

Abstract
The deubiquitinating enzyme CYLD has recently been implicated in the regulation of signal transduction, but its physiological function and mechanism of action are still elusive. In this study, we show that CYLD plays a pivotal role in regulating T cell activation and homeostasis. T cells derived from Cyld knockout mice display a hyperresponsive phenotype and mediate the spontaneous development of intestinal inflammation. Interestingly, CYLD targets a ubiquitin-dependent kinase, transforming growth factor–β-activated kinase 1 (Tak1), and inhibits its ubiquitination and autoactivation. Cyld-deficient T cells exhibit constitutively active Tak1 and its downstream kinases c-Jun N-terminal kinase and IκB kinase β. These results emphasize a critical role for CYLD in preventing spontaneous activation of the Tak1 axis of T cell signaling and, thereby, maintaining normal T cell function.
T cells serve as a central component of the adaptive immune system. A defect in T cell activation results in severe immune deficiencies, whereas deregulated T cell activation is associated with chronic inflammations and autoimmunity (1). Thus, the process of T cell activation is subject to tight regulation by positive and negative mechanisms. One recently discovered mechanism of T cell regulation is ubiquitination, which plays an important role in both the development and activation of T cells (2). Protein ubiquitination is a reversible process that is counter-regulated by ubiquitin-conjugating enzymes and deubiquitinating enzymes (DUBs; reference 3). Although the ubiquitin-conjugating enzymes have been extensively studied, little is known about how the DUBs participate in immune regulation, particularly in the regulation of T cell function. We have recently shown that a DUB, CYLD, regulates TCR-proximal signaling in thymocytes and is required for thymocyte development (4), although its role in peripheral T cell function is enigmatic.
CYLD was originally identified as a tumor suppressor that is mutated in familial cylindromatosis (5). Because patients carry a heterozygous mutation of the Cyld gene with a loss of heterozygosity occurring only in tumor cells, the physiological function of CYLD was not resolved by the patient studies. The DUB function of CYLD was first revealed by in vitro work showing that CYLD inhibits the ubiquitination of certain TNF receptor–associated factors and the regulatory subunit of IκB kinase ([IKK] IKKγ; references 6–8). When transfected in cell lines, CYLD inhibits the activation of NF-κB and mitogen-activated protein kinases stimulated by innate immune receptors, such as the Toll-like receptors and TNF receptors (6–11). However, recent studies using Cyld knockout (Cyld−/−) mice suggest that the signaling function of CYLD is complex and may vary among different cell types and stimulation conditions (4, 12, 13). As such, it is still not clear how CYLD regulates NF-κB and other signaling pathways under physiological conditions.
NF-κB represents a family of transcription factors that plays a central role in regulating the activation and homeostasis of T cells (14). The NF-κB factors are normally sequestered in the cytoplasm through physical interaction with inhibitory proteins, predominantly IκBα (15). Activation of NF-κB by diverse immunologic stimuli typically involves the degradation of IκBα, which, in turn, is triggered through IκBα phosphorylation by the IKK (15). IKK is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit termed IKKγ or NF-κB essential modulator. IKKβ plays the primary role in mediating the canonical pathway of NF-κB activation, whereas IKKα regulates an alternative pathway involving processing of the NF-κB2 precursor protein p100 (15–17).
Cross-linking of the TCR and CD28 costimulatory molecule in T cells leads to activation of the canonical NF-κB pathway; this signaling cascade involves several intermediate molecules, including CARMA1, Bcl10, and MALT1 (14). Recent genetic evidence suggests that IKK activation by the TCR signal requires TGF-β–activated kinase 1 (Tak1; references 18–20), a ubiquitin-dependent kinase (21) known to phosphorylate and activate IKKβ (22). In addition to activating IKK, Tak1 also mediates the TCR-stimulated activation of c-Jun N-terminal kinase (JNK; references 18–20). These findings suggest that Tak1 is a master kinase that mediates the activation of both IKK and JNK, although how Tak1 is regulated in T cells remains elusive. We show in this study that CYLD physically interacts with Tak1 and inhibits its ubiquitination and catalytic activity. The loss of CYLD in T cells results in the constitutive activation of Tak1 and its downstream kinases JNK and IKKβ. Consistently, the Cyld−/− T cells display a hyperresponsive phenotype and mediate the spontaneous development of intestinal inflammation in Cyld−/− mice. These findings establish CYLD as a specific DUB that prevents spontaneous activation of the Tak1 axis of TCR signaling and, thereby, maintains normal T cell responses.
RESULTS
Cyld-deficient T cells are hyperresponsive to TCR stimulation in vitro
To examine how CYLD regulates peripheral T cell activation, we began by analyzing the activation of Cyld−/− T cells in vitro. Interestingly, the loss of CYLD resulted in the hyperproliferation of lymph node T cells when stimulated with agonistic anti-CD3 and -CD28 antibodies (Fig. 1 A). Compared with the wild-type T cells, the Cyld−/− T cells also produced markedly larger amounts of cytokines, including IFN-γ (Fig. 1 B) and IL-2 (Fig. 1 C). Similar results were obtained with splenic T cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20062694/DC1). To determine whether the hyperresponsive phenotype of Cyld−/− T cells occurred in the naive or memory population, we purified these cells by flow cytometric cell sorting. Remarkably, the loss of CYLD resulted in hyperproliferation and aberrant cytokine production in both naive and memory T cells (Fig. 1, D and E). These results suggest a pivotal role for CYLD in negatively regulating peripheral T cell activation.
Figure 1.

Hyperresponsiveness of Cyld−/− T cells. (A–C) Wild-type (+/+) and Cyld−/− mesenteric lymph node T cells were either not treated (NT) or were stimulated with the indicated amounts of plate-bound anti-CD3 plus soluble anti-CD28. Cell proliferation (A) and cytokine production (B and C) were measured by thymidine incorporation and ELISA, respectively. Data are presented as means ± SD (error bars) of three independent experiments. (D and E) Purified naive and memory T cells were stimulated with plate-bound anti-CD3 (2.5 μg/ml for naive and 1 μg/ml for memory T cells) and anti-CD28 (2.5 μg/ml for naive and 1 μg/ml for memory T cells) for 48 h followed by measuring cell proliferation and IFN-γ production as in A and B.
Cyld−/− mice spontaneously develop autoimmune symptoms and colonic inflammation
Because abnormal T cell responses are often associated with chronic inflammations and autoimmunity (1), we examined whether Cyld−/− mice develop immunological abnormalities. Even at early ages (8 wk), the Cyld−/− mice exhibited prominent lymphocyte infiltration into the periportal region of the liver (Fig. 2 A). More prominently, the colons of the Cyld−/− mice contained markedly more and larger lymphoid follicles or colonic patches (Fig. 2 B, bottom; arrows). Colonic patch hypertrophy is a hallmark of hapten-induced colitis and is implicated in the development of inflammation in human inflammatory bowel disease (IBD; reference 23). Of note, the lymphocytes from the colonic patches often breached into the mucosal layer and were associated with crypt damage and focal architectural distortion (Fig. 2 B, bottom; arrows). Moreover, as seen in animal models of IBD and human IBD, inflammatory cell infiltration was also detected in the lamina propria of the colon in areas that lack the colonic patches (Fig. 2 C, arrow 1). Crypt damage (Fig. 2 C, arrow 2) and thickening of the muscularis layer (Fig. 2 C, arrow 3) were also readily detected in these inflamed areas. Consistent with the involvement of CD4 T cells in IBD, we detected abundant infiltrating CD4 T cells in both the colonic patches and the inflamed mucosa (Fig. 2 D). These IBD-like pathological phenotypes were not detected in the control mice (Fig. 2, A–D). Indeed, blind analyses of the colonic sections revealed that the Cyld−/− mice had substantially higher IBD histological scores than the background scores of their wild-type littermates (Fig. 2 E).
Figure 2.

Spontaneous development of autoimmune symptoms and colonic inflammation in Cyld−/− mice. (A–C) Hematoxylin-eosin staining of tissue sections of the liver (A) and the distal portion of the colon (B and C) from 8-wk-old control (+/+) and Cyld−/− mice. Inflammatory cell infiltration in the periportal vein of the liver (A), colonic patches in the colon (B; arrows), and inflammatory cell infiltration in the colonic mucosa (C; arrow 1) are indicated. In C, arrows 2 and 3 indicate crypt damage and muscularis layer thickening, respectively. (D) Immunohistochemistry staining of colon sections with anti-CD4 showing massive CD4 T cell infiltration in the Cyld−/− colon. Data in A–D are representative of four different experiments, each with four wild-type and four Cyld−/− mice. (E) Histological scores of mucosal inflammation in wild-type and Cyld−/− mice. Data were obtained from three wild-type and three Cyld−/− mice (8 wks of age), each with two colon sections. Similar results were obtained from three additional experiments. (F) RNase protection assay showing the constitutive expression of several proinflammatory genes in the colon of Cyld−/− mice (#1 and #3) but not wild-type mice (#2 and #4). The housekeeping genes L32 and Gapdh were included as loading controls. (G) Body weight of wild-type and Cyld−/− mice showing the reduced body weights of male and female Cyld−/− mice (8 wks old). *, P < 0.05. (H) Adjacent localization of Cyld and NOD2 genes on human chromosome 16. The nucleotide number was based on the sequence of Homo sapiens chromosome 16 clone RP11-327F22 (GenBank/EMBL/DDBJ accession no. AC007728). The map was drawn to scale, and the transcriptional direction of the genes is indicated by arrows. Note that the two genes have the same transcriptional direction and are separated by only 8,988 nucleotides. Error bars represent SD. Bars (A and D), 200 μm; (B) 500 μm; (C) 400 μm.
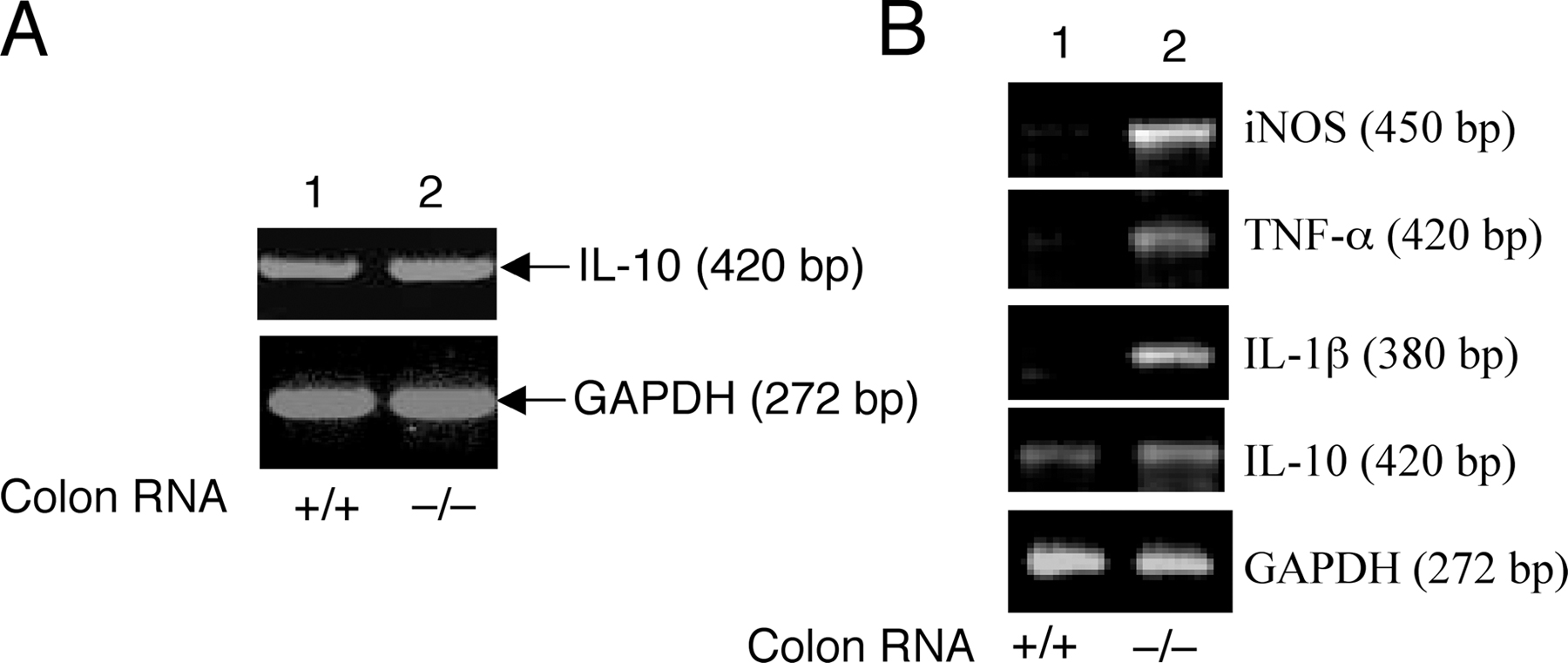
A molecular hallmark of IBD is the expression of proinflammatory mediators in the colon, which contributes to inflammation and destruction of the colonic tissue (24). To further examine the similarity between the intestinal inflammation of Cyld−/− mice and IBD, we analyzed cytokine gene expression in the colons of Cyld−/− and wild-type mice. The constitutive expression of several proinflammatory genes was detected in the colons of Cyld−/− mice but not in the colons of wild-type mice (Fig. 2 F). The Cyld−/− mice also expressed larger amounts of T cell–derived cytokines, including IL-4 and -12 (Fig. 2 F). The level of an antiinflammatory cytokine, IL-10, was moderately enhanced in the Cyld−/− colon (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20062694/DC1). The elevated expression of IL-10 has also been detected in other models of mouse colitis as well as in human IBD patients (25–27), although the underlying functional significance in IBD pathogenesis is unclear. Nevertheless, our results suggest that the intestinal inflammation in Cyld−/− mice is associated with the aberrant expression of proinflammatory cytokines. In addition to the histological and biochemical features of IBD, the Cyld−/− mice had substantial weight loss (Fig. 2 G), a characteristic clinical symptom of IBD patients (28). Of note, these results were obtained with mice housed under pathogen-free conditions with wild-type littermates being used as controls. Thus, the Cyld−/− mice develop spontaneous colonic inflammation and extraintestinal abnormalities that resemble IBD.
By analyzing the human genome sequence, we found that the human Cyld gene is located in a major IBD susceptibility locus (IBD-1) on chromosome 16 adjacent to a known IBD regulatory gene, NOD2 (Fig. 2 H). This finding suggests an intriguing possibility that CYLD may also be involved in the regulation of human IBD.
Adoptive transfer of Cyld−/− T cells induces autoimmune symptoms and colonic inflammation in recipient mice
Given the hyperresponsive phenotype of Cyld−/− T cells, it was important to examine whether these mutant T cells contribute to the development of IBD-like symptoms. We adoptively transferred T cells from either Cyld−/− or wild-type mice into RAG1−/− mice that lack endogenous lymphocytes. The RAG1−/− recipients of both Cyld−/− and wild-type T cells had an efficient T cell repopulation in the spleen 6 wk after the adoptive transfer (Fig. 3 A). However, the RAG1−/− recipients of Cyld−/− T cells but not those of wild-type T cells displayed lymphocyte infiltration into the periportal regions of the liver (Fig. 3 B). Moreover, the RAG1−/− recipients of Cyld−/− T cells spontaneously developed colitis, which was characterized by inflammatory cell infiltration, thickening of the mucosa, and goblet cell depletion (Fig. 3 C). The IBD histology scores of these mice were markedly higher than the background detected in recipients of wild-type T cells (Fig. 3 D). In addition to these histological features, the recipients of Cyld−/− T cells had substantial weight loss (Fig. 3 E) and expressed high levels of proinflammatory cytokines in the colon (Fig. S2 B). Severe bowel thickening was also observed in some of the recipients of Cyld−/− T cells but not in the controls (Fig. 3 F). These findings demonstrate that the T cells from Cyld knockout mice are sufficient to induce IBD-like features.
Figure 3.

Induction of autoimmunity and colitis by adoptively transferred Cyld−/− T cells. RAG1−/− mice (8 wks old) were intravenously injected with T cells isolated from the mesenteric lymph nodes of wild-type (+/+) or Cyld−/− mice. (A–C) After 6 wk, the recipient mice were killed for flow cytometry analyses of transferred T cells in the spleen (A), histology analyses of lymphocyte infiltration in the liver (B), and inflammation of the colon (C). The numbers in A indicate the percentages of CD8 (top left quadrants) and CD4 (bottom right quadrants) T cells. An arrow in C (bottom) indicates a colonic patch detected in a recipient of Cyld−/− T cells. Data are representative of three mice per group. (D) Histology scores measuring the degree of colonic inflammation in the recipient wild-type (+/+) and Cyld−/− T cells. Data were obtained from three mice per group. (E) Body weights of recipient mice. Body weights were measured 6 wk after T cell transfer, showing the substantial weight loss in the recipients of Cyld−/− T cells. (F) Image to compare the colons of recipients of wild-type (+/+) and Cyld−/− T cells. Error bars represent SD. Bars (B), 200 μm; (C) 1 mm.
Constitutive activation of JNK and NF-κB in Cyld−/− T cells
To understand the molecular mechanism mediating the abnormal T cell responses in Cyld−/− mice, we examined TCR signaling. We have previously shown that CYLD positively regulates LCK function and TCR-proximal signaling in thymocytes (4). However, because LCK is largely dispensable for peripheral T cell activation (29), it was intriguing to examine whether the loss of CYLD affected the TCR-proximal signaling of peripheral T cells. In naive T cells, the Cyld deficiency had no appreciable effect on the TCR/CD28-mediated phosphorylation of ZAP-70 and the mitogen-activated protein kinase extracellular-regulated kinase (ERK; Fig. 4 A). Interestingly, in the Cyld−/− T cells, JNK became constitutively activated (Fig. 4 A, lane 4). The constitutive activation of JNK was also detected using Cyld−/− total T cells (unpublished data). Thus, in peripheral T cells, CYLD is not required for TCR-proximal signaling but serves as a crucial negative regulator of JNK.
Figure 4.

Constitutive activation of JNK and NF-κB in Cyld−/− T cells. (A) Purified naive lymph node T cells from control (+/+) and Cyld−/− mice were stimulated with 1 μg/ml anti-CD3 and 1 μg/ml anti-CD28 for the indicated times. Immunoblotting (IB) assays were performed using the indicated phosphospecific (α-P) and pan-antibodies to determine the phosphorylation of ZAP-70, ERK, and JNK. (B) Hyperactivation of NF-κB in Cyld−/− T cells. Wild-type (+/+) and mutant (−/−) total lymph node T cells were stimulated with 1 μg/ml of plate-bound anti-CD3 and 1 μg/ml of soluble anti-CD28 for the indicated times. Nuclear extracts were subjected to EMSA to determine the activity of NF-κB. Actin IB was included as a loading control. (C) Nuclear extracts were isolated from purified naive and memory T cells and were subjected to EMSA to determine the constitutive activity of NF-κB. An EMSA of NF-Y was included as a control. (D) EMSA was performed using the nuclear extract isolated from untreated Cyld−/− T cells in the presence of the indicated antibodies. The antibody-shifted complexes are indicated by arrows. (E) Whole cell extracts were isolated from wild-type (+/+) and Cyld−/− mesenteric T cells and subjected to IB assays to detect the indicated NF-κB members. (F) RT-PCR was performed to detect mRNA of relB and Gapdh using total RNA isolated from wild-type (+/+) or Cyld−/− mesenteric lymph node cells.
To further investigate the signaling basis for the hyperresponsive property of Cyld−/− T cells, we examined the activation of transcription factor NF-κB. As expected, cross-linking of TCR/CD28 led to activation of the nuclear DNA-binding activity of NF-κB (Fig. 4 B). Remarkably, as seen with JNK, high levels of active NF-κB were detected in Cyld−/− T cells even under unstimulated conditions (Fig. 4 B, lane 5). Of note, the constitutive activation of NF-κB occurred in both the naive and memory Cyld−/− T cells (Fig. 4 C), further arguing for an intrinsic role for this DUB in negatively regulating the JNK and NF-κB signaling axes in T cells. Antibody supershift assays revealed that the constitutively activated NF-κB complex in Cyld−/− T cells was predominantly composed of the canonical NF-κB members p50 and RelA (Fig. 4 D). Parallel immunoblotting (IB) assays revealed the markedly enhanced expression of noncanonical NF-κB members RelB and p100 at the levels of both protein (Fig. 4 E) and RNA (Fig. 4 F and not depicted). Because the genes encoding these two proteins are regulated by NF-κB (30, 31), this result further supported the conclusion that CYLD deficiency causes the constitutive activation of NF-κB. We noticed that p100 remained largely unprocessed (not converted to p52; Fig. 4 E) despite its up-regulation. Because p100 is a specific inhibitor of RelB, this finding explained why RelB was not detected in the active nuclear NF-κB complex (Fig. 4 D). Thus, the loss of CYLD is associated with the constitutive activation of canonical NF-κB and JNK, a result that provides a mechanistic insight into the hyperresponsive phenotype of Cyld−/− T cells.
Constitutive activation of IKKβ in Cyld−/− T cells and Cyld knockdown Jurkat T cells
To understand the mechanism of constitutive NF-κB activation associated with Cyld deficiency, we examined the level and fate of the primary NF-κB inhibitor IκBα. In Cyld−/− T cells, the steady-state level of IκBα was substantially lower than in wild-type T cells (Fig. 5 A, top). This abnormality was not caused by reduced expression of the IκBα gene. In fact, the Cyld−/− T cells expressed a markedly higher level of IκBα mRNA than the wild-type T cells (Fig. 5 B). Interestingly, despite their lower level of total IκBα protein, the mutant T cells had more phosphorylated IκBα (Fig. 5 A, middle). These findings suggested that IκBα might be undergoing chronic degradation and resynthesis in the Cyld-deficient T cells. Indeed, inhibition of protein synthesis by cycloheximide led to the rapid depletion of IκBα in Cyld−/− T cells but only had a weak effect on the IκBα level in wild-type T cells (Fig. 5 C, compare lane 2 with lane 4). The chronic degradation of IκBα appeared to be dependent on IKKβ because this event was blocked by a selective IKKβ inhibitor, PS1145 (Fig. 5 C, lane 7). Consistent with these results, the Cyld−/− T cells displayed a high level of constitutive IKKβ catalytic activity (Fig. 5 D, top; lanes 1 and 2). Thus, the loss of CYLD results in constitutive activation of the NF-κB signaling pathway in T cells.
Figure 5.

Cyld deficiency results in the constitutive activation of IKKβ and chronic degradation of IκBβ. (A and B) Chronic degradation and resynthesis of IκBα in Cyld−/− T cells. Wild-type (+/+) and mutant (−/−) total T cells were subjected to IB assays (A) or RT-PCR (B) to detect the total and phosphorylated IκBα (P-IκBα) and the IκBα messenger RNA, respectively. (C) IκBα degradation in Cyld−/− T cells is mediated by IKKβ. Wild-type and mutant T cells were incubated for 50 min in the absence (–) or presence (+) of 10 μg/ml cycloheximide (CHX). In lane 7, the cells were preincubated with 10 μM of the IKKβ inhibitor PS1145 for 60 min before the start of cycloheximide treatment. The expression of IκBα as well as CYLD and tubulin was analyzed by IB. (D) Chronic activation of IKKβ in Cyld−/− T cells. IKKβ was isolated by immunoprecipitation (IP; using anti-IKKβ) from untreated wild-type (+/+) and Cyld −/− lymph node T cells and thymocytes, and the activity of IKKβ was measured by in vitro kinase assays using both GST-IκBα and GST-IKKβ substrates (top two panels). IB was performed to monitor the IKKβ protein level (bottom). (E) Whole cell extracts isolated from purified naive T cells (untreated) were subjected to IB. (F) Whole cell extracts isolated from untreated Jurkat-pSUPER and Jurkat-shCYLD cells were subjected to IKKβ kinase assay (KA; top) followed by IB to detect the IKKβ protein on the kinase assay membrane (second blot). Cell lysates were subjected to IB to detect the indicated proteins (third to fifth blots).
To determine whether the constitutive activation of IKKβ occurred in naive Cyld−/− T cells, we began by analyzing the IKKβ kinase activity in thymocytes. As seen with peripheral T cells, Cyld−/− thymocytes exhibited constitutive IKKβ activity (Fig. 5 D). Next, we purified naive peripheral T cells by flow cytometric cell sorting. Because the low number of highly purified naive T cells precluded us from performing in vitro kinase assays, we attempted to detect the in vivo phosphorylation of IKKβ based on its band shift on SDS gels. Indeed, when fractionated on a low-percentage gel, IKKβ exhibited a clear band shift in Cyld−/− but not control naive T cells (Fig. 5 E). Consistently, the naive T cells had a reduced level of IκBα and an up-regulated level of RelB and p100 (Fig. 5 E). Thus, IKKβ is chronically activated in naive T cells of Cyld−/− mice.
We next used an alternative approach to confirm that the loss of CYLD in T cells causes the constitutive activation of IKKβ. By infecting Jurkat T cells with a retroviral vector encoding a Cyld-specific small hairpin RNA (shCYLD), we were able to generate a bulk of cells with a greatly reduced level of CYLD (Fig. 5 F, bottom). As seen with the primary T cells, the Cyld knockdown Jurkat cells displayed the hyperactivation of IKKβ (Fig. 5 F, top), and this abnormality was associated with the reduction in IκBα protein level (Fig. 5 F, third blot). Collectively, these results suggest that CYLD plays a crucial role in preventing the spontaneous activation of IKKβ or its upstream signaling steps.
CYLD controls the activity of Tak1
The involvement of CYLD in the regulation of both JNK and IKKβ suggests a role for this DUB in controlling the activity of a more upstream molecule. However, because Cyld deficiency did not cause the activation of ERK, the target of CYLD might be an intermediate signaling factor specifically mediating the activation of JNK and IKKβ. In this regard, recent genetic studies highlighted a critical role for Tak1 in mediating the TCR-stimulated activation of IKK and JNK (18–20). Therefore, we examined whether Tak1 is under the control of CYLD. Indeed, the high constitutive activity of Tak1 was detected in both peripheral T cells and thymocytes of Cyld−/− mice (Fig. 6 A). Moreover, RNAi-mediated Cyld knockdown in Jurkat T cells also resulted in the constitutive activation of Tak1 (Fig. 6 A). These results establish a pivotal role for CYLD in controlling Tak1 function and explain how CYLD negatively regulates IKKβ and JNK.
Figure 6.

CYLD physically interacts with Tak1 and inhibits the ubiquitination and autoactivation of Tak1. (A) Tak1 was isolated by IP from lymph node T cells, thymocytes, or Jurkat T cells followed by kinase assays (KA) using recombinant MKK6 as a substrate (top). The kinase assay membrane was further subjected to IB to detect the Tak1 protein (bottom). (B) The CYLD complex was isolated by IP from wild-type (+/+) or Cyld−/− thymocytes followed by IB to detect CYLD and its associated Tak1. The Tak1 level in cell lysates was monitored by IB (bottom). (C) 293 cells were transfected with 150 ng CYLD either in the absence (–) or presence (+) of 100 ng Tak1. The Tak1 complex was isolated by IP followed by IB to detect Tak1 and associated CYLD. The CYLD expression level was analyzed by IB (bottom). (D) 293 cells were transfected with 100 ng Tak1 either in the absence (–) or presence (+) of CYLD (150 ng in lane 3 and 300 ng in lane 4) along with 200 ng hemagglutinin (HA)-tagged ubiquitin. Tak1 was isolated by IP followed by IB (with antihemagglutinin) to examine its ubiquitin conjugation. (E) 293 cells were transfected with 100 ng Tak1 in the absence (–) or presence (+) of two doses of CYLD (150 and 300 ng). The cells were also transfected with 100 ng of the Tak1 partner protein Tab1. Tak1 was isolated by IP followed by kinase assays using recombinant MKK6 as substrate (top). The kinase assay membrane was further subjected to IB to detect the precipitated Tak1 (middle). CYLD expression in lysates was monitored by IB (bottom). (F) Tak1 and IKKγ were isolated by IP from wild-type (+/+) and CYLD-deficient (–/–) T cells and were subjected to IB (using antiubiquitin) to detect the ubiquitinated Tak1 and IKKγ. The membrane was reprobed with anti-Tak1 to monitor the precipitated Tak1 protein (bottom). Because IKKγ comigrates with the Ig heavy chain, direct IB was performed to monitor its expression level (bottom).
Previous in vitro studies suggest that Tak1 is a ubiquitin-dependent kinase (21). Although how the catalytic activity of Tak1 is regulated in vivo is not well understood, a recent study suggests that the polyubiquitination of Tak1 mediates its autoactivation (32). Thus, it was logical to examine whether CYLD targets Tak1 and regulates its ubiquitination. Interestingly, endogenous CYLD and Tak1 indeed formed a complex in T cells, which was readily detected by co-immunoprecipitation (IP) assays using the anti-CYLD antibody (Fig. 6 B). This molecular interaction was specific because Tak1 was not precipitated by anti-CYLD from the Cyld−/− T cells (Fig. 6 B, lane 2). Specific CYLD–Tak1 interaction was also readily detected in transfected cells (Fig. 6 C). To determine whether Tak1 is a functional target of CYLD, we analyzed the effect of CYLD on Tak1 ubiquitination. As expected (32), transfected Tak1 underwent constitutive polyubiquitination (Fig. 6 D). Importantly, the ubiquitination of Tak1 was efficiently inhibited by CYLD. Furthermore, consistent with the constitutive Tak1 activation in Cyld−/− T cells, transfected CYLD potently suppressed the catalytic activity of Tak1 (Fig. 6 E).
To further assess the role of CYLD in regulating Tak1 ubiquitination, we analyzed the ubiquitination of endogenous Tak1. Remarkably, the loss of CYLD resulted in a considerable elevation of Tak1 ubiquitination in T cells (Fig. 6 F), thus providing evidence for the involvement of CYLD in modulating Tak1 ubiquitination in vivo. We also analyzed the effect of Cyld deficiency on the ubiquitination of IKKγ because this molecular event can be inhibited by transfected CYLD (6–8). A considerable basal level of IKKγ ubiquitination was detected in wild-type T cells, which was moderately enhanced in Cyld−/− T cells (Fig. 6 F). Thus, Tak1 appears to be a primary target of CYLD in T cells, although the involvement of CYLD in regulating other signaling components of the NF-κB pathway cannot be excluded.
DISCUSSION
In this study, we demonstrate a critical role for CYLD in controlling the catalytic activity of Tak1 and its downstream kinases in T cells. This function of CYLD was demonstrated using different cell systems: total T cells, naive T cells, and Cyld knockdown Jurkat T cells. Consistently, the Cyld−/− T cells are hyperresponsive to TCR/CD28 stimulation, and the Cyld−/− mice spontaneously develop intestinal inflammation. Furthermore, the adoptive transfer of Cyld−/− T cells into RAG1 knockout mice is sufficient to induce the inflammatory phenotype, thus suggesting that the abnormal response of T cells in Cyld−/− mice plays a primary role in the spontaneous development of intestinal inflammation.
Genetic and biochemical evidence has established Tak1 as a pivotal kinase that mediates the activation of IKK and JNK by diverse cellular stimuli (18–20, 22, 33–35). Using T cell–specific Tak1 knockout mice, several groups have recently shown that Tak1 is required for the TCR-stimulated activation of IKK and JNK and T cell development (18–20). Because of the lack of Tak1-deficient peripheral T cells in these mutant animals, the signaling function of Tak1 in naive T cells could not be assessed. In vitro deletion of Tak1 in preactivated T cells (similar to effector T cells) indicates that it is essential for JNK activation but functionally redundant for IKK activation under these conditions (20). Despite the potential variations in the necessity of Tak1, its potent activity in IKK/JNK activation suggests the requirement for a tight mechanism of regulation. Our data emphasize a critical role for CYLD in Tak1 regulation. The loss of CYLD in both primary T cells and the Jurkat T cell line causes the constitutive activation of Tak1 as well as its downstream targets JNK and IKK. Because ERK is not affected by the Cyld deficiency, it is unlikely that CYLD negatively regulates a further upstream component in the TCR signaling pathway. Indeed, our data suggest that CYLD directly targets Tak1 and inhibits Tak1 ubiquitination. In keeping with a previous finding that Tak1 ubiquitination mediates its autoactivation (32), we have shown that CYLD potently inhibits the catalytic activity of Tak1 in transfected cells. Thus, CYLD has a pivotal role in preventing the spontaneous activation of Tak1 and its downstream kinases JNK and IKK.
Previous studies suggest that A20 is a DUB that functions to prevent the prolonged activation of IKK in innate immune cells (36–38). To understand the functional difference between CYLD with A20, we examined whether CYLD regulates the kinetics or magnitude of the signal-induced activation of IKKβ in innate immune cells. Surprisingly, the loss of CYLD did not result in the basal activation of IKKβ or Tak1 in macrophages (Fig. S3, A and B, available at http://www.jem.org/cgi/content/full/jem.20062694/DC1). This result suggests a cell type–specific function of CYLD or functional redundancy of CYLD with other DUBs in certain cell types such as macrophages. The Cyld deficiency also did not alter the level of the LPS-stimulated activation of IKKβ or prolong the activation of IKKβ after an extended time course of LPS stimulation (Fig. S3 C). Thus, unlike A20 (37), CYLD does not play an essential role in controlling the basal or inducible activation of IKKβ in macrophages. Another major difference between CYLD and A20 is that CYLD does not seem to play a role in turning off the inducible IKK activation. Despite its essential role in suppressing the constitutive activity of IKKβ in T cells, the loss of CYLD in Jurkat T cells did not appreciably prolong the IKK activation induced by TNF-α or mitogens (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20062694/DC1). Consistently, the level of CYLD protein was not enhanced by the mitogens (Fig. S4 B) or TNF-α (not depicted). Thus, whereas A20 regulates the kinetics of inducible IKK activation in innate immune cells, CYLD is critical for regulating the spontaneous (and probably initially inducible) activation of Tak1 and IKK in T cells.
We have previously shown that CYLD positively regulates the late stage of thymocyte development (4). CYLD physically interacts with LCK and facilitates the binding of active LCK to its target ZAP-70, thereby enhancing the TCR-induced tyrosine phosphorylation of ZAP-70. We found in this study that the naive Cyld−/− peripheral T cells did not show a defect in TCR-proximal signaling. This finding is consistent with a previous study that LCK is required for thymocyte development but is largely dispensable for peripheral T cell activation (29). Thus, the constitutive activation of Tak1 coupled with the diminished effect of CYLD on TCR-proximal signaling contributes to the hyperresponsive phenotype of Cyld−/− T cells. Because Tak1 is also activated in Cyld−/− thymocytes, a question is raised with regard to its effect on thymocyte development. Based on the results of Tak1 knockout studies (18–20), the activation of Tak1 should promote thymocyte development. However, the Cyld−/− mice even produce reduced numbers of mature thymocytes (4). Although this phenotype is clearly caused by the attenuation of LCK function and TCR-proximal signaling, it is also likely that the constitutive activation of Tak1 may contribute to the thymocyte defect by causing excessive negative selection. Future studies will examine this possibility by crossing Cyld−/− mice with TCR transgenic mice.
One surprising finding with the Tak1 conditional knockout mice (Tak1flox/flox) was their spontaneous development of intestinal inflammation at old ages despite the critical role for Tak1 in NF-κB activation (19, 20). However, this phenotype has been attributed to the requirement of Tak1 in regulatory T cell (T reg cell) development. The Tak1flox/flox mice produce a scarce number of peripheral T cells, which turn out to be leaked cells that have escaped from Cre-mediated Tak1 locus deletion (19). Thus, the effector T cells that mediate inflammation are actually Tak1-expressing cells activated as a result of the lack of T reg cells (19). In contrast, the T cell abnormality of Cyld−/− mice is intrinsic to the mainstream (responder) T cells. Both naive and memory Cyld−/− T cells are hyperresponsive to TCR/CD28 stimulation, and the adoptively transferred T cells mediate colitis in RAG1−/− mice. Furthermore, our preliminary studies did not detect any defect in T reg cell development in Cyld−/− mice (unpublished data). Thus, the loss of T reg cell function or abnormality in responder T cells may contribute to the development of intestinal inflammation.
The spontaneous development of the colonic inflammation of Cyld−/− mice suggests the intriguing possibility that CYLD may serve as an important regulator of IBD. A recent study reveals that Cyld−/− mice are also more sensitive to dextran sodium sulfate–induced colitis (13). In addition to colonic inflammation, we have shown that the Cyld−/− mice display other features of IBD, such as weight loss, lymphocyte infiltration into the liver, and early onset of inflammatory symptoms. In concert with the animal studies, the Cyld gene is located within a major IBD susceptibility locus (Fig. 2 G). This locus, IBD-1, also contains the NOD2 gene that encodes an intracellular pattern recognition molecule that is known to mediate NF-κB activation in macrophages and intestinal epithelial cells (39). Genetic mutations of the human NOD2 gene increase the risk of, but are insufficient for, developing IBD (40, 41). Consistently, ablation or mutation of the NOD2 gene in mice does not cause spontaneous intestinal inflammation (42–44), although the NOD2 mutation sensitizes mice to dextran sodium sulfate–induced intestinal inflammation (44). These findings suggest the involvement of specific environmental factors or secondary genetic factors for the development of IBD in patients carrying the NOD2 gene mutations.
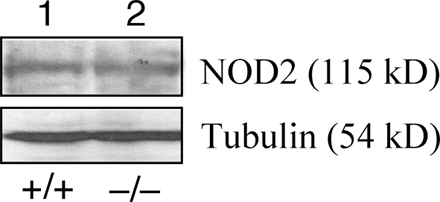
Notably, the Cyld gene is located adjacent to the NOD2 gene (Fig. 2 G). At least in mice, the loss of CYLD has no effect on the expression of NOD2 (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20062694/DC1). It remains to be examined whether mutations in the Cyld gene occur in some of the IBD patients who are epidemiologically linked with the IBD1 locus. Because the NOD2 gene is located immediately upstream of the Cyld gene, it is also possible that certain genetic mutations located at the 3′ region of the NOD2 gene may affect the expression of the Cyld gene. Although these possibilities need to be examined by further studies, a recent study does suggest the association of human IBD with the reduced expression of CYLD (45). Given that NOD2 and CYLD regulate the innate and adaptive immune responses, respectively, it is intriguing to examine whether the combined genetic mutations of these two genes have a synergistic effect on the development of colitis in mice.
MATERIALS AND METHODS
Mice.
Cyld knockout mice (in a C57BL6/DBA genetic background) were generated as described previously (4). Cyld+/– mice were intercrossed to generate Cyld−/− and Cyld+/+ littermates. Genotyping was performed by PCR using tail DNA and the following primers: Cyld forward primer, CCAGGCACTTTGAATTGCTGTC; Cyld reverse primer 1, CGTTCTTCCCAGTAGGGTGAAG; and Cyld reverse primer, GCATGCTCCAGACTGCCTTGG. When the three primers were used together, the PCR yielded a 209-bp product for Cyld+/+ mice, a 209- and 255-bp product for Cyld+/– mice, and a 255-bp product for Cyld−/− mice.
RAG1−/− mice (in a C57BL6 genetic background) were purchased from Taconic. All mice were housed in specific pathogen-free cages and monitored periodically (every 3 mo) for the lack of common pathogens. Animal experiments were performed in accordance with protocols approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee.
Plasmids, antibodies, and reagents.
Glutathione S-transferase (GST)–IKKβ was constructed by cloning a cDNA fragment encoding amino acids 166–197 of human IKKβ into the pGex-4T-3 vector (GE Healthcare). pCMV-hemagglutinin-Tak1 and pCMV-flag-Tab1 were provided by K. Matsumoto (Nagoya University, Nagoya, Japan; reference 46). Anti–mouse CD3ɛ (145-2C11), anti–mouse CD28, anti–CD4-PE-CY5.5 (L3T4), anti–CD44-FITC (IM7), and anti–CD25-PE (PC61.5) were purchased from eBioscience. Anti–CD19-PE-Cy7 (1D3) and other conjugated antibodies used for flow cytometric analyses and the unconjugated anti-CD4 (L3T4) were purchased from BD Biosciences. Goat anti–hamster Ig was purchased from Southern Biotechnology Associates, Inc. The anti-IKKβ (H470), anti-RelB (N-17), anti–lamin B (H-90), and anti-actin (C2) were purchased from Santa Cruz Biotechnology, Inc. Anti-ubiquitin was provided by V. Chau (Pennsylvania State College of Medicine, Hershey, PA), and anti-Tak1 was provided by K. Matsumoto and J. Ninomiya-Tsuji (North Carolina State University, Raleigh, NC). Recombinant MKK6 was purchased from Upstate Biotechnology, and cycloheximide was obtained from Sigma-Aldrich. All other antibodies and reagents have been described previously (4, 47, 48).
RNAi-mediated CYLD knockdown.
Jurkat T cells were infected with either the empty pSUPER-retro-puromycin vector (Oligoengine) or the same vector encoding Cyld small hairpin RNA followed by puromycin selection as reported previously (48). The bulk of infected cells, named Jurkat-pSUPER and Jurkat-shCYLD, were used in experiments.
Histology and immunohistochemistry.
Colons were removed from killed mice and flushed with Iscove's media. Distal and proximal halves of the colons were opened longitudinally, fixed in 10% neutral buffered formalin, embedded in paraffin, and sectioned for hematoxylin-eosin staining. Slides were analyzed blindly and scored for the degree of inflammation (0–40 scale) as described previously (49). Liver, lung, and salivary gland sections were prepared similarly, and pictures were taken from typical liver and colon sections.
For immunohistochemistry, the distal and proximal portions of colons were freshly frozen in Tissue-Tek optimal cutting temperature compound (VWR) using liquid nitrogen–prechilled 2-methlbutane. The frozen tissues were stored at −70°C until sectioning. 4–6-μm cryostat sections were prepared and stained with rat anti–mouse CD4 (eBioscience), and the bound anti-CD4 was detected by biotinylated rabbit anti–rat Ig and peroxidase-conjugated streptavidin with diaminobenzidine as chromagen (VECTASTAIN Elite ABC kit; Vector Laboratories).
Flow cytometry.
Splenic and lymph node cell suspensions were prepared by gentle homogenization using a Dounce homogenizer (Wheaton). Mononuclear cells were isolated by centrifugation over lymphocyte separation media (Cellgro) and were subjected to flow cytometry analyses as previously described (4). To isolate naive and memory T cells, unfixed mesenteric lymph node cells were stained with anti–CD44-FITC, anti–CD25-PE, and anti–CD19-PE-Cy7. Naive (CD44lo-medCD25−CD19−) and memory (CD44hiCD25−CD19−) T cells were purified using a cell sorter (Moflo; DakoCytomation). The purity of the isolated populations was >98%.
Cell proliferation and ELISA.
Purified T cells were stimulated in five replicate wells in 96-well plates (105 cells/well) with the indicated amounts of plate-bound anti-CD3 and anti-CD28 (1 μg/ml). After the indicated times of stimulation, cell culture supernatants were collected and subjected to ELISA assays (eBioscience) to measure the concentration of cytokines, whereas the cells were labeled for 6 h with [3H]thymidine for proliferation assays based on thymidine incorporation.
T cell adoptive transfer.
Wild-type and Cyld−/− T cells were purified using CD90-conjugated magnetic beads (Miltenyi Biotec) from mesenteric lymph nodes, and 7 × 106 cells were injected intravenously into RAG1−/− mice. Recipient mice were monitored for weight loss and were killed after 6 wk. Colon, liver, and salivary glands were removed for histology analyses. The efficiency of adoptive transfer was assessed by flow cytometry analyses of T cells in the spleen.
Ubiquitination assays.
293 cells were transfected in 12-well plates with the indicated plasmid expression vectors. Ubiquitination of transfected and endogenous proteins was analyzed as previously described (48).
IB, electrophoresis mobility shift assay, and in vitro kinase assays.
Mesenteric lymph node T cells were purified by using CD90-labeled magnetic beads and incubated on ice for 15 min with the indicated concentrations of anti–mouse CD3 and anti–mouse CD28. The cells were washed once with 500 μl of cold Iscove's media and stimulated by cross-linking the receptor-bound anti-CD3 and -CD28 for the indicated times in a 37°C water bath with 45 μg/ml of goat anti–hamster Ig. Jurkat cells were collected by centrifugation and resuspend in fresh media followed by stimulation with the indicated inducers. Untreated and stimulated thymocytes, lymph node T cells, and Jurkat cells were lysed in a kinase cell lysis buffer supplemented with phosphatase inhibitors and subjected to in vitro kinase assays and IB as previously described (50). For electrophoresis mobility shift assay (EMSA), thymocytes and T cells were either not treated or were stimulated with plate-bound anti-CD3 plus soluble anti-CD28 for the indicated times. Nuclear extracts were prepared and subjected to EMSA using a 32P-radiolabeled κB oligonucleotide as previous described (51).
RNase protection assay and RT-PCR.
Total cellular RNA was isolated from the colon using the TRI reagent (Molecular Research Center, Inc.). RNase protection assay was performed using the RiboQuant reagents (BD Biosciences) and a custom template set according to the manufacturer's instructions. Semiquantitative RT-PCR was performed using specific primers for the indicated mouse genes.
Statistics.
Statistical significance was determined by Student's t test.
Online supplemental material.
Fig. S1 shows the hyperresponsiveness of Cyld−/− splenic T cells, and Fig. S2 shows the enhanced expression of inflammatory genes in colons of RAG1 knockout mice adoptively transferred with Cyld−/− T cells. Data presented in Fig. S3 suggest that the loss of CYLD does not result in the constitutive activation of IKKβ and Tak1 nor affects the kinetics of inducible IKK activation in macrophages. Fig. S4 shows that CYLD is not important for turning off the IKK activation signal in Jurkat T cells. Fig S5 shows the normal expression of NOD2 in Cyld-deficient cells. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20062694/DC1.
Supplemental Material
Acknowledgments
We thank Kang Li, Nate Sheaffer, and Anne Stanley of the Pennsylvania State College of Medicine core facilities for assistance with tissue sections, flow cytometry, and oligonucleotide synthesis. We also thank Vincent Chau, Kunihiro Matsumoto, and Jun Ninomiya-Tsuji for reagents.
This study was supported by grants from the National Institutes of Health (NIH; AI064639 and CA94922 to S.C. Sun, AI057555 to S.C. Sun and M. Zhang, and AI056094 to C.C. Norbury) and by an award from the Carlino Account Funds of the Section of Colon and Rectal Surgery (Pennsylvania State College of Medicine) to S.C. Sun, L. Fitzpatrick, and M. Zhang. A. Wright is a recipient of the Ruth L. Kirschstein National Research Service Award. E.F. Tewalt is a trainee of a predoctoral/postdoctoral training grant (2T32CA60395-11) from the NIH. All animals were housed in a facility constructed with support from Research Facilities Improvement (grant C06 RR-15428-01) from the National Center for Research Resources (NIH).
The authors have no conflicting financial interests.
Abbreviations used: DUB, deubiquitinating enzyme; EMSA, electrophoresis mobility shift assay; ERK, extracellular-regulated kinase; GST, glutathione S-transferase; IB, immunoblotting; IBD, inflammatory bowel disease; IKK, IκB kinase; IP, immunoprecipitation; JNK, c-Jun N-terminal kinase; Tak1, TGF-β–activated kinase 1.
W. W. Reiley and W. Jin contributed equally to this paper.
References
- 1.Siegel, R.M., M. Katsumata, S. Komori, S. Wadsworth, L. Gill-Morse, S. Jerrold-Jones, A. Bhandoola, M.I. Greene, and K. Yui. 1990. Mechanisms of autoimmunity in the context of T-cell tolerance: insights from natural and transgenic animal model systems. Immunol. Rev. 118:165–192. [DOI] [PubMed] [Google Scholar]
- 2.Liu, Y.C. 2004. Ubiquitin ligases and the immune response. Annu. Rev. Immunol. 22:81–127. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson, K.D. 2000. Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Semin. Cell Dev. Biol. 11:141–148. [DOI] [PubMed] [Google Scholar]
- 4.Reiley, W.W., M. Zhang, W. Jin, M. Losiewicz, K.B. Donohue, C.C. Norbury, and S.C. Sun. 2006. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat. Immunol. 7:411–417. [DOI] [PubMed] [Google Scholar]
- 5.Bignell, G.R., W. Warren, S. Seal, M. Takahashi, E. Rapley, R. Barfoot, H. Green, C. Brown, P.J. Biggs, S.R. Lakhani, et al. 2000. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 25:160–165. [DOI] [PubMed] [Google Scholar]
- 6.Brummelkamp, T.R., S.M. Nijman, A.M. Dirac, and R. Bernards. 2003. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 424:797–801. [DOI] [PubMed] [Google Scholar]
- 7.Kovalenko, A., C. Chable-Bessia, G. Cantarella, A. Israel, D. Wallach, and G. Courtois. 2003. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 424:801–805. [DOI] [PubMed] [Google Scholar]
- 8.Trompouki, E., E. Hatzivassiliou, T. Tsichritzis, H. Farmer, A. Ashworth, and G. Mosialos. 2003. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 424:793–796. [DOI] [PubMed] [Google Scholar]
- 9.Regamey, A., D. Hohl, J.W. Liu, T. Roger, P. Kogerman, R. Toftgard, and M. Huber. 2003. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor κB activation by tumor necrosis factor. J. Exp. Med. 198:1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reiley, W., M. Zhang, and S.-C. Sun. 2004. Tumor suppressor negatively regulates JNK signaling pathway downstream of TNFR members. J. Biol. Chem. 279:55161–55167. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida, H., H. Jono, H. Kai, and J.D. Li. 2005. The tumor suppressor CYLD acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 and TRAF7. J. Biol. Chem. 280:41111–41121. [DOI] [PubMed] [Google Scholar]
- 12.Massoumi, R., K. Chmielarska, K. Hennecke, A. Pfeifer, and R. Fassler. 2006. Cyld inhibits tumor cell proliferation by blocking bcl-3-dependent NF-kappaB signaling. Cell. 125:665–677. [DOI] [PubMed] [Google Scholar]
- 13.Zhang, J., B. Stirling, S.T. Temmerman, C.A. Ma, I.J. Fuss, J.M. Derry, and A. Jain. 2006. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J. Clin. Invest. 116:3042–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin, X., and D. Wang. 2004. The roles of CARMA1, Bcl10, and MALT1 in antigen receptor signaling. Semin. Immunol. 16:429–435. [DOI] [PubMed] [Google Scholar]
- 15.Hacker, H., and M. Karin. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE. 10.1126/stke.3572006re13. [DOI] [PubMed]
- 16.Senftleben, U., Y. Cao, G. Xiao, G. Kraehn, F. Greten, Y. Chen, Y. Hu, A. Fong, S.-C. Sun, and M. Karin. 2001. Activation of IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 293:1495–1499. [DOI] [PubMed] [Google Scholar]
- 17.Xiao, G., E.W. Harhaj, and S.C. Sun. 2001. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell. 7:401–409. [DOI] [PubMed] [Google Scholar]
- 18.Liu, H.H., M. Xie, M.D. Schneider, and Z.J. Chen. 2006. Essential role of TAK1 in thymocyte development and activation. Proc. Natl. Acad. Sci. USA. 103:11677–11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato, S., H. Sanjo, T. Tsujimura, J. Ninomiya-Tsuji, M. Yamamoto, T. Kawai, O. Takeuchi, and S. Akira. 2006. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int. Immunol. 18:1405–1411. [DOI] [PubMed] [Google Scholar]
- 20.Wan, Y.Y., H. Chi, M. Xie, M.D. Schneider, and R.A. Flavell. 2006. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 7:851–858. [DOI] [PubMed] [Google Scholar]
- 21.Wang, C., L. Deng, M. Hong, G.R. Akkaraju, J.-I. Inoue, and Z.J. Chen. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 412:346–351. [DOI] [PubMed] [Google Scholar]
- 22.Ninomiya-Tsuji, J., K. Kishimoto, A. Hiyama, J. Inoue, Z. Cao, and K. Matsumoto. 1999. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 398:252–256. [DOI] [PubMed] [Google Scholar]
- 23.Dohi, T., K. Fujihashi, P.D. Rennert, K. Iwatani, H. Kiyono, and J.R. McGhee. 1999. Hapten-induced colitis is associated with colonic patch hypertrophy and T helper cell 2-type responses. J. Exp. Med. 189:1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sartor, R.B. 2006. Mechanisms of disease: pathogenesis of Chron's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3:390–407. [DOI] [PubMed] [Google Scholar]
- 25.Niessner, M., and B.A. Volk. 1995. Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT-PCR). Clin. Exp. Immunol. 101:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melgar, S., M.M. Yeung, A. Bas, G. Forsberg, O. Suhr, A. Oberg, S. Hammarstrom, A. Danielsson, and M.L. Hammarstrom. 2003. Over-expression of interleukin 10 in mucosal T cells of patients with active ulcerative colitis. Clin. Exp. Immunol. 134:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenner, O., D. Levanon, V. Negreanu, O. Golubkov, O. Fainaru, E. Woolf, and Y. Groner. 2004. Loss of Runx3 function in leukocytes is associated with spontaneously developed colitis and gastric mucosal hyperplasia. Proc. Natl. Acad. Sci. USA. 101:16016–16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Podolsky, D.K. 2002. Inflammatory bowel disease. N. Engl. J. Med. 347:417–429. [DOI] [PubMed] [Google Scholar]
- 29.Molina, T.J., K. Kishihara, D.P. Siderovski, W. van Ewijk, A. Narendran, E. Timms, A. Wakeham, C.J. Paige, K.U. Hartmann, A. Veillette, et al. 1992. Profound block in thymocyte development in mice lacking p56lck. Nature. 357:161–164. [DOI] [PubMed] [Google Scholar]
- 30.Lombardi, L., P. Ciana, C. Cappellini, D. Trecca, L. Guerrini, A. Migliazza, A.T. Maiolo, and A. Neri. 1995. Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res. 23:2328–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bren, G.D., N.J. Solan, H. Miyoshi, K.N. Pennington, L.J. Pobst, and C.V. Paya. 2001. Transcription of the RelB gene is regulated by NF-kappaB. Oncogene. 20:7722–7733. [DOI] [PubMed] [Google Scholar]
- 32.Thiefes, A., A. Wolf, A. Doerrie, G.A. Grassl, K. Matsumoto, I. Autenrieth, E. Bohn, H. Sakurai, R. Niedenthal, K. Resch, and M. Kracht. 2006. The Yersinia enterocolitica effector YopP inhibits host cell signalling by inactivating the protein kinase TAK1 in the IL-1 signalling pathway. EMBO Rep. 7:838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato, S., H. Sanjo, K. Takeda, J. Ninomiya-Tsuji, M. Yamamoto, T. Kawai, K. Matsumoto, O. Takeuchi, and S. Akira. 2005. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 6:1087–1095. [DOI] [PubMed] [Google Scholar]
- 34.Shim, J.H., C. Xiao, A.E. Paschal, S.T. Bailey, P. Rao, M.S. Hayden, K.Y. Lee, C. Bussey, M. Steckel, N. Tanaka, et al. 2005. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19:2668–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Omori, E., K. Matsumoto, H. Sanjo, S. Sato, S. Akira, R.C. Smart, and J. Ninomiya-Tsuji. 2006. TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. J. Biol. Chem. 281:19610–19617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee, E.G., D.L. Boone, S. Chai, S.L. Libby, M. Chien, J.P. Lodolce, and A. Ma. 2000. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 289:2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boone, D.L., E.E. Turer, E.G. Lee, R.C. Ahmad, M.T. Wheeler, C. Tsui, P. Hurley, M. Chien, S. Chai, O. Hitotsumatsu, et al. 2004. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5:1052–1060. [DOI] [PubMed] [Google Scholar]
- 38.Wertz, I.E., K.M. O'Rourke, H. Zhou, M. Eby, L. Aravind, S. Seshagiri, P. Wu, C. Wiesmann, R. Baker, D.L. Boone, et al. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 430:694–699. [DOI] [PubMed] [Google Scholar]
- 39.Strober, W., P.J. Murray, A. Kitani, and T. Watanabe. 2006. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat. Rev. Immunol. 6:9–20. [DOI] [PubMed] [Google Scholar]
- 40.Hugot, J.P., M. Chamaillard, H. Zouali, S. Lesage, J.P. Cezard, J. Belaiche, S. Almer, C. Tysk, C.A. O'Morain, M. Gassull, et al. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 411:599–603. [DOI] [PubMed] [Google Scholar]
- 41.Ogura, Y., D.K. Bonen, N. Inohara, D.L. Nicolae, F.F. Chen, R. Ramos, H. Britton, T. Moran, R. Karaliuskas, R.H. Duerr, et al. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 411:603–606. [DOI] [PubMed] [Google Scholar]
- 42.Pauleau, A.L., and P.J. Murray. 2003. Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol. Cell. Biol. 23:7531–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi, K.S., M. Chamaillard, Y. Ogura, O. Henegariu, N. Inohara, G. Nunez, and R.A. Flavell. 2005. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 307:731–734. [DOI] [PubMed] [Google Scholar]
- 44.Maeda, S., L.C. Hsu, H. Liu, L.A. Bankston, M. Limura, M.F. Kagnoff, L. Eckmann, and M. Karin. 2005. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science. 307:734–738. [DOI] [PubMed] [Google Scholar]
- 45.Costello, C.M., N. Mah, R. Hasler, P. Rosenstiel, G.H. Waetzig, A. Hahn, T. Lu, Y. Gurbuz, S. Nikolaus, M. Albrecht, J. Hampe, R. Lucius, G. Kloppel, H. Eickhoff, H. Lehrach, T. Lengauer, and S. Schreiber. 2005. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med. 2:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shibuya, H., K. Yamaguchi, K. Shirakabe, A. Tonegawa, Y. Gotoh, N. Ueno, K. Irie, E. Nishida, and K. Matsumoto. 1996. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science. 272:1179–1182. [DOI] [PubMed] [Google Scholar]
- 47.Waterfield, M., J. Wei, W. Reiley, M.Y. Zhang, and S.-C. Sun. 2004. IKKb Is an essential component of the Tpl2 signaling pathway. Mol. Cell. Biol. 24:6040–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reiley, W., M. Zhang, X. Wu, E. Graner, and S.-C. Sun. 2005. Regulation of the deubiquitinating enzyme CYLD by IKKg-dependent phosphorylation. Mol. Cell. Biol. 25:3886–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams, K.L., C.R. Fuller, L.A. Dieleman, C.M. DaCosta, K.M. Haldeman, R.B. Sartor, and P.K. Lund. 2001. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology. 120:925–937. [DOI] [PubMed] [Google Scholar]
- 50.Uhlik, M., L. Good, G. Xiao, E.W. Harhaj, E. Zandi, M. Karin, and S.-C. Sun. 1998. NF-kappaB-inducing kinase and IkappaB kinase participate in human T-cell leukemia virus I Tax-mediated NF-kappaB activation. J. Biol. Chem. 273:21132–21136. [DOI] [PubMed] [Google Scholar]
- 51.Sun, S.-C., P.A. Ganchi, C. Beraud, D.W. Ballard, and W.C. Greene. 1994. Autoregulation of the NF-κB transactivator Rel A (p65) by multiple cytoplasmic inhibitors containing ankyrin motifs. Proc. Natl. Acad. Sci. USA. 91:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}