

Abstract
Members of the Runx family of transcriptional regulators are required for the appropriate expression of CD4 and CD8 at discrete stages of T cell development. The roles of these factors in other aspects of T cell development are unknown. We used a strategy to conditionally inactivate the genes encoding Runx1 or Runx3 at different stages of thymocyte development, demonstrating that Runx1 regulates the transitions of developing thymocytes from the CD4−CD8− double-negative stage to the CD4+CD8+ double-positive (DP) stage and from the DP stage to the mature single-positive stage. Runx1 and Runx3 deficiencies caused marked reductions in mature thymocytes and T cells of the CD4+ helper and CD8+ cytotoxic T cell lineages, respectively. Runx1-deficient CD4+ T cells had markedly reduced expression of the interleukin 7 receptor and exhibited shorter survival. In addition, inactivation of both Runx1 and Runx3 at the DP stages resulted in a severe block in development of CD8+ mature thymocytes. These results indicate that Runx proteins have important roles at multiple stages of T cell development and in the homeostasis of mature T cells.
Development of T lymphocytes is initiated after migration of bone marrow–derived progenitors into the thymus (1). Thymocyte progenitors then differentiate through a series of stages into mature T cells that express TCRαβ and have distinct functional properties to allow for immune responses against foreign antigens while avoiding reactivity to self-antigens (2). T cell precursors in the thymus are classified into four major populations based on expression of the two coreceptor molecules, CD4 and CD8. The most immature thymocytes, which express neither CD4 nor CD8 (double-negative [DN] thymocytes), are further subdivided into four subpopulations designated as DN1–DN4 based on expression of the CD44 and CD25 markers (3). At the DN2 (CD44+CD25+) and the DN3 (CD44−CD25+) developmental stages, precursors initially rearrange and express Tcrb to form pre–T cell receptors (preTCRs) (4). Signals transduced through the preTCR after successful Tcrb rearrangement drive precursors into a robust proliferation phase (5). This process, named β selection, is followed by down-regulation of CD25 in DN4 cells and subsequent induction of CD4 and CD8 expression as cells transit to the double-positive (DP) stage, at which thymocytes rearrange Tcra and express TCRαβ heterodimers that interact with peptide–MHC complexes expressed on thymic epithelial cells. The few cells bearing TCRαβ with appropriate avidity for peptide–MHC undergo positive selection, whereas those with lower or higher avidities are eliminated by apoptotic processes known, respectively, as “death by neglect” and negative selection (2). Cells with TCRαβs specific for MHC class I are selected to become CD4−CD8+ single-positive (CD8SP) thymocytes that give rise to cytotoxic T cells, whereas those with MHC class II–restricted receptors are selected to become CD4+CD8− (CD4SP) thymocytes and helper T cells.
The transcription factor ThPOK/cKrox, encoded by Zbtb7b, plays a crucial role in generation of CD4SP mature thymocytes (6, 7). In spontaneous mutant mice with a loss of function mutation of ThPOK, termed hd/hd (helper-deficient) mice, all selected thymocytes differentiate into the CD8SP lineage regardless of the MHC specificity of the TCR. Conversely, transgenic overexpression of ThPOK in DP thymocytes causes redirection of thymocytes selected through interaction of TCR and MHC class I from the CD8 to the CD4 lineage. The transcription factor GATA3 is also important for generation of CD4SP thymocytes (8, 9). GATA3-deficient DP thymocytes fail to develop to the CD4SP stage after positive selection, and GATA3 overexpression blocks differentiation of CD8SP thymocytes after positive selection by MHC class I, whereas misexpression of GATA3 does not result in lineage redirection. It remains unclear whether there is a key transcription factor essential for commitment to the CD8SP lineage or if CD8SP development is a default pathway that can be altered only when ThPOK and GATA3 are expressed.
Regulation of CD4 and CD8 expression is critical for the appropriate selection of the TCRαβ repertoire during thymocyte differentiation (10). Whereas the Cd8a and Cd8b genes that encode CD8 are regulated by multiple stage-specific enhancers during T cell development, Cd4 is primarily regulated by its unique cis-acting element, the Cd4 silencer, located in the first intron (11). Targeted deletion of the Cd4 silencer resulted in CD4 derepression in DN and CD8SP thymocytes, indicating that the silencer is necessary for negative regulation of CD4 expression at both of these stages (12). Interestingly, conditional inactivation of the Cd4 silencer in committed CD4−CD8+ cells did not restore CD4 expression (13). This and other results have demonstrated that the Cd4 silencer is required for the establishment but not for the maintenance of CD4 silencing in CD8SP cells, and that the silencing is maintained by epigenetic mechanisms (12). Epigenetic CD4 silencing is thus tightly linked to development of the CD8SP lineage of T cells.
We have identified the Runx proteins as factors binding to the Cd4 silencer and negatively regulating CD4 expression in DN and CD8SP mature thymocytes (14). The Runx family of transcription factors consists of three members, Runx1/AML1/Cbfa2/PEBP2αB, Runx2/AML3/Cbfa1/Pebp2αA, and Runx3/AML2/Cbfa3/Pebp2αC (15). All three Runx proteins share a highly conserved DNA-binding runt domain and the C-terminal VWRPY motif. Association of Runx proteins with the common cofactor CBFβ by way of the runt domain increases binding to target DNA sequences. Members of the Groucho family of corepressors associate with the Runx C-terminal motif, which is required for Runx3 to mediate CD4 silencing (16, 17). Runx1 and Runx3 are differentially required for CD4 silencing at the DN and CD8SP stages of thymocyte differentiation, respectively (14). Runx1 also contributes to CD8 expression at the DP stage (14), whereas Runx3 binds to CD8 enhancers (18) and may contribute to CD8 expression in CD8SP and mature CD8 lineage T cells. Because Runx proteins regulate CD4 and CD8 expression, they may also have important roles in thymocyte selection and CD4 versus CD8 lineage-commitment processes. The Runx3 protein plays a crucial role in CD4 silencing that is linked to commitment to the CD8SP lineage (14, 19), and it is expressed in CD8SP thymocytes but not in CD4SP thymocytes (20). These findings suggest that Runx3 is a potential key regulator for CD8SP lineage specification. However, it remains unclear whether Runx proteins play any role in the process of lineage commitment of positively selected thymocytes.
To clarify the functions of Runx proteins during T cell development, we analyzed thymocyte and T cell development in T cell lineage-specific Runx1 and Runx3 conditional knockout mice and report that Runx1 plays an important role in thymocyte selection and maturation of selected T cells. Whereas Runx3 inactivation at the DP stage caused a mild reduction in the number of CD8+ mature thymocytes and CD8+ T cells, compound mutation of Runx1 and Runx3 resulted in a severe reduction of positively selected thymocytes and almost a complete loss of CD8SP mature thymocytes. Runx1 was found to be important for the survival of CD4 lineage T cells, in part by regulating expression of IL-7Rα. These results demonstrate that Runx proteins have multiple functions at different stages of T cell development and in T cell homeostasis.
RESULTS
Runx1 is required for the DN to DP transition in TCRαβ T cell development
We previously showed that Runx1 inactivation by the Lck-cre transgene at the DN2 and DN3 stages resulted in CD4 derepression and thymic hypocellularity (14). To elucidate the role of Runx1 in early thymocyte differentiation, we performed a detailed analysis in Runx1 F/F;Lck-cre mice. As a result of Runx1 inactivation at the DN stage, we observed a reduced number of DP and mature thymocytes, accompanied by a relative increase in the proportion of DN thymocytes (Fig. 1 A and see Fig. 2 E). Total thymocyte numbers were reduced by approximately sixfold compared with those in the littermate Runx1 F/F or control Lck-cre mice (unpublished data). At the DN stage, defined as TCRβ−CD8−CD4−/lo, we consistently observed accumulation of CD25hi DN2 and DN3 thymocytes, which had higher expression of CD25 than control DN2 and DN3 cells (Fig. 1 A, bottom). The absolute numbers of DN2 and DN3 thymocytes were similar between Runx1 F/F;Lck-cre and control mice, suggesting that thymocyte development in Runx1 F/F;Lck-cre mice was severely compromised during transition from the DN3 to DN4 stages (Fig. 1, B and E). TCRγδ cell development in the thymus was unaffected in Runx1 F/F;Lck-cre mice (Fig. 1 C).
Figure 1.

Runx1 requirement in the differentiation of DN thymocytes. (A) CD4 and CD8 expression in total thymocytes (top) and CD25 and CD44 expression in CD4lo/−CD8−TCRβ− thymocytes (bottom) from Runx1 F/F;Lck-cre mice and littermate Runx1 F/F control mice. Percentages of cells are shown in the indicated gates. (B and C) CD25 and icTCRβ (B) and icTCRβ and TCRγδ (C) in CD4lo/−CD8−TCRβ− thymocytes are shown. Gates used for population definition and frequencies of the gated population in CD4lo/−CD8−TCRβ− thymocytes are indicated. (D) Incorporation of BrdU by the thymocyte subpopulations defined in B. Numbers indicate the percentages of BrdU+ cells defined by the interval gates. (E) Absolute numbers of thymocytes in immature thymocyte subpopulations from Runx1 F/F;Lck-cre mice (closed bars) and littermate Runx1 F/F mice (open bars) are shown. Data were averaged from five mice with standard deviations.
Figure 2.

Runx1 is required for positive selection and maturation of CD4SP thymocytes. (A) Expression of CD4, CD8, TCRβ, CD69, and HSA (CD24) in thymocytes from Runx1 F/F;Cd4-cre and littermate Runx1 F/F mice (top) is shown. Percentages of cells are shown in the indicated gates. (B) TCRβ expression of total thymocytes from Runx1 F/F;Cd4-cre (open histogram) and littermate Runx1 F/F (shaded histogram) mice. (C) CD4 and CD8 expression in thymocytes gated for TCRβhiHSAhi (top) and TCRβhiHSAlo (bottom) from Runx1 F/F;Cd4-cre and littermate Runx1 F/F mice. Percentages of CD4+CD8− thymocytes in the gated rectangles are shown. (D) Development of CD4SP and CD8SP thymocytes in Runx1 F/F;Cd4-cre and littermate Runx1 F/F;Cd4-cre; b2m −/− mice. The percentages of CD4SP and CD8SP mature thymocytes are shown. (E and F) The absolute numbers of total thymocytes and DP thymocytes (E) and mature thymocyte subpopulations (F) are shown as the mean and standard deviation from 4–10 mice with the indicated genotypes. Statistical differences were tested by the Student's t test. *, P < 0.05; **, P < 0.01.
Developmental arrest at the DN3 stage often coincides with impaired β selection caused by either loss of rearranged TCRβ protein expression or defects in preTCR signaling. To determine whether TCRβ expression and/or β selection was affected, staining for intracellular TCR β chain (icTCRβ) protein and cell-cycle analysis were performed (Fig. 1, B and D; and not depicted). In DN thymocytes from control Runx1 F/F mice, ∼25% of CD25+ cells expressed icTCRβ protein, and 30% of the total DN thymocytes had down-regulated CD25 to become icTCRβ+CD25− DN4 thymocytes. 40–50% of icTCRβ+ cells, which were either CD25+ or CD25−, were in the S/G2/M phases in cell-cycle analysis and actively incorporated BrdU during a 2-h pulse labeling (Fig. 1, B and D; and not depicted), indicating that thymocytes selected through β selection underwent robust proliferation after they expressed icTCRβ and were stimulated by preTCR signaling. In DN thymocytes from Runx1 F/F;Lck-cre mice, a similar or slightly smaller fraction of CD25+ cells expressed icTCRβ protein. However, the number of CD25lo/−icTCRβ+ cells was severely reduced (Fig. 1 B). The CD25+icTCRβ+ cells exhibited an active cell-cycling status, as determined by both DAPI staining for DNA content and BrdU incorporation, whereas the residual CD25lo/−icTCRβlo DN4 cells displayed little proliferation (Fig. 1 D; and not depicted). These findings indicate that Runx1-deficient CD25hi cells could be primed by preTCR signals during β selection similar to control DN3 cells. However, Runx1-deficient DN3 cells failed to continue proliferating during transition from the DN3 to DN4 stages and, subsequently, to the DP stage, which caused a reduction in total thymocyte cellularity.
Runx1 is required for positive selection and maturation of CD4SP thymocytes
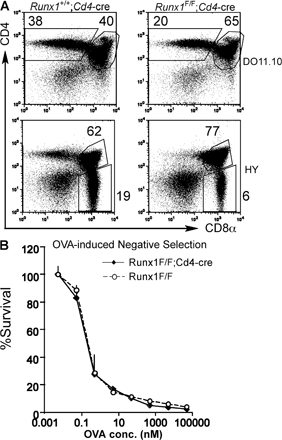
The block in thymocyte development in Runx1 F/F;Lck-cre mice prevented us from analyzing the role of Runx1 at later stages of T cell development. Therefore, we used the Cd4-cre mice to inactivate the Runx1 gene at the DP stage. Although we observed the intact Runx1 floxed allele in mature CD4+ and CD8+ T cells from Runx1 F/F;Lck-cre mice, suggesting selection of cells that escaped deletion (14), Cre-mediated deletion in CD4+ and CD8+ T cells from Runx1 F/F;Cd4-cre was almost complete at the DNA and protein levels (unpublished data). Total thymocyte numbers and DP thymocyte development were intact in Runx1 F/F;Cd4-cre as compared with control Runx1 F/F mice, except that the level of TCRβ in Runx1 F/F;Cd4-cre DP thymocytes was lower than in controls (Fig. 2, B and E), whereas the TCRβ expression level in CD4SP was slightly lower and CD8SP cells had normal TCRβ expression (unpublished data). A fraction of DP thymocytes from Runx1 F/F;Cd4-cre mice and the majority of DP thymocytes from Runx1 F/F;Lck-cre mice expressed CD25 at a low level (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070133/DC1). These results suggest that Runx1 is not necessary for the survival or maintenance of DP thymocytes but is necessary for completely silencing CD25 expression and for full TCRβ expression. In the thymus from Runx1 F/F;Cd4-cre mice, however, there was a reduction in SP mature thymocytes in both the CD4 and CD8 lineages (Fig. 2, A and F). During positive selection, thymocytes initially show a TCRβintCD69+ transitional phenotype immediately after TCR stimulation by self-MHC and peptide regardless of MHC class I– or class II–restricted selection and become TCRβhi cells, followed by down-regulation of CD69 and CD24 (heat-stable antigen [HSA]). In the Runx1 F/F;Cd4-cre thymocytes, there was a threefold reduction in the number of TCRβintCD69+ cells, and the number of TCRβhiHSAlo thymocytes was reduced by fourfold (Fig. 2 A, middle and bottom). The number of CD4SP mature thymocytes defined as TCRhiHSAloCD4+CD8− was reduced by eightfold compared with control mice, whereas the CD8 mature thymocyte number was less severely reduced (Fig. 2 C, bottom; and Fig. 2 F). The CD4/CD8 ratio at the TCRβhi HSAlo stage was reduced to 0.3 in Runx1 F/F;Cd4-cre mice from 2.4 in control mice (Fig. 2 C). Reduced mature thymocyte numbers were also observed in MHC class I–restricted HY TCR transgenic mice and MHC class II–restricted DO.11.10 TCR transgenic mice bred to Runx1 F/F;Cd4-cre mice, as compared with the TCR transgenic mice in the wild-type Runx1 background (Fig. S2 A).
These results suggested that Runx1 could play an important role for both thymocyte positive selection and commitment to the CD4SP lineage. However, CD8SP mature thymocytes failed to be generated in β2m−/−;Runx1 F/F;Cd4-cre mice (Fig. 2 D). Together with the finding that the frequency of CD4+8− thymocytes was similar between Runx1 F/F;Cd4-cre and control mice in the TCRβhiHSAhi transitional population (Fig. 2 C, top), the reduction of CD4SP thymocytes in the absence of Runx1 was therefore not caused by redirection of CD4SP thymocytes to the CD8SP lineage. Instead, it is likely that maturation or survival of committed CD4SP thymocytes is dependent on Runx1. The number of Foxp3+ regulatory T cells was slightly reduced but was not as severely affected as the number of Foxp3− CD4+ T cells (unpublished data).
To determine whether Runx1 is involved in negative selection, Runx1 F/F;Cd4-cre mice were crossed to DO11.10 TCR transgenic mice in the I-Ad MHC background, and thymocyte apoptosis was examined upon incubation with OVA peptide and antigen-presenting cells. No difference in the number of cells undergoing apoptosis was detected between Runx1 F/F;Cd4-cre and control thymocytes, indicating that Runx1 is not required for negative selection (Fig. S2 B).
Runx1 is required for homeostasis of mature CD4+ T cells but not of CD8+ T cells
We next examined the development of TCRαβ cells in the periphery of Runx1 F/F;Cd4-cre mice. In the spleen and lymph nodes, the CD4/CD8 ratio was further reduced to 0.15 in Runx1 F/F;Cd4-cre mice as compared with 1.6 in control mice (Fig. 3 A). The absolute number of CD4+ T cells in the spleen and lymph nodes from Runx1 F/F;Cd4-cre mice remained severely reduced, whereas the CD8+ T cell number was unaffected (Fig. 3 B). Interestingly, the frequency of Foxp3+ regulatory T cells was markedly increased in Runx1 F/F;Cd4-cre mice (Fig. 3 C). Approximately 50% of peripheral CD4+ T cells from Runx1 F/F;Cd4-cre mice were Foxp3+, compared with 15% of CD4+ T cells from control mice (Fig. 3 C). Runx1-deficient Foxp3+ cells expressed a slightly lower level of Foxp3 and a normal level of CD25 compared with control mice, whereas a majority of Foxp3−CD4+ T cells from Runx1 F/F;Cd4-cre mice expressed CD25 at an intermediate level. Because the absolute number of Foxp3+ regulatory T cells was only slightly reduced compared with control mice (unpublished data), the overall reduction of CD4+ T cells in Runx1 F/F;Cd4-cre mice was most likely caused by compromised homeostasis of Foxp3− conventional T cells.
Figure 3.

Runx1 regulates homeostasis of CD4+ T cells in the periphery. (A) TCRβ and CD19 expression (top) and CD4 and CD8 expression in gated TCRβ+ cells (bottom). The percentages of populations gated in the rectangles are shown. (B) Numbers of TCRβ+ cells, CD4+ T cells, and CD8+ T cells from spleens are shown as the means and standard deviations from 4–10 mice of each indicated genotype. (C) Foxp3 and CD25 expression in CD4+ T cells. Peripheral lymphocytes were stained for TCRβ, CD4, CD25, and intracellular Foxp3, and Foxp3 and CD25 expression in TCRβ+CD4+CD8− cells is shown. The percentages of Foxp3+ cells gated by the rectangles are shown. (D) BrdU incorporation by peripheral lymphocytes in Runx1 F/F;Cd4-cre and littermate Runx1 F/F mice. Mice were administered BrdU for 72 h in drinking water, and percentages of BrdU+ cells in gated B cells, CD4+ T cells, and CD8+ T cells are shown. Data shown represent three experiments with similar results. Statistical differences were tested by the Student's t test. *, P < 0.05; **, P < 0.01.
To determine if the reduced CD4+ T cell number was caused by impaired peripheral expansion of mature CD4+ T cells, we examined the proportion of peripheral T cell that entered S phase by measuring BrdU incorporation over a 72-h period (Fig. 3 D). BrdU incorporation by CD19+ B lymphocytes was similar between Runx1 F/F;Cd4-cre and control mice. In control Runx1 F/F mice, an approximately similar proportion of CD4+ and CD8+ T cells was labeled by BrdU. However, approximately threefold more CD4+ T cells were labeled in Runx1 F/F;Cd4-cre mice than in control mice. This difference was confined to CD4 lineage T cells, indicating that Runx1-deficient peripheral CD4+ T cells undergo homeostatic expansion more actively than CD8+ T cells but fail to restore this cellular compartment. These results suggest that the reduced CD4+ T cell number is caused by accelerated cell death rather than compromised homeostatic proliferation.
To determine if the reduction of CD4+ T cells was mainly caused by a specific impairment in maturation and homeostasis in the CD4 lineage and to quantitate defects caused by Runx1 inactivation during T cell development, we analyzed mixed bone marrow chimeric mice that had been reconstituted with a 1:1 mixture of CD45.1 wild-type and CD45.2 Runx1 F/F;Cd4-cre or control Runx1 F/F bone marrow cells (Fig. 4). In DN thymocytes, in which Runx1 was not inactivated by Cd4-cre, and in DP thymocytes, contribution of CD45.2+ cells was ∼50%, indicating that Runx1 is not required for maintenance of DP thymocytes. In TCRβintCD69+ thymocytes, found immediately after positive selection, the contribution of Runx1-deficient CD45.2+ cells was reduced to 30%, whereas control CD45.2+ cells were found in numbers equivalent to CD45.1+ wild-type cells. The contribution of CD45.2+ Runx1 F/F;Cd4-cre cells to CD4SP thymocytes and to peripheral CD4+ T cells was further decreased to 8 and 2%, respectively. Development of CD8SP thymocytes and mature CD8+ T cells was also affected by the ablation of Runx1 expression, but not nearly as severely as the development of CD4 lineage cells (Fig. 4 B). Mature CD8SP thymocytes generated from Runx1 F/F;Cd4-cre DP cells were similar to the control CD45.1+ cells in their ability to repopulate the peripheral T cell pool. In contrast, the contribution of Runx1-deficient CD4SP cells to the peripheral CD4 lineage T cell pool continued to erode during progression toward maturity (Fig. 4, B and C).
Figure 4.

Quantitative analysis of precursor activity of Runx1-deficient thymocytes by a competitive repopulation assay. (A) Strategy for generation of mixed bone chimeras with CD45.2+ bone marrow cells from Runx1 F/F;Cd4-cre (Mut) or littermate control Cd4-cre (WT) mice. CD45.2+ bone marrow cells mixed with wild-type CD45.2− (CD45.1; Comp) competitor bone marrow cells at the ratio of 1:1 were transferred into lethally irradiated Rag2−/− hosts. T cell development was analyzed 10–12 wk after transplantation. (B) Contribution of CD45.2+ bone marrow cells at different stages of T cell development. Reconstituted thymocytes or lymph node cells were analyzed for frequency of CD45.2+-expressing cells in populations defined by CD4, CD8, TCRβ, and CD69 expression. (C) Relative T cell precursor potential of Runx1-deficient thymocytes, as determined in the competitive repopulation assay. The values for T cell precursor potential in the absence of Runx1 were normalized with the precursor potential of thymocytes derived from CD45.2+ control bone marrow cells, as described in Materials and methods.
Expression of IL-7Rα is impaired in thymocytes and CD4+ T cells in the absence of Runx1
Earlier studies have suggested that both suboptimal TCR–MHC interactions and cytokine signals mediated by IL-7 are indispensable for the survival of naive T cells in the periphery (21, 22) . We hypothesized that the reduction of CD4+ mature thymocytes and T cells in the absence of Runx1 could be caused by compromised survival because of loss or reduction of survival signals. To test this hypothesis, we examined expression of IL-7Rα in the Runx1-deficient thymocytes and peripheral T cells (Fig. 5 A). In control mice, IL-7Rα expression was detected in a fraction of TCRβintCD69+ and in all TCRβhiCD69+ thymocytes after positive selection. Uniform expression of IL-7Rα was also observed in both CD4 and CD8 lineages as thymocytes further matured to the TCRβhiCD69− stage and into peripheral naive T cells. IL-7Rα expression was slightly decreased in TCRβintCD69+ thymocytes from Runx1 F/F;Cd4-cre mice compared with control mice. However, in TCRβhiCD69+ thymocytes, in which uniform IL-7Rα up-regulation was observed in wild-type mice, IL-7Rα expression was considerably compromised in the absence of Runx1. In the HSAlo/CD69− mature thymocyte population, IL-7Rα expression in the absence of Runx1 remained low in CD4+ cells but was almost normal in CD8+ T cells. Similarly, lymph node CD4+ T cells from Runx1 F/F;Cd4-cre mice expressed a lower level of IL-7Rα than CD4+ T cells from control mice, but CD8 lineage cells were unaffected (Fig. 5 B).
Figure 5.

IL-7Rα expression in developing thymocytes and T cells from Runx1F/F;Cd4-cre mice. (A) IL-7Rα (CD127) expression in thymocyte populations defined by rectangles (top) from Runx1 F/F (dotted histograms) and Runx1 F/F;Cd4-cre (open histograms) mice. (B) IL-7Rα expression in CD4+ and CD8+ lymph node T cells from Runx1 F/F (dotted histograms) and Runx1 F/F;Cd4-cre (open histograms) mice. CD4+ T cells were further subdivided into Foxp3+ and Foxp3− populations, and IL-7Rα expression in each subpopulation was compared between Runx1 F/F (dotted histograms) and Runx1 F/F;Cd4-cre (open histograms) mice. Shaded histograms in A and B are isotype staining controls. (C) Real-time PCR analysis of Il7r mRNA expression in mature thymocytes from Runx1 F/F;Cd4-cre and control Runx1 F/F mice. Il7r mRNA expression levels are normalized to Actb mRNA levels in individual samples. The relative Il7r mRNA expression is shown as the mean and standard deviation from three independent samples. Statistical differences were verified by the Student's t test. Flow cytometry data shown are representative of three to five mice.
In the absence of Runx1, reduced expression of IL-7Rα was observed in Foxp3−CD4+ T cells but not in the Foxp3+ population. This result indicates that the IL-7Rα down-regulation in the total CD4+ T cell fraction from Runx1 F/F;Cd4-cre mice was not caused by a relative increase in Foxp3+ T cells, which normally express a lower level of IL-7Rα compared with conventional T cells. DN2 and DN3 thymocytes from Runx1 F/F;Lck-cre mice also expressed lower levels of IL-7Rα compared with control mice (unpublished data). IL-7Rα expression was slightly lower in CD8+ T cells from Runx3 F/F;Cd4-cre mice compared with control mice, suggesting that Runx1 and Runx3 together regulate its expression in these cells (unpublished data). Real-time RT-PCR analysis showed that the amount of Il7r mRNA in CD4SP thymocytes was reduced by approximately fourfold in Runx1 F/F;Cd4-cre mice compared with control mice, whereas the transcript level was slightly lower but not statistically different in CD8SP thymocytes of Runx1 F/F;Cd4-cre versus control mice (Fig. 5 C). This finding suggests that the reduced level of IL-7Rα in Runx1-deficient CD4+ T cells is caused, at least in part, by its attenuated transcription.
To examine the ability of CD4+ T cells from Runx1 F/F;Cd4-cre mice to respond to IL-7, we initially examined whether recombinant IL-7 was able to support survival of mature T cells from Runx1 F/F;Cd4-cre and control mice (Fig. S3, A and B, available at http://www.jem.org/cgi/content/full/jem.20070133/DC1). Approximately 60 or 80% of CD4+ and CD8+ T cells from wild-type mice survived in media for 48 h, and their survival was improved to nearly 100% in the presence of IL-7. CD8+ T cells from Runx1 F/F;Cd4-cre mice survived similarly to wild-type CD8+ T cells either in the presence or absence of IL-7. In contrast, the viability of CD4+ T cells from Runx1 F/F;Cd4-cre mice was substantially lower compared with wild-type CD4+ T cells or Runx1-deficient CD8+ T cells. Only 50% of input Runx1-deficient CD4+ T cells survived after 2 h of incubation, suggesting that a large fraction of CD4+ T cells from Runx1 F/F;Cd4-cre mice were already predisposed to die in vivo or at a very early point in the culture. The frequency of Foxp3+ cells among total CD4+ T cells from Runx1 F/F;Cd4-cre mice remained unchanged over the first 2 h of incubation (unpublished data). In control samples, the Foxp3+ cell number declined by twofold at 2 h, whereas the number of Foxp3−CD4+ cells remained unchanged. These results suggest that the differential survival of CD4+ T cells between Runx1F/F;Cd4-cre and control mice is caused by a combination of preferential cell death of Foxp3− cells and potentially normal cell death of the enriched Foxp3+ cells. At 48 h, only 20% of the input number of CD4+ T cells survived in the absence of Runx1. In addition, an exogenous IL-7 addition to the culture had little effect on the survival of CD4+ T cells from Runx1 F/F;Cd4-cre mice. After overnight incubation in the presence of IL-7, bcl-2 was up-regulated in a dose-dependent manner in CD4+ and CD8+ T cells from wild-type mice and in CD8+ T cells from Runx1 F/F;Cd4-cre mice (Fig. S3 C). In contrast, there was little up-regulation of bcl-2 in Runx1-deficient CD4+ T cells. These results indicate that incomplete IL-7Rα expression results in compromised in vitro survival of CD4+ T cells from Runx1 F/F;Cd4-cre mice. Breeding of Runx1 F/F;Cd4-cre to IL-7Rα transgenic mice (23) resulted in an increased CD4/CD8 ratio, but the increase in CD4+ T cells was modest, whereas there was a reduction in CD8+ T cells, presumably because of competition for cytokines (Fig. S3, D and E). This suggests that the level of IL-7R is not the only factor limiting the number of CD4+ T cells in the absence of Runx1.
T cell–specific Runx3 inactivation causes a reduction in CD8SP cells
To further study the function of Runx3 during thymocyte development, we next examined Runx3 conditional knockout mice crossed to Cd4-cre transgenic mice. After inactivation of Runx3 at the DP stage, CD4 was derepressed in CD8+ mature thymocytes, as previously observed in CD8+ mature thymocytes derived from Runx3-deficient hematopoietic progenitors (Fig. 6 A) (14). CD8 expression in CD8+ mature thymocytes, but not in DP thymocytes, was slightly reduced, suggesting that Runx3 contributes to CD8 expression in positively selected thymocytes transiting from the CD4+8lo to the CD8SP stage (Fig. 6 B). CD103/integrin αE expression was lost in mature CD8+ thymocytes (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20070133/DC1), consistent with previous results using T cells derived from Runx3-deficient fetal liver cells (24). The CD8+ mature thymocyte number was decreased by 25%, whereas the number of CD4SP thymocytes did not significantly change compared with control mice (Fig. 6 A and Fig. 3 B). The number of CD4SP thymocytes remained unchanged in Runx3 F/F;Cd4-cre;I-A −/− mice compared with Runx3 F/+;Cd4-cre;I-A −/− mice, indicating that the reduced CD8SP thymocyte number was not caused by lineage redirection when Runx3 alone was inactivated (unpublished data).
Figure 6.

Runx3 inactivation at the DP stage results in reduced CD8SP thymocytes. (A) CD4 and CD8 expression in total thymocytes (top) and gated TCRβhiHSAlo thymocytes (bottom) from Runx3 F/F;Cd4-cre and littermate Runx3 F/F mice. Frequencies of mature thymocyte populations are shown. (B) CD8 expression in TCRβhiHSAlo thymocytes from Runx3 F/F;Cd4-cre (open histogram) and littermate Runx3 F/F (shaded histogram) mice.
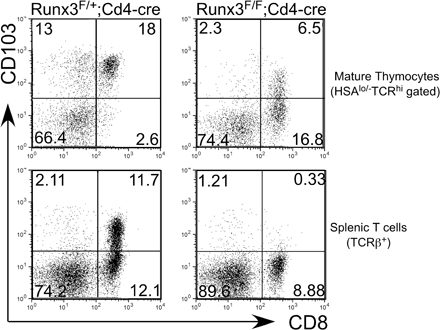
Runx1 and Runx3 have a high degree of sequence similarity, particularly within the DNA-binding runt domains and the C-terminal domain containing the VWRPY motif. A previous report demonstrated that CD4 derepression was enhanced when one Runx1 allele was inactivated in Runx3-deficient mice, suggesting that Runx1 and Runx3 may have at least partially redundant roles (19). Runx1 and Runx3 mRNAs are both expressed in DP and CD8SP thymocytes, although Runx3 expression is low in DP compared with CD8SP thymocytes or CD8+ T cells (14). It is therefore possible that phenotypes observed upon conditional inactivation of Runx1 or Runx3 are attenuated by compensation of Runx3 and Runx1, respectively. To test this hypothesis, we generated mice with Runx1 and Runx3 conditional alleles and the Cd4-cre transgene, thus inactivating both Runx1 and Runx3 at the DP stage (Fig. 7). After inactivation of both Runx1 and Runx3, TCRβ expression in DP thymocytes was further down-regulated compared with Runx1 F/F;Cd4-cre mice (Fig. 7 C). The number of CD69+ thymocytes was more substantially reduced in Runx1 F/F;Runx3 F/F;Cd4-cre mice than in Runx1 F/F;Cd4-cre mice (unpublished data). HSAlo mature thymocytes in the double-deficient mice were decreased by 20-fold and 5-fold as compared with wild-type and Runx1 F/F;Cd4-cre mice, respectively (Fig. 2 E and Fig. 7 A). Reduction of CD8+ mature thymocytes was much more striking in the double-deficient mice than in Runx1 F/F;Cd4-cre or Runx3 F/F;Cd4-cre animals (Fig. 7 B). CD8+ TCRβhiHSAlo thymocytes were considerably reduced in Runx1 F/F;Runx3 F/F;Cd4-cre mice as compared with Runx1 F/+ Runx3 F/F;Cd4-cre mice. Furthermore, the remaining CD8+ mature thymocytes and CD8+ peripheral T cells from the double-deficient mice had normal CD4 silencing, and many of them expressed CD103, whereas almost all CD8+ mature thymocytes derepressed CD4 expression and lacked CD103 expression in Runx1 F/+;Runx3F/F;Cd4-cre mice (Fig. 7, B and D). These CD4−CD8+ mature thymocytes from Runx1 F/F Runx3 F/F;Cd4-cre mice retained the intact Runx3-floxed allele (Fig. 7 E), suggesting that these cells escaped from Cre-mediated excision of the Runx3 alleles and that development of the CD8SP phenotype cells requires functional Runx protein activity supplied by either Runx1 or Runx3.
Figure 7.

Runx proteins are required for the development of CD8+ mature thymocytes. (A and B) CD4 and CD8 expression in total thymocytes (A, top) and gated TCRβhiHSAhi (B, top) or TCRβhiHSAlo (B, bottom) thymocytes, and TCRβ and HSA expression in total thymocytes (A, bottom) from mice with the indicated genotypes. Percentages within the gated populations are shown. (C) Surface TCRβ expression in total thymocytes from Runx1 F/F (shaded histogram), Runx1 F/F;Cd4-cre (dotted histogram), and Runx1 F/F;Runx3 F/F;Cd4-cre (open histogram) mice. (D) CD103 expression in CD8+ mature thymocytes from wild-type (shaded histogram), Runx1 F/+;Runx3 F/F;Cd4-cre (dotted histogram), and Runx1 F/F;Runx3 F/F;Cd4-cre (open histogram) mice. The gated CD8+ populations are defined in B (bottom). (E) Genomic PCR analysis to detect Cre-mediated recombination of the Runx3-floxed allele in CD4SP and CD8SP thymocytes from Runx1 F/F;Runx3 F/F;Cd4-cre mice. Genomic DNA was prepared from sorted mature thymocyte populations, and PCR was performed using primers for the intact Runx3-floxed allele and the recombined Runx3 allele. Positions of individual PCR products are indicated as lines on the right side.
Runx3 expression in CD8SP cells is regulated at both transcriptional and posttranscriptional levels
In peripheral TCRαβ cells, Runx3 mRNA levels were similar in CD4+ and CD8+ T cells (14). However, Runx3 protein was predominantly expressed in CD8SP medullary thymocytes and CD8+ T cells (Fig. 8, A and C) and was not detectable in CD4SP or CD4+ T cells. All three Runx genes have distal and proximal promoters, although Runx2 has multiple transcription and translation initiation sites for both of the promoters (25). For Runx1 and Runx3, transcripts from the distal and proximal promoters encode protein products that have unique 19– or 6–amino-acid N-terminal sequences, respectively (Fig. 8 B). We performed RT-PCR analysis using primers that distinguish the two transcripts for Runx3 to determine whether Runx3 promoter usage was different between CD4SP and CD8SP cells (Fig. 8 E). We found that the Runx3 transcript derived from the distal promoter was exclusively expressed in the CD8SwP population, but not in the CD4SP population or DN and DP thymocytes. To examine if Runx3 protein detected in CD8+ T cells is derived from the distal transcript, we generated an antiserum against the N-terminal 19 amino acids and performed immunoblotting using lysates of CD4+ and CD8+ T cells. We detected Runx3 protein derived from the distal promoter in CD8+ T cells (Fig. 8 D). Although we cannot rule out the possibility that a fraction of Runx3 protein expressed in CD8+ T cells is derived from the proximal transcript, our data suggest that CD8+ T cells, but not CD4+ T cells, specifically use the distal promoter of Runx3 and that the transcript produced is then preferentially translated.
Figure 8.

CD8SP cells use the distal promoter of Runx3. (A) Runx3 protein is predominantly expressed in CD8SP thymocytes in the thymic medulla. Frozen sections from a wild-type thymus were stained for CD4 (blue), CD8 (red), and Runx3 (green). Arrowheads indicate cells expressing both CD8 and Runx3. (B) Genomic organization of exons encoding the different mouse Runx3 isoforms. White boxes and filled boxes depict coding and noncoding sequences, respectively. (C) Western blot analysis of total Runx3 expression in CD4+ and CD8+ T cells from lymph nodes. Immunoblot against HMG1 was used as a loading control. (D) Western blot analysis to detect the distal promoter-derived Runx3 protein in CD8+ T cells. (E) RT-PCR analysis of promoter-specific Runx3 mRNA expression in different subsets of thymocytes and T cells. Numbers shown below each lane are the relative amount of Actb cDNA determined by real-time PCR.
DISCUSSION
Differential expression of the CD4 and CD8 glycoproteins during thymocyte differentiation marks different precursor stages and mature populations with different effector functions. Because Cd4 expression is primarily dependent on its silencer activity (11, 12), we had hypothesized that factors that contribute to regulation of the Cd4 silencer could also play important roles in T cell differentiation programs, particularly during specification of the CD8SP lineage. Members of the Runx family of transcription factors are thus far the only molecules known to regulate the function of the Cd4 silencer. Runx1 inactivation early in thymocyte differentiation resulted not only in partial derepression of Cd4 but also in arrest of thymocyte differentiation between the DN3 and DN4 stages (14). The DN3–DN4 transition requires a robust proliferation phase that is induced by preTCR signals during β selection. After Runx1 inactivation with the Lck-cre transgene, DN3 cells expressing icTCRβ were only slightly reduced in numbers compared with the wild type and displayed an active cell-cycle status, suggesting that expression of TCRβ and preTCR signaling components were unlikely to be defective. However, proliferation was decelerated at the DN4 stage, during which wild-type thymocytes continue to proliferate. This finding is consistent with the reduced thymocyte cellularity observed in Runx1 F/F;Lck-cre mice but not in Runx1 F/F;Cd4-cre mice.
The loss of proliferation of DN4 thymocytes in Runx1 F/F;Lck-cre mice was accompanied by reduced expression of icTCRβ. Such a reduction was also observed in DP thymocytes from Runx1 F/F;Cd4-cre mice and, more remarkably, in those from Runx1/Runx3 doubly conditional knockout mice bred to Cd4-cre. Runx binding sites are found in the Tcrb enhancer (Eβ) (26, 27), and Runx1 inactivation may result in incomplete Tcrb expression that is most remarkable at the DN4 stage. TCRβ expression cannot be maintained in DN4 cells without Eβ activity even after successful V to DJ rearrangement occurs and TCRβ protein is expressed at the DN2 and DN3 stages (28). After inactivation of Runx1 by the Lck-cre transgene, whose expression is initiated in DN2 cells, expression of the protein may persist long enough to initiate Tcrb rearrangement and allow icTCRβ expression in DN2/DN3, but not in DN4, thymocytes. It is also possible that Runx1 regulates other targets that are necessary for the survival or proliferation of DN4 thymocytes.
After Cd4-cre–mediated Runx1 inactivation at the DP stage, we observed a moderate reduction of TCRβintCD69+ thymocytes undergoing positive selection. A substantially more severe reduction of TCRβintCD69+ thymocytes was observed in Runx1/Runx3 double-deficient thymocytes with considerably low expression of TCRβ at the DP stage. These findings suggest that impaired positive selection may be caused by insufficient TCR levels. We were unable to detect differences in the expression of proximal signaling molecules, including Lck, ζ-associated protein of 70 kD, Vav-1, and phospholipase C γ1 in DP thymocytes from Runx1 F/F;Cd4-cre mice (unpublished data). Bim induction after TCR stimulation of DP thymocytes was not increased (unpublished data).
During thymocyte maturation, CD4SP lineage cells were specifically lost between the TCRβhiHSAhi and TCRβhiHSAlo stages after Runx1 inactivation at the DP stage, but maturation of CD8SP cells was less severely affected. This observation suggests that the specific reduction of CD4SP cells most likely occurs after positive selection. In CD8SP cells, Runx3 may compensate for the Runx1 deficiency or replace Runx1 requirements after positive selection.
The IL-7/IL-7R signal is essential for early thymocyte development (22, 29–32). It has been broadly recognized that IL-7Rα is also required for the survival of mature thymocytes and naive and memory T cells (21, 33). Development of CD8SP thymocytes is considered to be more sensitive to IL-7R signaling than that of CD4SP cells (34–36). However, the earlier studies did not rule out that IL-7R signaling is required for CD4SP development. Regulation of IL-7Rα expression is critical for efficient utilization of IL-7, a cytokine that is not abundantly expressed in the microenvironment (23). Exogenous IL-7 treatment of mature T cells leads to IL-7Rα down-regulation that is thought to contribute to more efficient distribution of the effect of IL-7, because loss of this negative feedback leads to a reduced number of peripheral mature T cells and to compromised thymocyte development (37).
The reduction of CD4SP thymocytes and of peripheral CD4+ T cells in Runx1 F/F;Cd4-cre mice coincided with the reduced expression of IL-7Rα, suggesting that Runx1-dependent IL-7Rα expression plays an important role in maturation and homeostasis of this lineage, while Runx3 could play a similar role in CD8SP cells. One potential explanation for the reduction of CD4SP cells relative to CD8SP cells in Runx1 F/F;Cd4-cre mice is that CD8SP cells with almost normal expression of IL-7Rα preferentially use the limiting amount of IL-7, resulting in their better survival. This could alter the CD4/CD8 ratio in HSAlo mature thymocytes and in the peripheral T cells despite the almost normal CD4/CD8 ratio in the intermediate HSAhi thymocytes. This hypothesis is consistent with our observation that the CD4/CD8 ratio in the periphery was not altered in CbfbF/F;Cd4-cre mice, in which there was reduced expression of IL-7Rα in both CD4+ and CD8+ T cells (unpublished data). The reduction of total CD4+ T cells in the absence of Runx1 was caused by a specific loss of Foxp3− conventional CD4+ T cells, which resulted in a high frequency, but a relatively normal number, of Foxp3+ regulatory T cells. IL-7Rα expression was normal in Runx1-deficient Foxp3+ cells in comparison to control Foxp3+ cells. This finding suggested that Foxp3+ T reg cells might be able to use IL-7 in the microenvironment to restore their numbers in the peripheral lymphoid organs similarly to CD8+ T cells, or that homeostasis of Foxp3+ cells might be dependent on other cytokines, such as IL-2. Because IL-7Rα expression was reduced in Foxp3+ cells from CbfbF/F;Cd4-cre mice (unpublished data), other Runx proteins appear to contribute to IL-7Rα expression in the population. Future experiments will be required to clarify how Runx proteins contribute to the development and homeostasis of Foxp3+ regulatory T cells. We also tested the possibility that enhanced activation-induced cell death might result in reduced CD4+ T cells in the periphery, but apoptosis after secondary in vitro TCR stimulation was not increased in CD4+ T cells from Runx1 F/F;Cd4-cre mice (unpublished data).
Runx3 inactivation at the DP stage caused reduction of CD8SP mature thymocytes, yet we did not observe remarkably altered IL-7Rα expression, possibly because of compensation from Runx1 expressed in the CD8 lineage cells. Runx3 inactivation alone did not result in redirection of MHC class I–selected thymocytes to the CD4SP phenotype (unpublished data).
During thymocyte differentiation, a high level expression of Runx3 is detected only in CD8SP thymocytes and not in CD4SP thymocytes, whereas Runx1 expression is detected in both CD4SP and CD8SP populations, as well as in DP thymocytes (14, 18). Because few CD8SP thymocytes that likely lost all Runx function developed in the Runx1/Runx3 doubly conditional knockout mice, Runx protein functions appear to be required for development of CD8SP thymocytes. Unfortunately, because of an escape of cells that failed to excise either Runx1 or Runx3, we were unable to determine the mechanism by which CD8SP development was impaired in the absence of any Runx protein function. The reduced CD8SP generation was possibly caused by redirection of MHC class I–selected thymocytes to the CD4SP population, to defective Runx protein function causing both CD4 derepression and lack of CD8 reactivation after positive selection, or to impaired survival or maturation of CD8SP thymocytes immediately after positive selection.
It has been reported that overexpression of a dominant-negative form of Runx caused lineage redirection in vitro (18). Expression of the dominant-negative Runx resulted in severe reduction of CD8SP thymocytes and generation of CD4SP cells instead after transient agonist stimulation of DP thymocytes. This result is also consistent with the importance of Runx protein function in the generation and commitment of CD8SP thymocytes.
Runx3 up-regulation and protein expression correlate with development of CD8SP cells and are indications tightly linked with CD8 lineage commitment. ThPOK expression, which is necessary and sufficient for commitment of CD4SP thymocytes, is robustly up-regulated in a CD4+CD8lo thymocyte population that contains cells with the potential to differentiate into both CD4 and CD8 lineages (7). Runx3 expression was also detected in this population, although its expression level was not as high as that in CD8 mature thymocytes (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20070133/DC1). Runx3 up-regulation thus seems to occur at a later stage than ThPOK up-regulation. This implies that MHC class I–selected CD4+CD8lo cells, which do not express a high level of ThPOK, remain uncommitted until they pass through the CD4+CD8lo stage and that CD4 and CD8 lineage commitment may then be initiated at different developmental stages. Alternatively, a genetic program for CD8 lineage commitment may be already initiated in the CD4+CD8lo transient population before Runx3 up-regulation and CD8α reactivation. The identification of factors that regulate Runx3 expression in developing CD8SP cells will allow us to propose a better working model for mechanisms of CD8 lineage commitment.
In conclusion, Runx proteins play important roles in thymocyte developmental programs. Among the three Runx transcription factors, Runx1 and Runx3 are required for stage- and lineage-specific functions. It will be important to identify target genes that contribute to the phenotypes observed in the Runx1 and Runx3 conditional knockout mice. Future studies will also clarify how Runx1 and Runx3 functions overlap and how different Runx proteins differentially control gene expression.
MATERIALS AND METHODS
Mouse strains.
Generation of Runx1 and Runx3 conditional knockout mice was previously described (14, 38). Runx1-flox and Runx3-flox mice were bred with Cd4-cre or Lck-cre transgenic mice (39) to inactivate Runx1 or Runx3 at different stages of T cell development. β2m- and I-A–deficient mice were purchased from Taconic. IL-7Rα transgenic mice were provided by A. Singer (National Cancer Institute, Bethesda, MD) (23). All mice used in the experiments were housed in a specific pathogen-free animal facility at the Skirball Institute, and experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee at the New York University School of Medicine.
Generation of mixed bone marrow chimera for quantification of precursor activity.
A mixture of 106 CD45.1+ wild-type and 106 of either CD45.2+ Runx1 F/F;Cd4-cre or CD45.2+ Runx1 +/+;Cd4-cre bone marrow cells was transferred to lethally irradiated Rag2 −/− hosts, as previously described (40). The precursor potential of developing thymocytes derived from CD45.2 bone marrow cells relative to those derived from CD45.1+ wild-type bone marrow was calculated based on contribution of CD45.2+ cells between the two stages of T cell differentiation as follows: P(A→B) = B2(100 − A2)/(A2[100 − B2]), where P(A→B) is a precursor potential of CD45.2+ cells proceeding from a developmental stage A to a stage B relative to CD45.2− cells, A2 is the percentage of CD45.2+ cells at stage A, and B2 is the percentage of 45.2+ cells at the subsequent stage B.
Flow cytometry.
Staining of surface antigens was performed as previously described (40). Antibodies were purchased from either BD Biosciences or eBioscience. For intracellular TCRβ staining, cells were stained for surface antigens, fixed with Cytofix/Cytoperm solution (BD Biosciences), and stained with the anti-TCRβ antibody diluted in Perm/Wash buffer (BD Biosciences). Intracellular Foxp3 staining was performed according to the manufacturer's instruction (eBioscience). For cell-cycle analysis, cells were fixed and stained with DAPI. Data were collected with a cytometer (LSRII; BD Biosciences) using FACSDiva software (BD Biosciences) and were analyzed with FlowJo software (TreeStar, Inc.). Cell sorting was performed on a cell sorter (MoFlo; DakoCytomation).
BrdU labeling.
For analysis of peripheral T cells, mice were injected i.p. with 1 mg BrdU (Sigma-Aldrich) and continuously administered BrdU in drinking water (0.8 mg/ml) for 72 h. For analysis of thymocytes, 1 mg BrdU was administered by i.p. injection 2 h before death. Surface staining and analysis were performed with a BrdU Flow Kit, according to the manufacturer's instruction (BD Biosciences).
Testing for cell survival.
Single-cell suspension of lymph node cells was prepared in RPMI 1640 supplemented with 10% fetal calf serum, sodium pyruvate, 2-mercaptoethanol, and antibiotics. Cells were cultured in media at 37°C with 5% CO2 either in the presence or absence of recombinant mouse IL-7 (PeproTech) at the concentrations described in the figure legends. After incubation for different times, cells were stained for CD4, CD8, and TCRβ on ice, incubated with propidium iodide and FITC–Annexin V (BD Biosciences) at room temperature for 15 min, and immediately analyzed by flow cytometry.
RT-PCR.
RNA and cDNA from sorted thymocyte and T cell populations were prepared using TRIzol and Superscript II reverse-transcriptase (Invitrogen). Input cDNA was quantitated based on the amount to Actb cDNA determined by Taqman PCR, using the primers and probe as previously described (40). For analysis of Il7r expression, real-time PCR was performed using iCycler and iQ SYBR reagent (Bio-Rad Laboratories). For analysis of Runx3 expression, PCR was performed with an Advantage GC2 PCR kit (CLONTECH Laboratories, Inc.). Primer sequences are as follows: Il7r-F, acacagtgcaaaccgctcg; Il7r-R, ctagccaggcatcttaagggtg; Runx3 distal F, cgacatggcttccaacag; Runx3 proximal F, atgcgtattcccgtagacc; and Runx3 common R, ccagctctccagagtcttc
Online supplemental material.
Fig. S1 shows ectopic CD25 expression in DP thymocytes from Runx1 F/F;Cd4-cre and Runx1 F/F;Lck-cre mice. Fig. S2 shows thymocyte positive and negative selection in Runx1 F/F;Cd4-cre mice with TCR transgenes. Fig. S3 shows in vitro survival of CD4+ and CD8+ T cells from Runx1 F/F;Cd4-cre mice. Fig. S4 shows CD103 expression in CD8SP cells from Runx3F/F;Cd4-cre mice. Fig. S5 shows ThPOK and Runx3 mRNA expression in developing thymocytes. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070133/DC1.
Supplemental Material
Acknowledgments
We would like to thank Dr. Alfred Singer for IL-7Rα transgenic mice, Dr. Joriene de Nooij and Dr. Thomas Jessell for anti-Runx3 antibody, and Mary Jean Sunshine, Oliver Zill, and Roy Min for technical support. We also thank Gretchen Diehl and Jun R. Huh for critical reading of the manuscript.
This study was supported by fellowships from the Human Frontier Science Program and the Leukemia and Lymphoma Society (to T. Egawa); by a Microbiology Training Grant from the National Institutes of Health (to R.E. Tillman); and by grants from PRESTO, the Japan Science and Technology Agency, and the Ministry of Education, Culture, Sports, Science and Technology of Japan (to I. Taniuchi). D.R. Littman is an Investigator of the Howard Hughes Medical Institute.
The authors have no conflicting financial interests.
Abbreviations used: DN, double negative; DP, double positive; HSA, heat-stable antigen; icTCR, intracellular TCR β chain; preTCR, pre–T cell receptor; SP, single positive.
References
- 1.Weerkamp, F., K. Pike-Overzet, and F.J. Staal. 2006. T-sing progenitors to commit. Trends Immunol. 27:125–131. [DOI] [PubMed] [Google Scholar]
- 2.von Boehmer, H., I. Aifantis, F. Gounari, O. Azogui, L. Haughn, I. Apostolou, E. Jaeckel, F. Grassi, and L. Klein. 2003. Thymic selection revisited: how essential is it? Immunol. Rev. 191:62–78. [DOI] [PubMed] [Google Scholar]
- 3.Godfrey, D.I., and A. Zlotnik. 1993. Control points in early T-cell development. Immunol. Today. 14:547–553. [DOI] [PubMed] [Google Scholar]
- 4.Aifantis, I., M. Mandal, K. Sawai, A. Ferrando, and T. Vilimas. 2006. Regulation of T-cell progenitor survival and cell-cycle entry by the pre-T-cell receptor. Immunol. Rev. 209:159–169. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman, E.S., L. Passoni, T. Crompton, T.M. Leu, D.G. Schatz, A. Koff, M.J. Owen, and A.C. Hayday. 1996. Productive T-cell receptor beta-chain gene rearrangement: coincident regulation of cell cycle and clonality during development in vivo. Genes Dev. 10:948–962. [DOI] [PubMed] [Google Scholar]
- 6.Sun, G., X. Liu, P. Mercado, S.R. Jenkinson, M. Kypriotou, L. Feigenbaum, P. Galera, and R. Bosselut. 2005. The zinc finger protein cKrox directs CD4 lineage differentiation during intrathymic T cell positive selection. Nat. Immunol. 6:373–381. [DOI] [PubMed] [Google Scholar]
- 7.He, X., V.P. Dave, Y. Zhang, X. Hua, E. Nicolas, W. Xu, B.A. Roe, and D.J. Kappes. 2005. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature. 433:826–833. [DOI] [PubMed] [Google Scholar]
- 8.Pai, S.Y., M.L. Truitt, C.N. Ting, J.M. Leiden, L.H. Glimcher, and I.C. Ho. 2003. Critical roles for transcription factor GATA-3 in thymocyte development. Immunity. 19:863–875. [DOI] [PubMed] [Google Scholar]
- 9.Hernandez-Hoyos, G., M.K. Anderson, C. Wang, E.V. Rothenberg, and J. Alberola-Ila. 2003. GATA-3 expression is controlled by TCR signals and regulates CD4/CD8 differentiation. Immunity. 19:83–94. [DOI] [PubMed] [Google Scholar]
- 10.Kioussis, D., and W. Ellmeier. 2002. Chromatin and CD4, CD8A and CD8B gene expression during thymic differentiation. Nat. Rev. Immunol. 2:909–919. [DOI] [PubMed] [Google Scholar]
- 11.Sawada, S., J.D. Scarborough, N. Killeen, and D.R. Littman. 1994. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell. 77:917–929. [DOI] [PubMed] [Google Scholar]
- 12.Taniuchi, I., M.J. Sunshine, R. Festenstein, and D.R. Littman. 2002. Evidence for distinct CD4 silencer functions at different stages of thymocyte differentiation. Mol. Cell. 10:1083–1096. [DOI] [PubMed] [Google Scholar]
- 13.Zou, Y.R., M.J. Sunshine, I. Taniuchi, F. Hatam, N. Killeen, and D.R. Littman. 2001. Epigenetic silencing of CD4 in T cells committed to the cytotoxic lineage. Nat. Genet. 29:332–336. [DOI] [PubMed] [Google Scholar]
- 14.Taniuchi, I., M. Osato, T. Egawa, M.J. Sunshine, S.C. Bae, T. Komori, Y. Ito, and D.R. Littman. 2002. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 111:621–633. [DOI] [PubMed] [Google Scholar]
- 15.van Wijnen, A.J., G.S. Stein, J.P. Gergen, Y. Groner, S.W. Hiebert, Y. Ito, P. Liu, J.C. Neil, M. Ohki, and N. Speck. 2004. Nomenclature for Runt-related (RUNX) proteins. Oncogene. 23:4209–4210. [DOI] [PubMed] [Google Scholar]
- 16.Javed, A., B. Guo, S. Hiebert, J.Y. Choi, J. Green, S.C. Zhao, M.A. Osborne, S. Stifani, J.L. Stein, J.B. Lian, et al. 2000. Groucho/TLE/R-esp proteins associate with the nuclear matrix and repress RUNX (CBF(alpha)/AML/PEBP2(alpha)) dependent activation of tissue-specific gene transcription. J. Cell Sci. 113:2221–2231. [DOI] [PubMed] [Google Scholar]
- 17.Yarmus, M., E. Woolf, Y. Bernstein, O. Fainaru, V. Negreanu, D. Levanon, and Y. Groner. 2006. Groucho/transducin-like Enhancer-of-split (TLE)-dependent and -independent transcriptional regulation by Runx3. Proc. Natl. Acad. Sci. USA. 103:7384–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato, T., S. Ohno, T. Hayashi, C. Sato, K. Kohu, M. Satake, and S. Habu. 2005. Dual functions of Runx proteins for reactivating CD8 and silencing CD4 at the commitment process into CD8 thymocytes. Immunity. 22:317–328. [DOI] [PubMed] [Google Scholar]
- 19.Woolf, E., C. Xiao, O. Fainaru, J. Lotem, D. Rosen, V. Negreanu, Y. Bernstein, D. Goldenberg, O. Brenner, G. Berke, et al. 2003. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc. Natl. Acad. Sci. USA. 100:7731–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayashi, K., N. Abe, T. Watanabe, M. Obinata, M. Ito, T. Sato, S. Habu, and M. Satake. 2001. Overexpression of AML1 transcription factor drives thymocytes into the CD8 single-positive lineage. J. Immunol. 167:4957–4965. [DOI] [PubMed] [Google Scholar]
- 21.Jameson, S.C. 2002. Maintaining the norm: T-cell homeostasis. Nat. Rev. Immunol. 2:547–556. [DOI] [PubMed] [Google Scholar]
- 22.Nosaka, T., J.M. van Deursen, R.A. Tripp, W.E. Thierfelder, B.A. Witthuhn, A.P. McMickle, P.C. Doherty, G.C. Grosveld, and J.N. Ihle. 1995. Defective lymphoid development in mice lacking Jak3. Science. 270:800–802. [DOI] [PubMed] [Google Scholar]
- 23.Park, J.H., Q. Yu, B. Erman, J.S. Appelbaum, D. Montoya-Durango, H.L. Grimes, and A. Singer. 2004. Suppression of IL-7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 21:289–302. [DOI] [PubMed] [Google Scholar]
- 24.Grueter, B., M. Petter, T. Egawa, K. Laule-Kilian, C.J. Aldrian, A. Wuerch, Y. Ludwig, H. Fukuyama, H. Wardemann, R. Waldschuetz, et al. 2005. Runx3 regulates integrin alpha E/CD103 and CD4 expression during development of CD4-/CD8+ T cells. J. Immunol. 175:1694–1705. [DOI] [PubMed] [Google Scholar]
- 25.Levanon, D., and Y. Groner. 2004. Structure and regulated expression of mammalian RUNX genes. Oncogene. 23:4211–4219. [DOI] [PubMed] [Google Scholar]
- 26.Kim, W.Y., M. Sieweke, E. Ogawa, H.J. Wee, U. Englmeier, T. Graf, and Y. Ito. 1999. Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J. 18:1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tripathi, R.K., N. Mathieu, S. Spicuglia, D. Payet, C. Verthuy, G. Bouvier, D. Depetris, M.G. Mattei, L.W. Hempe, and P. Ferrier. 2000. Definition of a T-cell receptor beta gene core enhancer of V(D)J recombination by transgenic mapping. Mol. Cell. Biol. 20:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Busse, C.E., A. Krotkova, and K. Eichmann. 2005. The TCRbeta enhancer is dispensable for the expression of rearranged TCRbeta genes in thymic DN2/DN3 populations but not at later stages. J. Immunol. 175:3067–3074. [DOI] [PubMed] [Google Scholar]
- 29.Sudo, T., S. Nishikawa, N. Ohno, N. Akiyama, M. Tamakoshi, and H. Yoshida. 1993. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc. Natl. Acad. Sci. USA. 90:9125–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park, S.Y., K. Saijo, T. Takahashi, M. Osawa, H. Arase, N. Hirayama, K. Miyake, H. Nakauchi, T. Shirasawa, and T. Saito. 1995. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. 3:771–782. [DOI] [PubMed] [Google Scholar]
- 31.DiSanto, J.P., W. Muller, D. Guy-Grand, A. Fischer, and K. Rajewsky. 1995. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc. Natl. Acad. Sci. USA. 92:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peschon, J.J., P.J. Morrissey, K.H. Grabstein, F.J. Ramsdell, E. Maraskovsky, B.C. Gliniak, L.S. Park, S.F. Ziegler, D.E. Williams, C.B. Ware, et al. 1994. Early lymphocyte expansion is severely impaired in interleukin 7 receptor–deficient mice. J. Exp. Med. 180:1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaech, S.M., J.T. Tan, E.J. Wherry, B.T. Konieczny, C.D. Surh, and R. Ahmed. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198. [DOI] [PubMed] [Google Scholar]
- 34.Akashi, K., M. Kondo, U. von Freeden-Jeffry, R. Murray, and I.L. Weissman. 1997. Bcl-2 rescues T lymphopoiesis in interleukin-7 receptor-deficient mice. Cell. 89:1033–1041. [DOI] [PubMed] [Google Scholar]
- 35.Kondo, M., K. Akashi, J. Domen, K. Sugamura, and I.L. Weissman. 1997. Bcl-2 rescues T lymphopoiesis, but not B or NK cell development, in common gamma chain-deficient mice. Immunity. 7:155–162. [DOI] [PubMed] [Google Scholar]
- 36.Chong, M.M., A.L. Cornish, R. Darwiche, E.G. Stanley, J.F. Purton, D.I. Godfrey, D.J. Hilton, R. Starr, W.S. Alexander, and T.W. Kay. 2003. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8+ T cell differentiation. Immunity. 18:475–487. [DOI] [PubMed] [Google Scholar]
- 37.Munitic, I., J.A. Williams, Y. Yang, B. Dong, P.J. Lucas, N. El Kassar, R.E. Gress, and J.D. Ashwell. 2004. Dynamic regulation of IL-7 receptor expression is required for normal thymopoiesis. Blood. 104:4165–4172. [DOI] [PubMed] [Google Scholar]
- 38.Naoe, Y., R. Setoguchi, K. Akiyama, S. Muroi, M. Kuroda, F. Hatam, D.R. Littman, and I. Taniuchi. 2007. Repression of interleukin 4 in T helper type 1 cells by Runx/Cbfß binding to the Il4 silencer. J. Exp. Med. 204:1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee, P.P., D.R. Fitzpatrick, C. Beard, H.K. Jessup, S. Lehar, K.W. Makar, M. Perez-Melgosa, M.T. Sweetser, M.S. Schlissel, S. Nguyen, et al. 2001. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 15:763–774. [DOI] [PubMed] [Google Scholar]
- 40.Egawa, T., G. Eberl, I. Taniuchi, K. Benlagha, F. Geissmann, L. Hennighausen, A. Bendelac, and D.R. Littman. 2005. Genetic evidence supporting selection of the Valpha14i NKT cell lineage from double-positive thymocyte precursors. Immunity. 22:705–716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}