

Abstract
The tumour suppressor PTEN is a PtdIns(3,4,5)P3 phosphatase that regulates many cellular processes through direct antagonism of PI 3-kinase signalling. Here we show that oxidative stress activates PI 3-kinase-dependent signalling via the inactivation of PTEN. We use two assay systems to show that cellular PTEN phosphatase activity is inhibited by oxidative stress induced by 1 mM hydrogen peroxide. PTEN inactivation by oxidative stress also causes an increase in cellular PtdIns(3,4,5)P3 levels and activation of the downstream PtdIns(3,4,5)P3 target, PKB/Akt, that does not occur in cells lacking PTEN. We then show that endogenous oxidant production in RAW264.7 macrophages inactivates a fraction of the cellular PTEN, and that this is associated with an oxidant-dependent activation of downstream signalling. These results show that oxidants, including those produced by cells, can activate downstream signalling via the inactivation of PTEN. This demonstrates a novel mechanism of regulation of the activity of this important tumour suppressor and the signalling pathways it regulates. These results may have significant implications for the many cellular processes in which PtdIns(3,4,5)P3 and oxidants are produced concurrently.
Keywords: oxidative stress/PTEN/phosphatase/phosphoinositide 3-kinase/redox signalling
Introduction
The phosphoinositide 3-kinase (PI 3-kinase) cellular signalling pathway plays a pivotal role in controlling numerous biological processes, including cellular survival, proliferation, growth and motility and is deregulated in numerous human tumour types (Cantrell, 2001; Katso et al., 2001; Vanhaesebroeck et al., 2001; Cantley, 2002). The pathway is characterized by the stimulated production of the second messenger, phosphatidylinositol (3,4,5) trisphosphate [PtdIns(3,4,5)P3] by Class I PI 3-kinase enzymes. PtdIns(3,4,5)P3 activates downstream signalling through proteins able specifically to recognize and to bind this lipid. The importance of deregulation of this pathway in tumour development was highlighted by the demonstration that the tumour suppressor, PTEN, is a PtdIns(3,4,5)P3 3-phosphatase, and in many cell types may mediate the principle route of metabolism of this signalling molecule (Maehama et al., 2001; Simpson and Parsons, 2001; Leslie and Downes, 2002). Thus, PTEN can inhibit entry into the cell cycle, metastasis and cell motility, promote apoptosis, and reduce cell growth and size (Furnari et al., 1998; Stambolic et al., 1998; Tamura et al., 1998; Sun et al., 1999; Backman et al., 2002; Davies et al., 2002). However, despite intensive research, a clear picture is yet to emerge of how cells regulate the activity of PTEN. The abundance of PTEN can be regulated transcriptionally (Patel et al., 2001; Virolle et al., 2001) and through phosphorylation-dependent modulation of protein stability (Vazquez et al., 2000; Torres and Pulido, 2001). PTEN can also be recruited into protein complexes through interaction with PDZ domain-containing proteins (Wu,X. et al., 2000; Wu,Y. et al., 2000) and may be selectively recruited onto cellular membrane surfaces that display a more acidic character, such as the inner leaflet of the plasma membrane (Das et al., 2003; McConnachie et al., 2003). However, the significance of much of this data with regard to the cellular regulation of the PI 3-kinase pathway and downstream processes is currently unclear (Leslie et al., 2001; Birle et al., 2002).
It has become clear that reactive oxygen species (ROS) are not simply damaging by-products of cellular metabolism, but also play important regulatory roles in many cellular processes (Finkel, 2000; Droge, 2002). For example, the stimulated production of oxidants appears to play a crucial role in the mitogenic response to many growth factors (Chen et al., 1995; Sundaresan et al., 1995; Bae et al., 1997). Recent work has indicated that members of the Protein Tyrosine Phosphatase family, which include the lipid phosphatase, PTEN, can be physiologically regulated through reversible oxidation and inactivation (Denu and Tanner, 1998; Lee et al., 1998; Meng et al., 2002). These enzymes contain a critical cysteine residue in the active site that must be in a reduced state in order to participate in catalysis (Denu and Tanner, 1998; Tonks and Neel, 2001). Consistent with this, PTEN was found to require reducing conditions for optimal activity in vitro (Maehama and Dixon, 1998; Myers et al., 1998). More recently, evidence has shown that inactivation by oxidation might represent a physiological mechanism of regulation of this class of enzymes, not only by oxidative stress, but by ROS produced in other cellular circumstances, including stimulation of cells by growth factors (Meng et al., 2002). A recent study extended these proposals to include PTEN, showing that oxidation of this enzyme in vitro by hydrogen peroxide (H2O2) caused inactivation and the formation of a disulfide bond between the active site cysteine (C124), and another nearby cysteine (C71). This study also showed that H2O2, either in vitro, or in cultured cells caused a reversible shift in the electrophoretic mobility of PTEN consistent with its oxidation (Lee et al., 2002), but did not investigate effects on downstream signalling. Here we show for the first time direct evidence for the oxidative inactivation of PTEN in cells, by both exogenous and endogenously produced oxidants. We also show that oxidants cause an increase in the levels of PtdIns(3,4,5)P3 and activate downstream signalling, and that these effects do not occur in cells lacking PTEN.
Results
PTEN is sensitive to oxidative inactivation in vitro
PtdIns(3,4,5)P3 is believed to be metabolized largely by two distinct cellular pathways; dephosphorylation of the 3 position by PTEN, to produce PtdIns(4,5)P2, or dephosphorylation of the 5 position by members of a family of 5-phosphatases, to produce PtdIns(3,4)P2. We therefore chose to analyse the sensitivity to oxidation of human PTEN by H2O2 in vitro in comparison with human SHIP-2, a structurally unrelated PtdIns(3,4,5)P3 5-phosphatase that uses a different mechanism of catalysis (Figure 1). These studies revealed that the sensitivity of PTEN to oxidation is such that washing the enzyme in ambient conditions in buffers lacking added reducing or oxidizing agents causes substantial loss of activity, relative to the full activity recovered after washing in reducing conditions (5 mM DTT). Addition of H2O2, even at 10 µM, caused an almost complete loss of activity. In contrast, SHIP-2 still displayed strong activity in the presence of 10 mM H2O2. The apparent sensitivity of PTEN to oxidation in vitro (Figure 1A) is somewhat conflicting with data presented by Lee et al. (2002), showing that a 50% reduction in PTEN phosphatase activity was caused by ∼200 µM H2O2. Although the exact redox potential is not known in these experiments, we obtained similar results to their data when directly diluting PTEN enzyme that is stored in buffers containing reducing agents (data not shown), suggesting that the sensitivity of PTEN to oxidation may be underestimated in these latter experiments through reaction of applied oxidants with reducing agents in these storage buffers.

Fig. 1. PTEN, but not SHIP-2, is highly sensitive to oxidative inactivation in vitro. Recombinant PTEN (A) and SHIP-2 (B) were separately purified by affinity chromatography and immunoprecipitation respectively. Enzymes were then washed in ambient conditions lacking additional reducing or oxidizing agents, and then assayed in the presence of either 5 mM DTT, no additional reducing or oxidizing agents, or of different concentrations of H2O2. Data in (A) show d.p.m. released labelled phosphate, and in (B) show production of labelled PtdIns(3,4)P2 determined by densitometry. Presented data points represent the mean of assays performed in duplicate and the range of these duplicates. The measured activity using boiled enzyme is presented as a negative control.
Oxidative stress inactivates cellular PTEN
In order to analyse the regulation of PTEN by oxidation in cells, we adapted a novel assay described by Meng et al. (2002). This assay uses lysis buffers containing iodoacetic acid (IAA) irreversibly to alkylate cysteine residues in cellular proteins and is represented diagrammatically in Figure 2A. Alkylation of the active site cysteine residue of PTP family members results in irreversible inactivation. However, oxidized cysteine residues are protected from alkylation and, since their oxidation is reversible, they can be recovered in the absence of IAA and assayed. Thus, the resultant activity data represents the proportion of the phosphatase that is oxidized (and inactive) at the moment of lysis (Meng et al., 2002). The following initial experiments supported the use of this assay (data not shown). Firstly, regardless of cellular oxidative stresses or H2O2 addition after cell lysis, in the absence of alkylating agents, if PTEN immunoprecipitates were washed and assayed in reducing conditions, the same activity was recovered as control samples, indicating that in these experiments oxidation is reversible. Secondly, if IAA was included in a reducing lysis buffer (5 mM DTT), no detectable activity was recovered, indicating alkylation. However, if IAA was included in an oxidizing lysis buffer (1 mM H2O2), and immunoprecipitated enzyme was later reduced in the absence of IAA, substantial activity was recovered, indicating that reversible oxidation can protect PTEN from alkylation.

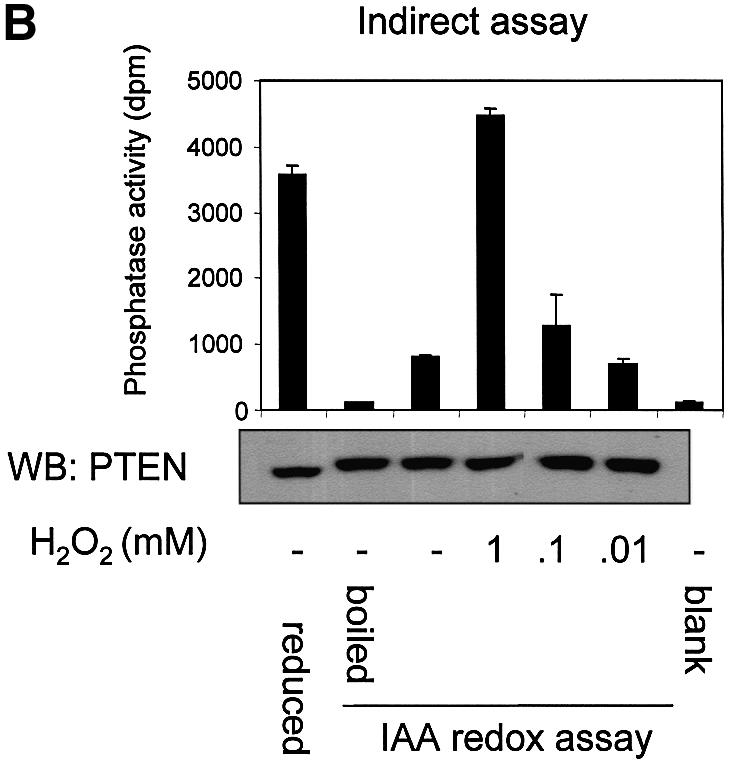

Fig. 2. Cellular PTEN is inactivated by oxidative stress. (A) The indirect alkylation method used to analyse the effects of oxidative stress on PTEN activity is shown. It is described in detail in the text and Materials and methods. Iodoacetic acid is abbreviated to IAA and immunoprecipitation to IP. The method causes PTEN that is oxidized and inactive at the moment of lysis to be recovered as activity. Reduced active cellular enzyme is alkylated and this activity is lost. This method is used in panel B. (B) For indirect redox analysis of PTEN, Swiss 3T3 cells were treated for 10 min with the indicated concentrations of H2O2 before being lysed in buffer containing the alkylating agent IAA. PTEN was immunoprecipitated and IAA washed away before PTEN was assayed in reducing conditions. Control cells were also lysed in buffer lacking alkylating agent, but with 5 mM DTT reducing agent and immunoprecipitated, washed and assayed in reducing conditions. A fraction of the first sample from each condition was analysed by western blotting (WB) using a different antibody raised against PTEN. (C) Direct analysis of PTEN activity. Swiss 3T3 cells were treated with or without 1 mM H2O2 for 10 min. Cells were then lysed, and endogenous PTEN was then immunoprecipitated and assayed, all in anaerobic conditions in the absence of additional reducing or oxidizing agents. As a positive control 5 µg of alkaline phosphatase (AP) was used. A fraction of each PTEN IP sample was analysed by western blotting using a different antibody raised against PTEN. Data is presented as mean activity (d.p.m. released phosphate) from duplicate samples ± the range of these duplicates, with the exception of the PTEN IPs from (C) which are derived from triplicate samples.
We applied this assay initially to analyse the oxidation of endogenous PTEN in cultured Swiss 3T3 fibroblasts exposed to oxidative stress induced by H2O2. In these experiments, very little PTEN activity was recovered from control cells in normal medium, indicating that most of the enzyme was in the reduced (active) state and was alkylated by IAA upon cell lysis. However, 1 mM H2O2 added to the medium was found to protect almost all cellular PTEN from alkylation, and allow its later recovery as active enzyme, indicating that PTEN was mostly oxidized in these cellular conditions (Figure 2B). In further analyses of the sensitivity of PTEN to exogenous H2O2, a steep response curve was evident, with a 10 min incubation in 1 mM H2O2 protecting most cellular PTEN from alkylation, whilst the protection induced by 100 µM H2O2 was barely detectable (Figure 2B). These experiments consistently showed a small decrease in the electrophoretic mobility of PTEN from lysates treated with IAA. It seems likely that this represents alkylation of the free cysteine residues of PTEN, as PTEN contains 10 cysteines including C71 and C124, and the gradient gels used are able to detect mobility differences of several hundred Daltons.
As a verification of the results from this indirect analysis of PTEN oxidative inactivation, cellular PTEN activity was immunoprecipitated and assayed under anaerobic conditions in the absence of additional reducing or oxidizing agents. This work showed that robust PTEN activity could be recovered in these conditions and that this activity was almost completely abolished by treatment of cells with 1 mM H2O2 for 10 min (Figure 2C). This result agrees closely with that found through oxidative protection from alkylation of PTEN (Figure 2B), and strongly validates the adapted method of Meng et al. (2002).
The activation of the PI 3-kinase pathway by oxidative stress requires PTEN
We have shown that PTEN can be inactivated by oxidative stress. In order to investigate whether this inactivation leads to an activation of downstream PI 3-kinase-dependent signalling, we chose first to analyse cellular phosphoinositide lipid levels. Therefore, we investigated the effects of oxidative stress on the levels of PtdIns(3,4,5)P3 and PtdIns(3,4)P2 in the PTEN-null glioblastoma cell line U87MG, with and without exogenous expression of wild-type PTEN (Figure 3A and C), or with expression of wild-type PTEN, or phosphatase-dead PTEN C124S (Figure 3B), using a viral delivery system. Cells were also treated with and without platelet-derived growth factor (PDGF), which is known to cause a strong activation of PI 3-kinase in these cells. These experiments showed, as would be expected, that in the absence of H2O2, wild-type PTEN expression reduced PtdIns(3,4,5)P3 levels. How ever, in all cases, the PtdIns(3,4,5)P3 levels of cells exposed to 1 mM H2O2 was similar in cells expressing wild-type PTEN compared with those lacking the enzyme activity, suggesting that H2O2 treatment oxidizes and inactivates any expressed PTEN. Most significantly however, these data also showed that in cells expressing PTEN, exposure to H2O2 led to a 3–4 fold increase in the levels of basal PtdIns(3,4,5)P3, whereas in cells lacking PTEN activity, exposure to H2O2 did not significantly change PtdIns(3,4,5)P3 levels. This strongly suggests that the principle mechanism of H2O2-induced PtdIns(3,4,5)P3 increase, is through oxidative inhibition of PTEN. In contrast, levels of PtdIns(3,4)P2 were greatly increased by oxidative stress regardless of cellular PTEN status. This suggests that other targets of oxidative stress exist that can regulate levels of this lipid, perhaps other oxidant sensitive PtdIns(3,4)P2 phosphatases.



Fig. 3. Oxidative stress increases cellular PtdIns(3,4,5)P3 levels only in cells expressing wild-type PTEN. Cellular levels of PtdIns(3,4,5)P3 (A and B) and PtdIns(3,4)P2 (C) were analysed in U87MG cells ± PTEN activity, stimulated with PDGF and/or H2O2. PTEN-null U87MG cells were labelled with [3H]inositol for 48 h in inositol-free culture medium. For the last 24 h of this incubation, cells were either left uninfected or infected with virus expressing wild-type GFP–PTEN (A and C), or infected with viruses expressing phosphatase-dead GFP–PTEN C124S or wild-type GFP–PTEN (B). Some cells were then stimulated as shown with 1 mM H2O2 or 50 ng/ml PDGF for 10 min before lysis. Cellular phosphoinositides were then purified, deacylated and analysed by HPLC. Data are presented as the mean of duplicate samples plus the range of these duplicates, with data for (A) and (C) being derived from the one experiment. These experiments were performed three times with similar results. The percentage of label incorporated into each lipid in unstimulated uninfected or C124S-infected cells corresponds to 1641, 1317 and 1462 d.p.m. above background in (A), (B) and (C), respectively.
To further analyse the activation of downstream PI 3-kinase-dependent signalling by oxidative stress, experiments were conducted investigating the activity and phosphorylation of the PtdIns(3,4,5)P3-dependent kinase, protein kinase B (PKB, also known as Akt) in the presence and absence of PTEN. These experiments showed that as with the PtdIns(3,4,5)P3 measurements, in cells lacking PTEN, PKB activity was not increased by oxidative stress, but was increased by PDGF. When PTEN was expressed in these cells, oxidative stress was then able to cause a substantial increase in PKB activity (Figure 4). In these experiments, a small decrease in PKB serine 473 phosphorylation was seen in response to H2O2 treatment, although a similar effect was not clearly evident in PKB kinase activity and its significance is unclear. These data indicate that oxidative stress is able to activate PKB only in cells expressing PTEN and therefore that this activation is mediated at least in part through oxidative inactivation of this phosphatase.

Fig. 4. Oxidative stress activates cellular PKB only in cells expressing PTEN. PTEN-null U87MG cells growing at low cell density were either left uninfected or infected with viruses encoding wild-type GFP–PTEN for 24 h. Cells were then stimulated with 1 mM H2O2 or 50 ng/ml PDGF for 10 min before cell lysis and determination of the activity (A) and phosphorylation (B) of PKB/Akt. (A) After lysis, cellular PKB was immunoprecipitated and assayed in vitro. Data is presented as the mean + SD d.p.m. of labelled phosphate incorporated into peptide substrate from triplicate samples. (B) After lysis, the phosphorylation of PKB was analysed by western blotting using antibodies specific for total PKB and for phosphoserine-473 PKB. These experiments were performed three times with similar results.
These data show by two distinct approaches, that cellular PTEN is inactivated by oxidative stress. Additionally, analysis of PtdIns(3,4,5)P3 levels and downstream signalling are consistent with the oxidative inactivation of PTEN and indicate that oxidative stress increases PtdIns(3,4,5)P3 levels and activates downstream signalling only in cells that express PTEN. This indicates that oxidative stress is able to regulate PI 3-kinase-dependent signalling through inactivation of PTEN.
Physiological redox regulation of PTEN in macrophages
It has become clear that H2O2 and other ROS are produced endogenously in many cell types in diverse processes (Finkel, 2000; Droge, 2002). It has also been proposed that ROS are active cellular signalling molecules (Finkel, 1998, 2000). In order to investigate the possibility that PTEN is inactivated by the endogenous production of ROS, we performed an analysis in the murine macrophage cell line, RAW264.7, in which the stimulated production of H2O2 and other ROS by the phagocytic NADPH oxidase complex has been well studied (Ahmed et al., 1997; Brune et al., 1997; Han et al., 2001; Pfeiffer et al., 2001). Significantly, it is known that the phagocytic NADPH oxidase complex can be activated by PtdIns(3,4,5)P3-dependent signals (Babior, 1999; Welch et al., 2002). This potential co-localization of the substrate of PTEN with an oxidant producing complex suggests a possible model for the localized inhibition of PTEN protecting a functionally significant pool of PtdIns(3,4,5)P3.
In RAW macrophages, the acute stimulation of oxidant production by lipopolysaccharide (LPS) and phorbol 12-myristate 13-acetate (PMA) has been described (Ahmed et al., 1997; Brune et al., 1997; Pfeiffer et al., 2001). It has also been shown that LPS treatment leads to the activation of the PtdIns(3,4,5)P3-regulated kinase PKB/Akt (Salh et al., 1998; Monick et al., 2001). Using the oxidant-sensitive fluorophore dichlorofluorescin diacetate, we were able to verify that LPS or PMA, but not insulin, rapidly induced the production of cellular oxidants in RAW264.7 macrophages (data not shown). Therefore, we investigated the oxidation of PTEN in RAW macrophages using the method described above and in Figure 2A. These experiments established that combined stimulation with LPS and PMA for 10 or 30 min induced a significant increase in the fraction of endogenous cellular PTEN protected from experimental alkylation. This was the case in cells both with and without 24 h priming with interferon γ (Figure 5A and B). The reliance of this effect upon cellular oxidant production was investigated using the cell permeant antioxidant N-acetyl cysteine (NAC, 10 mM) and a frequently used inhibitor of NADPH oxidase, diphenyleneiodonium chloride (DPI, 10 µM). Both NAC and DPI interfered with the protection of PTEN from alkylation, again indicating that this reflects oxidation (Figure 5D). Since stimulation did not affect the recovered activity of PTEN in reducing conditions (Figure 5C), these data indicate that these stimuli caused the oxidation and inactivation of a fraction of cellular PTEN that correlated with increased downstream signalling, indicated by phosphorylation of PKB (Figure 5E). The data correspond to an increase in the oxidized inactive fraction of cellular PTEN from ∼5% in unstimulated cells to ∼16% in cells stimulated for 10 min. We also found that stimulation with LPS or PMA alone appeared to result in a rise in the oxidized fraction of cellular PTEN, although this increase was not always statistically significant (LPS, 0 out of 2 experiments significant, PMA 1 out of 2) (data not shown). H2O2 (1 mM) was also found to lead to the oxidation of most cellular PTEN, and substantially to activate PKB in RAW macrophages (data not shown and Figure 6). We also assessed the oxidation of PTEN directly using a method previously used to analyse the p53 protein (Wu and Momand, 1998; Seo et al., 2002) that relies upon an initial cysteine-directed alkylation of proteins in a cell lysate, subsequent reduction, and second treatment with a biotinlyated alkylating agent. This allows the analysis of cysteine residues that were protected in an oxidized form in the initial lysate. This method also showed that in the RAW264.7 macrophages, H2O2 treatment and to a lesser extent stimulation with LPS and PMA lead to a substantial detection of oxidized PTEN (Figure 5F).



Fig. 5. Inactivation of cellular PTEN by stimulation of RAW264.7 macrophages. RAW264.7 macrophages were either grown in normal medium (A), or primed with 100 ng/ml interferon γ for 24 h (B), before some cells were stimulated with 100 ng/ml LPS and 1 µM PMA for the indicated times. Cellular PTEN was then immunoprecipitated in the presence of the alkylating agent IAA (as Figure 2A and B), and the fraction of cellular PTEN protected from alkylation by oxidation was determined. Data in (A) are presented as the mean of five samples ± the SEM. This experiment was performed three times with similar results. Data in (B) are presented as the mean of six samples ± SEM. This experiment was performed on three occasions with similar results. In (A) and (B), the mean fully reduced control PTEN activity were 3362 and 5515 d.p.m. respectively. (C) RAW264.7 macrophages were also stimulated for 10 min with 100 ng/ml LPS alone or 100 ng/ml LPS and 1 µM PMA, and subsequently cells were lysed and PTEN immunoprecipitated and assayed all in reducing conditions in the absence of alkylating agents. (D) The oxidant dependence of the protection of PTEN from alkylation in stimulated macrophages (as panel A) was investigated. The stimulated production of ROS in RAW macrophages was antagonized by a 5 min pre-treatment with either 10 µM DPI or 10 mM NAC before stimulation with 100 ng/ml LPS and 1 µM PMA for 10 min. Cells were then lysed and the protection of PTEN from alkylation was investigated as above (Figures 2 and 5A and B). Data are presented as the mean of three samples ± SEM. (E) PKB phosphorylation in lysates used for the investigation of the oxidation of cellular PTEN (see panels A and B) was analysed in duplicate samples by Western blotting using antibodies specific for phosphoserine-473 PKB and total PKB. The presence of the alkylating agent IAA in the lysis buffer did not appear to interfere with this analysis. (F) The oxidation of PTEN was analysed using a biotinlyated alkylating agent as described in Materials and methods. RAW264.7 macrophages were treated with or without either 1 mM H2O2 or with 100 ng/ml LPS and 1 µM PMA. Cells were lysed in the presence of alkylating agent and proteins sequentially reduced and alkyated with a second biotinylated alkylating agent. PTEN was then immunoprecipitated and analysed either by western blotting with different antibodies against the protein, or with streptavidin–HRP.

Fig. 6. Stimulation of RAW264.7 macrophages induces an oxidant-dependent activation of PKB. RAW264.7 macrophages were left untreated (–) or pre-treated for 5 min with 10 mM NAC (N) or DPI (D). Cells were then left untreated (control) or stimulated with 100 ng/ml LPS, 1 µM PMA, 10 µM insulin or 1 mM H2O2, each for 10 min. Cells were lysed and the activity state of cellular PKB was analysed using an in vitro kinase assay of immunoprecipitated PKB (A), or PKB phosphorylation analysed using western blotting with antibodies specific for phosphoserine-473 PKB, total PKB and PTEN (B). Data in (A) are presented as the mean activity ± SEM from a minimum of three samples. These experiments were performed three times with similar results.
Experiments measuring PtdIns(3,4,5)P3 levels in RAW macrophages proved unsuccessful, as levels of this lipid appeared to be unusually low (<0.001% of that of phosphatidylinositol in pilot experiments, N.Leslie and S.Safrany unpublished data). Therefore, PKB activation was analysed in these cells, and the reliance of this activation upon oxidant production investigated using NAC and DPI, as described above. Pilot experiments showed that in comparison with other cell types RAW cells express PKBγ and low levels of PKBα that can be activated by cellular stimulation with a number of stimuli including LPS, PMA, interferon γ, H2O2 and insulin, the latter possibly acting through an IGF1 receptor. While stimulation with LPS, PMA or insulin all increased cellular PKB activity several fold, the activation by LPS and PMA was significantly inhibited by either NAC or DPI, indicating a requirement for cellular oxidant production in the activation of PKB by these stimuli (Figure 6). In contrast, the activation of PKB by insulin was not significantly affected. These data suggest that LPS and PMA both activate PKB by an oxidant-dependent mechanism, but insulin does so by an oxidant-independent pathway. The extent of the oxidant-independent activation of PKB might be expected to indicate the relative importance of stimulated production of PtdIns(3, 4,5)P3 by PI 3-kinase, agreeing with previous data that insulin and LPS both activate PI 3-kinase. These data indicate that the full activation of PKB in macrophages by LPS or PMA is dependent upon cellular oxidants, and supports the observed oxidative inhibition of PTEN as a mechanism that plays a role in the activation of PKB.
Discussion
Here we show for the first time that oxidative stress can inactivate the PTEN tumour suppressor phosphatase in cells, and present evidence that this inactivation leads to an increased level of cellular PtdIns(3,4,5)P3 and the activation of signalling pathways downstream of this lipid second messenger. We also show that the endogenous production of cellular oxidants can inactivate a fraction of cellular PTEN, and that this correlates with an oxidant-dependent activation of downstream signalling. This is significant, as this is the first identification of a mechanism of acute physiological regulation of the activity of this important tumour suppressor and also provides a novel route of activation of PtdIns(3,4,5)P3-dependent signalling, not by activation of PI 3-kinase, but by inhibition of PTEN.
In previous work and that presented here we and others have found that oxidative stress increases cellular PtdIns(3,4,5)P3 levels and PKB activity. Significantly, we show here that these effects are only seen in cells expressing PTEN (either endogenous or recombinantly expressed). Our data from U87MG cells shows that these PTEN-null cells do not activate PKB/Akt in response to oxidative stress. However, when PTEN is expressed, 10 min exposure to 1 mM H2O2 stimulates an increase in PKB activity of ∼4-fold. This agrees well with the ∼5-fold activation of PKB seen in Swiss 3T3 fibroblasts and RAW macrophages that express endogenous PTEN (van der Kaay et al., 1999). These data indicate that the predominant activation of downstream PtdIns(3,4,5)P3-dependent signalling by oxidative stress occurs via inactivation of PTEN. Signalling downstream of PtdIns(3,4,5)P3 and PKB is known to promote many cellular processes, particularly proliferation, cell growth and survival, and therefore it might be expected that the oxidative stress-induced inactivation of PTEN would act to promote these functions, although other stress-induced pathways may act in opposition.
It is known that both PtdIns(3,4,5)P3 and the NADPH oxidase complex (Babior, 1999) are found in the plasma membrane and, indeed, evidence supports the precise co-localization with and activation of the complex by PtdIns(3,4,5)P3 (Welch et al., 2002). This proximity of its substrate with a complex producing an inhibitor of PTEN indicates that only a small, functionally significant pool of this largely cytoplasmic enzyme might need to be inhibited to affect downstream signalling. Our data showing the inactivation of ∼10% of cellular PTEN in stimulated macrophages and an oxidant-dependent activation of downstream signalling support this conclusion. ROS have been shown to be produced by many different cell types in numerous circumstances (Finkel, 2000; Droge, 2002) including cellular stimulation with many growth factors. This raises the possibility that some growth factors increase PtdIns(3,4,5)P3 levels not only by increasing its rate of production by PI 3-kinase, but also by reducing the metabolism of PtdIns(3,4,5)P3 by inhibition of PTEN. As with the production of oxidants in stimulated macrophages, it seems likely that only a small fraction of cellular PTEN in close proximity to sites of PtdIns(3,4,5)P3 production and PKB activation would need to be inhibited substantially to affect the sensitivity of the signalling pathway.
In macrophages stimulated to produce ROS, PTEN was shown to be oxidatively inactivated, and there was a concomitant oxidant-dependant activation of PKB. These data not only strongly support the hypothesis that the oxidative inactivation of PTEN is a physiological mechanism of regulating the PI 3-kinase pathway, but also have significant implications for the regulation of apoptosis in macrophages and other oxidant producing leukocytes. The activation of the PI 3-kinase pathway and PKB/Akt have been shown to be potent survival signals in many cell types including macrophages (Liu et al., 2001). It is clear that phagocytes must possess mechanisms to protect them from the ROS that they produce (Splettstoesser and Schuff-Werner, 2002) and our data strongly suggest that the activation of this potent cellular survival signal by oxidants may represent such a mechanism. Additionally, the deregulation of the PI 3-kinase signalling pathway is implicated in the development of many human tumour types. Since mutation of the PTEN gene has only been found with high frequency in the rather uncommon glioblastoma and endometrial tumour types and the significance of evidence for the loss of PTEN protein in the absence of mutation is unclear, it seems likely that most human tumours retain wild-type PTEN expression (Ali et al., 1999). It is possible that the oxidative inhibition of PTEN by ROS, that are present at abnormally elevated levels in many tumours (Benhar et al., 2002), could functionally impair the tumour suppressor activity of the enzyme, enhancing tumour development.
As previously referred to, there is significant evidence that the stimulated increase in PtdIns(3,4,5)P3 levels plays a role in the activation of the NADPH oxidase complex in phagocytic and non-phagocytic cells. It appears that in fMLP-stimulated neutrophils the initial burst of PtdIns(3,4,5)P3 production by PI 3-kinase γ may be transient and very rapid, occuring within the first two min of stimulation (Cadwallader et al., 2002). Although in pilot experiments, we did not detect a similar early peak of PKB activity in LPS-stimulated RAW macrophages (data not shown), it is known that LPS can induce the activation of PI 3-kinase in RAW264.7 cells. If, as our data suggest, the production of oxidants leads to an inactivation of PTEN and decreased metabolism of PtdIns(3,4,5)P3, this suggests a role for PI 3-kinase signalling in initiating a process that itself magnifies and/or sustains the further activation of downstream signalling. Since many of the stimuli that activate cellular ROS production also strongly activate PI 3-kinase, it is clear that the oxidative inactivation of PTEN would be only one factor regulating PtdIns(3,4,5)P3 levels and downstream signalling.
These data may have significant implications for the signalling processes controlling ageing. In organisms from mice to nematodes, it appears that signalling from insulin-like growth factor 1 (and possibly insulin itself), through their receptor kinases and the PI 3-kinase/PKB pathway, to transcription factors of the forkhead family, can accelerate many factors of the ageing process (Gems and Partridge, 2001; Burgering and Kops, 2002; Holzenberger et al., 2003). In mice, it has also been shown that deletion of the adaptor protein, p66Shc, reduces cellular oxidant levels, the sensitivity of cells to oxidative stress-induced apoptosis, signalling to forkhead transcription factors, and extends life span by ∼30% (Migliaccio et al., 1999; Nemoto and Finkel, 2002; Trinei et al., 2002). The oxidative inactivation of PTEN could provide a mechanism for the observed effects of p66Shc function and cellular oxidants on the PKB/forkhead pathway, and provide the likely connection between these two pathways, both of which are involved in regulating life span (Nemoto and Finkel, 2002).
Materials and methods
Tissue culture
U87MG cells and RAW264.7 macrophages were from the European Collection of Animal Cell Cultures. Most tissue culture media and additives were purchased from Gibco Life Technologies. Fetal bovine serum (FBS) was purchased from HyClone/Perbio. Swiss 3T3 murine fibroblasts and RAW264.7 murine macrophages were grown in DMEM + 10% FBS. PTEN was expressed in cultured U87MG cells by a modified baculovirus as previously used (Leslie et al., 2001). By microscopic visualization of GFP–PTEN in cells, it is evident that expression is achieved in well over 90% of these cells. LPS, PMA, NAC, DPI and bovine liver catalase were purchased from Sigma. The abundance of cellular phosphorylated inositol-derived lipids was determined as previously described (Leslie et al., 2001).
In vitro phosphatase assays of PTEN and SHIP-2
Wild-type human PTEN was expressed in Escherichia coli as a fusion protein with glutathione S-transferase (GST) and purified as previously described (Walker et al., 2001). Substrate preparation and in vitro assays against 3-[33P]PtdIns(3,4,5)P3 were also as described (Walker et al., 2001). SHIP-2 was immunoprecipitated from U87MG cells overexpressing human SHIP-2. SHIP-2 assays were performed in 50 mM Tris pH 7.4, 10 mM MgCl2 using 100 µM phosphatidylserine, 1 µM PtdIns(3,4,5)P3 substrate vesicles prepared by sonication, containing ∼100 000 d.p.m. 3-[33P]PtdIns(3,4,5)P3. After 30 min incubation at 37°C, a Bligh and Dyer phase split was performed, dried down lipids were separated by acidic TLC and the labelled PtdIns(3,4)P2 product was quantitated using a phosphorimager. For the in vitro comparison of the sensitivity to oxidation of PTEN and SHIP-2, immobilized purified enzyme was washed three times at 4°C in 25 mM HEPES pH 7.4, 100 mM NaCl before assaying in their respective assay buffers, with addition of either 5 mM DTT, water, or different concentrations of H2O2.
Assay of PTEN redox state
The indirect method relying upon the oxidative protection from alkylation was adapted from that of Meng et al. (2002) and is displayed diagrammatically in Figure 2A. Cells for redox analysis were rapidly washed once in PBS at 4°C and lysed in buffer A (25 mM HEPES pH 7.4, 150 mM sodium chloride, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 10 mM β-glycerophosphate, 50 mM sodium fluoride, 10 µg/ml leupeptin, 100 µM PMSF and 1 mM benzamidine) with 10 mM IAA and 100 µg/ml catalase. Control cells were washed and lysed in buffer A with 5 mM DTT. PTEN was then immunoprecipitated with 5 µg of sheep polyclonal anti-PTEN antibody pre-bound to Protein G Sepharose for 1 h at 4°C. For redox analysis, immune complexes were then washed twice in buffer A containing 1 mM H2O2, whereas reduced controls were washed twice in more buffer A containing 5 mM DTT. PTEN immune complexes were finally washed three times in buffer B (10 mM HEPES pH 7.4, 150 mM NaCl, 2 mM EGTA, 5 mM DTT). PTEN phosphatase activity against 3-[33P]PtdIns(3,4,5)P3 was determined as previously described (Walker et al., 2001).
The effects of oxidative stress upon PTEN activity were also analysed directly by lysing cells, immunopurifying and assaying PTEN in anaerobic conditions. In this method, Swiss 3T3 fibroblasts were incubated in modified Krebs–Henseleit buffer (118 mM NaCl, 4.69 mM KCl, 1.18 mM MgSO4, 1.29 mM CaCl, 1.18 mM KH2PO4, 11.67 mM glucose, 25 mM HEPES pH 7.4) for 30 min before addition to some samples of 1 mM H2O2 for 10 min. Cells were then transferred into an anaerobic/nitrogen cabinet, washed three times in ice cold degassed modified Krebs–Henseleit buffer before lysis on ice in cold degassed buffer A containing 100 µg/ml catalase with no added oxidizing or reducing agents. Lysates were then pre-cleared for 15 min at 14 000 g at 4°C before addition of 4 µg PTEN antibody and Protein G Sepharose beads that had been anaerobically equilibrated for 90 min. Immunoprecipitation was allowed to proceed with tumbling at 4°C for 1 h under nitrogen. Immune complexes were then washed once in degassed lysis buffer and three times in degassed buffer B without DTT at 4°C before PTEN was assayed under nitrogen against 3-[33P]PtdIns(3,4,5)P3 using a method adapted from that of Walker et al. (2001). Briefly lipid substrate vesicles were prepared by bath sonication under nitrogen in degassed lipid resuspension buffer (25 mM HEPES pH 7.4, 2 mM EGTA), and mixed with PTEN immune complexes in degassed reaction buffer (buffer B lacking DTT or other reducing agents). Assays were then incubated for 30 min with shaking at 37°C under nitrogen before released [33P]phosphate was determined as previously described.
The oxidation state of PTEN was also investigated without assaying the enzyme, using biotinylated alkylating agents, following the methods of Seo et al. (2002) and Wu and Momand (1998). RAW264.7 macrophages were left untreated or stimulated for 10 min with 1 mM H2O2 or with 100 ng/ml LPS and 1 µM PMA before lysis in SEE buffer (100 mM Na2HPO4, 5 mM EDTA, 5 mM EGTA pH 7.5) with 0.1% Triton X-100, 20 mM NEM and protease inhibitors. Lysates were then dialysed at 4°C in three changes of SEE buffer with protease inhibitors before treatment for 1 h with 20 mM DTT, further dialysis, and treatment for 1 h with 20 µg/ml (N-maleimidopropionyl)biocytin. PTEN was then immunoprecipitated from 3 mg of each treated lysate, and analysed by western blotting with the mouse monoclonal PTEN antibody and with horseradish peroxidase (HRP)-conjugated streptavidin.
Antibodies and western blot analysis
An antibody recognizing phosphoserine-473 PKB was purchased from Cell Signalling and those recognizing total PKB and PTEN (mouse monoclonal, A2B1) were purchased from Santa Cruz. For blotting analysis, cells were lysed in buffer A containing 0.1% 2-mercaptoethanol and proteins separated by polyacrylamide gel electrophoresis and blotted onto PVDF membranes (Polyscreen NEN/Perkin Elmer). Most reagents and protocols for electrophoresis and blotting were from Novex, and standard protocols were followed.
Assay of PKB/Akt activity
For PKB assays, RAW macrophages were seeded at low cell density an hour prior to stimulation. U87MG cells were seeded at very low cell density (∼10% confluence) for 24 h, and some infected with wild-type PTEN expression virus during a further 24 h before stimulation. Cells were treated and lysed in ice cold buffer A with 0.1% (v/v) 2-mercaptoethanol. Immnuoprecipitation and kinase assay using the peptide substrate Crosstide have been previously described (Alessi et al., 1996; Leslie et al., 2001). For experiments with RAW macrophages, immunoprecipitations were performed using previously described antibodies against both PKBα and PKBγ (Walker et al., 1998).
Acknowledgments
Acknowledgements
We wish to thank Rod Herbert for advice and assistance regarding experiments in anaerobic conditions and Steven Walker for provision of labelled PtdIns(3,4,5)P3. We wish to thank Paul Crocker, Simon Morton and Simon Rousseau for advice and reagents for experiments using macrophages and Richard Greir for assistance with tissue culture. Work in the laboratory of C.P.D is supported by the Medical Research Council and the Division of Signal Transduction Therapy Consortium (AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, NovoNordisk and Pfizer).
References
- Ahmed N., Kansara,M. and Berridge,M.V. (1997) Acute regulation of glucose transport in a monocyte-macrophage cell line: glut-3 affinity for glucose is enhanced during the respiratory burst. Biochem. J., 327, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi D.R., Andjelkovic,M., Caudwell,B., Cron,P., Morrice,N., Cohen,P. and Hemmings,B.A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J., 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Ali I.U., Schriml,L.M. and Dean,M. (1999) Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J. Natl Cancer Inst., 91, 1922–1932. [DOI] [PubMed] [Google Scholar]
- Babior B.M. (1999) NADPH oxidase: an update. Blood, 93, 1464–1476. [PubMed] [Google Scholar]
- Backman S., Stambolic,V. and Mak,T. (2002) PTEN function in mammalian cell size regulation. Curr. Opin. Neurobiol., 12, 516–522. [DOI] [PubMed] [Google Scholar]
- Bae Y.S., Kang,S.W., Seo,M.S., Baines,I.C., Tekle,E., Chock,P.B. and Rhee,S.G. (1997) Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem., 272, 217–221. [PubMed] [Google Scholar]
- Benhar M., Engelberg,D. and Levitzki,A. (2002) ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep., 3, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birle D., Bottini,N., Williams,S., Huynh,H., deBelle,I., Adamson,E. and Mustelin,T. (2002) Negative feedback regulation of the tumor suppressor PTEN by phosphoinositide-induced serine phosphoryl ation. J. Immunol., 169, 286–291. [DOI] [PubMed] [Google Scholar]
- Brune B., Gotz,C., Messmer,U.K., Sandau,K., Hirvonen,M.R. and Lapetina,E.G. (1997) Superoxide formation and macrophage resistance to nitric oxide-mediated apoptosis. J. Biol. Chem., 272, 7253–7258. [DOI] [PubMed] [Google Scholar]
- Burgering B.M. and Kops,G.J. (2002) Cell cycle and death control: long live Forkheads. Trends Biochem. Sci., 27, 352–360. [DOI] [PubMed] [Google Scholar]
- Cadwallader K.A., Condliffe,A.M., McGregor,A., Walker,T.R., White,J.F., Stephens,L.R. and Chilvers,E.R. (2002) Regulation of phosphatidylinositol 3-kinase activity and phosphatidylinositol 3,4,5-trisphosphate accumulation by neutrophil priming agents. J. Immunol., 169, 3336–3344. [DOI] [PubMed] [Google Scholar]
- Cantley L.C. (2002) The phosphoinositide 3-kinase pathway. Science, 296, 1655–1657. [DOI] [PubMed] [Google Scholar]
- Cantrell D.A. (2001) Phosphoinositide 3-kinase signalling pathways. J. Cell Sci., 114, 1439–1445. [DOI] [PubMed] [Google Scholar]
- Chen Q., Olashaw,N. and Wu,J. (1995) Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J. Biol. Chem., 270, 28499–28502. [DOI] [PubMed] [Google Scholar]
- Das S., Dixon,J.E. and Cho,W. (2003) Membrane-binding and activation mechanism of PTEN. Proc. Natl Acad. Sci. USA, 100, 7491–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M.A., Kim,S.J., Parikh,N.U., Dong,Z., Bucana,C.D. and Gallick,G.E. (2002) Adenoviral-mediated expression of MMAC/PTEN inhibits proliferation and metastasis of human prostate cancer cells. Clin. Cancer Res., 8, 1904–1914. [PubMed] [Google Scholar]
- Denu J.M. and Tanner,K.G. (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry, 37, 5633–5642. [DOI] [PubMed] [Google Scholar]
- Droge W. (2002) Free radicals in the physiological control of cell function. Physiol. Rev., 82, 47–95. [DOI] [PubMed] [Google Scholar]
- Finkel T. (1998) Oxygen radicals and signaling. Curr. Opin. Cell Biol., 10, 248–253. [DOI] [PubMed] [Google Scholar]
- Finkel T. (2000) Redox-dependent signal transduction. FEBS Lett., 476, 52–54. [DOI] [PubMed] [Google Scholar]
- Furnari F.B., Huang,H.J. and Cavenee,W.K. (1998) The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res., 58, 5002–5008. [PubMed] [Google Scholar]
- Gems D. and Partridge,L. (2001) Insulin/IGF signalling and ageing: seeing the bigger picture. Curr. Opin. Genet. Dev., 11, 287–292. [DOI] [PubMed] [Google Scholar]
- Han Y.J., Kwon,Y.G., Chung,H.T., Lee,S.K., Simmons,R.L., Billiar,T.R. and Kim,Y.M. (2001) Antioxidant enzymes suppress nitric oxide production through the inhibition of NF-κB activation: role of H2O2 and nitric oxide in inducible nitric oxide synthase expression in macrophages. Nitric Oxide, 5, 504–513. [DOI] [PubMed] [Google Scholar]
- Holzenberger M., Dupont,J., Ducos,B., Leneuve,P., Geloen,A., Even,P.C., Cervera,P. and Le Bouc,Y. (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature, 421, 182–187. [DOI] [PubMed] [Google Scholar]
- Katso R., Okkenhaug,K., Ahmadi,K., White,S., Timms,J. and Waterfield,M.D. (2001) Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis and cancer. Annu. Rev. Cell. Dev. Biol., 17, 615–675. [DOI] [PubMed] [Google Scholar]
- Lee S.R., Kwon,K.S., Kim,S.R. and Rhee,S.G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem., 273, 15366–15372. [DOI] [PubMed] [Google Scholar]
- Lee S.R., Yang,K.S., Kwon,J., Lee,C., Jeong,W. and Rhee,S.G. (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem., 277, 20336–20342. [DOI] [PubMed] [Google Scholar]
- Leslie N.R. and Downes,C.P. (2002) PTEN: the down side of PI 3-kinase signalling. Cell Signal, 14, 285–295. [DOI] [PubMed] [Google Scholar]
- Leslie N.R., Bennett,D., Gray,A., Pass,I., Hoang-Xuan,K. and Downes,C.P. (2001) Targeting mutants of PTEN reveal distinct subsets of tumour suppressor functions. Biochem. J., 357, 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Perlman,H., Pagliari,L.J. and Pope,R.M. (2001) Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages. Role of Mcl-1, independent of nuclear factor (NF)-κB, Bad, or caspase activation. J. Exp. Med., 194, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T. and Dixon,J.E. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidyl inositol 3,4,5-trisphosphate. J. Biol. Chem., 273, 13375–13378. [DOI] [PubMed] [Google Scholar]
- Maehama T., Taylor,G.S. and Dixon,J.E. (2001) PTEN and myotubularin: novel phosphoinositide phosphatases. Annu. Rev. Biochem., 70, 247–279. [DOI] [PubMed] [Google Scholar]
- McConnachie G., Pass,I., Walker,S.M. and Downes,C.P. (2003) Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN: evidence for activation by anionic phospholipids. Biochem. J., 371, 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng T.C., Fukada,T. and Tonks,N.K. (2002) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell, 9, 387–399. [DOI] [PubMed] [Google Scholar]
- Migliaccio E., Giorgio,M., Mele,S., Pelicci,G., Reboldi,P., Pandolfi,P.P., Lanfrancone,L. and Pelicci,P.G. (1999) The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature, 402, 309–313. [DOI] [PubMed] [Google Scholar]
- Monick M.M., Carter,A.B., Robeff,P.K., Flaherty,D.M., Peterson,M.W. and Hunninghake,G.W. (2001) Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of β-catenin. J. Immunol., 166, 4713–4720. [DOI] [PubMed] [Google Scholar]
- Myers M.P., Pass,I., Batty,I.H., Van der Kaay,J., Stolarov,J.P., Hemmings,B.A., Wigler,M.H., Downes,C.P. and Tonks,N.K. (1998) The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc. Natl Acad. Sci. USA, 95, 13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S. and Finkel,T. (2002) Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science, 295, 2450–2452. [DOI] [PubMed] [Google Scholar]
- Patel L., Pass,I., Coxon,P., Downes,C.P., Smith,S.A. and Macphee,C.H. (2001) Tumor suppressor and anti-inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Curr. Biol., 11, 764–768. [DOI] [PubMed] [Google Scholar]
- Pfeiffer S., Lass,A., Schmidt,K. and Mayer,B. (2001) Protein tyrosine nitration in cytokine-activated murine macrophages. Involvement of a peroxidase/nitrite pathway rather than peroxynitrite. J. Biol. Chem., 276, 34051–34058. [DOI] [PubMed] [Google Scholar]
- Salh B., Wagey,R., Marotta,A., Tao,J.S. and Pelech,S. (1998) Activation of phosphatidylinositol 3-kinase, protein kinase B and p70 S6 kinases in lipopolysaccharide-stimulated Raw 264.7 cells: differential effects of rapamycin, Ly294002 and wortmannin on nitric oxide production. J. Immunol., 161, 6947–6954. [PubMed] [Google Scholar]
- Seo Y.R., Kelley,M.R. and Smith,M.L. (2002) Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc. Natl Acad. Sci. USA, 99, 14548–14553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L. and Parsons,R. (2001) PTEN: life as a tumor suppressor. Exp. Cell Res., 264, 29–41. [DOI] [PubMed] [Google Scholar]
- Splettstoesser W.D. and Schuff-Werner,P. (2002) Oxidative stress in phagocytes–‘the enemy within’. Microsc. Res. Tech., 57, 441–455. [DOI] [PubMed] [Google Scholar]
- Stambolic V. et al. (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell, 95, 29–39. [DOI] [PubMed] [Google Scholar]
- Sun H. et al. (1999) PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl Acad. Sci. USA, 96, 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan M., Yu,Z.X., Ferrans,V.J., Irani,K. and Finkel,T. (1995) Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science, 270, 296–299. [DOI] [PubMed] [Google Scholar]
- Tamura M., Gu,J., Matsumoto,K., Aota,S., Parsons,R. and Yamada,K.M. (1998) Inhibition of cell migration, spreading and focal adhesions by tumor suppressor PTEN. Science, 280, 1614–1617. [DOI] [PubMed] [Google Scholar]
- Tonks N.K. and Neel,B.G. (2001) Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol., 13, 182–195. [DOI] [PubMed] [Google Scholar]
- Torres J. and Pulido,R. (2001) The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem., 276, 993–998. [DOI] [PubMed] [Google Scholar]
- Trinei M. et al. (2002) A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene, 21, 3872–3878. [DOI] [PubMed] [Google Scholar]
- van der Kaay J., Beck,M., Gray,A. and Downes,C.P. (1999) Distinct phosphatidylinositol 3-kinase lipid products accumulate upon oxidative and osmotic stress and lead to different cellular responses. J. Biol. Chem., 274, 35963–35968. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B., Leevers,S.J., Ahmadi,K., Timms,J., Katso,R., Driscoll,P.C., Woscholski,R., Parker,P.J. and Waterfield,M.D. (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem., 70, 535–602. [DOI] [PubMed] [Google Scholar]
- Vazquez F., Ramaswamy,S., Nakamura,N. and Sellers,W.R. (2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol., 20, 5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virolle T., Adamson,E.D., Baron,V., Birle,D., Mercola,D., Mustelin,T. and de Belle,I. (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol., 3, 1124–1128. [DOI] [PubMed] [Google Scholar]
- Walker K.S., Deak,M., Paterson,A., Hudson,K., Cohen,P. and Alessi,D.R. (1998) Activation of protein kinase B β and γ isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase Bα. Biochem. J., 331, 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S.M., Downes,C.P. and Leslie,N.R. (2001) TPIP: a novel phosphoinositide 3-phosphatase. Biochem. J., 360, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch H.C., Coadwell,W.J., Ellson,C.D., Ferguson,G.J., Andrews,S.R., Erdjument-Bromage,H., Tempst,P., Hawkins,P.T. and Stephens,L.R. (2002) P-Rex1, a PtdIns(3,4,5)P3- and Gβγ-regulated guanine-nucleotide exchange factor for Rac. Cell, 108, 809–821. [DOI] [PubMed] [Google Scholar]
- Wu H.H. and Momand,J. (1998) Pyrrolidine dithiocarbamate prevents p53 activation and promotes p53 cysteine residue oxidation. J. Biol. Chem., 273, 18898–18905. [DOI] [PubMed] [Google Scholar]
- Wu X. et al. (2000) Evidence for regulation of the PTEN tumor suppressor by a membrane- localized multi-PDZ domain containing scaffold protein MAGI-2. Proc. Natl Acad. Sci. USA, 97, 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Dowbenko,D., Spencer,S., Laura,R., Lee,J., Gu,Q. and Lasky,L.A. (2000) Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J. Biol. Chem., 275, 21477–21485. [DOI] [PubMed] [Google Scholar]