOI] [PubMed] [Google Scholar]
- 55.Carr, J.M., M.J. Carrasco, J.E. Thaventhiran, P.J. Bambrough, M. Kraman, A.D. Edwards, A. Al-Shamkhani, and D.T. Fearon. 2006. CD27 mediates interleukin-2-independent clonal expansion of the CD8+ T cell without effector differentiation. Proc. Natl. Acad. Sci. USA. 103:19454–19459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwamoto, S., M. Ishida, K. Takahashi, K. Takeda, and A. Miyazaki. 2005. Lipopolysaccharide stimulation converts vigorously washed dendritic cells (DCs) to nonexhausted DCs expressing CD70 and evoking long-lasting type 1 T cell responses. J. Leukoc. Biol. 78:383–392. [DOI] [PubMed] [Google Scholar]
- 57.Iwamoto, S., M. Ishida, S. Tamaoki, T. Hagiwara, H. Sueki, and A. Miyazaki. 2004. A human Langerhans cell-like cell line, ELD-1, promotes CD8 T cells to produce IFN-γ through CD70-dependent alternative pathway. Cell. Immunol. 232:49–56. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez, P.J., J.A. McWilliams, C. Haluszczak, H. Yagita, and R.M. Kedl. 2007. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J. Immunol. 178:1564–1572. [DOI] [PubMed] [Google Scholar]
- 59.Schnurr, M., Q. Chen, A. Shin, W. Chen, T. Toy, C. Jenderek, S. Green, L. Miloradovic, D. Drane, I.D. Davis, et al. 2005. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood. 105:2465–2472. [DOI] [PubMed] [Google Scholar]
- 60.Hendriks, J., L.A. Gravestein, K. Tesselaar, R.A.W. van Lier, T.N.M. Schumacher, and J. Borst. 2000. CD27 is required for generation and long-term maintenance of T cell immunity. Nat. Immunol. 1:433–440. [DOI] [PubMed] [Google Scholar]
- 61.Matter, M., B. Odermatt, H. Yagita, J.M. Nuoffer, and A.F. Ochsenbein. 2006. Elimination of chronic viral infection by blocking CD27 signaling. J. Exp. Med. 203:2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao, Y., J. Hendriks, P. Langerak, H. Jacobs, and J. Borst. 2004. CD27 is acquired by primed B cells at the centroblast stage and promotes germinal center formation. J. Immunol. 172:7432–7441. [DOI] [PubMed] [Google Scholar]
- 63.Belz, G.T., C.M. Smith, L. Kleinert, P. Reading, A. Brooks, K. Shortman, F.R. Carbone, and W.R. Heath. 2004. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc. Natl. Acad. Sci. USA. 101:8670–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Granelli-Piperno, A., A. Pritsker, M. Pack, I. Shimeliovich, J.-F. Arrighi, C.G. Park, C. Trumpfheller, V. Piguet, T.M. Moran, and R.M. Steinman. 2005. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin/CD209 is abundant on macrophages in the normal human lymph node and is not required for dendritic cell stimulation of the mixed leukocyte reaction. J. Immunol. 175:4265–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heath, A.W., W.W. Wu, and M.C. Howard. 1994. Monoclonal antibodies to murine CD40 define two distinct functional epitopes. Eur. J. Immunol. 24:1828–1834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}