

Abstract
Cytolysis, interferon γ and tumor necrosis factor (TNF) α secretion are major effector mechanisms of memory CD8+ T cells that are believed to be required for immunological protection in vivo. By using mutants of the intracellular bacterium Listeria monocytogenes, we found that none of these effector activities is sufficient to protect against secondary infection with wild-type (WT) bacteria. We demonstrated that CCL3 derived from reactivated memory CD8+ T cells is required for efficient killing of WT bacteria. CCL3 induces a rapid TNF-α secretion by innate inflammatory mononuclear phagocytic cells (MPCs), which further promotes the production of radical oxygen intermediates (ROIs) by both MPCs and neutrophils. ROI generation is the final bactericidal mechanism involved in L. monocytogenes clearance. These results therefore uncover two levels of regulation of the antibacterial secondary protective response: (a) an antigen-dependent phase in which memory CD8+ T cells are reactivated and control the activation of the innate immune system, and (b) an antigen-independent phase in which the MPCs coordinate innate immunity and promote the bactericidal effector activities. In this context, CCL3-secreting memory CD8+ T cells are able to mediate “bystander” killing of an unrelated pathogen upon antigen-specific reactivation, a mechanism that may be important for the design of therapeutic vaccines.
Memory CD8+ T cells represent the major effector arm of the adaptive immune system to maintain long-lived protective immunity against intracellular bacteria, protozoa, and viruses. In secondary infected individuals, pathogen-specific memory CD8+ T lymphocytes are required for both the elimination of infected cells and host survival (1). Upon antigen-driven reactivation, memory CD8+ T cells differentiate into cytolytic, IFN-γ–, and TNF-α–secreting effector cells. The current paradigm is that these effector functions, or at least some of them, allow for the control of the growth and the clearance of intracellular pathogens.
However, previous experiments using the Gram-positive intracellular bacterium Listeria monocytogenes have challenged this view. Mice primary immunized with L. monocytogenes develop long-lasting memory CD8+ T cells that mediate protective responses against secondary challenge with an otherwise lethal dose of bacteria (2, 3). Upon primary immunization, mice lacking perforin, CD95/CD95L, or IFN-γ control secondary infection with WT bacteria as efficiently as WT mice, suggesting that none of these molecules by themselves is absolutely required for protection (4–7). Although TNF-α–deficient mice control secondary infection with WT bacteria as efficiently as WT mice (8), other data using TNF-α–blocking reagents argue for a critical role of TNF-α in the control of secondary L. monocytogenes infection (9–12). Therefore, the actual role of TNF-α during secondary infection needs to be clarified.
Other experiments have addressed which effector functions of CD8+ T cells are necessary to confer protective immunity to naive recipient animals that are subsequently infected (4, 6, 13, 14). These studies have shown that primary effector CD8+ T cells purified from IFN-γ–, TNF-α–, perforin-, CD95-deficient, or WT infected animals confer protection to naive mice, albeit to a lesser extent for perforin-deficient CD8+ T cells that are fivefold less efficient on a per-cell basis as compared with those from WT mice (5, 15). Despite this defect, perforin-deficient CD8+ T cells provide immunity equally well when transferred into WT and CD95-deficient recipient mice but fail to do so in the absence of TNF-α (13). This latter result further suggested that bacteria elimination is dependent on TNF-α secretion but does not absolutely require CD8+ T cells to express TNF-α, IFN-γ, perforin, or CD95L. In contrast, primary effector CD8+ T cells purified from macrophage inflammatory protein 1α (MIP-1α/CCL3)–deficient animals fail to confer protective immunity to naive mice, demonstrating that CCL3 from primary effector CD8+ T cells is required for protection in this experimental setting (14). Of note, these studies all relied on the adoptive transfer of primary effector CD8+ T cells differentiating from naive T cells. However, very little is known on the functional activities that need to be expressed by the secondary effector CD8+ T cells differentiating from memory T cells to protect mice upon rechallenge.
Therefore, in this study, we have investigated the molecular mechanisms of secondary effector CD8+ T cell–mediated bacterial clearance in mice rechallenged with WT bacteria. To address this issue, we have taken advantage of two mutants of L. monocytogenes, secA2 − (ΔSecA2) (16) and actA − (ΔActA) (17), that exhibit the same LD50 of 107 in WT mice (18). Both mutants escape primary phagocytic vacuoles and multiply in the cytosol of infected cells at similar rates. However, in contrast to actA − mutants that induce long-term protective memory, secA2 − mutants do not (18, 19).
RESULTS
The secA2 − mutant of L. monocytogenes fails to induce protective immunity
In a first set of experiments, we have compared the immune response induced by secA2 − and actA − bacteria (Fig. 1). Mice were primary infected with 0.1 × LD50 of mutant or WT bacteria. Control animals were injected with PBS, heat-killed L. monocytogenes (HKLM) (20), or 0.1 × LD50 of llo − (ΔLLO) (21), three protocols that do not induce protective memory. Mice were challenged 1 mo later with 10 × LD50 of WT bacteria and monitored for bacterial titers in the spleen and liver. As expected, control animals immunized with PBS or HKLM rapidly died and exhibited, respectively, 1,000- and 240-fold more bacteria in the spleen and liver than mice immunized with WT bacteria (not depicted) that survived and cleared infection (18). Interestingly, mice immunized with the secA2 − mutant or the llo − control mutant failed to control secondary infection and exhibited 662- and 912-fold more bacteria in the spleen and liver, respectively, as compared with those immunized with WT or actA − bacteria. Therefore, while the actA − mutant induced protective memory, the secA2 − mutant did not.
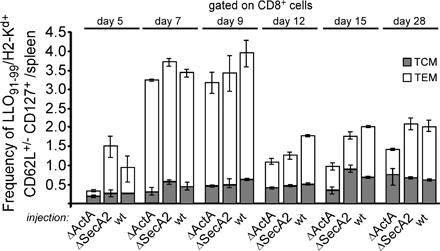
Figure 1.

The secA2− mutant does not induce long-term protective immunity. BALB/c mice (five per group) were injected with PBS or immunized with 109 HKLM or 0.1 × LD50 of the indicated bacteria (WT, 3 × 103; llo −, 5 × 108; secA2 − and actA −, 106). Mice were secondary challenged with 3 × 105 WT bacteria 1 mo later and killed at the indicated times after the infection. Data show the number of bacteria (mean ± SD) in the spleen (empty bars) and liver (filled bars) in a pool of three replicate experiments. p-values were calculated between groups of mice immunized with secA2 − versus actA − bacteria (with n = 9 mice).
Cytolytic and IFN−γ−/TNF-α–secreting activities of secondary effector CD8+ T cells are not sufficient to mediate protection against recall infection
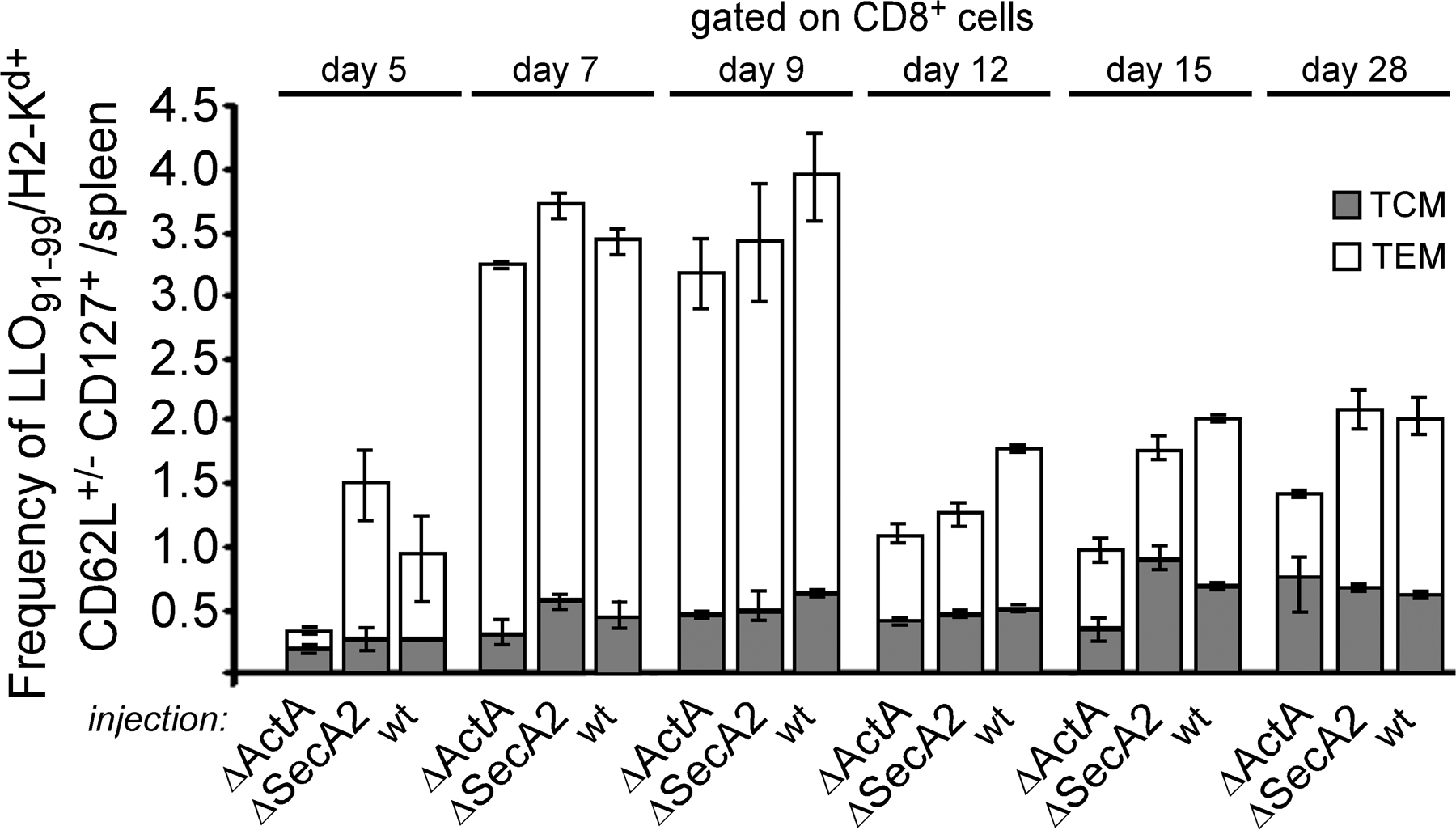
To investigate why the secA2 − mutant failed to induce protective immunity, we have characterized the CD8+ T cell response in groups of mice immunized with secA2 −, actA −, or WT bacteria (primary response) and further challenged the same groups of mice 1 mo later with WT L. monocytogenes (secondary response). Spleen cells were harvested at different times after infection and analyzed by FACS after staining with LLO91-99/H2-Kd tetramers, anti-CD62L, a cell surface marker that is lost upon activation and acquisition of effector functions (2, 22), and anti-CD127 (IL-7 receptor), a marker of long-living memory T cells (23, 24). Mice from all groups exhibited similar frequencies of CD62Llow LLO91-99/H2-Kd tetramer+ CD8+ T cells at the peak of primary and secondary responses (Fig. 2 A) (25). Moreover, LLO91-99/H2-Kd tetramer+ CD8+ T cells expanded with the same kinetics upon secondary challenge in mice immunized with the secA2 − and actA − mutant (Fig. 2 B). Therefore, the absence of a protective secondary response in mice immunized with the secA2 − mutant did not result from a decreased ability of Listeria-specific memory CD8+ T cells from these mice to proliferate upon secondary infection. We also found that mice primary immunized with secA2 − bacteria were not impaired in the generation of CD127high CD62Llow effector–memory (TEM) cells as compared with mice immunized with WT and actA − bacteria (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070204/DC1). Thus, the inability of mice primary immunized with secA2 − bacteria to control secondary infection did not result from a reduced number of TEM cells (26).
Figure 2.

Analysis of primary and secondary CD8+ T cell responses in mice immunized with secA2−, actA−, or WT L. monocytogenes. BALB/c mice were immunized with PBS or 0.1 × LD50 of the indicated bacteria. (A) Mice (three per group) were either killed 8 d after the first immunization (top, primary) or challenged with 3 × 105 WT bacteria 30 d later and co-treated with 25 mg ampicillin and killed after 5 d (bottom, secondary). Spleen cells were analyzed by FACS upon staining with H2-Kd/LLO91-99 tetramers, anti-CD62L, and anti-CD8 mAbs. Data show representative FACS dot plots after gating on CD8+ T cells in one out of three experiments. The frequency of cells in each upper quadrant is indicated. (B) Mice (three per group) primary immunized with PBS, secA2 −, actA −, or WT bacteria were secondary challenged with 3 × 105 WT bacteria 30 d later, treated with ampicillin as above, and killed at the indicated times. Spleen cells were analyzed by FACS as described in A. Data show the frequencies (mean ± SE) of CD62Llow H2-Kd/LLO91-99 tetramer+ cells among CD8+ T lymphocytes in a representative (out of three) experiment.
We next assessed whether Listeria −specific CD8+ T cells from mice immunized with the secA2 − mutant were impaired in effector functions such as cytolytic activity, expression of granzyme B, CD107a (27) or CD95 ligand, and secretion of IFN-γ or TNF-α. To measure cytolytic activity, spleen cells were pulsed or not with LLO91-99, labeled with high or low concentrations of CFSE, respectively, and injected into mice primarily immunized with secA2 −, actA −, or WT bacteria that were secondary challenged with WT bacteria. Spleen cells were recovered 3 h or 1 or 4 d after secondary infection and analyzed by FACS to determine the specific lysis of peptide-pulsed CFSEhigh cells. Lysis of these target cells was similar among all groups (Fig. 3 A and not depicted). In addition, CD8+ T cells from mice immunized with the secA2 − mutant were not impaired in the expression of granzyme B, CD107a, and CD95 ligand (not depicted). To monitor the presence of IFN-γ- and TNF-α–secreting cells, spleen cells from secondary challenged mice were incubated with LLO91-99 peptide–pulsed splenocytes and analyzed by intracellular cytokine staining. The total numbers of IFN-γ– and TNF-α–secreting CD8+CD3+ T cells per spleen, as well as the frequencies of IFN-γ– and TNF-α–secreting cells among CD8+CD3+ T cells, were similar in mice primarily immunized with secA2 −, actA −, or WT bacteria and challenged with WT bacteria at early and late (2- or 3-d) time points (Fig. 3 B and not depicted). Collectively, although Listeria-specific reactivated memory CD8+ T cells from secA2 − immunized mice did not confer protective immunity, they exhibited effector functions such as cytolytic activity, expression of granzyme B, CD107a and CD95 ligand, and secretion of IFN-γ and TNF-α. Thus, the inability of mice primary immunized with secA2 − bacteria to control secondary infection was neither due to ineffective memory CD8+ T cell development nor to impaired known effector activities of these cells.
Figure 3.

Cytolytic and IFN-γ–/TNF-α–secreting activities of CD8+ T cells in mice primary immunized with WT and mutants of L. monocytogenes. BALB/c mice (five per group) primarily immunized with PBS or 0.1 × LD50 of the indicated bacteria (PBS, secA2 −, actA −, or WT) were secondary challenged 30 d later with either PBS, 3 × 105, or 104 (A, bottom) WT bacteria. (A) 3 h (top) or 4 d (bottom) after secondary challenge, animals were transferred with 107 CFSElow unpulsed and CFSEhigh LLO91-99 peptide–pulsed target cells. Spleen cells from individual mice were analyzed by FACS 15 h later. Data show representative FACS histograms in one out of three experiments. The numbers indicate the percentage of specific lysis of peptide-pulsed CFSEhigh cells. (B) At specified times, spleen cells from individual mice (three per group) were restimulated in vitro with LLO91-99 and analyzed by FACS for intracellular staining of IFN-γ and TNF-α. Data show the total numbers (mean ± SE) of IFN-γ– (left) and TNF-α– (right) secreting CD8+CD3+ T cells per spleen in a representative (out of three) experiment.
CCL3 secreted by secondary effector CD8+ T cells is required for protective immunity
CCL3 was the only effector activity that was known to be required for primary activated CD8+ T cells to protect naive recipients (14). We have therefore investigated whether Listeria-specific secondary effector CD8+ T cells from mice primary immunized with the secA2 − mutant were impaired in the secretion of CCL3 during secondary infection. We observed that the total number of CCL3-secreting CD8+CD3+ T cells per spleen was twofold lower in mice immunized with the secA2 − mutant or PBS as compared with those immunized with actA − or WT bacteria (Fig. 4 A). Furthermore, CD8+CD3+ T cells from mice immunized with the secA2 − mutant exhibited a lower mean fluorescence intensity upon staining with anti-CCL3 serum than those from mice immunized with actA − or WT bacteria (27.8 for secA2 − vs. 44 and 62 for actA − and WT, respectively) (Fig. 4 A). Likewise, CD8+CD3+ T cells from mice immunized with the secA2 − mutant or PBS secreted less CCL3 when restimulated with LLO91-99 than those from the two other groups (Fig. 4 B). Therefore, the secretion of CCL3 was the only known effector function of secondary effector CD8+ T cells that was significantly altered in mice immunized with the secA2 −mutant as compared with those immunized with the actA − mutant.
Figure 4.

CCL3 from CD8+ T cells is required for secondary protective immunity. BALB/c mice primarily immunized with PBS or 0.1 × LD50 of the indicated bacteria (PBS, secA2 −, actA −, or WT) were secondary challenged 30 d later with WT bacteria. (A and B) At the indicated times, spleen cells from individual mice were restimulated in vitro with LLO91-99 peptide. (A, left) Data show a representative FACS profile of intracellular CCL3 staining after gating on CD3+ T cells. The total numbers (filled bars) and the mean fluorescence intensity (empty bars) (mean ± SD) of CCL3-secreting CD8+CD3+ T cells per spleen were shown 6 h after challenge (right). (B) Data show the concentration of CCL3 (mean ± SD) measured by ELISA in the supernatant of restimulated cells. (A and B) Data result from the pool of three replicate experiments. p-values were calculated between groups of mice immunized with secA2 − versus actA − bacteria (with n = 7 mice). (C) Mice (three per group) primary immunized with PBS or WT bacteria were treated with anti-CCL3 or normal goat IgG control and secondary challenged. Data show the number of bacteria (mean ± SE) in the spleen 2 d later in a representative (out of two) experiment. p-value was estimated accordingly to the rules of SE bars in the experiment shown (reference 54). (D) PBS-injected or primary immunized WT or CCL3−/− mice (8–12 per group) were secondary challenged. Purified CD8+ T cells were transferred into CCL3−/− or WT mice. Recipient animals were further treated with anti-CCL3 or control serum and challenged. Data show bacteria titers (mean ± SD) in the spleen 2 d later and result from the pool of three replicate experiments (n = 7). p-values are indicated.
To determine whether CCL3 was required during the effector phase of secondary protective response against L. monocytogenes, mice were immunized with PBS or WT bacteria, injected 30 d later with anti-CCL3 or control goat serum, and challenged with WT bacteria. PBS-injected mice exhibited the same number of bacteria when treated with control or anti-CCL3 serum (Fig. 4 C and Fig. S2 A, which is available at http://www.jem.org/cgi/content/full/jem.20070204/DC1). In contrast, mice immunized with WT bacteria exhibited 100- and 12-fold more bacteria in the spleen and liver, respectively, when treated with anti-CCL3 serum as compared with control serum. Although mice injected with control serum survived to secondary infection, those treated with anti-CCL3 serum rapidly died (Fig. S3). Therefore, in vivo neutralization of CCL3 during secondary infection abrogated protective immunity.
To investigate if the CCL3 secreted by secondary effector CD8+ T cells was responsible for protection, WT CD8+ T cells from secondary challenged mice were transferred into CCL3−/− recipient animals that received anti-CCL3 or control serum and were subsequently infected. In this experiment, 2 × 107 CD8+ T cells, accounting for ∼40,000 LLO91-99/H-2Kd–specific CD8+ T cells (Fig. 3 B), were purified from WT mice and transferred into naive CCL3−/− or control WT recipients that were treated or not with anti-CCL3 serum and infected with 3 × 105 WT bacteria (Fig. 4 D and Figs. S2 B and S4, which are available at http://www.jem.org/cgi/content/full/jem.20070204/DC1). Protection was similar whether recipient mice were WT or CCL3−/− with 4.4- (spleen) and 5.7- (liver) fold less bacteria in mice infused with CD8+ T from secondary infected animals as compared with mice transferred with CD8+ T cells from PBS-injected mice (Fig. 4 D and Figs. S2 B and S4). Although the level of protection achieved in these latter experiments was relatively low, it should be noticed that we have transferred 10–15-fold less epitope-specific T cells than other authors (6, 14, 28). Furthermore, the level of protection is also known to be highly dependent on the infecting dose of bacteria. Indeed, when mice transferred with CD8+ T cells from secondary infected mice were challenged with 5 × 104 bacteria (instead of 3 × 105), 15–27 less bacteria were enumerated in their organs as compared with those infused with CD8+ T cells from PBS-injected animals (Fig. S5).
Most importantly, protection transferred to CCL3−/− mice by WT CD8+ T cells from mice immunized with WT bacteria was completely abrogated upon treatment with anti-CCL3 serum, further showing that CCL3 derived from WT memory CD8+ T cells was required for secondary protection. We also found that CCL3 from memory CD8+ T cells accounted for the majority of the CCL3-mediated protective effect in the liver of CCL3−/− recipient mice transferred with WT CD8+ T cells from secondary infected WT animals (Figs. S2 B and S4).
To independently demonstrate that CCL3 from secondary effector CD8+ T cells was responsible for protection, CCL3−/− or WT control mice were primary and secondary infected with WT bacteria, and CD8+ T cells from these mice were transferred into naive WT mice (Fig. 4 D and Figs. S2 B and S4). Although the transfer of WT CD8+ T cells resulted in 4.4-fold less bacteria in the spleen, CCL3−/− CD8+ T cells only conferred a marginal protective effect in the spleen and liver, further confirming a critical role of the CCL3 derived from secondary effector CD8+ T cells. Collectively, our data demonstrated that CCL3, and not other cytokines/chemokines from reactivated memory CD8+ T cells (and not from other cell types), is mediating secondary protective immunity.
Mononuclear phagocytes secrete TNF-α during secondary protective response
Although CCL3 mediates the chemotaxis of monocytes and T and NK cells (29, 30), this chemokine also promotes TNF-α secretion from macrophages in vitro (31, 32). Because TNF-α has been shown to be critical for L. monocytogenes clearance and host survival during secondary infection (9–11), we hypothesized that mice immunized with the secA2 − mutant did not clear secondary infection because their CD8+ T cells were impaired in their ability to secrete CCL3 and subsequently to induce myeloid cells to release TNF-α. In agreement with previous reports, mice secondary challenged with WT bacteria rapidly died (Fig. S3) and exhibited 10,000- and 300-fold more bacteria in the spleen and liver, respectively (Fig. 5 A and not depicted), when treated with soluble recombinant p75 TNF-α receptor (Enbrel) as compared with control animals. TNF-α was produced by myeloid cells that expressed high levels of CD11b (Fig. 5 B). Further characterization of the TNF-α–secreting cells demonstrated that 90% of these cells were monocytic-derived cells (F4/80+CD11c+/−CD11bmed/highLy-6ChighGr1low) and 10% were neutrophils (F4/80−CD11c−CD11bhighLy-6Cmed/highGr1high) (Fig. 5 B and not depicted). These monocytic-derived cells expressed MHC class II molecules, CD80, and CD86 and produced inducible nitric oxide synthase (iNOS) (Fig. S6, A and B, available at http://www.jem.org/cgi/content/full/jem.20070204/DC1). They also contained bacterial antigens (Fig. 5 C) as well as live replicating bacteria (not depicted) and exhibited ultrastructural characteristics of phagocytic mononuclear cells (Fig. S6, C and D). They were therefore designated TP-MPCs for TNF-α–producing mononuclear phagocytic cells.
Figure 5.

MPCs produce TNF-α during secondary protective response. (A) BALB/c mice (two per group) primary immunized with PBS or 0.1 × LD50 of WT bacteria were treated 30 d later with the soluble p75 TNF-α receptor (Enbrel) and secondary challenged with 3 × 105 WT bacteria. Data show the number of bacteria (mean ± SE) in the spleen 2 d later in a representative (out of three) experiment. p-value was estimated accordingly to the rules of SE bars in the experiment shown (54). (B) Mice (two per group) were primary immunized and secondary challenged with WT bacteria (B and C) or PBS (C). 10 h later, spleen cells from individual mice were restimulated with HKLM (B) or not (C) and analyzed by FACS for surface staining of CD11b, Ly-6C, CD11c, and control isotype and for intracellular staining of L. monocytogenes–derived antigens and TNF-α. Data show representative FACS histograms after gating on the indicated cell population in a representative (out of three) experiment. (D) Mice primarily immunized with PBS, secA2 −, actA −, or WT bacteria were secondary challenged. At the indicated times, spleen cells from individual mice were restimulated with HKLM and analyzed by FACS for surface CD11b, Ly-6C, and intracellular TNF-α. The number (mean ± SD) of TNF-α–producing CD11bmed/highLy6Chigh mononuclear phagocytic cells (TP-MPCs) per spleen is indicated and result from the pool of three replicate experiments (n = 9). The p-values between the numbers of TP-MPCs in secA2 −- versus actA − -infected mice are indicated.
Both the number (Fig. 5 D) and the frequency (Fig. S7 A, available at http://www.jem.org/cgi/content/full/jem.20070204/DC1) of TP-MPCs in the spleen increased more slowly and to lower levels in mice immunized with the secA2 − mutant or PBS and secondary challenged as compared with those immunized with actA − or WT bacteria. In contrast, the frequency of TNF-α–secreting neutrophils in the spleen remained identical in mice immunized with secA2 − and actA − bacteria (not depicted). Moreover, the frequency of MPCs that secreted TNF-α among total MPCs was twofold lower at 6 and 10 h after challenge in mice immunized with the secA2 − mutant compared with those immunized with actA − or WT bacteria (Fig. S7 B). Therefore, the kinetics of TNF-α secretion by MPCs, but not by neutrophils, was strongly altered in mice that received a primary immunization with the secA2 − mutant.
CCL3 induces MPCs to secrete TNF-α
To assess whether CCL3 promotes TNF-α secretion by MPCs, we measured the number of TP-MPCs in primary and secondary infected mice treated or not with anti-CCL3 serum. In mice primary immunized with WT bacteria, neutralization of CCL3 before secondary infection resulted in a lower number of TP-MPCs in the spleen (Fig. 6 A, left). In contrast, the number of TNF-α–secreting neutrophils remained the same (not depicted). A similar reduction in the number of TP-MPCs, but not neutrophils, was observed in mice depleted of CD8+ T cells before the secondary infection (Fig. 6 A, right, and not depicted). Furthermore, recombinant CCL3 induced MPCs to secrete TNF-α in vitro as efficiently as HKLM (Fig. 6 B). As expected, anti-CCL3 prevented TNF-α release, demonstrating that this effect was dependent on CCL3. CCR1 and CCR5 are the two major receptors of CCL3 (33). Although we could not detect the expression of CCR5 at the surface of TP-MPCs, 50% of these cells expressed CCR1, a result in agreement with their responsiveness to CCL3 (Fig. 6 C). Collectively, these data support the hypothesis that CCL3 secreted by CD8+ T cells during a secondary infection induces MPCs to rapidly release TNF-α.
Figure 6.

CCL3 promotes TNF-α secretion by MPCs. (A) Mice primary immunized with PBS or 0.1 × LD50 of WT bacteria were treated or not 30 d later with the indicated blocking (anti-CCL3), depleting (anti-CD8β), or control antibodies. 10 h after secondary challenge with 3 × 105 WT bacteria, spleen cells from individual mice were restimulated with HKLM and analyzed by FACS for surface expression of CD11b, Ly-6C, and intracellular TNF-α. Data show the number (mean ± SD) of TP-MPCs per spleen and result from the pool of two to three replicate experiments (n = 7). p-values are indicated. (B and C) Mice were infected with 3 × 105 WT bacteria and killed 24 h later. (B) Spleen cells from individual mice (five per group) were pooled, and flow-sorted MPCs were incubated in vitro with or without HKLM or with 1 μg/ml of recombinant CCL3 (endotoxin <1.2 pg/ml) with or without 10 μg/ml anti-CCL3 serum. Cells were analyzed by FACS for intracellular TNF-α. Data show the frequency of TNF-α–secreting cells among MPCs (± SE) and result from the pool of three experiments. p-value was estimated accordingly to the rules of SE bars from the pool of the three experiments (54). (C) Spleen cells from individual mice (two per group) were analyzed for intracellular TNF-α and surface expression of CD11b, Ly-6C, CCR1 (filled histograms), and CCR5 (filled histograms) or isotype control (empty histograms). Data show representative FACS profiles of CCR1 (left) and CCR5 (right) expression after gating on TP-MPCs in a representative (out of two) experiment.
TNF-α induces mononuclear phagocytes and neutrophils to produce reactive oxygen intermediates (ROIs)
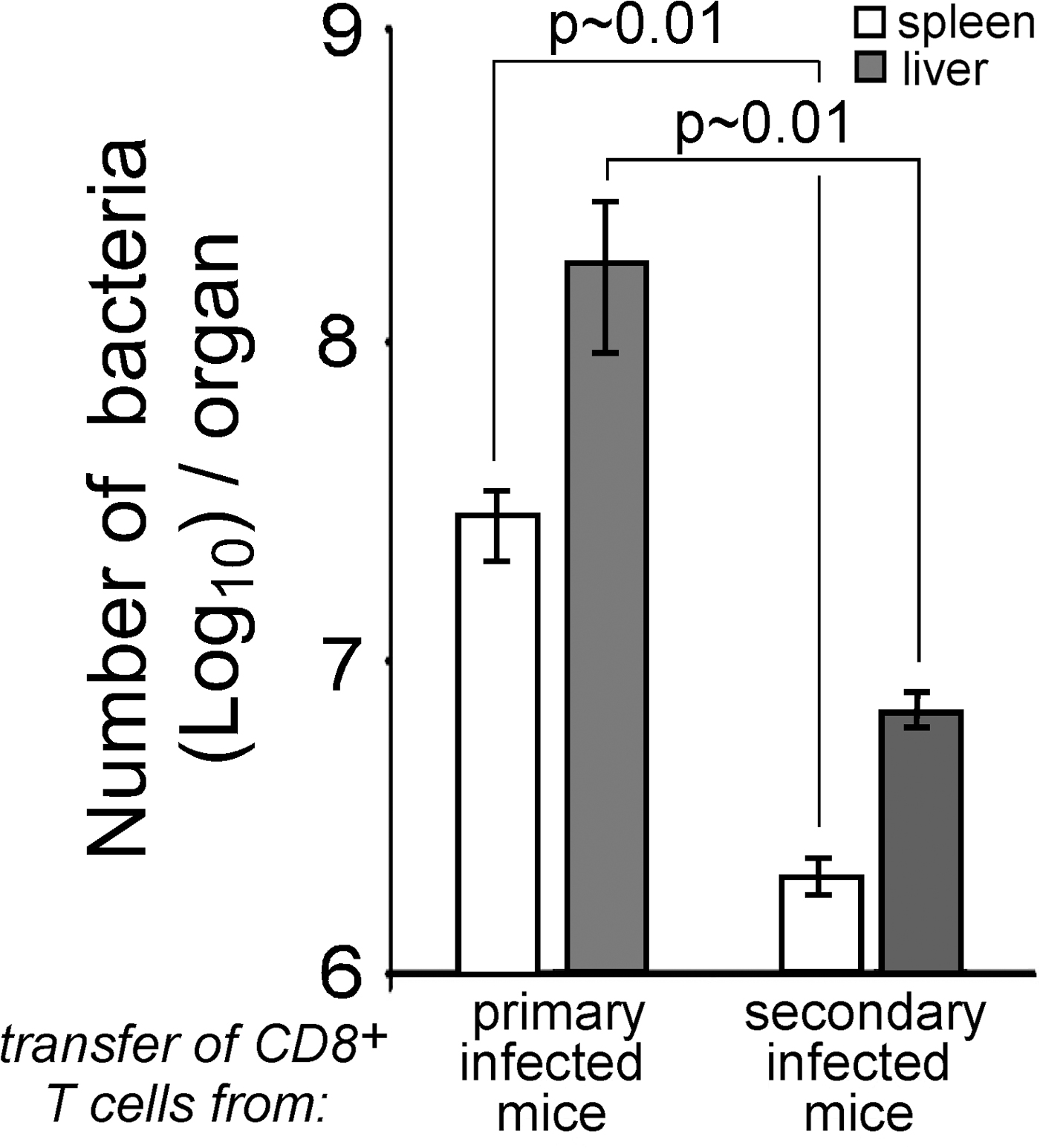
TNF-α is an in vitro inducer of ROIs that directly allows infected myeloid cells to kill bacteria (34, 35). Therefore, the rapid release of TNF-α by MPCs may be critical for the production of high levels of ROI and subsequent clearance of bacteria. To test this hypothesis, mice received a primary immunization with PBS, WT, or mutant bacteria and were further challenged with WT L. monocytogenes. Spleen cells were analyzed for ROI production upon staining with anti-CD11b and anti–Ly-6C mAbs. In both PBS- and bacteria-immunized mice, challenge with WT bacteria induced ROI production (Fig. 7 A). At each time, the frequency of ROI-producing MPCs was 1.3- to 1.5-fold lower in mice primarily immunized with the secA2 − mutant or PBS as compared with those immunized with actA − or WT bacteria. In contrast, although neutrophils accounted for the majority (∼80%) of ROI-producing CD11b+ cells (Fig. 7, A and B, Fig. S8, which is available at http://www.jem.org/cgi/content/full/jem.20070204/DC1), their numbers did not differ between mice primarily immunized with secA2 −, actA −, or WT bacteria. In PBS-injected animals, however, the number of ROI-producing neutrophils was significantly lower compared with mice primarily immunized with WT bacteria (Fig. S8). Both the neutralization of CCL3 or TNF-α and the depletion of CD8+ T cells before the secondary challenge in mice primarily immunized with WT bacteria reduced the number of ROI-producing MPCs and neutrophils to similar numbers as those measured in the PBS-immunized control group (Fig. 7 C). Collectively, these data reveal a causal relationship between the secretion of CCL3 and the production of TNF-α by MPCs, and between the secretion of TNF-α and the production of ROIs by both MPCs and/or neutrophils.
Figure 7.

ROI production is induced by TNF-α during secondary response. (A and B) Mice primary immunized with (A) PBS or 0.1 × LD50 of secA2 −, actA −, or (A and B) WT bacteria were challenged 30 d later with 3 × 105 WT bacteria. At the indicated times (A) or at 10 h (B) after secondary challenge, spleen cells from individual mice were restimulated with HKLM in the presence of hydroethidine. Cells were analyzed by FACS after staining with anti-CD11b and anti–Ly-6C mAbs. (A) Data show the number (mean ± SD) of ROI-producing MPCs (left) and neutrophils (right) per spleen. (B) Data show a representative histogram on Ly-6C (gray) or control isotype (empty) after gating on ROI-producing cells. Numbers indicate the frequency of MPCs (CD11bmed/highLy6Chigh) and neutrophils (CD11bhighLy6Cmed) in the indicated gate. (C) Mice primary immunized with PBS or WT bacteria were treated or not 30 d later with the indicated antibodies or sera. 10 h after secondary challenge, spleen cells from individual mice were treated and analyzed as above. Data show the number (mean ± SD) of ROI-producing MPCs (top) and neutrophils (bottom) per spleen. (A and C) Data result from the pool of two to three replicate experiments. p-values are indicated. A, n = 7; C, right and left panels, n = 7; C, middle panel, n = 9.
ROIs produced by mononuclear phagocytes and/or neutrophils are necessary for bacterial clearance
To determine whether ROIs were required for bacterial clearance upon secondary infection, mice that were unable to generate ROIs as the result of a targeted deletion of the p47phox gene (36) were immunized and challenged or not with WT bacteria. It has been reported that p47phox−/− mice are able to clear primary L. monocytogenes infection (37). In contrast, upon immunization and subsequent acute challenge with WT bacteria, p47phox−/− mice rapidly died and exhibited 3,000- and 9,000-fold more bacteria in the spleen and liver, respectively, as compared with WT animals (Fig. 8 and Figs. S3 and S9, which are available at http://www.jem.org/cgi/content/full/jem.20070204/DC1).
Figure 8.

CCL3-producing memory CD8+ T cells promote ROI generation by phagocytic cells for secondary bacterial clearance. Indicated mice (three per group) were primary immunized with PBS (filled bars) or 3 × 103 WT bacteria (empty bars). When specified, p47phox−/− mice were reconstituted 1 mo later with 4 × 105 MPCs or 4 × 106 neutrophils purified from WT or p47phox−/− mice. Mice were then treated or not with anti-CCL3 or anti-CD8β (right) and secondary challenged with 3 × 105 bacteria. Mice were killed 2 d later, and the number of bacteria in the liver (mean ± SE) was measured. Data are representative of one out of two to three experiments. p-values were estimated accordingly to the SE bars rules in the experiment shown (54).
To further confirm that the lack of protection in p47phox−/− mice actually resulted from the inability of innate cells to generate ROIs and not from a defective memory response, we compared primary and secondary CD8+ T cell responses in WT and p47phox−/− mice. This was done using HKLM to restimulate CD8+ T cells because p47phox−/− mice were on the 129 genetic background in which Listeria epitopes have not been characterized. Upon primary infection, the spleens of p47phox−/− mice contained similar numbers of granzyme B–expressing and IFN-γ–secreting CD8+ T cells reacting against HKLM as those from WT mice. Upon secondary infection, these numbers were three- to fivefold increased as expected for a recall response (Fig. S10, available at http://www.jem.org/cgi/content/full/jem.20070204/DC1). Most importantly, this memory response was similar in p47phox−/− and WT mice, suggesting that the lack of protective immunity in secondary infected p47phox−/− mice did not result from defective CD8 T cell memory.
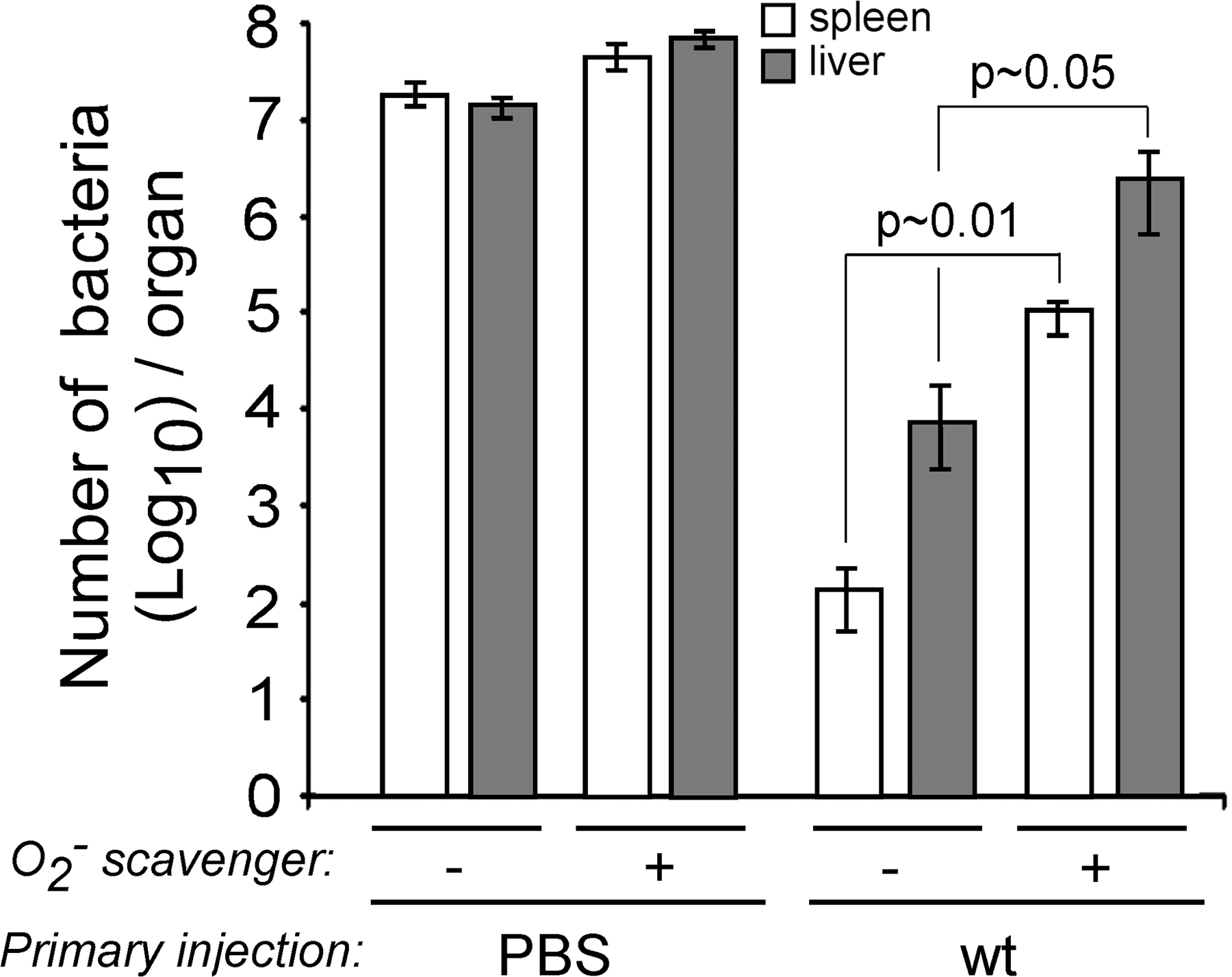
To independently confirm that ROIs were critical for secondary protection against L. monocytogenes, we used a different experimental approach in which WT BALB/c mice primarily immunized with WT bacteria were treated with a superoxyde scavenger (38) before secondary challenge to neutralize the oxidative burst in vivo (Fig. S11, available at http://www.jem.org/cgi/content/full/jem.20070204/DC1). As compared with untreated animals, mice treated with a superoxyde scavenger exhibited 1,073- and 230-fold more bacteria in the spleen and liver, respectively. Therefore, collectively, these results on two distinct genetic backgrounds demonstrate that the generation of ROIs is required for both bacterial clearance and survival of secondary infected mice.
To further clarify the role of MPCs and neutrophils in ROI production and bacteria elimination, 4 × 105 MPCs or 4 × 106 neutrophils from WT or p47phox−/− naive mice were transferred into p47phox−/− mice primary immunized with WT bacteria or PBS and secondary challenged with WT bacteria (Fig. 8 and Fig. S9). Mice reconstituted with MPCs from WT mice exhibited 170- and 10-fold less bacteria in the spleen and liver, respectively, than those reconstituted with MPCs from p47phox−/− mice. Similarly, mice reconstituted with neutrophils from WT mice exhibited 1,000- and 80-fold less bacteria in the spleen and liver, respectively, than those reconstituted with neutrophils from p47phox−/− mice. Also, PBS-immunized p47phox−/− recipient mice failed to clear infection with WT bacteria even when reconstituted with MPCs or neutrophils from WT mice. Collectively, these results show that both MPCs and neutrophils from WT mice, but not those from p47phox−/− mice, were able to restore a protective secondary response when transferred into p47phox−/− mice. Because protection was only observed in p47phox−/− mice primary immunized with WT bacteria, these data strongly suggested that the presence of memory cells was required to activate ROI production and bacteria killing.
We next verified that the restoration of protective immunity by MPCs or neutrophils in secondary challenged p47phox−/− mice was dependent on CCL3 and CD8+ T cells. p47phox−/− mice were primary immunized with WT bacteria, reconstituted with MPCs or neutrophils from WT mice, treated or not with anti-CCL3 or with anti-CD8, and secondary challenged with WT bacteria. Neutralization of CCL3 or CD8+ T cell depletion prevented bacterial clearance both in mice reconstituted with MPCs or neutrophils (Fig. 8 and Fig. S9). Therefore, secretion of CCL3 by reactivated memory CD8+ T cells during secondary infection induces the accelerated release of TNF-α by MPCs that in turn promotes the production of ROIs by MPCs and neutrophils, leading to subsequent clearance of the bacteria (Fig. 9).
Figure 9.

Schematic representation of memory CD8+ T cell–mediated antibacterial immunity. The cartoon shows (A) release of CCL3 by secondary effector CD8+ T cells, binding of CCL3 to its receptor CCR1 at the surface of MPC that results in MPC activation, (B) secretion of TNF-α by MPCs that leads to neutrophil and MPC activation, and (C) TNF-α–induced production of ROIs by neutrophils and MPCs, and subsequent bacteria killing.
Secondary effector Listeria-specific CD8+ T cells mediate CCL3-dependent bystander killing of the intracellular parasite Leishmania major
One important prediction of our findings is that CCL3-secreting memory CD8+ T cells should promote an antigen-independent antimicrobial effector phase by innate immune cells upon antigen-specific restimulation. Therefore, we have postulated that any ROI-sensitive pathogen would be killed upon restimulation of memory cells in a bystander fashion. To test this hypothesis, BALB/c mice were immunized with WT L. monocytogenes and infected 3 wk later with the intracellular ROI-sensitive L. major promastigotes (39). Parasite-containing mice were challenged 2 wk later either with L. monocytogenes or LPS-matured DCs pulsed or not with the dominant Listeria-derived LLO91-99 peptide (Fig. 10 and not depicted). Animals challenged with L. monocytogenes or peptide-pulsed DCs exhibited 10,000- and 1,000-fold less L. major parasites in their spleen than those injected with PBS and control LPS-matured DCs, respectively. This dramatic decrease in parasite burden was completely abrogated upon injection of a CCL3-neutralizing, but not of a control, serum (not depicted). Therefore antigen-driven restimulation of L. monocytogenes–specific memory CD8+ T cells can induce antigen-independent CCL3-mediated bystander killing of an unrelated intracellular parasite.
Figure 10.

CCL3 from secondary effector CD8+ T cells mediate bystander killing in vivo. BALB/c mice (three per group) primarily immunized with 0.1 × LD50 of WT L. monocytogenes (Lm) or control PBS were infected i.v. 3 wk later with the intracellular parasite L. major (2 × 106/mouse). 2 wk later, mice were treated or not with anti-CCL3 serum and injected with 3 × 105 WT Lm, 2 × 107 LPS-matured splenic DCs pulsed with the Lm-derived LLO91-99 dominant epitope, or PBS (control). Spleens were harvested 2 d later, and cells were incubated in L. major growth medium (M199). (A) Schematic representation of the experimental protocol. (B) The number of live parasites per well (± SD) was determined after 4 d of culture. Data show the results of the pool of two replicate experiments (with n = 6 mice). p-value is indicated.
DISCUSSION
Several studies have demonstrated that neither cytolytic activity nor secretion of IFN-γ or TNF-α by CD8+ T cells was absolutely required for resistance to L. monocytogenes in mice (4, 6, 13). In contrast, CCL3-deficient CD8+ T cells isolated from primary infected mice did not confer protective immunity upon transfer into recipient mice (14). Although this suggested that secretion of CCL3 by primary effector CD8+ T cells is required for protection, it was not known whether CCL3 derived from secondary effector CD8+ T cells could confer protection to primary infected animals. In this study, we show that protection conferred by reactivated memory CD8+ T cells depends on their ability to secrete CCL3. This was independently shown by experiments in which CD8+ T cells from WT or CCL3−/− mice were injected into CCL3−/− mice, as well as in neutralization experiments using anti-CCL3 mAb. In agreement with these data, we found that secondary effector CD8+ T cells from mice immunized with secA2 − bacteria did not secrete CCL3 as compared with those from mice immunized with actA − or WT bacteria. Although these differences were modest, they were significant and reproducible. How could relatively small differences account for such a dramatic effect in protection? At least two explanations can account for these observations. First, the CCL3-secreting cells that were present in mice immunized with actA − or WT bacteria may be localized in strategic areas of the spleen that are critical for the development of protective immunity. A small difference in the number of CCL3-secreting cells could therefore have a major impact on the outcome of the recall infection. Second, several cytokines such as TNF-α (40), IFN-α/β (41), or IL-12 (42) have been shown to be biologically active and/or to exert a distinct effect above a specific threshold. Likewise, small differences in the amount of secreted CCL3 could lead to a distinct biological response, i.e., protective versus nonprotective.
CCL3 is an inflammatory molecule that activates macrophages to secrete TNF-α (31, 32) and exhibits chemotactic activities on several cell types (29, 30). Neutralization of CCL3 in protected animals reduces both the total number of MPCs in the spleen and the number of these cells that secrete TNF-α. It remains unclear whether CCL3 induces activation or recruitment of MPCs or both. We have found (a) that recombinant CCL3 induces MPCs to secrete TNF-α in vitro, and (b) that the total number of MPCs in the spleen is identical in mice immunized with secA2 − bacteria as compared with those immunized with actA − bacteria, whereas the frequency of TP-MPCs among MPCs is twofold lower. Thus, although it remains to be determined whether CCL3 also promotes MPC recruitment, it does activate MPCs to secrete TNF-α.
We and others have demonstrated that TNF-α blockade using mAbs or soluble receptor prevents protective immunity after secondary infection, therefore demonstrating a critical role of this cytokine in this process (9, 10). These results are in apparent contrast to those obtained in TNF-α−/− mice or in mice exhibiting TNF-α–deficient macrophages and neutrophils that are able to develop long-term protective immunity (8). How can these data be reconciled? Regarding TNF-α−/− mice, it is well established that these animals do not regulate properly the development and the organization of splenic follicular architecture and exhibit defects in their lymphoid organs and germinal centers (43, 44). Furthermore, these mice are unable to regulate the homeostasis and the extent and duration of inflammatory processes. Therefore, it is conceivable that the development of protective immunity in TNF-α−/− mice results from compensatory mechanisms that do not reflect what is happening in WT mice.
In a recent study, Grivennikov et al. (8) have taken advantage of the lysozyme M (lys-M) promoter to generate mice in which macrophages and neutrophils did not express TNF-α. These mice were protected against secondary infection, further suggesting that lys-M–expressing cells were not required for protection. We have found that at least 50% of the MPCs that are the main source of TNF-α during secondary infection do not express lys-M in the lys-M–GFP+/− knock-in mouse (Fig. S12, available at http://www.jem.org/cgi/content/full/jem.20070204/DC1) (45). Therefore, this cell-specific TNF-α knockout strategy did not completely abrogate the production of TNF-α from all CD11b-expressing cells, further providing an explanation for the survival of these mice after a secondary challenge.
Our results also highlight that the fine-tuning of TNF-α production is crucial for efficient control of secondary bacterial growth. Although TNF-α is produced by several cell types including CD8+ T cells, this is the TNF-α derived from non–CD8+ T cells that is required for secondary protection (12). As mentioned before, 80–90% of TNF-α–secreting cells during secondary infection are MPCs, and depletion of CD8+ T cells or neutralization of CCL3 abrogated both the release of TNF-α by MPCs and mice resistance. This result, together with the lys-M experiments described above, strongly suggests that TNF-α produced by MPCs, but not by other cells, is critical for secondary protective immunity.
A recent study has identified an inflammatory subset of DCs that produce large amounts of TNF-α and are necessary for mice survival upon primary infection. These cells were designated Tip-DCs for TNF-α/iNOS-producing DCs (46). Tip-DCs and MPCs express the same DC-specific surface markers, and both produce iNOS that generates reactive nitric intermediates (RNIs). However, in contrast to Tip-DCs, MPCs express the macrophage marker F4/80, contain live and dead bacteria, and produce ROIs. It remains to be determined whether Tip-DCs and MPCs are related or not.
A previous study has suggested that RNIs are not necessary for bacterial clearance during a secondary response (47). Our results further show that ROI generation is the final bacteria killing effector mechanism required for secondary protection. In contrast, RNIs, but not ROIs, are necessary for bacterial clearance and mice survival during primary infection, (48). RNI depends on iNOS synthesis that is regulated at the transcriptional level (49). The generation of ROIs is dependent on phosphorylation of the cytosolic regulatory protein p47phox. This event leads to activation of the NADPH oxidase, a TNF-α–induced activity that is far more rapid than the regulation at the transcriptional level (50). All the mechanisms involved in this regulation loop are rapid, including secretion of CCL3 by memory CD8+ T cells, secretion of TNF-α by MPCs, phosphorylation of p47phox upon binding of TNF-α to its surface receptor, activation of NADPH oxidase, and ROI production. The ability of TNF-α to induce both MPCs and neutrophils to produce ROIs results in an amplification loop that allows rapid clearance of high doses of bacteria, a mechanism that does not occur in naive mice because of the lack of memory CD8+ T cells.
The aim of this study was to elucidate the molecular and cellular events that orchestrate the control of bacterial growth in secondary infected mice. However, the molecular signals that are required for the priming of fully functional memory CD8+ T cells remain to be identified. Although both secA2 − and actA − bacteria escape from phagocytic vacuoles, multiply in cytosol, and exhibit the same LD50, these mutants nevertheless exhibit distinct defects. ActA − bacteria cannot mobilize actin (17, 19). SecA2 − bacteria lack an ATPase that is critical for an auxiliary protein secretion system important for the secretion of at least 17 bacterial proteins (16). It is conceivable that egress in the cytosol of one or more of the SecA2-dependent proteins is required for the differentiation of bacteria-specific naive CD8+ T cells into fully functional memory cells.
The measure of cytolytic IFN-γ– and TNF-α–secreting activity of memory CD8+ T cells is widely used to predict the ability of an immunized individual to develop a protective immune response. Although these activities may accurately reflect the ability of the host to resist to a secondary challenge in some infectious diseases, this study suggests that it may not always be the case and that another effector activity such as CCL3 secretion could be an important marker to predict vaccine efficacy. Importantly, we have shown that it is also possible to induce, in an antigen-dependent fashion, a CCL3-mediated bystander killing of an unrelated pathogen, a mechanism that could potentially be used for therapeutic immunologically based protocols. In fact, some microbial infections cannot be treated with conventional medications anymore because the pathogens have become resistant to multiple drugs. For instance, this is the case for some strains of Mycobacterium tuberculosis or Staphylococcus aureus. If such patients were previously vaccinated with attenuated L. monocytogenes bacteria, it should be possible to specifically reactivate the CCL3-secreting memory CD8+ T cells to mediate the bystander elimination of multidrug-resistant pathogens without the need of any other medication. We believe that taking advantage of this mechanism may have important therapeutic implications for curative medicine.
MATERIALS AND METHODS
Mice
6–8-wk-old BALB/cByJ female mice (Janvier Laboratories) were used in all experiments unless otherwise indicated. p47phox−/− mice (36) were obtained from European Mouse Mutant Archive, and CCL3−/− (51) were obtained from The Jackson Laboratory. Aged-matched 129S2/SvPasCrl control mice were purchased from Charles River Laboratories. lys-M GFP-expressing knock-in mice (lys-M-GFP+/−) (45) were provided by T. Graf (Albert Einstein College of Medicine, New York, NY). L9.6 TCR transgenic mice have been described (2). p47phox −/−, CCL3−/−, lys-M–GFP+/−, and L9.6 mice were housed and bred in our specific pathogen-free animal facility. All mice were used in accordance with Animal Care and Use regional Côte d'Azur Committee procedures.
Bacteria and parasites
The L. monocytogenes 10403s WT strain was used in all experiments. The actA −, llo −, and secA2 − mutant strains were on the 10403s WT strain background and have been described. The WT, actA −, llo −, and secA2 − strains exhibit an LD50 of 3 × 104, 5 × 109, 107, and 107, respectively, in BALB/c mice (16, 19, 21). HKLM was prepared as described previously (2). All bacteria were prepared from clones grown from organs of infected mice. Stocks of bacteria were kept frozen at −80°C. DsRed-expressing L. major promastigotes (World Health Organization strain WHOM/IR/-/173) (52) were grown in M199 medium containing 20% FCS.
Infection of mice and measure of protective immunity and mice survival
For L. monocytogenes infections, bacteria were grown to a logarithmic phase (OD600 = 0.05–0.15) in broth heart infusion (BHI) medium (Sigma-Aldrich) diluted in PBS and injected i.v. into the lateral tail vein. In all experiments, mice were primary immunized with a 0.1 × LD50 of bacteria (3 × 103). Secondary infections were performed 1 mo later with 10 × LD50 of WT bacteria (3 × 105) except when specified otherwise. To measure bacterial titers in the spleen and liver, organs were harvested and dissociated on metal screens in 10 ml of 0.1% Triton X-100 (Sigma-Aldrich). Serial dilutions were performed in the same buffer, and 100 μl was plated onto BHI media plates. Mice survival was monitored twice a day.
For L. major infections, mice were injected with PBS or infected with 0.1 × LD50 of WT bacteria and infected i.v with 2 × 106 stationary phase promastigotes 3 wk later. Mice were secondary challenged after 2 wk with WT bacteria or LPS-matured DCs pulsed with the Listeria-derived LLO91-99 peptide and co-treated or not with anti-CCL3. To determine the parasite burden in the spleens, organs were harvested 48 h after challenge and dissociated on nylon screens in 5 ml of M199 medium containing 20% FCS, 100 μg/ml penicillin, and 100 U/ml streptomycin. Serial dilutions were performed in the same buffer, and cells were incubated in flat-bottom 96-well plates at 27°C for 3–6 d. The number of parasite was determined using a FACS Array (Becton Dickinson) to monitor the red fluorescence of the live parasite.
Antibodies and reagents
The following mAbs were purchased from BD Biosciences: anti–CD8α (53–6.7)-FITC, -PE, –peridinin chlorophyll protein (PcP), or -allophycocyanin (APC), anti–CD3ε-FITC (145-2C11), anti–Ly-6C–FITC (AL-21), anti–CD107a-FITC (1D4B), anti–CD11c-PE (HL3), anti–CD11b-PE, -PcP, or -APC (M1/70); CD80-PE (16-10A1), anti–CD86-PE (GL1), anti–I-Ad–PE (AMS-32.1), anti–CCR5-PE (C34-3448), anti–CD62L-APC (MEL-14), anti–CD127-APC (A7R34), anti–IFN-γ–APC (XMG1-2), anti–TNF-α–APC (MP6-XT22), and control rat IgG1 mAb. Anti–NOS-2 (M-19) polyclonal rabbit and anti–CCR-1 (C-20) antibodies were purchased from Santa Cruz Biotechnology, Inc. Difco Listeria O rabbit polyserum was purchased from Fischer. Goat and donkey anti–rabbit Alexa 647 and CFSE were purchased from Invitrogen. Goat polyclonal anti-CCL3 antibody and recombinant mouse CCL3 (rCCL3) was purchased from R&D Systems. Endotoxin content of 1 μg rCCL3 from Gram-negative bacteria was <1.2 pg/ml as determined using the Limulus Amebocyte Lysate QCL-1000 (Cambrex). PE-conjugated LLO91-99/H2-Kd tetramers were obtained from the National Institutes of Health (NIH) tetramer core facility.
Cell suspensions
Organs were cut in small pieces and incubated at 37°C for 20 min in HBSS medium (Invitrogen) containing 4,000 U/ml collagenase I (Invitrogen) and 0.1 mg/ml DNase I (Roche). Red blood cells were lysed for 2–3 min in 170 mM NH4Cl, 17 mM Tris HCl, pH 7.4. All steps of intracellular CCL3 staining as well as purification of CD8+ T cells for transfer experiments were performed in the presence of 2 μg/ml Golgi plug (BD Biosciences).
Antibody staining and flow cytometry
Cells were stained with the specified antibodies in 100 μl of PBS containing 0.5% BSA (FACS buffer). For intracellular staining, splenocytes were incubated at 37°C and 5% CO2 for 3–4 h in RPMI 1640 (Invitrogen) containing 5% FCS and 2 μg/ml Golgi Plug (BD Biosciences) with or without 5 × 108 HKLM/ml, 1 nM LLO91-99 (Mimotopes), or 1 μg/ml of recombinant mouse CCL3 (R&D Systems). Cells were incubated for 20 min on ice with the indicated cell surface marker mAbs, fixed in 1% paraformaldehyde FACS buffer for 20 min on ice, and permeabilized for 30 min in 1XPerm/Wash (BD Biosciences). For intracellular staining of TNF-α or IFN-γ, cells were incubated for 20 min on ice in FACS buffer containing anti–IFN-γ, anti–TNF-α, or control rat IgG1. For intracellular staining of iNOS or Listeria antigens, cells were incubated for 20 min on ice in FACS buffer containing anti-iNOS rabbit polyclonal, anti-Listeria Difco Listeria O rabbit polyserum, or control normal goat IgG, and staining was revealed using goat anti–rabbit Alexa 647 mAb. For intracellular staining of CCL3, cells were incubated for 20 min on ice in FACS buffer containing anti-CCL3 goat polyclonal antibodies, and staining was revealed using donkey anti–goat Alexa 647 mAbs. In all cases, cells were washed, fixed for 30 min in 1% paraformaldehyde FACS buffer, and analyzed on a FACSCalibur cytofluorometer (Becton Dickinson). When indicated, cells were sorted on a FACSVantage SE cell sorter (Becton Dickinson).
Assay for CCL3 secretion
Supernatants were analyzed for CCL3 content by ELISA using the DuoSet ELISA kit (R&D Systems). The sensitivity of the assay was 2 pg/ml.
In vivo killing assay
Spleen cells were pulsed or not with 1 nM LLO91-99 for 1 h at 37°C. Pulsed and unpulsed cells were incubated for 10 min at 37°C in PBS containing 5 or 0.5 μM CFSE, respectively. Cells were washed in PBS, and 107 of peptide-pulsed and 107 of unpulsed cells were co-injected i.v. into recipient mice. Mice were killed 15 h later, and spleen cells were analyzed by FACS. Cytolytic activity against peptide-pulsed target cells was determined by measuring the frequency of CFSEhigh and CFSElow cells and by calculating the percentage of specific lysis using the following formula: 100 − ([(% peptide pulsed in infected/% unpulsed in infected) / (% peptide pulsed in uninfected/% unpulsed in uninfected)] × 100) (53).
In vivo neutralization and depletion experiments
For neutralization of TNF-α, mice were injected with 1.5 mg of soluble p75 TNF-α receptor (Enbrel) i.p. and infected 1 d later. For superoxyde neutralization (MnTBAP; A.G. Scientific, Inc.), mice were injected with 10 μg/g of body weight (38) and infected 1 d later. For neutralization of CCL3, mice were treated with 75 μg anti-CCL3 or goat IgG control i.v at the time of the secondary infection and 24 h later. For depletion of CD8+ T cells, mice were injected three times daily with 100 μg of the anti-CD8β H-35 mAb or its control isotype i.p., and then infected 1 d after.
Cell transfer experiments
CD8+ T cell transfers.
WT or CCL3−/− mice were PBS injected or primary immunized with WT bacteria and secondary challenged 1 mo later with 3 × 105 WT bacteria. Mice were killed 6 h later, and splenic CD8+ T cells were negatively enriched using 5 μg/ml of homemade anti–MHC-II (MKD6), anti-CD4 (GK1.5), anti-B220 (RA3), and anti-CD19 (1D3) mAbs in 5% FCS RPMI 1640 containing 5% FCS and 2 μg/ml Golgi Plug. CD8+ T cells were further flow cell sorted, and 20 × 106 cells (∼40,000 LLO91-99/H2-Kd–specific CD8+ T cells) with a purity of >90% were transferred per naive WT or CCL3−/− recipients. Mice were subsequently treated with anti-CCL3 or control goat serum and infected with 3 × 105 or 5 × 104 WT bacteria.
Monocyte and neutrophil transfers.
Naive WT or p47phox−/− mice were killed, and spleen and bone marrow cells were first positively enriched using anti-CD11b–specific MACS beads (Miltenyi Biotec) according to the manufacturer's standard protocol and further flow cell sorted based on their expression of CD11b and Ly-6C cell surface markers, with monocytic-derived cells being CD11bmed/highLy-6Chigh and neutrophils being CD11bhighLy-6Cmed/high. PBS-injected or WT-immunized p47phox−/− mice were reconstituted with 4 × 105 MPCs (purity >87%) or 4 × 106 neutrophils (purity >93%). 1 d later, mice were treated with anti-CCL3 or control goat serum and infected with 3 × 105 WT bacteria.
DC transfers.
Spleen cells of naive BALB/c mice were positively enriched using anti-CD11c–specific MACS beads (Miltenyi Biotec) according to the manufacturer's standard protocol. For LPS stimulation, cells were plated at 106 cells/ml in RPMI 1640 medium containing 20% FCS supplemented with 0.1 μg/ml LPS (Sigma-Aldrich). 20 h later, cells were washed and pulsed with 1 μM of the LLO91-99 peptide for 1 h, and 20 × 106 of these peptide-pulsed mature DCs were transferred per recipient mouse.
Electron microscopy
Flow-sorted CD11bmed/highLy-6Chigh cells were fixed with 1.6% glutaraldehyde in 100 mM phosphate buffer, pH 7.5. Cells were incubated at 4°C in 100 mM cacodylate buffer, pH 7.0, containing 1% osmium tetroxyde. Cellular pellets were washed with distilled water and incubated for 2 h with 0.5% uranyl acetate buffer at room temperature in the dark. Cells were washed in water, and pellets were dehydrated in increasing acetone series and embedded in epoxy resins. Blocks were thin-sectioned using standard procedures and contrasted for the observation on an electron microscope (CM12; Philips).
Assay for the production of ROIs
5-10 × 106 splenocytes were incubated for 3 h at 37°C and 5% CO2 in 5% FCS RPMI 1640 with 5 × 108 HKLM/ml and 160 μM hydroethidine (Polysciences). Hydroethidine is oxidized by ROIs in red fluorescent ethidium bromure, which allows for the detection of ROI-producing cells. Cells were washed in FACS buffer and stained for expression of cell surface markers.
Statistical analysis
Statistical significance was calculated using an unpaired Mann-Whitney test and Instat software. All p-values of ≤0.05 were considered significant and are referred to as such in the text. In experiments for which we did not have n > 6 subjects to run a Mann-Whitney test, we have applied the rules of SE bars (54) and accordingly provided an estimated p-value for all panels shown in the study.
Online supplemental material
Fig. S1 shows the frequency of tetramer+CD127+CD62L− (TEM) and tetramer+CD127+CD62L+ (TCM) (± SE) among CD8+ T cells. Fig. S2 shows that CCL3 from CD8+ T cells is required for protection against secondary infection in the liver. Fig. S3 shows the survival kinetics of WT mice treated or not with anti–TNF-α or anti-CCL3 and of p47phox−/− mice during secondary infection. Fig. S4 shows that CCL3 from CD8+ T cells is required for secondary protective immunity in the spleen and liver (as in Fig. 4 D and Fig. S2), but data are expressed in the percentage of the maximum protective effect achieved by transferring WT CD8+ T cells from secondary infected animals. Fig. S5 demonstrates that the protection achieved by transferring WT CD8+ T cells increases when recipient animals are challenged with lower numbers of bacteria (5 × 104 instead of 3 × 105). Fig. S6 provides a phenotypic characterization of the TP-MPCs and shows that they are infected using electron microscopy. Fig. S7 shows the frequencies of TP-MPCs in the spleen and among total MPCs during secondary infection in mice primarily immunized with PBS or 0.1 × LD50 of the indicated bacteria. Fig. S8 monitors the numbers of ROI-producing MPCs and neutrophils in primary and secondary infected mice. Fig. S9 shows that the CCL3-producing memory CD8+ T cells promote ROI generation by phagocytic cells for secondary bacterial clearance in the liver of infected mice. Fig. S10 shows the number of granzyme B–expressing and IFN-γ–secreting CD8+ T cells in WT and p47phox−/− 129 mice after primary and secondary infection. Fig. S11 demonstrates that ROIs are required for mediating secondary protection against L. monocytogenes infection in WT mice treated with an O2 − scavenger before secondary challenge. Fig. S12 provides an analysis of lys-M expression in TP-MPCs during secondary infection. The online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070204/DC1.
Supplemental Material
Acknowledgments
We thank C. Schrike and N. Guy for their help with mice, E. Pamer for providing L9.6 TCR transgenic mice, T. Graf for providing the lys-M–GFP knock-in mice, D. Portnoy for the L. monocytogenes–targeted deletion mutants, O. Brocq and A. Bernard for Enbrel, O. Leo for the H35 anti-CD8β hybridoma, and T. Aebischer for the DsRed-expressing L. major strain. H2-Kd/LLO91-99 tetramer was obtained through the NIH Tetramer Facility.
This work was supported by grants from INSERM (Avenir), Human Frontier Science Program (CDA), Agence Nationale de la Recherche (ANR-IRAP-2005), and Fondation pour la Recherche Médicale (FRM). E. Narni-Mancinelli and L. Campisi are both recipients of a MENRT fellowship.
The authors have no conflicting financial interests.
Abbreviations used: HKLM, heat-killed Listeria monocytogenes; iNOS, inducible nitric oxide synthase; lys-M, lysozyme M; MPC, mononuclear phagocytic cell; RNI, reactive nitric intermediate; ROI, reactive oxygen intermediate; TEM, effector–memory; Tip-DC, TNF-α/iNOS-producing DC; TP-MPC, TNF-α–producing MPC.
References
- 1.Harty, J.T., A.R. Tvinnereim, and D.W. White. 2000. CD8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 18:275–308. [DOI] [PubMed] [Google Scholar]
- 2.Lauvau, G., S. Vijh, P. Kong, T. Horng, K. Kerksiek, N. Serbina, R.A. Tuma, and E.G.P. Am. 2001. Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science. 294:1735–1739. [DOI] [PubMed] [Google Scholar]
- 3.Mielke, M.E., G. Niedobitek, H. Stein, and H. Hahn. 1989. Acquired resistance to Listeria monocytogenes is mediated by Lyt-2+ T cells independently of the influx of monocytes into granulomatous lesions. J. Exp. Med. 170:589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badovinac, V.P., and J.T. Harty. 2000. Adaptive immunity and enhanced CD8+ T cell response to Listeria monocytogenes in the absence of perforin and IFN-gamma. J. Immunol. 164:6444–6452. [DOI] [PubMed] [Google Scholar]
- 5.Badovinac, V.P., A.R. Tvinnereim, and J.T. Harty. 2000. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science. 290:1354–1358. [DOI] [PubMed] [Google Scholar]
- 6.Harty, J.T., and M.J. Bevan. 1995. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 3:109–117. [DOI] [PubMed] [Google Scholar]
- 7.Jensen, E.R., A.A. Glass, W.R. Clark, E.J. Wing, J.F. Miller, and S.H. Gregory. 1998. Fas (CD95)-dependent cell-mediated immunity to Listeria monocytogenes. Infect. Immun. 66:4143–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grivennikov, S.I., A.V. Tumanov, D.J. Liepinsh, A.A. Kruglov, B.I. Marakusha, A.N. Shakhov, T. Murakami, L.N. Drutskaya, I. Forster, B.E. Clausen, et al. 2005. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity. 22:93–104. [DOI] [PubMed] [Google Scholar]
- 9.Neighbors, M., X. Xu, F.J. Barrat, S.R. Ruuls, T. Churakova, R. Debets, J.F. Bazan, R.A. Kastelein, J.S. Abrams, and A. O'Garra. 2001. A critical role for interleukin 18 in primary and memory effector responses to Listeria monocytogenes that extends beyond its effects on interferon γ production. J. Exp. Med. 194:343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samsom, J.N., J.A. Langermans, H.F. Savelkoul, and R. van Furth. 1995. Tumour necrosis factor, but not interferon-gamma, is essential for acquired resistance to Listeria monocytogenes during a secondary infection in mice. Immunology. 86:256–262. [PMC free article] [PubMed] [Google Scholar]
- 11.White, D.W., V.P. Badovinac, X. Fan, and J.T. Harty. 2000. Adaptive immunity against Listeria monocytogenes in the absence of type I tumor necrosis factor receptor p55. Infect. Immun. 68:4470–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White, D.W., V.P. Badovinac, G. Kollias, and J.T. Harty. 2000. Cutting edge: antilisterial activity of CD8+ T cells derived from TNF-deficient and TNF/perforin double-deficient mice. J. Immunol. 165:5–9. [DOI] [PubMed] [Google Scholar]
- 13.White, D.W., and J.T. Harty. 1998. Perforin-deficient CD8+ T cells provide immunity to Listeria monocytogenes by a mechanism that is independent of CD95 and IFN-gamma but requires TNF-alpha. J. Immunol. 160:898–905. [PubMed] [Google Scholar]
- 14.Cook, D.N., O. Smithies, R.M. Strieter, J.A. Frelinger, and J.S. Serody. 1999. CD8+ T cells are a biologically relevant source of macrophage inflammatory protein-1 alpha in vivo. J. Immunol. 162:5423–5428. [PubMed] [Google Scholar]
- 15.Messingham, K.A., V.P. Badovinac, and J.T. Harty. 2003. Deficient anti-listerial immunity in the absence of perforin can be restored by increasing memory CD8+ T cell numbers. J. Immunol. 171:4254–4262. [DOI] [PubMed] [Google Scholar]
- 16.Lenz, L.L., S. Mohammadi, A. Geissler, and D.A. Portnoy. 2003. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc. Natl. Acad. Sci. USA. 100:12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kocks, C., E. Gouin, M. Tabouret, P. Berche, H. Ohayon, and P. Cossart. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 68:521–531. [DOI] [PubMed] [Google Scholar]
- 18.Muraille, E., E. Narni-Mancinelli, P. Gounon, D. Bassand, N. Glaichenhaus, L.L. Lenz, and G. Lauvau. 2007. Cytosolic expression of SecA2 is a prerequisite for long-term protective immunity. Cell. Microbiol. 9:1445–1454. [DOI] [PubMed] [Google Scholar]
- 19.Goossens, P.L., and G. Milon. 1992. Induction of protective CD8+ T lymphocytes by an attenuated Listeria monocytogenes actA mutant. Int. Immunol. 4:1413–1418. [DOI] [PubMed] [Google Scholar]
- 20.von Koenig, C.H., H. Finger, and H. Hof. 1982. Failure of killed Listeria monocytogenes vaccine to produce protective immunity. Nature. 297:233–234. [DOI] [PubMed] [Google Scholar]
- 21.Berche, P., J.L. Gaillard, and P.J. Sansonetti. 1987. Intracellular growth of Listeria monocytogenes as a prerequisite for in vivo induction of T cell-mediated immunity. J. Immunol. 138:2266–2271. [PubMed] [Google Scholar]
- 22.Sallusto, F., D. Lenig, R. Forster, M. Lipp, and A. Lanzavecchia. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 401:708–712. [DOI] [PubMed] [Google Scholar]
- 23.Huster, K.M., V. Busch, M. Schiemann, K. Linkemann, K.M. Kerksiek, H. Wagner, and D.H. Busch. 2004. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc. Natl. Acad. Sci. USA. 101:5610–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaech, S.M., J.T. Tan, E.J. Wherry, B.T. Konieczny, C.D. Surh, and R. Ahmed. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198. [DOI] [PubMed] [Google Scholar]
- 25.Busch, D.H., I.M. Pilip, S. Vijh, and E.G.P. Am. 1998. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 8:353–362. [DOI] [PubMed] [Google Scholar]
- 26.Huster, K.M., M. Koffler, C. Stemberger, M. Schiemann, H. Wagner, and D.H. Busch. 2006. Unidirectional development of CD8+ central memory T cells into protective Listeria-specific effector memory T cells. Eur. J. Immunol. 36:1453–1464. [DOI] [PubMed] [Google Scholar]
- 27.Betts, M.R., J.M. Brenchley, D.A. Price, S.C. De Rosa, D.C. Douek, M. Roederer, and R.A. Koup. 2003. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods. 281:65–78. [DOI] [PubMed] [Google Scholar]
- 28.Mackaness, G.B. 1962. Cellular resistance to infection. J. Exp. Med. 116:381–406. [PubMed] [Google Scholar]
- 29.Salazar-Mather, T.P., J.S. Orange, and C.A. Biron. 1998. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1α (MIP-1α)–dependent pathways. J. Exp. Med. 187:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castellino, F., A.Y. Huang, G. Altan-Bonnet, S. Stoll, C. Scheinecker, and R.N. Germain. 2006. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 440:890–895. [DOI] [PubMed] [Google Scholar]
- 31.Fahey, T.J., III, K.J. Tracey, P. Tekamp-Olson, L.S. Cousens, W.G. Jones, G.T. Shires, A. Cerami, and B. Sherry. 1992. Macrophage inflammatory protein 1 modulates macrophage function. J. Immunol. 148:2764–2769. [PubMed] [Google Scholar]
- 32.Dorner, B.G., A. Scheffold, M.S. Rolph, M.B. Huser, S.H. Kaufmann, A. Radbruch, I.E. Flesch, and R.A. Kroczek. 2002. MIP-1alpha, MIP-1beta, RANTES, and ATAC/lymphotactin function together with IFN-gamma as type 1 cytokines. Proc. Natl. Acad. Sci. USA. 99:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maurer, M., and E. von Stebut. 2004. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 36:1882–1886. [DOI] [PubMed] [Google Scholar]
- 34.Muller, M., R. Althaus, D. Frohlich, K. Frei, and H.P. Eugster. 1999. Reduced antilisterial activity of TNF-deficient bone marrow-derived macrophages is due to impaired superoxide production. Eur. J. Immunol. 29:3089–3097. [DOI] [PubMed] [Google Scholar]
- 35.Goossens, V., J. Grooten, K. De Vos, and W. Fiers. 1995. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc. Natl. Acad. Sci. USA. 92:8115–8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson, S.H., J.I. Gallin, and S.M. Holland. 1995. The p47phox mouse knock-out model of chronic granulomatous disease. J. Exp. Med. 182:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Endres, R., A. Luz, H. Schulze, H. Neubauer, A. Futterer, S.M. Holland, H. Wagner, and K. Pfeffer. 1997. Listeriosis in p47(phox−/−) and TRp55−/− mice: protection despite absence of ROI and susceptibility despite presence of RNI. Immunity. 7:419–432. [DOI] [PubMed] [Google Scholar]
- 38.Houstis, N., E.D. Rosen, and E.S. Lander. 2006. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 440:944–948. [DOI] [PubMed] [Google Scholar]
- 39.Murray, H.W. 1982. Cell-mediated immune response in experimental visceral leishmaniasis. II. Oxygen-dependent killing of intracellular Leishmania donovani amastigotes. J. Immunol. 129:351–357. [PubMed] [Google Scholar]
- 40.Xanthoulea, S., M. Pasparakis, S. Kousteni, C. Brakebusch, D. Wallach, J. Bauer, H. Lassmann, and G. Kollias. 2004. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 200:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Theofilopoulos, A.N., R. Baccala, B. Beutler, and D.H. Kono. 2005. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 23:307–336. [DOI] [PubMed] [Google Scholar]
- 42.Langenkamp, A., M. Messi, A. Lanzavecchia, and F. Sallusto. 2000. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 1:311–316. [DOI] [PubMed] [Google Scholar]
- 43.Marino, M.W., A. Dunn, D. Grail, M. Inglese, Y. Noguchi, E. Richards, A. Jungbluth, H. Wada, M. Moore, B. Williamson, et al. 1997. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA. 94:8093–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasparakis, M., L. Alexopoulou, V. Episkopou, and G. Kollias. 1996. Immune and inflammatory responses in TNF α–deficient mice: a critical requirement for TNF α in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 184:1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faust, N., F. Varas, L.M. Kelly, S. Heck, and T. Graf. 2000. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 96:719–726. [PubMed] [Google Scholar]
- 46.Serbina, N.V., W. Kuziel, R. Flavell, S. Akira, B. Rollins, and E.G.P. Am. 2003. Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity. 19:891–901. [DOI] [PubMed] [Google Scholar]
- 47.Samsom, J.N., J.A. Langermans, P.H. Groeneveld, and R. van Furth. 1996. Acquired resistance against a secondary infection with Listeria monocytogenes in mice is not dependent on reactive nitrogen intermediates. Infect. Immun. 64:1197–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiloh, M.U., J.D. MacMicking, S. Nicholson, J.E. Brause, S. Potter, M. Marino, F. Fang, M. Dinauer, and C. Nathan. 1999. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 10:29–38. [DOI] [PubMed] [Google Scholar]
- 49.Xie, Q.W., H.J. Cho, J. Calaycay, R.A. Mumford, K.M. Swiderek, T.D. Lee, A. Ding, T. Troso, and C. Nathan. 1992. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. 256:225–228. [DOI] [PubMed] [Google Scholar]
- 50.Dang, P.M., A. Stensballe, T. Boussetta, H. Raad, C. Dewas, Y. Kroviarski, G. Hayem, O.N. Jensen, M.A. Gougerot-Pocidalo, and J. El-Benna. 2006. A specific p47-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Invest. 116:2033–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cook, D.N., M.A. Beck, T.M. Coffman, S.L. Kirby, J.F. Sheridan, I.B. Pragnell, and O. Smithies. 1995. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science. 269:1583–1585. [DOI] [PubMed] [Google Scholar]
- 52.Sorensen, M., C. Lippuner, T. Kaiser, A. Misslitz, T. Aebischer, and D. Bumann. 2003. Rapidly maturing red fluorescent protein variants with strongly enhanced brightness in bacteria. FEBS Lett. 552:110–114. [DOI] [PubMed] [Google Scholar]
- 53.Aizu, K., W. Li, T. Yajima, T. Arai, K. Shimoda, Y. Nimura, and Y. Yoshikai. 2006. An important role of Tyk2 in APC function of dendritic cells for priming CD8+ T cells producing IFN-gamma. Eur. J. Immunol. 36:3060–3070. [DOI] [PubMed] [Google Scholar]
- 54.Cumming, G., F. Fidler, and D.L. Vaux. 2007. Error bars in experimental biology. J. Cell Biol. 177:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}