

Abstract
Cerebrovascular dysfunction contributes to the pathology and progression of Alzheimer's disease (AD), but the mechanisms are not completely understood. Using transgenic mouse models of AD (TgCRND8, PDAPP, and Tg2576), we evaluated blood–brain barrier damage and the role of fibrin and fibrinolysis in the progression of amyloid-β pathology. These mouse models showed age-dependent fibrin deposition coincident with areas of blood–brain barrier permeability as demonstrated by Evans blue extravasation. Three lines of evidence suggest that fibrin contributes to the pathology. First, AD mice with only one functional plasminogen gene, and therefore with reduced fibrinolysis, have increased neurovascular damage relative to AD mice. Conversely, AD mice with only one functional fibrinogen gene have decreased blood–brain barrier damage. Second, treatment of AD mice with the plasmin inhibitor tranexamic acid aggravated pathology, whereas removal of fibrinogen from the circulation of AD mice with ancrod treatment attenuated measures of neuroinflammation and vascular pathology. Third, pretreatment with ancrod reduced the increased pathology from plasmin inhibition. These results suggest that fibrin is a mediator of inflammation and may impede the reparative process for neurovascular damage in AD. Fibrin and the mechanisms involved in its accumulation and clearance may present novel therapeutic targets in slowing the progression of AD.
Alzheimer's disease (AD) is characterized by abnormal senile plaque and neurofibrillary tangle formation. Derived from the amyloid-β (Aβ) precursor protein (AβPP), Aβ peptide is the major component of senile plaques in the brain and cerebrovasculature. Aβ is thought to be a major contributor to the neurodegenerative process, acting either through neurotoxic mechanisms or local inflammatory processes (1). However, Alzheimer's pathology is not limited to neurons, as one of the earlier manifestations of the disease is abnormal cerebral vasculature. This neurovascular pathology may accelerate other Aβ-mediated pathologies or affect neuronal damage directly (2–4).
Fibrinogen is a 340-kD macromolecule classically known for its role as the protein component of blood clots and is normally excluded from the brain parenchyma by the blood–brain barrier. Neurovascular damage can allow fibrinogen access to the central nervous system. Fibrinogen is present in the brains of AD patients (5), but the pathologic significance is not known.
Fibrin deposition increases in the context of deficiency in the tissue plasminogen activator/plasminogen (tPA/plg) protease cascade (6). Plasminogen-deficient mice accumulate extravascular fibrin and have impaired wound healing and high mortality, both of which are corrected in mice deficient for both plasminogen and fibrinogen (7). Fibrin accumulation can also be reduced genetically or pharmacologically with administration of the fibrinogen-depleting protease ancrod. This serine protease derived from the venom of the Malayan pit viper Agkistrodon rhodostoma has been used to alleviate fibrin-mediated pathology in various systems (8, 9).
In the brains of AD patients and mouse models of the disease, clearance of fibrin by the tPA/plasmin system is expected to be reduced as tPA activity is diminished (10, 11). Because inflam mation is universal in AβPP transgenic mice and can be observed as early as 13 wk of age (12), we hypothesized that extravasated fibrin might initiate or exacerbate the observed neuroinflammation. We further hypothesized that this exaggerated fibrin-induced inflammatory process could inflate the damage to the vasculature, thus promoting this process of disease progression.
We sought to examine these possibilities by measuring blood–brain barrier permeability and fibrin deposition in three mouse models of AD. Our results demonstrate blood–brain barrier damage and the presence of fibrin in the parenchyma in AD mice. Modulation of fibrinogen levels affects the pathology of AD mice: reducing fibrinolysis, thereby increasing fibrin deposition, worsens the pathology, whereas fibrinogen depletion attenuates microgliosis and neurovascular damage. These results indicate a role for fibrin in aggravating and accelerating the inflammatory component of disease progression in transgenic AβPP-overexpressing mice.
RESULTS
Fibrin is deposited through a disrupted neurovasculature in transgenic mouse models of AD
When the blood–brain barrier is compromised, macromolecules in circulation can accumulate in the brain parenchyma (13). Because Evans blue dye binds to albumin in the blood, extravasation of the dye serves as a marker for blood–brain barrier permeability and neurovascular damage. We compared Alzheimer's mouse models Tg2576, PDAPP, and TgCRND8 to nontransgenic littermates for defects in the neurovasculature. Because these mice bear AβPP with different familial AD mutations and are driven by different promoters, they exhibit differing ages of onset of Aβ-associated pathology. Therefore, we compared extravasation of Evans blue dye in mice at 6 and 12 mo of age.
As shown in Fig. 1 A, brains of all three Alzheimer's mouse models were considerably more permeable to the dye. Nontransgenic littermates showed increased blood–brain barrier permeability as age increased. However, in all three Alzheimer's mouse models, the brain was considerably more permeable to the dye at these ages. These data are consistent with previously observed microvascular damage in the Tg2576 mouse (14), although the TgCRND8 mice show earlier onset.
Figure 1.

Blood–brain barrier permeability and neurovascular damage is increased in three mouse models of AD. (A) Evans blue assay indicates increased blood–brain barrier permeability in the Tg2576, PDAPP, and TgCRND8 transgenic mice compared with nontransgenic (NTg) littermates. Data are given as Evans blue extravasation calculated from A620 of perfused brain homogenates and normalized to plasma Evans blue levels. For 6-mo mice, n = 9 for NTg (three each from Tg2576, PDAPP, and TgCRND8 litters), n = 3 for Tg2576, n = 6 for PDAPP, and n = 7 for TgCRND8; for 12-mo mice, n = 9 for NTg (three each from Tg2576, PDAPP, and TgCRND8 litters), n = 4 for Tg2576, n = 4 for PDAPP, and n = 4 for TgCRND8. Error bars represent the mean ± SEM. *, P < 0.05; **, P < 0.001, relative to nontransgenic littermates. (B) Laser-scanning micrograph of microvasculature in hippocampus of 6-mo TgCRND8 mouse and NTg littermate injected with Evans blue (red) 6 h before and perfused at the time of killing with 2,000-kD dextran (green). Fluorescence intensity across a cross section (indicated by white arrow; 100 μm) of a capillary is scanned for the distribution of each fluorochrome.
To visually observe extravascular deposition of Evans blue, mice treated with Evans blue for 6 h were perfused with fluorescent dextran at the time of killing. This 2,000-kD dextran is impermeable to both healthy and damaged blood vessels, and therefore serves as an outline of intravascular space. The nontransgenic littermate blood–brain barrier retained both dyes within the blood vessel as shown in Fig. 1 B (left). The damaged TgCRND8 blood vessel in Fig. 1 B (right) showed diffuse accumulation of Evans blue around the contained dextran. As shown underneath each micrograph, the distribution of each fluorochrome can be analyzed for fluorescence intensity across a cross section of a capillary, which reveals Evans blue with a broader distribution than the dextran in the AD mouse (15). Together with the quantitative extravasation assay, these comprehensive estimates indicated increased blood–brain barrier permeability in the TgCRND8 mouse.
To gain insight into blood vessel health, mice brain sections were stained for platelet/endothelial cell adhesion molecule-1 (PECAM-1). Images of perfused and stained microvasculature were obtained from the cortex of TgCRND8 mice and nontransgenic littermates at 6 mo of age (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070304/DC1). Healthy endothelial cells constitutively express this surface marker (16), but sections of TgCRND8 brains showed diminished signal intensity, and vessels appeared tortuous and fragmented.
Because the AD mouse blood–brain barrier was permeable to albumin-bound Evans blue dye, we hypothesized that fibrinogen could gain access to the brain's extravascular space. Given this, and because tPA activity is reduced in the AD mouse brain (11), we reasoned that fibrin could deposit and accumulate over the lifespan of the mouse. Perfused TgCRND8 brains contained elevated levels of fibrin as determined by ELISA. 3–9-mo-old mice showed that Aβ accumulated in an age-dependent manner, and fibrin levels correlated with soluble Aβ1-40 and Aβ1-42 levels, as measured by ELISA from the same tissue homogenates (Fig. 2).
Figure 2.

Fibrinogen accumulates through the damaged neurovasculature. (A) Fibrin deposition parallels age-dependent Aβ accumulation. TgCRND8 cortex and hippocampus were isolated, and homogenates were assayed each for fibrinogen and Aβ1-40 by ELISA. Each point represents one TgCRND8 mouse. Correlation coefficients are indicated. (B) The same cortex and hippocampus homogenates were assayed each for fibrinogen and Aβ1-42 by ELISA.
Neuroinflammation and microvascular injury are diminished by pharmacologic depletion of fibrinogen
Because fibrin is a proinflammatory molecule and could aggravate pathology upon exiting the vasculature (17, 18), we asked if fibrinogen depletion might reduce the inflammation in AβPP transgenic mice. TgCRND8 mice are appropriate because of the early onset of neurovascular dysfunction and neuroinflammation, as seen by microgliosis at 13 wk (12). We therefore reduced circulating fibrinogen levels using a recombinant form of the Malayan pit viper protease ancrod. Ancrod is a thrombin-like protease that cleaves fibrin and prevents its polymerization, allowing degradation by the liver and removal from circulation (19, 20). TgCRND8 mice were treated with either ancrod or saline for 4 wk before killing at 6 mo of age. Fibrinogen levels were reduced by 50–75% in circulation by ancrod. Accordingly, sections of perfused brains after ancrod treatment showed diminished fibrin immuno reactivity (see Fig. 7 A).
Figure 7.

Fibrin deposition and vascular damage are modified by manipulation of fibrinogen levels and fibrinolysis. (A) Representative images of perfused brains from each treatment group stained for fibrin. Images were tiled together using a motorized stage on a confocal microscope. (B) Evans blue extravasation from ancrod-, saline-, and tranexamic acid–treated 6-mo TgCRND8 mice. Data are represented as mean ± SEM of each treatment group, where n = 3 for each group. *, P < 0.05, relative to saline-treated mice. Fibrinogen depletion with ancrod reduces vascular damage, whereas plasmin inhibition enhances permeability. (C) High-magnification images of fibrin deposition (green) with Aβ plaques (red). Bar, 20 μm.
Perfused sections of transgenic brains from each treatment group were stained with CD11b, an integrin receptor present on microglia, and inflammatory foci were visualized. The total area of inflammatory foci can be quantified within the regions of interest, as shown in Fig. 3 A. Areas of inflammation were compared between ancrod and saline. Ancrod treatment reduced the area of inflammation by ∼64% (Fig. 3 C; P = 0.00001). In saline-treated mice, microglia were identified by their amoeboid morphology. As shown in Fig. 3 D, these aggregated microglia formed inflammatory foci and appeared to concentrate around plaques with reduced number after fibrinogen depletion. Because ancrod is a protease and could be acting directly on Aβ levels, we quantified levels of plasma Aβ1-40 and cortical Aβ, neither of which were substantially different in ancrod-treated mice (Fig. 3 B), indicating that depletion of fibrinogen, rather than deposited Aβ, is responsible for the reduced microgliosis.
Figure 3.

Fibrinogen depletion and inhibition of fibrinolysis in the 6-mo TgCRND8 mouse have opposite effects on neuroinflammation. (A) Representative immunofluorescent images of brains colabeled for Aβ (left) and CD11b, a marker for activated microglia (right). Regions of interest are outlined to demark the cortical region quantified in each image. Images show increased density of inflammatory foci in the cortex of plasmin-inhibited transgenic mice, whereas fibrinogen-depleted transgenics show decreased density relative to age-matched saline-treated TgCRND8 mice. Bar, 200 μm. (B) Analysis of total Aβ staining within marked regions of interest in the cortex; the differences were not significant (P > 0.05). (C) Analysis of CD11b staining shows increased inflammation density in the cortex of plasmin-inhibited transgenic mice, whereas fibrinogen-depleted transgenics show a decrease. Error bars present the mean ± SEM of four images for each of five ancrod-, four tranex-, and two saline-treated mice. *, P < 0.05; **, P < 0.001, relative to saline-treated mice. (D) High-magnification images of microglia (green) and Aβ plaques (red) in each treatment group. Bar, 50 μm.
As inflammation can contribute to blood–brain barrier permeability, we assayed ancrod-treated mice for Evans blue extravasation and found a reduction when compared with saline-treated mice (see Fig. 7 B). This attenuation of vascular damage prompted analysis of the microvasculature from both cortex and hippocampus. Identical areas of the brain (Fig. 4 A) were quantified for vascular density as a percentage of image area (Fig. 4 B) and indicated that ancrod treatment partially prevents blood vessel loss.
Figure 4.

Fibrinogen depletion and inhibition of fibrinolysis in the TgCRND8 mouse have opposite effects on neurovascular damage. (A) Neurovasculature in ancrod-, saline-, and tranexamic acid–treated TgCRND8 mice. Images of brains labeled for PECAM-1 (black) are shown with higher magnification images of the regions shown in red to the right. Regions defining cortex (orange) and hippocampus (blue) were used for quantification of vascular density. The images show decreased vascular density in the cortex (cx) and hippocampus (hp) of plasmin-inhibited transgenic mice, whereas fibrinogen-depleted transgenics show increased vascular density over age-matched saline-treated TgCRND8 mice at 6 mo. (B) Semiquantitative analysis of PECAM-1 staining in the cortex and hippocampus shows decreased vascular density in plasmin-inhibited transgenic mice, whereas fibrinogen-depleted transgenics show an increase. Bars represent the percentage of image area reported as the mean ± SEM of four images of the cortex and hippocampus of four mice in each treatment group. *, P < 0.05; **, P < 0.001, relative to saline-treated mice.
Neurovascular pathology is promoted by pharmacologic inhibition of fibrinolysis
To complement fibrinogen-depletion experiments, we tested whether absence of plasmin-mediated clearance of fibrin accelerates pathology. We treated TgCRND8 mice with a plasmin inhibitor, tranexamic acid, for 4 wk before killing at 6 mo of age alongside littermates treated with saline or ancrod. Inflammatory foci were again visualized using microglia staining with CD11b (Fig. 3 A). Plasmin inhibition by tranexamic acid treatment significantly increased microgliosis in treated mice as compared with control mice (P = 0.014; Fig. 3 C).
We also reasoned that the inhibition of plasmin-mediated clearance of fibrin and subsequent inflammation could aggravate neurovascular damage in TgCRND8 mice. We observed that administration of tranexamic acid increases damage to the blood–brain barrier (see Fig. 7). Decreased blood–brain barrier integrity after 4-wk tranexamic acid treatment prompted analysis of the microvasculature. Tranexamic acid–treated TgCRND8 mice showed a reduction in microvascular density, and vessels appeared damaged (Fig. 4).
With the increased pathology observed in the tranexamic acid–treated animals, we asked if inflammation and Aβ were sufficient to promote neurodegeneration. Active caspase-3 staining did not reveal apoptotic cells in treatment or control groups. Samples also were negative for neurodegeneration by Fluoro-Jade B staining (unpublished data).
Neurovascular pathology in the transgenic mouse model is modulated by genetic deficiency in plasminogen or fibrinogen
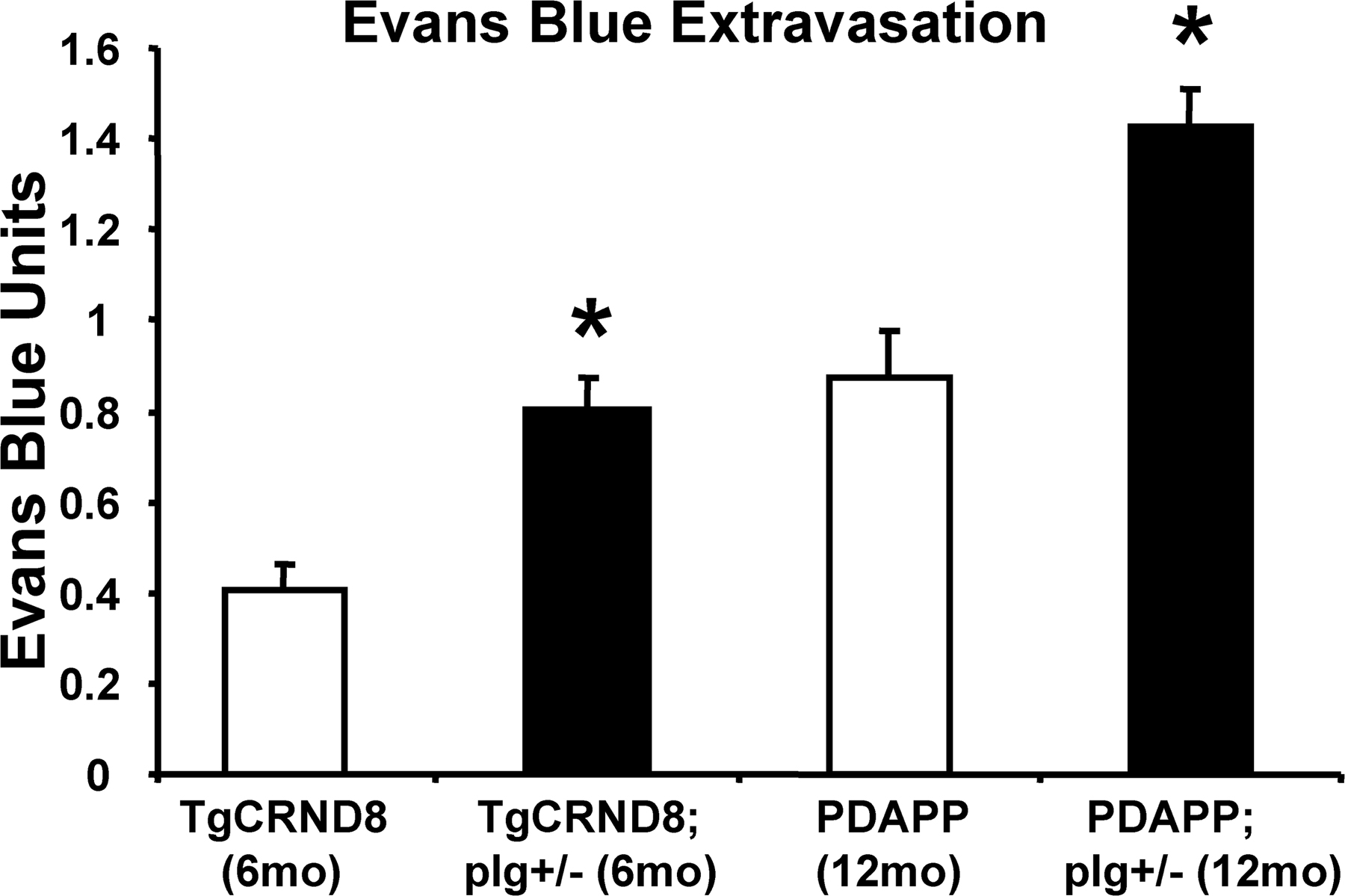
We crossed transgenic AD mice to mice deficient for fibrinogen (fib−/−) to obtain TgCRND8;fib+/− mice bearing only one copy of the fibrinogen gene. Additionally, because accumulated fibrin in the extravascular space can cause damage, we asked if a reduction in plasminogen levels on a background of the AβPP transgene could promote neurovascular pathology. Similar to the fibrinogen cross, we generated TgCRND8;plg+/− mice and compared them to TgCRND8 littermates. We examined the N1 generation from mice crossed to TgCRND8 mice because pathology presents at an earlier age than PDAPP and Tg2576. Heterozygosity for plasminogen deficiency in TgCRND8 mice produced a significant increase in Evans blue extravasation (Fig. 5; P = 0.042). Conversely, TgCRND8;fib+/− mice showed reduced neurovascular pathology at 6 mo (P = 0.003). Plg+/− and fib+/− controls showed little permeability to the dye, suggesting that a product of the AβPP transgene is necessary for neurovascular pathology. To control for the possible effects of different genetic backgrounds on the production and metabolism of the AβPP transgene, PDAPP mice were backcrossed >10 generations onto the C57BL/6 background before crossing with plg−/− mice, which share the C57 background. The results shown in Fig. S2 (available at http://www.jem.org/cgi/content/full/jem.20070304/DC1) indicate increased blood–brain barrier pathology in PDAPP;plg+/− mice when compared with PDAPP littermates, consistent with the results shown in Fig. 5.
Figure 5.

Genetic plasminogen and fibrinogen deficiency modulate defects in the AD mouse blood–brain barrier. 6-mo-old AD mice deficient for plasminogen (TgCRND8;plg+/−) and fibrinogen (TgCRND8;fib+/−) were assayed for Evans blue extravasation alongside TgCRND8 littermates. TgCRND8;plg+/− mice showed increased blood–brain barrier permeability, whereas TgCRND8;fib+/− mice showed a decrease. Data are given as Evans blue extravasation calculated from A620 of perfused brain homogenates and normalized to plasma Evans blue levels. n = 8 for NTg; n = 8 for TgCRND8; and n = 3 for TgCRND8;plg+/−, TgCRND8;fib+/−, fib+/−, and plg+/− mice. Bars represent the mean ± SEM. **, P < 0.001, relative to nontransgenics; †, P < 0.05, relative to TgCRND8.
Because Evans blue dye is fluorescent, the entire hemisphere can be visualized for dye extravasation to determine which areas of the brain are most affected. Images of cerebral hemispheres of each genotype were compared (Fig. 6). The cortex and hippocampus were affected with the highest levels of neurovascular damage, consistent with the observation that hippocampus and cortex show the most Aβ deposition. The data indicated that the removal of one copy of the plasminogen gene accelerates the loss of microvascular integrity in TgCRND8 mice, whereas reduction of one copy of the fibrinogen gene slowed pathogenesis. At 3 mo of age, mice homozygous for plasminogen deficiency showed blood–brain barrier damage (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070304/DC1) but did not show substantial neuroinflammation. Because these mice died early, it cannot be determined if the inflammation would have developed to levels comparable to those of older AD mice. Nonetheless, this finding indicates that the presence of Aβ exaggerates fibrin-related neuroinflammation.
Figure 6.

Localization of vascular pathology in AD mice deficient for fibrinogen or fibrinolysis. (A–F) Composite images of cerebral hemispheres of 6-mo-old mice perfused with Evans blue for 6 h. TgCRND8;fib+/− (D) and TgCRND8;plg+/− (F) mice are compared with TgCRND8 mice (E) and fib+/− (A), NTg (B), and plg+/− (C) controls. Pathologic dye accumulation is most apparent in the cortex (cx), hippocampus (hp), and thalamus (th).
Fibrinogen depletion protects against the deleterious effects of plasmin inhibition
Suppressing plasmin activity with tranexamic acid leads to increased blood–brain barrier breakdown and inflammation, as shown in Fig. 7 B and Fig. 3 C, respectively. As fibrin is the primary target of plasmin proteolysis, this treatment also led to increased fibrin deposition (Fig. 7 A). However, because plasmin is a potent protease that could have many substrates, we investigated whether the vascular damage and inflammation were due to fibrin accumulation or some other effect of plasmin inhibition. Therefore, we implanted pumps with either saline or ancrod in two groups of mice for a pretreatment period of 1 wk. After pretreatment, both fibrinogen-depleted and control groups received tranexamic acid, to inhibit plasmin activity for 1 wk. All mice were assayed for blood–brain barrier damage and inflammation as before. As expected, plasmin inhibition increased Evans blue extravasation and microglial staining in the control group. By comparison, the fibrinogen-depleted group showed considerably less pathology (Fig. 8). Levels for fibrinogen-depleted mice were similar to those of untreated mice (Fig. 1 A), suggesting that fibrinogen depletion protected mice from the increased vascular damage and inflammation induced by plasmin inhibition. This result indicates that fibrin deposition is a critical pathologic consequence of reduced plasmin activity in the brains of AD mice.
Figure 8.

Ancrod treatment protects AD mice from increased pathology induced by tranexamic acid. (A) Two groups of 6-mo TgCRND8 mice were treated as indicated. (B) Evans blue extravasation. (C) Analysis of CD11b staining. In both cases, pretreatment with ancrod reduced the effect of tranexamic acid. Error bars present the mean ± SEM of four images for each of four ancrod- and four saline-treated mice. **, P < 0.001.
DISCUSSION
Neurovascular dysfunction in the AβPP transgenic mouse
The Tg2576 mouse has deficits in vascular integrity shown by blood–brain barrier damage (21, 22), decreased blood flow, and increased susceptibility to ischemia (23). In addition, vascular density is reduced in these animals, as measured by glucose transporter type 1 beginning at 12 mo (24). Subsequently, it was shown that the blood–brain barrier compromise is rescued in the Tg2576 mouse with passive Aβ1-40 peptide immunization (14). The present study explores the contribution of fibrin to neurovascular damage as part of the Aβ disease process and broadens the knowledge of vascular deficits to other AβPP-transgenic mouse models.
The Aβ peptide causes endothelial and smooth muscle cell dysfunction and cell death in vitro, thus disrupting two major components of the blood–brain barrier (25–31). Therefore, the initial insult to the microvasculature likely arises from increased Aβ levels. Cerebral amyloid angiopathy is a hallmark of AD, and it will be important to consider the specific vascular amyloid burden in these mice.
However, the neurovascular damage in the plasminogen-deficient mouse suggests that decreased fibrinolysis might promote the damage to blood vessels and subsequent fibrin deposition. The necessity of Aβ to initiate the inflammatory process is indicated by the low level of neuroinflammation in the plasminogen-deficient mouse.
AD and the tPA/plasmin fibrinolytic system
tPA/plasmin fibrinolytic activity is regulated by serine protease inhibitors (serpins), such as plasminogen activator inhibitor-1 (PAI-1). In mice, PAI-1 is up-regulated in the presence of Aβ, which agrees with the clinically observed elevation of PAI-1 levels in the cerebrospinal fluid of AD patients (11, 27, 32) and decreased plasmin activity in the AD brain (10).
The tPA/plasmin system is down-regulated in AD, in accord with reductions in other naturally occurring Aβ-degrading proteases (10, 33, 34). The direct consequences of this general lack of protease activity might be the diminished clearance of Aβ peptide (34). The present study proposes that decreases in clearance of fibrin can also contribute to the progression of Aβ pathology.
It is important to consider the effects of the loss of a single copy of the plasminogen gene. Heterozygosity decreases the fibrinolytic activity to 55% of normal in pooled plasma obtained from wild-type mice according to clot lysis assays (35). We did not attempt to generate TgCRND8;plg−/− mice because plg−/− mice develop several thrombotic complications, which could complicate the central nervous system pathology. Additionally, plg−/− mice exhibit early mortality, which would preclude comparison at later ages. TgCRND8;fib−/− mice are also difficult to generate because of breeding restrictions and limited survival (36).
These genetic constraints are addressed with the experiments using pharmacologic reduction of fibrinolysis and fibrinogen depletion by ancrod. The convergence of the results of both genetic and pharmacologic approaches forms solid evidence that manipulation of fibrin levels in Alzheimer's mouse models modulates inflammation and vascular integrity.
Fibrinogen and inflammation
Fibrinogen has several active roles in normal and abnormal physiology, including cellular responses in clotting and inflammation that are mediated by fibrinogen receptors. Of interest in the present study are the integrin receptors expressed on leukocytes, monocytes, and macrophage/microglia. Three notable inflammatory integrins are α5β1, αvβ3, and αMβ2 (Mac-1 or CD11b/CD18; reference 37). Binding of the fibrinogen dimer to microglial αMβ2/CD11b elicits activation of the NF-κB pathway, causing increased expression of cytokine genes (38, 39).
The involvement of inflammation has been studied in AD. Clinical and epidemiological data concerning anti-inflammatory medications are provocative and require further studies to determine their potential in controlling disease progression (40, 41). The present study suggests that fibrin may be an upstream effector of neuroinflammation, and the damaging effect of decreased fibrinolysis on the neurovasculature is intriguing. Fibrin-induced microgliosis could be toxic to endothelial cells, as microglia have been shown to increase cell death in primary endothelial cells after oxygen-glucose deprivation, which can be reduced by inhibiting microglial activation with minocycline (42).
How inflammation may contribute to neurodegeneration in AD is still largely unknown. As blood vessels from AD brains are directly toxic to neurons (43), we asked if the compromised neurovasculature and inflammation in these experimental mice could promote neurodegeneration. Although we did not observe neuronal death, studies involving older TgCRND8 mice with enhanced inflammation and vascular damage may reveal detectable levels of neurodegeneration.
As fibrinogen is polymerized after activation by thrombin, it is important to note that thrombin injection into the rat cortex induces microgliosis (44). Additionally, in human patients with advanced AD, plasma prothrombin can be found in the extravascular space (45), which provides an environment where fibrin is likely to deposit.
Once polymerized in the parenchyma, fibrin is covalently cross-linked by tissue-resident transglutaminase factor XIII (46). Cross-linked fibrin is stabilized by covalent bonds between γ chains. Because the capturing antibody used in the ELISA recognizes the γ chain specifically (47), some insoluble fibrin may have escaped detection, and the ELISA results likely underestimate the total fibrin deposition. However, the immunofluorescence uses a polyclonal antibody, which detects all forms of fibrinogen. Similarly, Triton X-100–soluble Aβ levels likely underestimate the total Aβ burden. However, Aβ levels measured by ELISA correlate with plaque burdens observed with immunofluorescence. Both soluble and insoluble fibrin activates microglia via the CD11b receptor and contributes to the observed inflammation in AβPP transgenic mice and AD patients.
This study begins to outline a role for fibrin in the neuroinflammation seen in AD. These results also indicate a role for fibrin deposition in accelerating the neurovascular damage observed in these mouse models and perhaps in reducing the brain's reparative capacity. Extravascular fibrinogen functioning as a restraint to mechanisms of tissue repair has been shown in the peripheral nervous system (48). The present study also demonstrates this effect on the progression of disease in mouse models of AD. Therefore, fibrin and the mechanisms involved in its clearance may present novel therapeutic targets in slowing the progression of AD.
MATERIALS AND METHODS
Animals.
The AD transgenic mice used, which develop Aβ-associated pathology, include Tg2576 (49), TgCRND8 (50), and PDAPP (51). The Tg2576 mice (APP695; K670N and M671L driven by the human prion protein promoter) are on a C57B6/SJL mixed strain background and develop cognitive deficits by 9 mo of age. The TgCRND8 mice (APP695; K670N, M671L, and V717F driven by the human prion protein promoter) are on a mixed background (C57XC3H/C57) and exhibit defects in memory as early as 3 mo of age (provided by A. Chishti and D. Westaway, University of Toronto, Toronto, Canada). The PDAPP mice (APP695, -751, and -770; V717F driven by the platelet-derived growth factor β promoter) used in this study were backcrossed to C57BL/6 mice and display memory defects by 8 mo (provided by R.B. Demattos and S.M. Paul, Eli Lilly Research Laboratories, Indianapolis, IN). Mice deficient for plasminogen (7) and fibrinogen (36, 52), both backcrossed onto the C57BL/6 background, were used for crosses with TgCRND8 and PDAPP mice. Littermates were used in all experiments whenever transgenic mice were compared with nontransgenic (wild-type) mice. Where multiple crosses or strains are presented in a single figure, nontransgenic littermates from each cross were averaged and presented as one bar for clarity and brevity. Mice were maintained in the Rockefeller University Laboratory Animal Research Center (LARC) and treated in accordance with protocols approved by LARC.
Evans blue extravasation assay.
A solution of 2% Evans blue/PBS was injected (4 ml/kg) via the tail vein. 6 h after injection, the mice were anesthetized and blood was drawn by cardiac puncture followed by transcardial perfusion with 0.9% saline-heparin (5 U/ml) to remove intravascular dye. One brain hemisphere was frozen for sectioning and microscopy studies, whereas the other hemisphere was weighed and homogenized in 400 μl dimethyl formamide to solubilize the Evans blue. To extract the dye, samples were centrifuged, and the supernatant was collected and analyzed for absorbance at 620 nm. The plasma was diluted 1:100 in dimethylformamide and analyzed exactly as the brain homogenate. Evans blue units of extravasation were calculated as the A620 of brain homogenate divided by the A620 of plasma.
Evans blue fluorescence profiling.
To visualize the extent of the blood–brain barrier damage, mice were injected via tail vein with Evans blue dye. After 6 h, mice were anesthetized and perfused with a solution containing large-fragment 2,000-kD FITC-conjugated dextran dissolved in PBS to outline the intraluminal space (53). Evans blue fluorescence from 50-μm coronal sections was visualized with a laser-scanning confocal imaging system (LSM 510 confocal system fitted on an Axiovert 200 inverted microscope; Carl Zeiss MicroImaging, Inc.). Optical slices were processed by AxioVision confocal imaging software, and reconstructed images were evaluated for Evans blue dye present outside the fluorescein-delineated intraluminal space. For views of the entire brain hemisphere, a composite of stitched 10× images (8 × 8) was produced during acquisition with a motorized stage.
ELISA (brain or plasma).
Brains were perfused, weighed, and homogenized in 0.1 M Tris, pH 7.2/0.2% Triton X-100 with 5 mM EDTA, 100 mM tranexamic acid, and protease inhibitor cocktail (Roche). Protein concentrations were measured by Lowry Assay (Bio-Rad Laboratories, Inc.). Quantification of fibrinogen was performed using a hamster anti–mouse fibrinogen-capturing antibody, 7E9, provided by M. Jirouskova (The Rockefeller University, New York, NY; reference 47) and HRP-conjugated rabbit anti–human fibrinogen-detecting antibody (DakoCytomation). Aβ levels were measured by ELISA according to the manufacturer's protocol (Biosource International).
Immunostaining and semiquantitative analysis.
To localize the leakage of Evans blue dye, one brain hemisphere was sectioned and fixed with ice-cold ethanol. To evaluate fibrinogen extravasation and deposition, sections from Evans blue dye–treated animals were processed for fibrinogen immunoreactivity with a FITC-conjugated anti-fibrinogen antibody (DakoCytomation). Microglial staining was performed with a biotin-conjugated anti-CD11b antibody (1:100; BD Biosciences) visualized with FITC- or Rhodamine-conjugated avidin (1:500). To analyze microvasculature, rat anti–PECAM-1 antibody (BD Biosciences) was used (1:50) and visualized with an FITC-conjugated goat anti–rat antibody (1:1,000). Aβ was detected with a rabbit anti–pan-Aβ antibody (1:100; Biosource International). Apoptotic cells were stained with a rabbit anti–active caspase-3 antibody (1:200; Cell Signaling Technology, Inc.).
Coronal sections (from bregma −1.5 to −2.0 mm) were processed and stained for the markers listed above. A composite (3 × 3) of 10× images was stitched together to include hippocampus and cortex during acquisition using a laser-scanning confocal imaging system equipped with a motorized stage (LSM 510 confocal system fitted on an Axiovert 200 inverted microscope). Composite images were converted 1-bit images using ImageJ (http://rsb.info.nih.gov/ij/). Using this bit depth, regions including hippocampus, cortex, or both were selected by hand as shown in Figs. 3 and Figs.4 and quantified for percentage of immunofluorescence.
Fluoro-Jade B staining.
Fluoro-Jade B staining was performed according to the protocol by Schmued and Hopkins (54) and viewed under fluorescence microscopy. In brief, sections were immersed sequentially in 1% NaOH/80% alcohol for 2 min, 70% alcohol for 2 min, water for 2 min, 0.06% potassium permanganate for 10 min, water for 2 min, and 0.0004% Fluoro-Jade B in 1% acetic acid for 20 min, and then rinsed in water three times. The sections were then dried, immersed in Histoclear, and mounted with neutral DPX polystyrene medium. The sections were viewed under blue-green excitation light with a fluorescent microscope.
Ancrod/tranexamic acid treatments.
To explore whether removal of fibrinogen from the circulation can affect the progression of Aβ pathology, we treated transgenic mice with Viprinex (ancrod; provided by D.E. Levy, Neuro biological Technologies, Inc., Emeryville, CA). Mini-osmotic pumps were subcutaneously implanted to deliver 4 U/day of ancrod activity over 4 wk with replacement at 2 wk while a control group received saline (DURECT Corp.). To measure the fibrinogen-depletion effect of ancrod, plasma samples were obtained weekly by tail prick, and fibrinogen was quantified by ELISA.
Deficiency in fibrinolysis was accomplished pharmacologically by implantation of a mini-osmotic pump for delivery of tranexamic acid. Because the drug is also orally active, this dosage was supplemented with tranexamic acid dissolved in drinking water at 20 mg/ml. The total daily dose was estimated to be 100 mg/day.
Online supplemental material.
Fig. S1 shows examples of neurovascular staining in untreated 6-mo NTg and TgCRND8 mice. Fig. S2 shows PDAPP;plg+/− mice compared with PDAPP littermates, and references the TgCRND8;plg+/− mice (Fig. 5). Fig. S3 shows that complete genetic plasminogen deficiency (plg−/−) produces early neurovascular damage.
Supplemental Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants NS50537 (S. Strickland), GM66699 (J. Paul), and AG20901 (J.P. Melchor); by the Institute for the Study of Aging; by Alzheimer's Association grant IIRG-04-1356 (S. Strickland); and by NIH Medical Scientist Training Program grant GM07739 (J. Paul).
We thank Anita Ramnarain and Yuliya Keptsi for assistance in mouse genotyping; Dr. Barry Coller, Dr. Marketa Jirouskova, and members of the Strickland laboratory for helpful discussions; and the Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Program for support. We also thank Dr. M. Azhar Chishti and Dr. David Westaway for the TgCRND8 mice and Drs. Ronald B. Demattos and Steven M. Paul for the PDAPP mice.
The authors have no conflicting financial interests.
Abbreviations used: Aβ, amyloid-β; AβPP, Aβ precursor protein; AD, Alzheimer's disease; PECAM-1, platelet/endothelial cell adhesion molecule-1; tPA, tissue plasminogen activator.
References
- 1.Hardy, J., and D.J. Selkoe. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 297:353–356. [DOI] [PubMed] [Google Scholar]
- 2.de la Torre, J.C. 2004. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 3:184–190. [DOI] [PubMed] [Google Scholar]
- 3.Farkas, E., and P.G. Luiten. 2001. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog. Neurobiol. 64:575–611. [DOI] [PubMed] [Google Scholar]
- 4.Vinters, H.V., Z.Z. Wang, and D.L. Secor. 1996. Brain parenchymal and microvascular amyloid in Alzheimer's disease. Brain Pathol. 6:179–195. [DOI] [PubMed] [Google Scholar]
- 5.Fiala, M., Q.N. Liu, J. Sayre, V. Pop, V. Brahmandam, M.C. Graves, and H.V. Vinters. 2002. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer's disease brain and damage the blood-brain barrier. Eur. J. Clin. Invest. 32:360–371. [DOI] [PubMed] [Google Scholar]
- 6.Tabrizi, P., L. Wang, N. Seeds, J.G. McComb, S. Yamada, J.H. Griffin, P. Carmeliet, M.H. Weiss, and B.V. Zlokovic. 1999. Tissue plasminogen activator (tPA) deficiency exacerbates cerebrovascular fibrin deposition and brain injury in a murine stroke model: studies in tPA-deficient mice and wild-type mice on a matched genetic background. Arterioscler. Thromb. Vasc. Biol. 19:2801–2806. [DOI] [PubMed] [Google Scholar]
- 7.Bugge, T.H., M.J. Flick, C.C. Daugherty, and J.L. Degen. 1995. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 9:794–807. [DOI] [PubMed] [Google Scholar]
- 8.Akassoglou, K., R.A. Adams, J. Bauer, P. Mercado, V. Tseveleki, H. Lassmann, L. Probert, and S. Strickland. 2004. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc. Natl. Acad. Sci. USA. 101:6698–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busso, N., V. Peclat, K. Van Ness, E. Kolodziesczyk, J. Degen, T. Bugge, and A. So. 1998. Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J. Clin. Invest. 102:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ledesma, M.D., J.S. Da Silva, K. Crassaerts, A. Delacourte, B. De Strooper, and C.G. Dotti. 2000. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep. 1:530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melchor, J.P., R. Pawlak, and S. Strickland. 2003. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J. Neurosci. 23:8867–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudal, S., P. Krzywkowski, J. Paquette, C. Morissette, D. Lacombe, P. Tremblay, and F. Gervais. 2004. Inflammation occurs early during the Abeta deposition process in TgCRND8 mice. Neurobiol. Aging. 25:861–871. [DOI] [PubMed] [Google Scholar]
- 13.Yepes, M., M. Sandkvist, E.G. Moore, T.H. Bugge, D.K. Strickland, and D.A. Lawrence. 2003. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J. Clin. Invest. 112:1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickstein, D.L., K.E. Biron, M. Ujiie, C.G. Pfeifer, A.R. Jeffries, and W.A. Jefferies. 2006. Abeta peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J. 20:426–433. [DOI] [PubMed] [Google Scholar]
- 15.Benchenane, K., V. Berezowski, C. Ali, M. Fernandez-Monreal, J.P. Lopez-Atalaya, J. Brillault, J. Chuquet, A. Nouvelot, E.T. MacKenzie, G. Bu, et al. 2005. Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein receptor-related protein-mediated transcytosis. Circulation. 111:2241–2249. [DOI] [PubMed] [Google Scholar]
- 16.Baldwin, H.S., H.M. Shen, H.C. Yan, H.M. DeLisser, A. Chung, C. Mickanin, T. Trask, N.E. Kirschbaum, P.J. Newman, S.M. Albelda, et al. 1994. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31): alternatively spliced, functionally distinct isoforms expressed during mammalian cardiovascular development. Development. 120:2539–2553. [DOI] [PubMed] [Google Scholar]
- 17.Adams, R.A., M. Passino, B.D. Sachs, T. Nuriel, and K. Akassoglou. 2004. Fibrin mechanisms and functions in nervous system pathology. Mol. Interv. 4:163–176. [DOI] [PubMed] [Google Scholar]
- 18.Akassoglou, K., K.W. Kombrinck, J.L. Degen, and S. Strickland. 2000. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J. Cell Biol. 149:1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burkhart, W., G.F. Smith, J.L. Su, I. Parikh, and H. LeVine III. 1992. Amino acid sequence determination of ancrod, the thrombin-like α-fibrinogenase from the venom of Akistrodon rhodostoma. FEBS Lett. 297:297–301. [DOI] [PubMed] [Google Scholar]
- 20.Bell, W.R., S.S. Shapiro, J. Martinez, and H.L. Nossel. 1978. The effects of ancrod, the coagulating enzyme from the venom of Malayan pit viper (A. rhodostoma) on prothrombin and fibrinogen metabolism and fibrinopeptide A release in man. J. Lab. Clin. Med. 91:592–604. [PubMed] [Google Scholar]
- 21.Ujiie, M., D.L. Dickstein, D.A. Carlow, and W.A. Jefferies. 2003. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 10:463–470. [DOI] [PubMed] [Google Scholar]
- 22.Kumar-Singh, S., D. Pirici, E. McGowan, S. Serneels, C. Ceuterick, J. Hardy, K. Duff, D. Dickson, and C. Van Broeckhoven. 2005. Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer's disease are centered on vessel walls. Am. J. Pathol. 167:527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang, F., C. Eckman, S. Younkin, K.K. Hsiao, and C. Iadecola. 1997. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J. Neurosci. 17:7655–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouznetsova, E., M. Klingner, D. Sorger, O. Sabri, U. Grossmann, J. Steinbach, M. Scheunemann, and R. Schliebs. 2006. Developmental and amyloid plaque-related changes in cerebral cortical capillaries in transgenic Tg2576 Alzheimer mice. Int. J. Dev. Neurosci. 24:187–193. [DOI] [PubMed] [Google Scholar]
- 25.Haass, C., E.H. Koo, A. Mellon, A.Y. Hung, and D.J. Selkoe. 1992. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 357:500–503. [DOI] [PubMed] [Google Scholar]
- 26.Blanc, E.M., M. Toborek, R.J. Mark, B. Hennig, and M.P. Mattson. 1997. Amyloid beta-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells. J. Neurochem. 68:1870–1881. [DOI] [PubMed] [Google Scholar]
- 27.Sutton, E.T., G.R. Hellermann, and T. Thomas. 1997. β-Amyloid-induced endothelial necrosis and inhibition of nitric oxide production. Exp. Cell Res. 230:368–376. [DOI] [PubMed] [Google Scholar]
- 28.Hase, M., S. Araki, and H. Hayashi. 1997. Fragments of amyloid beta induce apoptosis in vascular endothelial cells. Endothelium. 5:221–229. [DOI] [PubMed] [Google Scholar]
- 29.Thomas, T., C. McLendon, E.T. Sutton, and G. Thomas. 1997. β-Amyloid-induced cerebrovascular endothelial dysfunction. Ann. N. Y. Acad. Sci. 826:447–451. [DOI] [PubMed] [Google Scholar]
- 30.Thomas, T., G. Thomas, C. McLendon, T. Sutton, and M. Mullan. 1996. β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 380:168–171. [DOI] [PubMed] [Google Scholar]
- 31.Melchor, J.P., and W.E. Van Nostrand. 2000. Fibrillar amyloid beta-protein mediates the pathologic accumulation of its secreted precursor in human cerebrovascular smooth muscle cells. J. Biol. Chem. 275:9782–9791. [DOI] [PubMed] [Google Scholar]
- 32.Sutton, R., M.E. Keohane, S.R. VanderBerg, and S.L. Gonias. 1994. Plasminogen activator inhibitor-1 in the cerebrospinal fluid as an index of neurological disease. Blood Coagul. Fibrinolysis. 5:167–171. [DOI] [PubMed] [Google Scholar]
- 33.Leissring, M.A., W. Farris, A.Y. Chang, D.M. Walsh, X. Wu, X. Sun, M.P. Frosch, and D.J. Selkoe. 2003. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 40:1087–1093. [DOI] [PubMed] [Google Scholar]
- 34.Selkoe, D.J. 2001. Clearing the brain's amyloid cobwebs. Neuron. 32:177–180. [DOI] [PubMed] [Google Scholar]
- 35.Ploplis, V.A., P. Carmeliet, S. Vazirzadeh, I. Van Vlaenderen, L. Moons, E.F. Plow, and D. Collen. 1995. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. 92:2585–2593. [DOI] [PubMed] [Google Scholar]
- 36.Degen, J.L., A.F. Drew, J.S. Palumbo, K.W. Kombrinck, J.A. Bezerra, M.J. Danton, K. Holmback, and T.T. Suh. 2001. Genetic manipulation of fibrinogen and fibrinolysis in mice. Ann. N. Y. Acad. Sci. 936:276–290. [DOI] [PubMed] [Google Scholar]
- 37.Ugarova, T.P., and V.P. Yakubenko. 2001. Recognition of fibrinogen by leukocyte integrins. Ann. N. Y. Acad. Sci. 936:368–385. [DOI] [PubMed] [Google Scholar]
- 38.Flick, M.J., X. Du, D.P. Witte, M. Jirouskova, D.A. Soloviev, S.J. Busuttil, E.F. Plow, and J.L. Degen. 2004. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J. Clin. Invest. 113:1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez, R.L., J.D. Ritzenthaler, and J. Roman. 1999. Transcriptional regulation of the interleukin-1beta promoter via fibrinogen engagement of the CD18 integrin receptor. Am. J. Respir. Cell Mol. Biol. 20:1059–1066. [DOI] [PubMed] [Google Scholar]
- 40.Koistinaho, M., and J. Koistinaho. 2005. Interactions between Alzheimer's disease and cerebral ischemia—focus on inflammation. Brain Res. Brain Res. Rev. 48:240–250. [DOI] [PubMed] [Google Scholar]
- 41.McGeer, P.L., M. Schulzer, and E.G. McGeer. 1996. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 47:425–432. [DOI] [PubMed] [Google Scholar]
- 42.Yenari, M.A., L. Xu, X.N. Tang, Y. Qiao, and R.G. Giffard. 2006. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 37:1087–1093. [DOI] [PubMed] [Google Scholar]
- 43.Grammas, P., U. Reimann-Philipp, and P.H. Weigel. 2000. Cerebro vasculature-mediated neuronal cell death. Ann. N. Y. Acad. Sci. 903:55–60. [DOI] [PubMed] [Google Scholar]
- 44.Lee, D.Y., K.W. Park, and B.K. Jin. 2006. Thrombin induces neuro degeneration and microglial activation in the cortex in vivo and in vitro: proteolytic and non-proteolytic actions. Biochem. Biophys. Res. Commun. 346:727–738. [DOI] [PubMed] [Google Scholar]
- 45.Zipser, B.D., C.E. Johanson, L. Gonzalez, T.M. Berzin, R. Tavares, C.M. Hulette, M.P. Vitek, V. Hovanesian, and E.G. Stopa. 2007. Micro vascular injury and blood-brain barrier leakage in Alzheimer's disease. Neurobiol. Aging. 28:977–986. [DOI] [PubMed] [Google Scholar]
- 46.Furie, B., and B.C. Furie. 1988. The molecular basis of blood coagulation. Cell. 53:505–518. [DOI] [PubMed] [Google Scholar]
- 47.Jirouskova, M., S.S. Smyth, B. Kudryk, and B.S. Coller. 2001. A hamster antibody to the mouse fibrinogen gamma chain inhibits platelet-fibrinogen interactions and FXIIIa-mediated fibrin cross-linking, and facilitates thrombolysis. Thromb. Haemost. 86:1047–1056. [PubMed] [Google Scholar]
- 48.Akassoglou, K., W.M. Yu, P. Akpinar, and S. Strickland. 2002. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron. 33:861–875. [DOI] [PubMed] [Google Scholar]
- 49.Hsiao, K., P. Chapman, S. Nilsen, C. Eckman, Y. Harigaya, S. Younkin, F. Yang, and G. Cole. 1996. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 274:99–102. [DOI] [PubMed] [Google Scholar]
- 50.Chishti, M.A., D.S. Yang, C. Janus, A.L. Phinney, P. Horne, J. Pearson, R. Strome, N. Zuker, J. Loukides, J. French, et al. 2001. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276:21562–21570. [DOI] [PubMed] [Google Scholar]
- 51.Games, D., D. Adams, R. Alessandrini, R. Barbour, P. Berthelette, C. Blackwell, T. Carr, J. Clemens, T. Donaldson, F. Gillespie, et al. 1995. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 373:523–527. [DOI] [PubMed] [Google Scholar]
- 52.Suh, T.T., K. Holmback, N.J. Jensen, C.C. Daugherty, K. Small, D.I. Simon, S. Potter, and J.L. Degen. 1995. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 9:2020–2033. [DOI] [PubMed] [Google Scholar]
- 53.Morris, D.C., Z. Zhang, K. Davies, J. Fenstermacher, and M. Chopp. 1999. High resolution quantitation of microvascular plasma perfusion in non-ischemic and ischemic rat brain by laser-scanning confocal microscopy. Brain Res. Brain Res. Protoc. 4:185–191. [DOI] [PubMed] [Google Scholar]
- 54.Schmued, L.C., and K.J. Hopkins. 2000. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 874:123–130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}