

Abstract
The Regulators of G protein Signaling (RGS) proteins were initially characterized as inhibitors of signal transduction cascades initiated by G-protein-coupled receptors (GPCRs) because of their ability to increase the intrinsic GTPase activity of heterotrimeric G proteins. This GTPase accelerating (GAP) activity enhances G protein deactivation and promotes desensitization. However, in addition to this signature trait, emerging data have revealed an expanding network of proteins, lipids, and ions that interact with RGS proteins and confer additional regulatory functions. This review highlights recent advances in our understanding of the physiological functions of one subfamily of RGS proteins with a high degree of homology (B/R4) gleaned from recent studies of knockout mice or cells with reduced RGS expression. We also discuss some of the newly-appreciated interactions of RGS proteins with cellular factors that suggest RGS control of several components of G-protein-mediated pathways as well as a diverse array of non-GPCR-mediated biological responses.
1. Introduction
Signal transduction mediated by heterotrimeric G proteins (Gαβγ) elicits responses in every organ system, evoking such diverse outcomes as neurotransmission, immunity, cardiovascular function, and hormone secretion (Gilman, 1987; Neer, 1995). G-protein-coupled receptors (GPCRs), which possess a heptahelical structure, catalyze guanosine triphosphate (GTP) exchange on guanosine diphosphate(GDP)-bound G protein alpha subunits (Wess, 1997). Gα-GTP and Gβγ both activate specific downstream effectors such as adenylyl cyclase, phospholipase Cβ, Rho GTPases, mitogen activated protein (MAP) kinases, and ion channels (Goldsmith and Dhanasekaran, 2007; Marinissen and Gutkind, 2001). These effectors in turn produce a number of cellular responses including proliferation, morphological changes, and gene transcription. Nearly 60% of all pharmaceutical agents in current use target GPCRs, making detailed understanding of their intracellular signaling routes critical for the treatment of human disease (Pierce, et al., 2002).
Numerous studies have suggested that cells undergo desensitization to continual GPCR stimulation due to hydrolysis of GTP by the alpha subunit, which allows Gα-GDP to re-unite with Gβγ and form an inactive heterotrimer (Tsang, et al., 1998). However, purified Gα subunits hydrolyze GTP too slowly in vitro to account for the rapid recovery from G-protein-mediated biological responses (Tsang, et al., 1998). Therefore, additional co-factors that could aid in desensitization to GPCR-induced activation were hypothesized.
One candidate group of cellular proteins that probably serve this function are the Regulator of G protein Signaling (RGS) proteins, which bind to activated Gα subunits and accelerate their intrinsic GTPase activity (Blumer, 2004; He, et al., 1998). Because RGS GAP activity hastens G protein deactivation, RGS proteins would be predicted to reduce signaling output elicited by GPCR activation. The first described RGS protein, Sst2p, inhibited the pheromone-evoked mating response of the yeast Saccharomyces cerevisiae by binding to the yeast Gα protein, Gpa1, and acting as its principal GAP (Dohlman, et al., 1998). Shortly thereafter, RGS orthologues were discovered, which complemented the phenotype of an SSt2- yeast strain and inhibited GPCR-evoked signaling in mammalian cells (Druey, et al., 1996). A plethora of subsequent overexpression studies supported the conclusion that RGS proteins were negative regulators of G-protein-mediated signaling in part due to their GAP activity (Huang, et al., 1997; Willars, 2006; Yan, et al., 1997).
However, this concept has since proven to be too simplistic. RGS proteins, which number greater than 30 in mammalian cells, can be subdivided into subfamilies based on primary sequence homology and the presence of additional domains (Abramow-Newerly, et al., 2006b) (Figure 1). All RGS proteins house a ~120 amino acid RGS domain (box), which, based on biochemical and crystallographic studies, mediates direct binding to Gαi and Gαq (Tesmer, et al., 1997). Members of the R4/B subfamily, which includes RGS1-5, 8, 13, 16, 18 and 21, are the smallest RGS proteins in size, containing only short peptide sequences flanking the RGS box with one notable exception (RGS3) (Figure 1). Nonetheless, these proteins appear to bind numerous cellular signaling factors in both G-protein-mediated and non-GPCR pathways.
Figure 1.
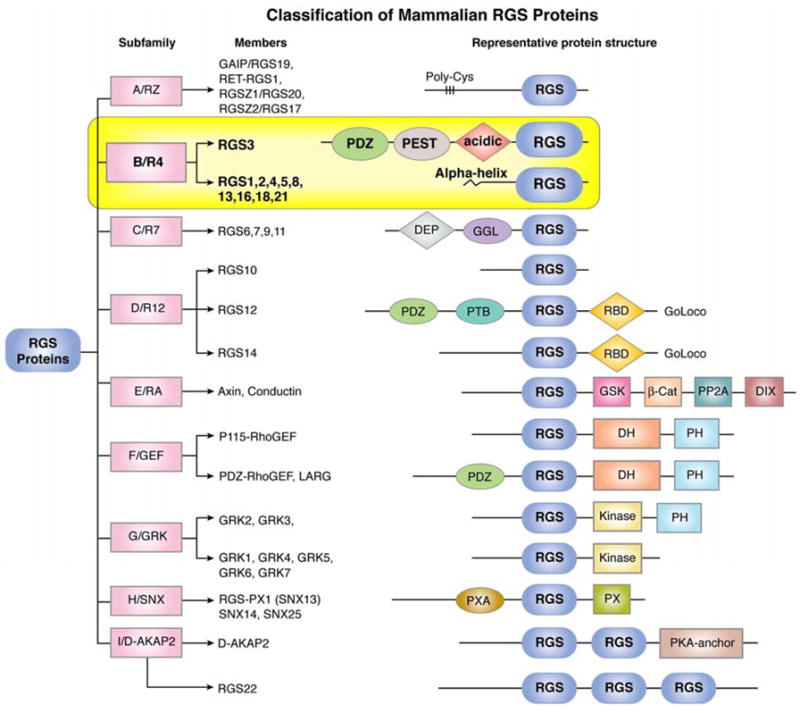
Classification of mammalian RGS proteins into different subfamilies and their representative protein structure showing identified motifs or domains. Abbreviations: β-Cat, β-catenin-binding; D, dimerization domain; D-AKAP, dual-specificity A-kinase anchoring protein; DEP, dishevelled/EGL-10/pleckstrin; DH, double homology; DIX, dishevelled homology domain; GAIP, G alpha interacting protein; GEF, guanine nucleotide exchange factor; GGL, Gγ-like; GoLoco, Gαi/o-Loco; GRK, G protein-coupled receptor kinase; GSK, glycogen synthase kinase 3β-binding; PDZ, PSD95/Dlg/Z0-1/2; PEST, proline, glutamine, serine, threonine-rich; PH, pleckstrin homology; PP2A, protein phosphatase 2A; PTB, phosphotyrosine-binding; PX, phosphatidylinositol-binding; PXA, PX-associated; RBD, Ras-binding domain; RGS, Regulator of G protein Signaling domain; SNX, sorting nexin.
The biochemical and molecular determinants of RGS expression and activity have been extensively studied and are the subject of several excellent recent reviews (Neitzel and Hepler, 2006; Tinker, 2006). However, because many studies relied on RGS overexpression in transformed cell lines or analysis of purified recombinant proteins in vitro, the physiological function(s) of most R4 RGS proteins have not been well-defined. In this review, we focus on studies of mice containing targeted deletions of R4 Rgs genes as well as RNA interference analyses that have begun to clarify biological roles of individual RGS proteins in native mammalian systems. We will first describe recent findings for each R4 RGS individually that illuminate physiological function(s) before detailing novel interactions of R4 RGS proteins with proteins or other factors that either affect RGS activity and/or specificity within GPCR signaling pathways or implicate them in non-GPCR-mediated cellular responses. These studies have challenged the notion that this subfamily acts strictly as negative modulators of G-protein-coupled signaling.
2. Physiological functions of individual RGS proteins
2.1. RGS1
The major portal of RGS1 expression appears to be the hematopoietic compartment including T and B lymphocytes (Agenes, et al., 2005; Moratz, et al., 2000), natural killer (NK) cells (Kveberg, et al., 2005), dendritic cells (Shi, et al., 2004), and monocytes (Denecke, et al., 1999). In B lymphocytes, RGS1 is upregulated by B-cell receptor activation by surface immunoglobulin, and its expression is concentrated in germinal center B cells (Hong, et al., 1993). Lymphoid organ germinal centers are the site of B lymphocyte differentiation and maturation during adaptive humoral immune responses. Studies of Rgs1 knockout mice revealed a role for Rgs1 in the control of B lymphocyte migration induced by chemokines (Moratz, et al., 2004). Rgs1-deficient B cells migrated to a greater extent after exposure to the chemokines CXCL12 and CXCL13 in vitro, whose receptors, CXCR4 and CXCR5, are required for germinal center formation (Allen, et al., 2004). Rgs1−/− B lymphocytes pretreated with CXCL12 remained responsive to subsequent challenge with either chemokine, suggesting that Rgs1 mediates desensitization of these cells to prolonged ligand exposure. Accordingly, spleens of Rgs1−/− mice contained increased numbers of germinal centers even in the absence of immunization, and immune challenge induced both elevated and persistent germinal center formation. In contrast, Peyer’s patches, which are lymphocyte-rich follicles in the gastrointestinal tract (GI) mucosa, were reduced in size following immunization, implying aberrant migration of lymphocytes during the immune response. Lastly, trafficking of antibody-secreting cells was abnormal in the absence of Rgs1.
Subsequent analysis of adoptively-transferred, fluorescently-labeled lymphocytes confirmed that B cell migration was substantially affected by the loss of Rgs1 (Han, et al., 2005). There were significantly more Rgs1-deficient B cells than WT cells in peripheral lymph nodes of recipient mice and decreased cell numbers in blood, suggesting enhanced homing of Rgs1−/− cells into lymphoid tissue. Intravital multiphoton microscopy of lymph nodes after cell injection revealed that Rgs1−/− cells adhered better to high endothelial venules (HEVs) of peripheral lymph nodes and moved with greater velocity within lymphoid follicles in relation to WT cells. Interestingly, B lymphocytes from Gnai2−/− mice, which lack Gαi2 expression, exhibited the opposite phenotype. Thus, Rgs1 regulates B cell homing to lymph nodes and motility within the lymph node microenvironment by regulating Gαi2 signaling pathways induced by chemokines.
Another recent study reported enrichment of both Rgs1 and Rgs16 in regulatory CD4+ T cells and activated T cells compared with naïve T lymphocytes (Agenes, et al., 2005). This differential RGS expression correlated inversely with the ability of these subpopulations to migrate in surgical parabiosis experiments. The specific chemokines involved were not determined (Agenes, et al., 2005). The consequences of abnormal lymphocyte migration associated with Rgs1 deficiency for immune responses of whole organisms await further clarification. Preliminary analysis indicated that Rgs1−/− mice produced a delayed antibody response to immunization with T-cell dependent antigens, but the ultimate antibody titer and affinity profile of the immunoglobulins were normal (Moratz, et al., 2004). These studies suggest that Rgs1 may play a major role in the chemokine-mediated homing of lymphocytes to secondary lymphoid organs as well as their localization within these spaces during the immune response.
2.2. RGS2
RGS2 is expressed widely in both mouse and human tissues (Kehrl and Sinnarajah, 2002). Preliminary studies of Rgs2−/− mice and RGS2 knockdown in both human and mouse cells have suggested that it regulates G protein-mediated responses in the immune system, brain, heart, lung, bone, and olfactory epithelium. In an initial report, Oliveira-dos-Santos et al. described a variety of abnormalities in Rgs2 knockout mice (Oliveira-dos-Santos et al. 2000). Rgs2−/− T lymphocytes proliferated less and produced less interleukin-2 (IL-2) after phorbol ester or T-cell receptor (CD3/CD28) stimulation. In whole animal studies, these mice exhibited reduced inflammation (footpad swelling) to virus (lymphocytic choriomeningitis virus) infection. Studies of central nervous system (CNS) function revealed that the brains of Rgs2−/− mice showed reduced density and basal electrical activity of hippocampal CA1 neurons (Oliveira-Dos-Santos, et al., 2000). These cellular abnormalities were accompanied by abnormal behavior such as increased anxiety (measured by light/dark preference) and decreased male aggression. Although no mechanistic insights were provided into how Rgs2 might control such a diverse group of biological parameters in these organ systems, the mice provided a preliminary blueprint for subsequent detailed investigation into the potential physiological functions of Rgs2. In fact, recent genetic quantitative trait analysis has confirmed that Rgs2 is a gene that controls anxiety in mice (Yalcin, et al., 2004).
Subsequent characterization of Rgs2−/− mice revealed that Rgs2 controls systemic blood pressure. Both Rgs2+/− and Rgs2−/− mice were found to be profoundly hypertensive with increased systemic and renal vascular resistance and hypertrophy of the renal arterial vasculature (Heximer, et al., 2003). This parameter correlated with excessive and prolonged Ca++ signaling to vascoconstrictors such as ATP acting on P2Y receptors. In addition, the hypertension of Rgs2-deficient mice appeared to be especially sensitive to acute angiotensin II receptor antagonism, suggesting elevated vascular tone due to heightened signaling responses to this hormone. Despite the hypertension, there was no cardiac hypertophy change in cardiac systolic contractility or that might have contributed to the elevated blood pressure in these mice.
Several additional abnormalities may also promote hypertension in Rgs2−/− mice. First, the mice displayed some characteristics of increased peripheral sympathetic tone, such as elevated urinary catecholamine secretion, relative resistance to the α1-adrenergic receptor antagonist prazosin, and reduced blood pressure decrease after environmental stress (Gross, et al., 2005). These defects could lead to a re-setting of the baroreceptor reflex and hypertension. However, additional tests such as heart rate response to β-adrenergic receptor blockade were normal, arguing against a significant increase in peripheral sympathetic outflow. Further studies will be required to determine whether altered autonomic outputs contribute to the hypertensive phenotype (Stauss, 2005).
Second, the mice displayed renovascular abnormalities including increased responsiveness to vasopressin, which could result in impaired water handling and changes in plasma volume (Zuber, et al., 2007). By RT-PCR, Rgs2 expression in the mouse kidney appeared to be limited to the principal cells of the connecting tubules and collecting duct, which serve to concentrate urine in response to anti-diuretic hormone (ADH or vasopressin). Vasopressin acts on Gs-coupled V2 receptors, which generate cAMP. Rgs2 localization mirrored expression of V2 receptors in the kidney, and vasopressin treatment lead to upregulated Rgs2 (mRNA and protein) in mouse collecting duct principal cells (Zuber, et al., 2007). In whole mice, water restriction, which induces vasopressin secretion, also upregulated Rgs2 expression. Collecting duct cells microdissected from kidneys of Rgs2−/− mice produced more cAMP after vasopressin treatment than WT cells, and the Rgs2 knockout mice exhibited abnormal patterns of urine excretion after water loading. A separate study recently described inhibition of angiotensin II-mediated aldosterone secretion by adrenal cells induced by RGS2 overexpression (Romero, et al., 2006). Thus, defective signaling pathways in the kidney of Rgs2−/− mice could impair water and solute processing. The authors of the original study of the hypertension of Rgs2−/− mice hypothesized that the differential acute changes in blood pressure between WT and Rgs2-deficient mice to vasoconstrictor antagonists argued against plasma volume changes in Rgs2−/− mice as the primary cause of hypertension (Heximer, et al., 2003). An even more recent study has found no difference in renal sympathetic activity in WT or Rgs2−/− mice (Tank, et al., 2007). Although the significance of the renal abnormalities in the absence of Rgs2 for regulation of plasma volume and systemic blood pressure is far from clear, its role in renal water and salt handling merits further study. Third, RGS2 exerts control over vascular tone through an interaction with a component of the nitric oxide (NO) pathway, protein kinase G (PKG)/cGMP-dependent protein kinase, which influences vascular relaxation mediated by NO (discussed in detail below)(Sun, et al., 2005; Tang, et al., 2003). Collectively, these studies paint a complex picture of the regulation of systemic blood pressure in mice by Rgs2.
Preliminary evidence points to a role for RGS2 in the pathogenesis of human hypertension. Analysis of peripheral blood mononuclear cells (PBMCs) and skin fibroblasts from 11 normals and 12 hypertensives revealed substantially reduced RGS2 expression and increased responsiveness (Ca++ mobilization and Erk phosphorylation) to angiotensin II in hypertensives compared to controls (Semplicini, et al., 2006). The reduction in RGS2 expression correlated with a polymorphism (C1114G) in the 3′ untranslated region of the RGS2 gene. An independent analysis of RGS2 polymorphisms in hypertensives found association between two haplotypes in the 3′ non-coding region in black patients. RGS2 protein expression levels were not assessed in this study (Riddle, et al., 2006). A third report of a Japanese population identified a rare coding mutation in RGS2 (Q2L) only in hypertensives (Yang, et al., 2005), which destabilized RGS2 expression. A separate study found that this mutant was expressed at reduced levels in HEK293T cells compared to WT and failed to inhibit angiotensin II-mediated signaling (Bodenstein, et al., 2007).
Conversely, RGS2 expression may be elevated in Bartter/Gitelman (BG) syndrome, a condition that manifests as normo/hypotension due to sodium and potassium wasting and volume depletion caused by an unknown genetic defect in kidney electrolyte transporters. Decreased plasma volume leads to hyperactivation of the renin/angiotensin system and elevated serum aldosterone levels. Several compensatory changes result in decreased vascular tone and hyporeactivity to angiotensin II signaling, which may be due to decreased expression of Gαq. Calo et al. (Calo, et al., 2004) described significantly increased RGS2 protein levels in peripheral blood mononuclear cells of 6 BG patients compared to 6 healthy controls, which could contribute to their angiotensin II resistance. Another group recently reported upregulation of Rgs2 in vascular smooth muscle cells, which appeared to be mediated by activation of Group VIA phospholipase A2 (iPLA2β) (Xie, et al., 2007). Although the detailed molecular mechanisms remain to be determined, these studies point to a potential role for RGS2 in the control of systemic blood pressure in humans through its regulation of GPCR-evoked signals governing vascular resistance and possibly water/solute processing in the kidney.
Analysis of mouse cells with reduced Rgs2 expression also suggests that it may regulate other responses in the cardiopulmonary system. Although no changes in cardiac contractility or evidence of cardiac hypertrophy were observed in Rgs2−/− mice, a recent study indicated that Rgs2 could contribute to heart failure in mice. In contrast to end-stage failing human hearts with upregulated RGS4 (Mittmann, et al., 2002; Owen, et al., 2001), models of mouse cardiac hypertrophy such as transverse aortic constriction (banding) or transgenic overexpression of constitutively active Gαq were associated with significantly decreased Rgs2 mRNA and protein levels prior to the development of hypertrophy with no concordant change in expression of Rgs3-5 (Zhang, et al., 2006). Knockdown of endogenous Rgs2 in ventricular myocytes by selective RNAi increased inositol phosphate (IP) formation and cellular hypertrophy in response to phenylephrine and endothelin-1, which act on Gq-coupled receptors. By contrast, activation of MAP kinase pathways (Erk, JNK, and p38), which presumably would be involved in hypertrophic responses, was not affected by reduced Rgs2 expression in these cells. Thus, decreased Rgs2 levels may lead to cardiac hypertrophy by way of amplified signaling responses to catecholamines and other vasoactive mediators. Finally, in the lung knockdown of RGS2 in human ciliated airway epithelial cells by antisense oligonucleotides led to increased Ca++ flux and ciliary beat frequency in response to purinergic receptor stimulation with ATP (Nlend, et al., 2002). It will be of interest to determine whether these in vitro abnormalities translate into increased susceptibility of Rgs2−/− mice to hypertrophic myocardial failure or resistance to microbial pulmonary infection due to increased clearance of microorganisms.
Finally, Rgs2 may have a function in bone formation by osteoblasts. Rgs2 is upregulated by parathyroid hormone (PTH) and PTH-related peptide (PTHrP) or forskolin, which induce cAMP formation. cAMP stimulates osteoblast proliferation and differentiation, as do ATP or phorbol ester (PMA), which activate Gq effectors such as protein kinase C (PKC) (Roy, et al., 2006b). Rgs2−/− osteoblasts displayed no differences from WT cells in PTHrP-stimulated cAMP formation or IP generation evoked by ATP or endothelin-1. However, upregulation of Rgs2 by forskolin resulted in reduced endothelin-induced inositol phosphate formation and ATP-evoked Ca++ mobilization in WT but not Rgs2-deficient osteoblasts. Similarly, pre-treatment of WT but not Rgs2−/− osteoblasts with ATP lead to diminished PTHrP-stimulated cAMP. The authors concluded that Rgs2 at basal levels does not regulate either Gq or Gs signaling in osteoblasts, but at higher expression levels Rgs2 may cross-desensitize both Gs and Gq signals. However, other abnormalities that could have accounted for the differences between these strains such as altered expression of GPCRs or other downstream signaling components (e.g., G proteins, phospholipase Cβ) were not examined with the exception of the sarco/endoplasmic reticulum calcium ATPase 2b, which controls calcium content in the ER. Nonetheless, these studies lend credence to the hypothesis that Rgs2 could regulate bone repair in conditions associated with elevated PTH (hyperparathyroidism) or stress, which is accompanied by increased catecholamines (e.g., infection or fractures).
2.3. RGS3
RGS3 exists as several isoforms that are splice variants of the same gene, RGS3. A short form, RGS3S, which contains little more than the RGS domain, was expressed in the nucleus and induced apoptosis when overexpressed (Dulin, et al., 2000). RGS3 has two longer isoforms, RGS3L and PDZ-RGS3 (Kehrl, et al., 2002), the latter of which has been linked to cell migration through interaction with Ephrin receptors and in Gβγ-evoked signaling (described below). Mice containing a targeted deletion of Rgs3 have not been reported, and few studies of its physiological function exist. Ribozyme-mediated knockdown of RGS3 in aortic smooth muscle cells lead to increased M3 muscarinic acetylcholine (mAch) receptor-induced MAP kinase activation but no effect on angiotensin II-evoked signaling (Wang, et al., 2002). A recent study reported upregulation of RGS3 in p53-mutated tumors, and RGS3 siRNA treatment of MCF-7 breast cancer cells resulted in enhanced sensitivity to chemotherapy (docetaxel)-induced apoptosis compared to control (Ooe, et al., 2007). Further studies will be required to determine the physiological significance of this finding and the signaling pathways involved.
2.4. RGS4
In both humans and rodents, RGS4 appears to be selectively enriched in the CNS and heart (Erdely, et al., 2004; Zhang, et al., 1998). During embryonic CNS development, Rgs4 is expressed transiently in the mouse locus coeruleus, sympathetic ganglionic neurons, and cranial sensory and motor neurons, and expression is linked to the homeodomain transcription factor Phox2B (Grillet, et al., 2003). Recently, homologous recombination techniques were utilized to introduce expression of green fluorescent protein (GFP) with an internal ribosomal entry sequence (IRES) into a bacterial artificial chromosome (BAC) construct containing the Rgs4 gene. This BAC was then expressed in mice to evaluate the expression of Rgs4 in the mouse brain (Ebert, et al., 2006). Although the extensive microanatomical localization of RGS4-GFP within brain regions cannot be detailed here, expression of the GFP reporter faithfully reproduced localization of endogenous Rgs4 mRNA detected by in situ hybridization. These studies revealed widespread Rgs4 expression in most cortical neuronal layers and at all stages of development. In all cases, RGS4-GFP was detected more in grey matter than in white matter, suggesting that Rgs4 is not expressed in glial cells. Another important observation was the striking overlap between Rgs4 expression patterns and that of acetylcholinesterase, which implies a potential physiological role for Rgs4 in the regulation of mAch receptor signaling. In subcortical regions, RGS4-GFP was most abundant in the striatum and amygdala. In human brain, in situ hybridization using RGS4 riboprobes revealed enrichment of RGS4 in several regions of the frontal cortex with lower levels in the thalamus and striatum (Erdely, et al., 2004).
Several functional studies have linked RGS4 to regulation of opioid, cholinergic, and serotonergic signaling in the brain. After morphine treatment, RGS4 levels increase in the rat locus coeruleus (LC) and decline rapidly after opiate withdrawal. Treatment of LC neurons with RGS4 reduced opioid-induced electrophysiological responses, suggesting a role in opioid tolerance (Gold, et al., 2003). In contrast, studies of Rgs4 knockout mice did not support a substantial role for Rgs4 in the control of opioid signaling in sensory neurons as knockout mice displayed normal antinocioceptive responses (pain sensitivity and analgesia evoked by morphine). Nonetheless, in shock tests, the threshold for pain response (flinch test) was significantly increased in knockout mice, suggesting abnormal central processing of painful stimuli. This response presumably involves postsynaptic afferent sensory cortical neurons, which express relatively high levels of Rgs4 (Garnier, et al., 2003; Grillet, et al., 2003).
A potential role for RGS4 in Parkinson disease (PD) was revealed by a recent study of cholinergic interneurons of the striatum, which are enriched in RGS4 (Ding, et al., 2006). In PD, loss of dopaminergic neurons in the striatum is accompanied by increased acetylcholine (Ach) release, which exacerbates motor symptoms of the disease. It was previously thought that decreased dopamine levels lead to reduced D2 receptor-mediated inhibition of synaptic Ca++ channels (Cav2) and subsequent increased Ach release. These investigators found that while D2-evoked inhibition of Cav2 activity was unchanged after chemical dopamine depletion in mouse cholinergic interneurons, M4 muscarinic receptor-induced suppression of channel electrical activity was markedly attenuated, which would lead to increased Ach release. It was hypothesized that the reduced M4-mediated signaling was attributable to upregulation of Rgs4 levels by dopamine depletion (reserpine), as had been previously demonstrated (Geurts, et al., 2003) and was confirmed in this study. As proof-of-principle, dialysis of recombinant RGS4 with cholinergic interneurons inhibited M4-evoked Ca++ channel activity. Interestingly, infusion of an RGS4 peptide lacking the amino-terminus (RGS4ΔN) reversed the attenuated M4 response of reserpine-treated neurons, suggesting that RGS4ΔN acted as a dominant negative inhibitor of endogenous Rgs4. Although these results await confirmation by studies of interneurons with reduced RGS4 expression or cells from Rgs4 knockout mice, this study implies a role for RGS4 in the control in motor symptoms of PD associated with increased striatal acetylcholine levels.
Over the past several years, considerable attention has been paid to RGS4 as a potential etiological factor for schizophrenia. Postmortem studies have identified reduced RGS4 expression in several areas of the frontal cortex of schizophrenics, such as the superior temporal gyrus (Bowden, et al., 2007). RGS4 mapped to the schizophrenia susceptibility locus (1q23), and several single nucleotide polymorphisms have been associated with the disease (Chowdari, et al., 2007). In particular, one haplotype was most recently linked to deficit-subtype schizophrenia, in which patients have a preponderance of negative symptoms such as social withdrawal and psychomotor retardation (Bakker, et al., 2007).
Functional studies have not clearly established whether RGS4 expression levels play a role in the development of schizophrenia. A report of human subjects with an RGS4 haplotype previously associated with psychosis found significant reductions in cortical grey matter volume and connectivity that may affect cognitive responses (Buckholtz, et al., 2007). Another study employing a rat model of schizophrenia (phencyclidine treatment) demonstrated downregulated Rgs4 expression in prefrontal cortical neurons (PFCs) after phencyclidine exposure (Gu, et al., 2007). Inhibition of RGS4 function by a specific antibody potentiated serotonergic receptor (5HT1A)-mediated regulation of NMDA receptor channels in PFCs while RGS4 overexpression inhibited the response. An RGS2-selective antibody had no effect on channel activity induced by serotonin. Single-cell studies of PFCs obtained from phencyclidine-treated rats confirmed that the heightened serotonin-evoked signaling response was limited to cells with reduced endogenous Rgs4 expression. In contrast, Rgs4 knockout mice were found to lack any substantial behavioral abnormalities such as decreased pre-pulse inhibition, which is widely considered a standard marker for schizophrenic behavior in rodents (Grillet, et al., 2005). These studies suggest that species variability in the expression and/or function of RGS4 in the cerebral cortex could exist. Thus, the exact role of RGS4 in the etiology of schizophrenia remains unclear.
As mentioned previously, RGS4 appears to be upregulated in failing human hearts due to dilated cardiomyopathy (Owen, et al., 2001). In murine models of cardiac hypertrophy induced by transgenic overexpression of activated Gq or in models of diabetic cardiomyopathy as a result of transgenic expression of the peroxisome proliferated-activated receptor alpha (PPARα) or treatment with streptozotocin, concomitant aortic banding markedly increased mortality in transgenic mice overexpressing RGS4, consistent with a maladaptive role of RGS4 in cardiomyopathy (Rogers, et al., 1999). However, in Rgs4LacZ/LacZ mice, which should display surrogate β-galactosidase activity in tissues expressing Rgs4, staining was detected in the large vessels of the heart including aorta and pulmonary artery trunk as well as coronary vasculature but not in cardiac muscle (Grillet, et al., 2005). It will be of interest to determine whether these mice display differences in mortality after induction of cardiac hypertrophy.
2.5. RGS5
Little is known about the physiological role of RGS5 in whole animals due to the lack of gene-targeted mice. Rgs5 is enriched in peri-endothelial cells or “pericytes” and vacular smooth muscle cells (Bondjers, et al., 2003; Cho, et al., 2003). In certain anatomical locations in the mouse, Rgs5 is enriched in pericytes of both capillaries and arterioles, and expression mirrors abundance of the tyrosine kinase receptor for platelet-derived growth factor beta (PDGFRβ) as well as its ligand PDGF. Receptor or ligand-null embryos lack Rgs5 expression and the presence of pericytes, suggesting Rgs5 as a pericyte-specific marker in microvasculature. In primate vascular smooth muscle, RGS5 was identified in larger arteries (e.g. aorta, carotid) and afferent glomerular arteioles but not coronary arteries or venous structures (Li, et al., 2004). Further, RGS5 mRNA was downregulated in certain regions of atherosclerotic placques (Adams, et al., 2006). In contrast, RGS5 was abnormally elevated in the vasculature of renal carcinomas as well as in neovascularized pancreatic islet cell carcinomas and astrocytomas (Berger, et al., 2005; Furuya, et al., 2004). Together, these studies imply a role for RGS5 in pericyte development and vascular smooth muscle activation associated with neovascularization of tumors, as well as in arterial smooth muscle cell development or function under physiological conditions.
Another site of RGS5 expression is the heart, and beta-adrenergic receptor hyper-function has been shown to upregulate RGS5 levels (Jean-Baptiste, et al., 2005). This finding suggests a potential physiological function in cardiac conditions associated with elevated sympathetic tone. Finally, a novel splice variant of RGS5, RGS5S, which lacks 104 amino acids of the amino-terminus, was recently described. This alternate splice form appears to be differentially expressed in human tissues with high expression in the ciliary body of the eye, kidney, brain, spleen, skeletal muscle and small intestine, and undetectable transcripts in the liver, lung, and heart (Liang, et al., 2005).
2.6. RGS8
This RGS isoform garnered considerable attention soon after its discovery because of its apparent paradoxical regulation of G-protein-gated potassium channels (see below). Increased RGS8 expression was associated not only with increased “off” kinetics, which might be expected from RGS8 GAP activity, but also faster activation kinetics (Saitoh, et al., 1997). Despite this early observation, no definitive mechanism has been defined to explain the ability of RGS8 to accelerate channel kinetics nor have the physiological implications of this finding been explored in whole animals. Several theories have been proposed including “kinetic” scaffolding (Ross and Wilkie, 2000), which suggests that increased G protein deactivation induced by RGS proteins or other GAPs actually improves efficiency of the GTPase cycle by facilitating protein-protein interactions within the GPCR-heterotrimeric G protein complex and mitigates against limiting G protein concentrations in the presence of a strong extracellular stimulus. A more recent study suggested that the RGS protein may be a component of a stable quaternary complex consisting of GPCR-G protein-RGS (Benians, et al., 2005). Fluorescence resonance energy transfer (FRET) experiments showed that RGS8-YFP protein associated with Gα regardless of the activation state or receptor-ligand interaction. In contrast, a separate set of experiments revealed that membrane association of an RGS protein (RGS2) was stable in the presence of a constitutively active, immobile G protein [Gαq(R183C)] but was only transiently recruited to the membrane by expression of a GPCR (M3R) (Clark, et al., 2007). The localization of RGS2 after ligand stimulation of the receptor was not examined in the latter study. Thus, in some cases, depending on the receptor or G protein involved, RGS proteins could act as a physical scaffold between Gαβγ and its receptor.
In fact, the work of several laboratories has recently determined that in contrast to prevailing dogma, some G protein heterotrimers do not physically dissociate in living cells (Bunemann, et al., 2003; Digby, et al., 2006). Instead, depending on the identity of the Gα subunit, receptor stimulation induces a subunit rearrangement rather than heterotrimer dissociation (Bunemann, et al., 2003; Frank, et al., 2005). Two distinct models of G protein activation have been postulated: “collision coupling”, which postulates ligand-dependent, random interactions between GPCRs and G proteins diffusing laterally within the membrane, or “precoupling” of stably-associated receptors and G proteins. Recent FRET studies of fluorescently-tagged GPCRs and G proteins found no evidence for precoupling in living cells (Hein, et al., 2005). In a separate study, RGS2 did not affect association of fluorescently-tagged GPCRs and G proteins detected by FRET in living cells (Clark, et al., 2007). Thus, it is unclear how RGS scaffolding might affect receptor-G protein and heterotrimer interactions during the GTPase cycle as these results do not support a role for RGS proteins act as physical scaffolds, at least for this particular GPCR-G protein combination. It will be interesting to examine these interactions in cells with reduced or absent expression of one or more endogenous RGS proteins.
Expression of RGS8 appears to be concentrated in the brain (Larminie, et al., 2004). RGS8 levels can be modulated by acute and chronic electroconvulsive seizures (Gold, et al., 2002). RGS8 mRNA and protein are enriched in Purkinje cells of the granule layer of the cerebellum (Saitoh, et al., 2003; Saitoh and Odagiri, 2003). RGS8 was found to exist as a distinct splice variant containing an alternate amino-terminus (RGS8S) (Itoh, et al., 2006). RGS8S demonstrated reduced interactions with M1 mAch receptors and impaired inhibition of M1-evoked signaling (see below). In the hematopoietic system, analysis of leukocyte subsets revealed selective expression of Rgs8 in rat natural killer (NK) cells (Kveberg, et al., 2005).
2.7. RGS13
RGS13 exhibits relatively restricted tissue expression in T and B lymphocytes, and subsequent studies have demonstrated even higher expression in mast cells (Shi, et al., 2002); Druey Rgs13. UCSD-Nature Molecule Pages 2005, doi:10.1038/mp.a000020.01) RGS13, like RGS1 and RGS16, is concentrated in germinal center B cells and in activated lymphocytes (treated with anti-CD40 plus IL-4), suggesting a function in adaptive immune responses (Estes, et al., 2004; Shi, et al., 2002). In addition, RGS13 is abundant in Burkitt lymphoma, a tumor thought to represent malignant germinal center B lymphocytes, but is absent in mantle cell lymphomas (Islam, et al., 2003). In the rat brain, Rgs13 mRNA was detected in the hippocampus and discrete thalamic nuclei (Grafstein-Dunn, et al., 2001).
Overexpression studies showed that RGS13 inhibits migratory responses and signaling induced by the chemokines CXCL12 and CXCL13 in B lymphocytes, which are required for germinal center formation in lymphoid organs (Shi, et al., 2002). These studies have recently been corroborated by RNAi in a Burkitt lymphoma cell line, where RGS13 knockdown enhanced chemokine responsiveness (Han, et al., 2006b). Rgs13 knockout mice were recently generated by our laboratory, and preliminary studies suggest that Rgs13 has a function in IgE-mediated mast cell responses and in B cell transcriptional activity (Bansal et al. and Xie et al., submitted). Of note, Rgs13 is also expressed in dendritic cells (Shi, et al., 2004) and is also abundant in neuroendocrine cells of the thymus, gastrointestinal, and respiratory tracts (our unpublished data).
2.8. RGS16
RGS16 was initially cloned from the retina (Chen, et al., 1996) and subsequent analysis demonstrated expression in the rat heart (Patten, et al., 2002) and brain (especially suprachiasmatic nucleus) (Grafstein-Dunn, et al., 2001), mouse liver (Kurrasch, et al., 2004), and hematopoietic cells. In blood cells, RGS16 has been found in NK cells (Kveberg, et al., 2005), platelets (Kim, et al., 2006), dendritic cells (Shi, et al., 2004), and T lymphocytes (Beadling, et al., 1999). RGS16 is upregulated in germinal center and activated T cells, suggesting a role for this RGS in adaptive immunity (Estes, et al., 2004). In the mouse liver, Rgs16 is restricted to periportal hepatocytes and demonstrates diurnally-regulated expression patterns. Rgs16 is downregulated during fasting and rapidly upregulated by refeeding (Huang, et al., 2006). These studies suggest a role for Rgs16 in circadian-regulated pathways involved in glucose and/or fat metabolism.
Rgs16 knockout mice have been generated, which are viable and fertile, but no phenotype has yet been determined (Druey, unpublished observations). In a megakaryocytic cell line, RNAi-mediated knockdown of RGS16 enhanced signaling responses to CXCL12, whereas overexpression reduced CXCL12-evoked signaling and migration (Berthebaud, et al., 2005). These preliminary results imply that RGS16 may control CXCR4-elicited migration of platelet precursors.
2.9. RGS18
Several groups recently cloned this novel RGS protein, whose expression appears to be relatively restricted to bone marrow-derived cells (Nagata, et al., 2001; Park, et al., 2001; Yowe, et al., 2001). RGS18 mRNA and protein are detectable in platelets and granulocytes, but not in lymphocytes or erythrocytes (Gagnon, et al., 2002). Few studies have addressed the transcriptional regulation of RGS expression. Interestingly, however, promoter analysis at the Rgs18 locus demonstrated highly restricted occupancy of both GATA-1 and GATA-2 transcription factors, which may partially explain its enrichment in the hematopoietic compartment (Johnson, et al., 2007). To date, most functional analysis has been limited to overexpression studies, which have shown that RGS18 is capable of inhibiting both Gi- and Gq-mediated signaling pathways. Interestingly, RGS18 overexpression had no effect on CXCR4-induced responses in a megakaryocytic cell line (Nagata, et al., 2001).
Recent work showed that RGS18 is also expressed in osteoclasts (Iwai, et al., 2007). Osteoclasts are multinucleated giant cells that promote bone resorption necessary for remodeling. Osteoclast differentiation is controlled by RANKL ligand (receptor activator of nuclear factor κB ligand), which acts on the RANK receptor to induce osteoclast differentiation. It had been previously shown that this process was mediated by upregulation of a GPCR that senses extracellular acidosis, OGR1. Proton stimulation of OGR1 activates the Gq-PLCβ pathway, which in turn increases nuclear factor of activated T cells (NFAT) activity and promotes osteoclastogenesis. RANKL reduced RGS18 expression in the osteoclast precursor cell line RAW246.7 and in primary bone marrow-derived osteoclast precursor monocytes. RGS18 knockdown in RAW246.7 cells (by RNAi) enhanced osteoclast differentiation evoked by RANKL, and this phenotype was neutralized by an anti-OGR1 blocking antibody. Further, RGS18 siRNA increased NFAT activation induced by extracellular acidosis, while RGS18 (but not RGS2 or GAIP) overexpression inhibited OGR1-mediated responses. Taken together, the studies indicate that RGS18 levels may control osteoclastogenesis mediated by RANKL through modulation of OGR1-evoked signaling.
2.10. RGS21
The latest R4 member to be identified, RGS21, is the smallest member of the B/R4 subfamily, with 152 amino acids. It was originally described as selectively expressed in taste tissue (von Buchholtz, et al., 2004). By RT-PCR RGS21 mRNA was detected only in sensory taste cells that express sweet taste receptors and the taste Gα subunit, gustducin. Although no functional studies of endogenous RGS21 have yet been reported, this RGS interacts with activated gustducin, suggesting a potential role in regulating taste transduction. Subsequent to the initial report, a second group cloned RGS21 from fetal brain cDNA; they reported that RGS21 was widely expressed in 16 tissues examined (Li, et al., 2005).
2. Interactions with GPCR signaling components
2.1. Role of RGS amino terminus
R4 RGS protein binding to G proteins depends strictly on the intact RGS domain (box) (Popov, et al., 1997). The RGS box is the most highly conserved region in these proteins, with 47–62% identity and 69–83% homology within the R4 subfamily. By contrast, the amino-terminal and carboxy-terminal flanking regions are less well-conserved. The amino-terminus of several R4 family members has a function in subcellular localization. Although R4 RGS proteins lack obvious membrane targeting motifs such as pleckstrin homology (PH) domains, most members contain an amphipathic α-helix at the amino-terminus that facilitates direct binding to membrane phospholipids. The amino-terminus of RGS4 (Bernstein, et al., 2000) and RGS8 (Saitoh, et al., 2001) mediates translocation to membranes and binding to phospholipids. Both the amphipathic α-helix and palmitoylation of amino-terminal cysteine residues play a role in membrane targeting of these RGS proteins (Bernstein, et al., 2000; Tu, et al., 2001).
In addition to mediating cellular localization, the amino-terminus also serves as a platform for additional protein-protein interactions unique to one or more R4 RGS proteins. For example, RGS16 but not RGS4 binds Gα13 through its amino-terminal 30 amino acids and inhibits Gα13-mediated gene transcription and cell morphological changes. (Johnson, et al., 2003). The epithelial Ca++ channel TRPV6, which has a function in Ca++ transport in placenta, pancreas, small intestine, and colon (den Dekker, et al., 2003), binds the RGS2 amino-terminus in a Ca++-independent fashion (Schoeber, et al., 2006). Electrophysiological studies using whole- cell patch clamp revealed that RGS2 overexpression inhibited Na+ and Ca++ currents in HEK293 cells co-transfected with TRPV6 but not TRPV5. Deletion of the amino-terminus of RGS2 disrupted binding to TRPV6 and restored electrophysiological properties of the channel in the absence of RGS2 (Schoeber, et al., 2006).
RGS2 also interacts directly with tubulin via a short polypeptide within its amino-terminus (amino acids 41-60) (Heo, et al., 2006). This region was necessary and sufficient for RGS2 to inhibit microtubule polymerization observed microscopically in Vero cells stably expressing GFP-tagged α-tubulin. Overexpression of RGS2 enhanced nerve growth factor-induced neurite outgrowth in PC12 cells whereas RGS2 knockdown suppressed neurite outgrowth. Thus, RGS2 may have a function in neuronal cell differentiation through direct regulation of tubulin. PDZ-RGS3 has been shown to bind phosphorylated EphrinB through its PDZ domain at the amino terminus (Su et al., 2004). These are just a few examples demonstrating the importance of the RGS protein amino-terminus for diverse protein-protein interactions. More examples of RGS binding partners and the implications for various signaling pathways are discussed below.
3.2. RGS domain-dependent G protein selectivity
R4 RGS proteins bind Gαq and Gαi through their RGS domains to increase the GTPase activity of Gα. This GAP activity reduces the lifetime of activated Gα and Gβγ, attenuating GPCR signaling. As noted above, the proteins of the R4 RGS subfamily have highly homologous primary structure, and most often a given cell or tissue expresses two or more members of this subfamily. Often these proteins display roughly equivalent GAP activity for Gq or Gi family members in vitro. Thus, the principal question one might pose is how does an individual RGS protein achieve selectivity to regulate specific GPCR signaling pathways? Emerging evidence points to a myriad of co-factors that either confer selectivity of a given RGS protein for certain GPCRs or endow additional functions outside GPCR signaling pathways. RGS protein selectivity for certain receptors or signaling routes might be accomplished by the following: 1) measurable differences in GAP activity toward different classes of Gα subunits, which may or may not depend on additional co-factors or modification of the RGS protein or Gα; 2) direct interaction with a subset of GPCRs; 3) formation of an RGS/G protein complex that sequesters the G protein from either the receptor or downstream effectors (i.e., independent of RGS GAP activity or 4) direct binding of RGS proteins to effectors within the GPCR signaling axis.
Assessments of the relative potency of RGS proteins in cells have been mainly limited to overexpression in transformed cell lines, as noted above, with some exceptions. For example, RGS2 was found to be five-fold more potent than RGS4 in inhibiting Gαq-stimulated IP3 formation (Heximer, et al., 1999); in contrast, RGS4 was 8-fold more potent that RGS2 in complementing SSt2p in a yeast mating pheromone assay (Heximer, et al., 1999). RGS2 inhibited Gαq-activated PLCβ1 activity with an IC50 of 30 nM, while RGS4 was 10-fold less potent than RGS2 with IC50 of 300 nM (Heximer, et al., 1997b). In ventricular myocytes, RGS2, RGS3, RGS4 and RGS5 inhibited Gq-mediated hypertrophic cell growth and IP3 formation with similar potency (Hao, et al., 2006). However, RGS3, RGS4 and RGS5, but not RGS2, reversed carbachol-evoked inhibition of cAMP production induced by isoproterenol, which is thought to be mediated by Gαi (Hao, et al., 2006). Comparison of RGS activity on the Gq/G11 subfamily of G-proteins revealed that RGS1, RGS2, RGS3 and RGS4 inhibited cell morphology change in a yeast system expressing both mammalian Gαq and Gα11, whereas RGS5 and RGS16 were much less effective against Gα11 than Gαq (Ladds, et al., 2007).
The differences in efficacy of RGS proteins toward Gα subunits could be due to variable affinity of a given RGS protein for Gα family members. Assessment of relative potency of RGS proteins has proven difficult as methods used to assess GAP activity in vitro are somewhat crude. Using purified recombinant proteins, GAP activity is measured in either a single turnover assay or phospholipid vesicles containing both receptor and heterotrimeric G protein, which allows measurement of steady-state GTP hydrolysis. In the single turnover assay, Gα is loaded with 32P-GTP in the absence of Mg++, which is required for GTPase activity. Mg++ is then added along with excess unlabeled GTP, which allows measurement of a single catalytic cycle of GTP hydrolysis.
There are a limited number of Gαi/q family members (Gαi1-3, Gαo, Gαz, transducin, Gαz, Gαq, 11, 14/15, and 16), and most R4 RGS proteins will accelerate the GTPase activity of these Gα subunits promiscuously in single turnover assays (Berman, et al., 1996; Watson, et al., 1996). A notable exception is RGS2, which appears to be a more potent GAP for Gαq than Gαi. RGS4 binds both Gαi/o/z and Gαq, whereas RGS2 was found to bind Gαq selectively in vitro (Heximer, et al., 1997b). In addition, significant GAP activity of RGS2 against Gi is only detectable in the presence of a receptor in phospholipids vesicles. Whether this reflects a requirement for receptor binding or reduced affinity of RGS2 for Gi family members remains to be definitively established (Ingi, et al., 1998). In this regard, RGS2-Gαi interaction may be unfavorable due to geometry of the binding pocket of RGS2 and the switch I region of Gαi. Three RGS2 residues within this region, Cys106, Asn184 and Glu191, were critical for Gαi binding (Heximer, et al., 1999). Mutation of amino acids in RGS2 to the analogous residues in RGS4 (C106S/N184D/E191K) rendered RGS2 able to increase GTPase activity of Gαo (Gαi subfamily) under conditions where WT RGS2 demonstrated no GAP activity (Heximer, et al., 1999). Reciprocal substitutions of these RGS2 residues into RGS4 (S85C, D163N and K170E) abolished the GAP activity of RGS4 on Gαo. Finally, RGS2, but none of the other R4 family members interacts with Gαs (see below). Although the significance of this finding has not been clarified, together these studies clearly indicate that variance in primary structure within the RGS fold determine the G-protein selectivity and specificity of some R4 RGS proteins.
In addition, recent advances in measurement of GAP activity or RGS/G protein interactions in vitro may illuminate more subtle differences in affinity of various RGS proteins for their substrates. Novel techniques utilizing high-throughput bead-based flow cytometry (Roman, et al., 2007) or quench-flow or spectroscopy of fluorescently-tagged GTP have been utilized to measure G protein binding and GTP hydrolysis in real time (Willard, et al., 2005). A selective small molecule inhibitor (CCG-4986) of RGS4 was discovered, which apparently does not affect activity of RGS8 (Roman, et al., 2007). By using a flow cytometry protein interaction assay, CCG-4986 inhibited RGS4 binding to Gαo in a dose-dependent manner with IC50 ~ 7 μM but did not affect the ability of RGS8 to bind Gαo even at 100 μM. Furthermore, incubation of permeabilized μ-opioid receptor-expressing rat C6 glioma cells with CCG-4968 reversed ~75% of the inhibitory effect of RGS4 on forskolin-induced cAMP production without affecting the activity of RGS8.
3.3. RGS-GPCR interactions
Receptor-dependent, rather than primarily G-protein-dependent, inhibition of GPCR signaling by RGS proteins has been demonstrated some time ago (Xu, et al., 1999). Recombinant RGS4 exhibited marked differences in its ability to inhibit three distinct Gq-coupled receptors mediating Ca++ flux and IP3 formation in pancreatic acinar cells. Recombinant RGS4 inhibited carbachol, bombesin and cholecystokinin (CCK)-induced Ca++ mobilization with IC50s of 35, 110 and 380 nM, respectively. In other words, cholinergic receptors were 3- and 10-fold more sensitive to RGS4 than bombesin and CCK receptors, respectively. By contrast, RGS1 and RGS16, but not RGS2, showed a similar ability to inhibit the carbachol- and CCK-stimulated Ca++ responses. The differential pattern of inhibition of these responses was observed in cells from mice lacking one or more individual Gαq family members. Thus, this study suggested that in certain instances R4 RGS selectivity may be determined primarily by interaction with the receptor complex rather than affinity for a specific Gq class α subunit. The amino-terminal region of R4 RGS proteins has been suggested to mediate interaction with receptors (Bernstein, et al., 2004; Itoh, et al., 2006; Zeng, et al., 1998), which is discussed below.
3.3.1. Muscarinic acetycholine (mAch) receptors
The aforementioned studies are consistent with the hypothesis that the amino terminus of certain R4 RGS proteins such as RGS4 may regulate GPCR signaling independent of GAP activity. The RGS4 amino-terminus inhibits Gq-mediated Ca++ signaling in pancreatic acinar cells (Zeng, et al., 1998). Recently, direct evidence of the interaction of RGS proteins with the 3rd intracellular loop (3iL) of certain GPCRs has been obtained (Bernstein, et al., 2004; Georgoussi, et al., 2006; Hague, et al., 2005; Itoh, et al., 2006), suggesting a mechanism for these findings. Recombinant GST-3iL of M1, M3 and M5 mAch receptors pulled down overexpressed RGS2 from CHO cell lysates (Bernstein, et al., 2004). In contrast, RGS4 expressed in these cells interacted with GST-3iL of M1 and M5, but not M3, and there was no binding of either RGS1 or RGS16 to any mAch receptors. The amino-terminal domain of RGS2 was required for the direct interaction with mAchR 3iL in this study, as well as the functional inhibition of M1 mAch receptor-stimulated PIP2 formation (Bernstein, et al., 2004). M1 3iL bound to recombinant RGS2 and Gαq in the presence of AMF (aluminum magnesium fluoride), which mimics the transition state between Gα–GTP and Gα-GDP, suggesting that RGS2 forms a stable heterotrimeric complex with active Gαq and the M1 3iL.
RGS8 was also found to interact directly with M1 mAch at 3iL through its first 9 amino acids (Itoh, et al., 2006). Two arginine residues in that region play a key role in M1 3iL binding, as mutation of both Arg8 and Arg9 (RGS8-R8A/R9A), reduced binding more than 60%. This double mutant displayed substantially reduced ability to inhibit acetylcholine-stimulated Ca++ current (M1 mAchR-mediated) in Xenopus laevis oocytes. The interaction of RGS8 and M1-mAch receptor was also observed in living 293T cells using bioluminescence energy transfer (BRET), (Itoh, et al., 2006), reaffirming the existence of the interaction in vivo. Collectively, these studies indicate receptor-selective interactions of individual R4 RGS proteins that may depend on less well-conserved regions outside of the RGS domain.
3.3.2. Adrenergic receptors (ARs)
Alpha-2 adrenergic receptors (α2AR) may directly interact with RGS1 and RGS16 based on comparison of the relative efficacy of three distinct agonists to stimulate a α2AR-Go1α fusion protein in the absence or presence of RGS proteins (Hoffmann, et al., 2001). Both the α2AR full agonist adrenaline and partial agonist UK14304 stimulated this fusion protein expressed in COS-7 cells to induce GTPase activity from membrane preparations. RGS1 and RGS16 significantly increased high-affinity GTPase activity of the α2AR-Go1α fusion protein induced by both adrenaline and UK14304. However, both RGS proteins reduced agonist potency for adrenaline to a much greater extent than for the partial agonist UK14304 and did not alter potency of the weak agonist oxymetazoline. This result suggests a possible interaction between RGS proteins and the α2AR which changes the conformation of the receptor and leads to altered intrinsic agonist activity. The RGS-G-protein interaction itself would not be predicted to affect receptor-agonist interactions that determine relative agonist potency. A related study found that in some cases recovery from agonist stimulation may depend less on G-protein deactivation than agonist unbinding from the receptor, which was selectively affected by RGS8 in a receptor and agonist-dependent manner (Benians, et al., 2003). If signal termination were dependent only on RGS8 interaction with G proteins and GAP activity, increasing RGS8 expression levels should increase deactivation independent of the receptor or agonist involved. However, since a subsequent study by the same group found no direct interaction between RGS8 and the GPCR in living cells by FRET, further experiments are needed to determine if a physical association between α2AR or other receptors and RGS proteins occurs in native mammalian cells (Benians, et al., 2005).
α1AAR was shown to bind RGS2 directly in purified recombinant protein pulldown assays. Expression of α1AAR in HEK293 cells recruited RGS2, but not RGS16, to plasma membrane from the nucleus, whereas co-expression of α1BAR had no effect on the localization of either RGS2 or RGS16 (Hague, et al., 2005). This result suggests a selective interaction between α1AAR and RGS2. Receptor selectivity of RGS2 toward α1AAR was further supported by the observation that RGS2 expression suppressed norepinephrine-stimulated α1AAR-, but not α1BAR, -mediated IP formation by HEK293 cells. The interaction appears to be mediated through 3iL of α1AAR since recombinant GST-α1AAR-3iL extracted HA-RGS2 from lysates of transfected HEK293 cells. Mutational studies indicated that the amino terminus of RGS2 was required for association with α1AAR and inhibition of signaling, and amino acids Lys219, Ser220, and Arg238 within the α1AAR 3iL were found to be essential for this interaction (Hague, et al., 2005).
β2ARs had been previously shown to specifically recruit RGS2, but not RGS4, to the cell plasma membrane when co-expressed in 293T cells (Roy, et al., 2003). This receptor-promoted association was not altered by agonist, implying a constitutive interaction of RGS2 with β2AR. Further studies by the same group demonstrated a direct interaction of RGS2 with β2AR (Roy, et al., 2006a). RGS2, but not the closely related RGS16, interacted with the 3iL of β2AR, and over expression of RGS2 in HEK293 cells significantly suppressed iosproterenol-β2AR-stimulated cAMP production. Thus, receptor binding by some RGS proteins may serve a negative regulatory function independent of GAP activity since RGS2 does not exhibit GAP activity toward Gαs (Roy, et al., 2003).
3.3.3. Opioid receptors
Recombinant RGS4 directly bound the carboxy-terminal domains of both μ-opioid and δ-opioid receptors, as well as the 3iL of δ-opioid receptor (Georgoussi, et al., 2006). This interaction was agonist-dependent since it was only observed in COS-7 cells stimulated with opioid receptor ligand. The interaction also required the presence of AMF, indicating that the presence of activated Gα was essential. Overexpression of RGS4 in 293T cells significantly reduced opioid receptor-mediated inhibition of forskolin-stimulated cAMP formation. Furthermore, Gβγ appeared to compete with RGS4 for binding to the C-terminal μ-opioid receptor, as the amount of RGS4 pulled down by C-terminal μ-opioid receptor deceased in the presence of increasing amounts of Gβγ. Thus, RGS4 is recruited to the plasma membrane after receptor activation to interact with the 3iL of opioid receptor and Gαi, resulting in increased Gα GTPase activity and inhibition of GPCR signaling. These studies stand in contrast with aforementioned studies of RGS8, in which constitutive interaction of RGS8-YFP with the heterotrimeric G protein complex throughout the GTPase cycle was observed. In other words, Gα-RGS interaction was not altered by Gα binding to βγ (Benians, et al., 2005).
3.4. Gαs and Adenylyl cyclase (AC)
Gαs-GTP stimulates AC to produce cAMP in response to activation of GPCRs such as β2AR. Although none of the R4 RGS proteins possesses GAP activity towards Gαs (Hollinger and Hepler, 2002; Roy, et al., 2003), expression of RGS1, RGS2 and RGS3, and RGS13 exerts a negative effect on Gαs/AC-stimulated cAMP production (Johnson and Druey, 2002; Roy, et al., 2003; Sinnarajah, et al., 2001). These observations suggested that RGS proteins may inhibit Gαs signaling downstream of the G protein independent of their GAP activity. Co-expression of Gαs with RGS2, but not RGS4, in HEK293 cells promoted plasma membrane translocation of RGS2 (Roy, et al., 2003) implying association of Gαs and RGS2. The interaction between RGS2 and Gαs was subsequently demonstrated by co-immunoprecipitation of proteins expressed in HEK293 cells as well as in vitro binding of recombinant proteins (Tseng and Zhang, 1998). BRET also revealed a significant interaction between Gαs and RGS2 in living cells (Roy, et al., 2006a).
In addition to Gαs, RGS2 interacted with several isoforms of AC. RGS2 inhibited forskolin-induced cAMP production in Sf9 insect cell membranes expressing ACIII, ACV and ACVI, but not ACI and ACII, consistent with an isoform-selective interaction (Sinnarajah, et al., 2001). RGS2 also suppressed forskolin- or Gαs-GTPγS -induced cAMP generation in olfactory membranes (Sinnarajah, et al., 2001), which express endogenous ACIII predominantly. BRET subsequently revealed an interaction between RGS2 and both ACII and ACVI in intact HEK293 cells, and this complexation was confirmed by co-immunoprecipitation (Roy, et al., 2006a). Further studies have recently shown that the first 19 amino acids of RGS2 are required for binding to ACV as deletion of the amino-terminal 19 amino acids of RGS2 abolished its ability to inhibit Gαs- and β2AR-stimulated cAMP formation (Salim, et al., 2003). Three residues within the RGS2 amino-terminus were required for the inhibition of AC function by RGS2 (Val9, Gln10 and His11).
Thus, RGS2 may form a signaling complex with Gαs and AC to negatively regulate Gs-mediated signaling. Blockade of endogenous RGS2 by anti-RGS2 antibody significantly increased the odorant-induced inward current in whole-cell voltage clamp studies of olfactory neurons, and pre-incubation of recombinant RGS2 blocked the antibody-induced enhancement of the current (Sinnarajah, et al., 2001). Thus, endogenous RGS2 regulates odorant-induced AC signaling in olfactory neurons. RGS2 expression in HEK293 cells reduced glucose-dependent insulinotropic receptor (GIP-R)-stimulated cAMP formation by 50%. This effect mirrored the response of cells pre-incubated with low concentration of GIP upon restimulation (Tseng and Zhang, 1998), suggesting a role for RGS2 in GIP-induced desensitization response. Expression of RGS2 in mouse insulinoma βTC3 cells attenuated GIP-induced insulin release. Collectively, these studies suggest that regulation of the Gαs-AC signaling axis by RGS2 has physiological significance.
3.5. PLCβ
PLCβ, an enzyme activated by activated Gαq and Gβγ, catalyzes the conversion of PIP2 to IP3 and diacylglycerol (DAG), which leads to the mobilization of intracellular Ca++ stores and activation of certain isoforms of protein kinase C (PKC). Interaction of PLCβ with RGS4 has been detected by surface plasmon resonance (Dowal, et al., 2001) and FRET techniques. Binding to RGS4 appears to require the carboxy-terminus of PLCβ1 (Dowal, et al., 2001). Further studies revealed that PLCβ1 can be regulated by RGS4 through protein kinase A (PKA) and protein kinase G (PKG) (Huang, et al., 2007). Both PKA and PKG phosphorylate RGS4, which increased its GAP activity. Phosphorylation of RGS4 by PKA or PKG also caused translocation of RGS4 from cytosol to plasma membrane. Pre-treatment of dispersed rabbit gastric smooth muscle cells expressing WT RGS4 with compounds that activate PKA or PKG suppressed PLCβ-induced IP formation in response to Ach stimulation compared to cells exposed to Ach alone (Huang, et al., 2007), indicating an increased inhibitory effect of phosphorylated RGS4. The phosphorylation site of RGS4 was identified at Ser52. Co-expression of a RGS4/S52A mutant resistant to PKA- and PKG-induced stimulation of GAP activity on Gq reversed the inhibition of Ach-stimulated IP formation induced by pre-activation of PKA and PKG.
Agonist-stimulated contraction of smooth muscle is initiated by activation of PLCβ1 via Gαq or PLCβ3 via Gαi, leading to generation of IP3 and Ca++ release from intracellular stores. Ca++ activates Ca++/calmodulin-dependent myosin light chain (MLC) kinase, which phosphorylates myosin light chain kinase 20 (MLC20), a prerequisite for smooth muscle contraction. Relaxation of contracted smooth muscle is mediated by PKA or PKG through the inhibition of IP3 formation. Thus, the increased ability of phosphorylated RGS4 to inhibit PLCβ activity provided a molecular mechanism by which PKA and PKG indirectly regulate PLCβ activity, which, in turn, affects smooth muscle contraction.
RGS proteins may also suppress GPCR-evoked signaling independent of GAP activity by modulating G-protein-effector interactions. RGS2, RGS3, RGS5 and RGS16 all inhibited carbachol-induced activation of PLCβ in COS-7 cells expressing muscarinic M3 receptors as assessed by IP formation (Anger, et al., 2004). However, RGS2 and RGS3, but not RGS5 and RGS16, inhibited IP formation stimulated by constitutively activated Gαq, which is inert to GTP hydrolysis. This finding implies a GAP-independent role for certain RGS proteins as effector antagonists of Gαq. In this regard, RGS4 was originally shown to inhibit the interaction between Gαq-GTPγS and PLCβ (Hepler, et al., 1997). RGS4 has also been suggested to reside within the Gαq/PLCβ complex acting as an effector antagonist against PLCβ with its amino-terminus bound to the receptor 3iL. This hypothesis is based on the previously noted observation that amino-terminal region of RGS4 (outside of RGS domain) inhibited carbachol-stimulated Ca++ signaling in rat pancreatic acinar cells, and deletion of the amino-terminus from full-length RGS4 not only reduced its inhibitory potency by 104-fold but also abolished the receptor selectivity of RGS4 for mAchR (Zeng, et al., 1998). In addition, as noted above, this construct was used to compete with endogenous RGS4 activity in neurons, although the mechanism for dominant inhibition is unclear (Ding, et al., 2006).
3.6. Spinophilin/neurabin
Spinophilin (SPL) and neurabin (NRB), structurally similar scaffolding proteins with more than 30 binding partners, are involved in the regulation of membrane and cytoskeletal functions (Sarrouilhe, et al., 2006). SPL, but not NRB, binds the 3iL of some GPCRs including the dopamine D2 receptor and α1BAR (Smith, et al., 1999; Wang, et al., 2007). RGS1, RGS2, RGS4, and RGS16 co-immunoprecipitated with SPL and NRB expressed in HEK293 cells. Moreover, recombinant RGS2 bound NRB/SPL in vitro, suggesting a direct interaction (Wang, et al., 2007). The interaction of RGS proteins with SPL was also demonstrated in Xenopus laevis oocytes (Wang, et al., 2007; Wang, et al., 2005) and prefrontal cortex neurons (Liu, et al., 2006). SPL binding to certain RGS proteins promotes GPCR-Gα-RGS interaction, which enhances the inhibitory effect of RGS on α1AR-stimulated, Gq-mediated Ca++ signaling (Liu, et al., 2006; Wang, et al., 2007). NRB competed with SPL for the binding to RGS proteins, reversing the enhancement of RGS activity by SPL. Overexpression of NRB in X. laevis oocytes increased α1BAR-induced Ca++ signaling, whereas overexpression of SPL decreased the Ca++ response, suggesting opposing roles for SPL and NRB (Wang, et al., 2007). Parotid duct cells from SPL−/− mice and NRB−/− mice displayed a phenotype opposite of cells overexpressing these proteins (Wang, et al., 2007). The amino-terminus of RGS proteins appears to be involved in the interaction with NRB as a mutant RGS2 lacking the amino-terminus, GST-ΔNRGS2, did not extract NRB from lysates of HEK293 cells expressing NRB exogenously. Furthermore, infusion of SPL into parotid cells from NRB−/− mice further enhanced its inhibition of epinephrine-stimulated Ca++ signaling compared to infusion of SPL into WT cells, suggesting a reciprocal effect of NRB and SPL. Thus, the following model has been proposed: SPL and NRB form a functional pair of opposing regulators that modulate the intensity of Ca++ signaling by GPCR-Gαq. SPL acts as a bridge between 3iL of GPCR and R4-RGS proteins. By binding to RGS proteins and 3iL of GPCRs, SPL recruits RGS proteins to the GPCR signaling complex. NRB competes with SPL for binding to RGS proteins to remove them from the complex, which would be predicted to increase the intensity of Ca++ signaling.
3.6. Gβγ
Although members of the R7-RGS subfamily, which contain a G-protein γ-like (GGL) domain, can bind to Gβ5 in the absence of Gγ, the R4-RGS protein RGS3, which lacks the GGL domain, was found to interact with Gβ1γ2 both in vivo and in vitro. RGS3 co-immunoprecipitated with Gβγ from COS-7 cells overexpressing RGS3 and Gβγ, and recombinant His-RGS3 pulled down purified Gβγ subunits (Shi, et al., 2001). RGS3 inhibited Gβ1γ2 -stimulated IP production and activation of MAPK and Akt. Although mutational studies mapped two regions on RGS3 for binding to Gβγ located between amino acids 313 to 390 and 391 to 458, which are located within the RGS domain, binding of RGS3 to Gβγ did not require the complete RGS domain. Since the intact RGS box is required for Gα binding, RGS3 regulation of Gβγ is therefore independent of RGS3 GAP activity (Shi, et al., 2001).
3.7. Homer-2
Homer proteins are scaffolds that interact with GPCRs, IP3 receptors, and ryanodine receptors involved in Ca++ signaling. Homer 2 was found to increase the GAP activity of RGS4 in M1 mAch receptor-stimulated steady state GTP hydrolysis assays, although direct interaction of RGS4 and Homer 2 was not demonstrated (Shin, et al., 2003). Pancreatic acinar cells from Homer 2−/− mice showed enhanced carbachol-stimulated Gq-mediated Ca++ mobilization. RGS4 infusion into acinar cells from WT mice completely abolished the carbachol-induced Ca++ response, but had no effect on cells from Homer 2−/− mice. Although the detailed mechanisms remain unclear, these data suggested that Homer 2 fine-tunes Gq-mediated Ca++ signaling intensity by regulating the GAP activity of RGS4.
4. Interactions with non-GPCR signaling components
4.1. 14-3-3 proteins
14-3-3 are small dimeric proteins containing seven highly conserved isoforms whose functions are largely similar. 14-3-3 acts predominantly as scaffolding regulatory proteins through their interactions with components of diverse signaling pathways. 14-3-3 exerts its effects by binding either of two conserved phosphorylated motifs: RSXpSXP or RXY/FXpSSxP (Muslin, et al., 1996), although some protein interactions are phosphorylation-independent. Most of the R4 RGS subfamily members bind 14-3-3 including RGS3, RGS4, RGS5, and RGS16 (Abramow-Newerly, et al., 2006a; Benzing, et al., 2000; Ward and Milligan, 2005).
In some cases, binding of 14-3-3 with a conserved phosphorylated Ser residue within the RGS domain inhibits RGS GAP activity. In RGS7, Ser434, which is conserved in all RGS proteins within the RGS domain, was found to be critical for the binding of 14-3-3 proteins. Furthermore, this binding diminished the GAP activity of RGS7 when measured by single turnover GTP hydrolysis assay (Benzing, et al., 2000). However, later studies contradicted some of these findings. The conserved serine in the RGS domain appeared to be dispensable for the interaction of RGS proteins with 14-3-3; mutation of the corresponding Ser residue in RGS3 (RGS3/S496A) or RGS16 (RGS16/S166A) did not prevent binding to 14-3-3 in pulldown experiments using GST-14-3-3 and lysates of 293T cells expressing the mutant RGS proteins (Ward and Milligan, 2005). However, mutants lacking the conserved Ser within the RGS domain (RGS3/S496A, RGS1/S173A and RGS16/S166A, respectively) were unable to increase adrenaline-stimulated GTPase activity by a α2AAR-Cys351Ile-Go1 fusion protein expressed in membranes of HEK293 cells, suggesting impaired GAP activity of this mutant. Interestingly, two groups (Niu, et al., 2002; Ward and Milligan, 2005) have subsequently identified a Ser in the amino-terminus of RGS3 (Ser264, which lies outside the RGS domain) responsible for 14-3-3 binding. Mutation of Ser264 in RGS3 (S264D or S264A), disrupted the interaction in GST-14-3-3 and RGS proteins expressed in HEK293 cell lysates (Ward and Milligan, 2005) or in co-immunoprecipitation studies of CHO cells overexpressing myc-RGS3 (Niu, et al., 2002).
14-3-3 binding to RGS3 reduced the inhibitory effect of RGS3 on endothelin 1-induced, Gq-mediated activation of the transcription factor Elk1 (Niu, et al., 2002). These authors suggested that 14-3-3 may sequester RGS3 from Gqα as incubation of CHO cell lysates expressing myc-RGS3 with exogenous MBP-14-3-3 recombinant protein reduced the amount of Gα co-immunoprecipitated by anti-myc antibody dramatically (Niu, et al., 2002). Binding of 14-3-3 to RGS proteins appears to be direct as recombinant His614-3-3α efficiently purified GST-RGS4, GST-RGS5, and GST-RGS16 (Abramow-Newerly, et al., 2006a).
Functional consequences of the interaction of 14-3-3 with RGS proteins vary depending on the 14-3-3 isoform studied. Both 14-3-3ε and β inhibited the GAP activity of RGS4 and RGS16 but not RGS5 in steady-state GTP hydrolysis assays (Abramow-Newerly, et al., 2006a). 14-3-3τ had no effect on GAP activity of several RGS proteins including RGS3, RGS7 and RGS16 when tested on an agonist-stimulated α2AAR-Cys351Ile-Go1 fusion protein expressed in HEK293 cells (Benzing, et al., 2000). When GAP activities of these RGS proteins were measured by high-affinity GTPase assay in adrenaline-stimulated membrane preparations of HEK293 cells expressing α2AAR-Cys351Ile-Go1 fusion protein, 14-3-3τ had no effect on GAP activity (Ward and Milligan, 2005). By contrast, in a separate set of experiments, 14-3-3τ inhibited RGS7 GAP activity towards Gαi1 in a single turnover assay (Benzing, 2000), suggesting distinct effects of 14-3-3τ on individual RGS proteins. Another recent study showed that 14-3-3ε competes with activated Gαo for RGS4 binding and that overexpression of 14-3-3ε reversed Gαi-induced translocation of RGS4 from cytosol to plasma membrane (Abramow-Newerly, et al., 2006a). Thus, in certain instances depending on the 14-3-3 isoform involved, 14-3-3 may sequester RGS proteins from Gα, resulting in mis-localization within the cell and thus indirect inhibition of GAP activity.
4.2. PKG1-α
Nitric oxide (NO) inhibits vascular contraction by activating cGMP-dependent protein kinase G (PKG), leading to vascular smooth muscle relaxation. PKG1α has been shown to attenuate thrombin receptor-stimulated Gq-mediated IP formation through activation of RGS2 (Tang, et al., 2003). PKG1α directly binds and phosphorylates RGS2 at Ser 46 and Ser 64 sites. Phosphorylation of RGS2 by PKG1α enhanced its GAP activity on Gq, therefore inhibiting thrombin-stimulated signaling. As noted, Rgs2−/− mice are markedly hypertensive, and their blood vessels exhibit enhanced contraction, especially in response to angiotensin II. These findings support a novel mechanism whereby NO inhibits vascular contraction and blood pressure through PKG-mediated activation of RGS2.
4.3. Receptor tyrosine kinases (RTKs)
RGS16 has been shown to interact with epidermal growth factor receptor (EGFR) by co-immunoprecipitation (Derrien and Druey, 2001). Tyrosine phosphorylation of RGS16 by EGFR enhanced RGS16 GAP activity on Gαi in single turnover assays. Whether binding of RGS16 and EGFR was direct or indirect was not determined. However, mutation of EGFR target tyrosine residues in RGS16 severely reduced the capacity of RGS16 to regulate Gi-mediated (M2 mAch) MAP kinase activation or inhibition of adenylyl cyclase in HEK293T cells. RGS4 interacted with ErbB3 (EGFR family member) in yeast and human cells by yeast two-hybrid assay and co-immunoprecipitation, respectively (Thaminy, et al., 2003). Yeast two-hybrid analysis of RGS4 truncations mutants indicated that the residues located at the carboxy-terminus of the RGS box were important for binding to ErbB3. RGS5, which, as previously mentioned, is expressed in a pattern similar to PDGF receptors in pericytes, inhibited PDGFRβ-induced ERK-2 phosphorylation when overexpressed in NIH3T3 cells in addition to inhibiting GPCR (endothelin-1 and angiotensin II)-evoked Erk activation (Cho, et al., 2003). Although the molecular mechanism for the inhibition of PDGFβ-induced MAP kinase activation by RGS5 has not yet been determined, these three studies suggest potential cross-talk between GPCR- and RTK-induced signaling routes through RGS proteins such as RGS4, 5 and 16.
Activating mutations of RTKs cause malignant transformation of cells leading to cancers. RGS2 expression has recently been shown to be repressed by internal tandem duplications of the RTK fetal liver tyrosine kinase 3 (Flt3) gene (Flt3-ITD), a mutation which may cause acute myeloid leukemia (AML) (Schwable, et al., 2005). RGS2 mRNA was reduced by overexpression of Flt3 with activating mutations in two myeloid cell lines. Furthermore, RGS2 levels detected by real time PCR were decreased in the bone marrow of Flt3-ITD+ AML cases compared to Flt3-ITD− AML. Overexpression of RGS2 inhibited Flt3-ITD–induced transformation of myeloid cells and inhibited Flt3-ITD-induced phosphorylation of Akt and glycogen synthase kinase 3β (GSK3β). Although the mechanism for these observations has not been elucidated, these data suggest that RGS2 has a function in oncogenic cellular transformation induced by an RTK.
4.4. Steroid hormone receptors
Steroid hormone receptors are nuclear hormone receptors that belong to the superfamily of ligand-inducible transcription factors. These receptors are structurally related and are composed of six major functional domains. Domain A/B in the amino-terminal region is the constitutive activation domain 1 (AF-1). Domain C, the DNA-binding domain, is arranged in two zinc-stabilized DNA-binding finger motifs. Region D contains a nuclear localization signal. The ligand binding and ligand-dependent transcriptional activation function 2 (AF-2) is located in domain E/F of the carboxy-terminus of the protein. A yeast two-hybrid system using domains C and D of the rat estrogen receptor α (rERα C/D) as bait recovered a cDNA encoding a protein that was termed steroid receptor-binding (SRB) protein bearing the RGS domain at the carboxy terminus (SRB-RGS) from a rat ovary cDNA library (Ikeda, et al., 2001). The sequence of SRB-RGS was identical to RGS3. SRB-RGS/RGS3 interacted with ER-α by co-immunoprecipitation of [35S]methionine-labeled in vitro translated proteins, even in the presence of ER-α–DNA binding. Gel-shift assays using nuclear extracts from rat uterine cells bound to Xenopus vitellogenin A2 estrogen response element (VRE) oligonucleotides demonstrated supershifting with an anti-RGS3 antibody, suggesting that SRB-RGS interacted with ER-α bound to VRE (Ikeda, et al., 2007). Expression of SRB-RGS suppressed ERα and β transcriptional activity of a VRE-chloramphenicol acetyltransferase (CAT) reporter in COS-7 cells. SRB-RGS also induced apoptosis of Hela cells in the presence or absence of the caspase inhibitor Z-VAD-FMK. These studies suggested that SRB-RGS/RGS3 regulates estrogen-induced transcription as well as caspase-independent apoptosis.
4.5. Ephrin B and Eph receptors
The ephrin-Eph signaling pathway plays important roles in developmental processes, including axon pathway selection, angiogenesis, rhombomere compartmentalization and migration (Kullander and Klein, 2002). Eph molecules resemble RTKs because they contain a cytoplasmic kinase domain. Ephrin-Eph signaling is bidirectional with a forward signal through the Eph receptor and a reverse signal through the ligand (Bruckner, et al., 1997; Holland, et al., 1996). Due to reverse signaling, cell-attached ephrins communicate cell contact signals not only to cells expressing Eph molecules, but also cells expressing the ephrins through binding of their cytoplasmic domain with intracellular effector proteins. A yeast two-hybrid screen using the intracellular domain of ephrin B as bait revealed PDZ-RGS3 as a binding partner (Lu, et al., 2001). RGS3 interacted with ephrin B in a GST pull-down assay and in co-immunoprecipitations of either transfected or endogenous proteins. Reverse signaling from soluble EphB2-Fc receptor inhibited the effect of CXCL12 on cerebellar granule cells, indicating a control over neuronal migration. These authors hypothesized that PDZ-RGS3 binding to the cytoplasmic domain of ephrin B via its PDZ domain localizes RGS3 at the membrane where it inhibits G-protein signaling through its RGS domain. This result provides evidence for direct cross-talk between ephrin-Eph and G-protein dependent pathways. Since CXCL12 and its receptor CXCR4 are required for normal granule cell migration, PDZ-RGS3 may have an essential function in neuronal development.
Recently, Src homology 2 (SH2) domain-containing Grb4 has also been shown to interact with the intracellular portion of ephrin B (Su, et al., 2004). The SH2 domain of Grb4 and PDZ domain of PDZ-RGS3 bound Ephrin B phosphorylated at Tyr304 simultaneously and independently, forming a three component molecular complex. PDZ-RGS3 also bound neuroligin, a postsynaptic adhesion molecule that associates with β-neurexin in the synaptic cleft and has a function in glutamatergic synapses (Meyer, et al., 2004). Thus, RGS3 may have several functions in neuronal signaling through its RGS domain as well as a PDZ domain that binds several receptors.
4.6. COPI
COPI, a cytosolic protein complex consisting of seven subunits (α, β, β′, γ, δ, ε and ζ-COP) plus the monomeric GTPase, ARF1 (ADP-ribosylation factor), is an integral component of intracellular transport carriers. COPI is important for maintaining Golgi integrity and normal intracellular transport. RGS4 and RGS2 directly bound β′-COP and and the intact COPI complex in cell lysates (Sullivan, et al., 2000). These associations reduced COPI binding to Golgi membranes in vitro and diminished COPI-Golgi association in intact cells as determined by immunofluorescence. RGS4 overexpression inhibited secretion of alkaline phosphatase in HEK293T cells without affecting Golgi integrity. RGS4 bound COPI through two dilysine motifs in the RGS domain, but at a site not involving Gα binding. Thus, the RGS4-COPI interaction is independent of RGS4 GAP activity. Although these data need to be confirmed in native systems, additional studies showing presence of Gα subunits on intracellular organelle membranes (Denker, et al., 1996; Leyte, et al., 1992; Maier, et al., 1995; Slessareva, et al., 2006) suggest an interplay between G protein signaling, RGS proteins and COPI-mediated intracellular trafficking.
4.7. GIRK channels
G-protein regulated inward-rectifier potassium channels (GIRK or Kir3) are members of a superfamily of inward-rectifier K+ channels that includes seven family members. Four GIRK subunits, designated GIRK1-4 (also designated Kir3.1-4), have been identified in mammals which co-assemble to form neuronal and cardiac GIRK channels (Yamada, et al., 1998). GIRK5 was identified in Xenopus oocytes. GIRK channels exist in vivo both as homotetramers and heterotetramers. In contrast to the other mammalian GIRK family members, GIRK1 cannot form functional channels by itself and must co-assemble with GIRK2, 3 or 4. GIRK channels are modulated by heterotrimeric G proteins, phosphatidylinositol 4,5-bisphosphate (PIP2), intracellular sodium, ethanol and mechanical stretch (reviewed in (Mark and Herlitze, 2000).
Kir3 channels are activated following stimulation of GPCRs that utilize the Gi/o family of G proteins. Gβγ binds to and activates Kir3 channels (Huang, et al., 1995; Reuveny, et al., 1994; Wickman, et al., 1994). Gα GTPase activity is required for terminating Kir3 activation. RGS GAP activity thus accelerates GIRK channel “off” kinetics by promoting Gα deactivation. Somewhat unexpectedly, co-expression of RGS1, 3, and 4 but not RGS2 with GIRK channels and M2 mAch receptors in X. laevis oocytes or CHO cells accelerated agonist (carbachol)-evoked GIRK currents (Doupnik, et al., 1997). Several studies since then have shown that both the “on” and “off” kinetics of endogenous channels observed in tissues can be re-capitulated by expression of RGS proteins and heterologously-expressed GIRK channels.
Coexpression of RGS2, 5 and 8 accelerated mAchR-mediated GIRK kinetics in Xenopus oocytes (Herlitze, et al., 1999; Saitoh, et al., 1999) whereas GABAb receptor-activated GIRK currents in HEK293T cells were modulated by RGS4 (Fowler, et al., 2007; Mutneja, et al., 2005). Channels coupled to serotonin 1A and muscarinic M2 receptors in CHO-K1 cells were differentially regulated by the alternatively spliced short RGS3 isoform (RGS3s) (Jaen and Doupnik, 2005). RGS3s dramatically reduced steady-state M2-receptor-activated GIRK currents, but not 5-HT1A receptor-coupled currents. Thus, RGS3s modulated GIRK channels in a GPCR-selective manner whereas RGS4 modulated M2 and 5-HT1A-evoked GIRK channel activity with equal potency. RGS4 co-immunoprecipitated with multiple GPCR-Kir3 channel complexes expressed in CHO-K1 cells whereas RGS3s did not (Jaen and Doupnik, 2006). Both the RGS4 amino terminus and the RGS domain were necessary for complexation with M2-Kir3 channel complexes (Jaen and Doupnik, 2006). Precoupling of RGS4 to the GPCR-Kir3 complex lead to greater acceleration of G protein-dependent Kir3 channel gating kinetics than that was observed for RGS3s, which does not associate with the GPCR-Kir channel complex.
In the case of RGS8, GAP activity was not required for modulation of GIRK activation. Pertussis toxin (PTX)-treated rat sympathetic neurons expressing GIRK channels activated by Gβγ and a GαoA mutant resistant to both PTX and RGS GAP activity displayed markedly abnormal activation and deactivation kinetics and failed to desensitize to prolonged agonist (noradrenaline) stimulation (Jeong and Ikeda, 2001). Heterologous expression of RGS8 in neurons expressing the PTX- and RGS-insensitive GαoA construct restored rapid GIRK current activation kinetics in response to noradrenaline. Expression of a mutant RGS8 protein containing only the amino-terminus affected channel activation kinetics similar to full length RGS8 whereas expression of the RGS8 RGS domain alone had no effect. These results suggested that amino-terminus of RGS8 modulated GIRK current activation kinetics independent of GAP activity.
4.8. Calcium channels
Voltage-gated calcium channels are found at presynaptic and postsynaptic terminals and play an essential role in neurosecretion. The activities of the L-, N-, P/Q- and R- type channels are modulated by G-proteins, mainly Gi/o and Gq, which in turn are inhibited by RGS proteins. R4 RGS proteins including RGS2, 3, and 4 accelerate the onset and turnoff of transmitter-mediated inhibition (Gαz, Gαq and Gαi/o-mediated) of presynaptic calcium channels (Abramow-Newerly, et al., 2006b) in recombinant as well as endogenous systems. Electrophysiological recordings of hippocampal neurons from Rgs2−/− mice demonstrated that endogenous RGS2 increases synaptic transmitter release and thus synaptic plasticity (Han, et al., 2006a).
The RGS3s isoform also modulated G-protein-mediated synaptic transmission in dorsal root ganglion neurons by directly binding Ca++ through an EF hand domain (Tosetti, et al., 2003). The direct interaction of Ca++ to RGS3s was confirmed by calcium-induced gel mobility-shift. Overxpression of either RGS3s or RGS3ss, two endogenous isoforms detected in these neurons, terminated G-protein-dependent inhibition of calcium channels with differing kinetics. RGS3s rapidly desensitized G-protein-dependent inhibition of calcium channels. In contrast, expression of the RGS3ss mutant, which lacks the EF hand domain, produced a slower Ca++-dependent signal termination that was attenuated by a calmodulin antagonist. This result suggested distinct mechanisms of regulation of G-protein dependent inhibition of calcium channels by these RGS3 isoforms.
4.9. Calcium/calmodulin and anionic phospholipids
Levels of RGS proteins may be regulated by diverse extracellular cues depending on the cell type. Calcium-mediated signaling pathways have been implicated in regulation of RGS2 levels, and in turn RGS2 regulates Ca++ signaling (Popov, et al., 2000). RGS2 mRNA expression was increased in human blood mononuclear cells treated with ionomycin (Heximer, et al., 1997a) and in cultured myometrial cells in response to oxytocin (Park, et al., 2002). In human adrenocortical cells, angiotensin II upregulated RGS2 mRNA, and this effect was reversed by both a calcium channel blocker and by calmodulin and calcium/calmodulin-dependent kinase (CaMK) antagonists (Romero, et al., 2006).
Calmodulin regulates membrane association of many intracellular signaling molecules (Popov, et al., 2000). Calmodulin binds several RGS proteins and indirectly regulates their GAP activity. RGS1, 2, 4, 10, 16 and GAIP bound calmodulin in the presence of calcium, but calcium/calmodulin did not directly affect GAP activity of RGS proteins in single turnover assays (Popov, et al., 2000). In contrast, PIP3, but not other membrane phosphatidylinositol phosphates or synthetic phospholipids, bound RGS4 and inhibited its GAP activity in a concentration-dependent manner (Popov, et al., 2000). PIP3 inhibited GAP activity of several RGS proteins including RGS1, 2, 10 and GAIP but not RGS16. Furthermore, PIP3 inhibition of GAP activity was reversed by calcium/calmodulin, suggesting calcium/calmodulin competition with PIP3 as an intracellular mechanism for feedback regulation of RGS activity and thus calcium signaling. There are two potential calmodulin binding regions in RGS4, one in the amino-terminus (amino acids 1-33) and another in the RGS fold (helices 4–5, amino acids 99-113). An independent study demonstrated that PIP3 and calcium/calmodulin competitively bind to RGS4 within the RGS domain (Ishii, et al., 2005a). Mutation of RGS4 residues Lys99 and Lys100 impaired the interaction between the RGS box and PIP3/CaM in vitro and in intact HEK293 cells (Ishii, et al., 2005a; Ishii, et al., 2005b). The physiological importance of PIP3 regulation of RGS activity was demonstrated by analysis of Ach-induced GIRK channel activity in cardiac atrial myocytes by whole cell patch-clamp technique. PIP3 blocked the effect of RGS4 on GIRK channel activity, which was reversed by Ca++/CaM (Ishii, et al., 2002).
Other anionic phospholipids such as phosphatidic acid (PA) also inhibit RGS4 GAP activity (Ou-Yang, et al., 2003; Ouyang, et al., 2003) in single turnover and steady-state GTP hydrolysis assays. Deletion of the RGS4 amino-terminus ablated PA binding as well as PA-mediated functional inhibition. Lys20 was essential for PA-mediated inhibition of RGS4 GAP activity, while mutation of either Arg22 or Leu23 in the amino-terminal amphipathic α-helix of RGS4 reduced PA-mediated inhibition of RGS4 GAP activity. Thus, interaction of PA and RGS4 involves basic-charged stretches within RGS4 distinct from those required for RGS-PIP3 binding. Since PA is a product of the activities of phospholipase D (PLD) and diacylglycerol kinase (DGK) downstream of G-protein dependent pathways, these results suggest feedback regulation of RGS4 activity induced by GPCR stimulation.
5. Conclusions/Future Perspectives
The importance of the RGS domain for R4 RGS protein regulation of G-protein-mediated signals is undisputed. Nearly ten years ago, crystallographic analysis of RGS4 complexed with its substrate Gαi1 demonstrated that the RGS fold mediates direct binding to the G protein through key contact residues (Tesmer, et al., 1997). Biochemical studies utilizing recombinant proteins or ectopic expression of RGS proteins in numerous mammalian cell types confirmed their ability to negatively influence GPCR effector activation through GAP activity. What this characteristic translates to in the whole organism has been slowly forthcoming. Newer studies utilizing gene-targeted mice or RNAi have now demonstrated that in addition to specifying agonist threshold responses or mediating recovery from prolonged external stimulation, RGS proteins may influence numerous components of the signaling response independent of their GAP activity.
The amino terminus of several RGS proteins, which was disordered in the crystal structure, may direct the RGS domain to specific GPCR-G protein complexes by binding the receptor itself. Thus, depending on the duration and intensity of the external cue and cellular localization of the RGS at the time of stimulation (which may be somewhat fluid), an RGS protein could actually participate in a stable complex with GPCR and heterotrimeric G protein throughout the signaling process. Depending on the effector involved, such “kinetic” (or perhaps even physical) scaffolding may enhance activation. In turn, this concept raises the possibility that RGS proteins may be required for certain cellular responses.
Small molecules that selectively inhibit the G protein interactions of an individual RGS protein are now a reality (Kimple, et al., 2007; Roman, et al., 2007). Such compounds might be predicted to profoundly affect the efficacy and potency of existing GPCR-targeted therapeutic agents. In addition, the studies summarized in this review clearly indicate that many regions of RGS proteins, particularly their amino termini, also mediate interactions with a host of factors within and outside of GPCR signaling pathways. Drugs targeting these “off” sites could represent another novel class of therapeutic agents for many processes influenced by this large and diverse family of regulatory molecules.
Table I.
Major Sites of R4 RGS Expression and Associated Loss-of-Function Phenotype(s)
| RGS | Major Sites of Expression | Phenotypes | References |
|---|---|---|---|
| RGS1 | B lymphocytes | ↑ B cell chemotaxis to CXCL12, 13*
↑ B cell LN homing and motility w/in LN |
(Moratz, et al., 2004)
(Han, et al., 2005) |
| RGS2 | Widespread | ↓ T cell proliferation, cytokine production, antiviral immunity; ↑ basal hippocampal neuron activity; ↓ male aggression; ↑ anxiety
↑ Hippocampal neuron NT release Hypertension Abnormal renal solute handling ↑ Gq-mediated IP formation in cardiac myocytes (phenylephrine, ET-1)* ↑ ATP-induced ciliary beat frequency in lung epithelium |
(Oliveira-Dos-Santos, et al., 2000)
(Han, et al., 2006a) (Gross, et al., 2005; Heximer, et al., 2003) (Zuber, et al., 2007) (Zhang, et al., 2006) (Nlend, et al., 2002) |
| RGS3 | Widespread | ↑ M3 mAchR-evoked MAPK activation in aortic smooth muscle cells**
↑ Docetaxel-induced apoptosis of breast cancer cells* |
(Wang, et al., 2002)
(Ooe, et al., 2007) |
| RGS4 | Brain | ↑ pain threshold
↑ 5HT1A-evoked inhibition of NMDA channels in prefrontal cortical neurons*** |
(Grillet, et al., 2005)
(Gu, et al., 2007) |
| RGS5 | Vascular smooth muscle
Pericytes |
↑ Angiotensin II-induced MAPK activation in aortic smooth muscle cells** | (Wang, et al., 2002)
(Cho, et al., 2003) |
| RGS8 | Cerebellum | N/A | (Saitoh, et al., 2003) |
| RGS13 | B lymphocytes
Mast cells Neuroendocrine cells T lymphocytes Dendritic cells |
↑ Chemotaxis to CXCL12,13 in B cell line* | (Han, et al., 2006b)
Druey, et al., unpublished observations (Estes, et al., 2004) (Shi, et al., 2004) |
| RGS16 | Platelets
Liver T lymphocytes |
↑ Ca++ response to CXCL12 in megakaryocyte cell line* | (Berthebaud, et al., 2005)
(Huang, et al., 2006) (Beadling, et al., 1999; Johnson, et al., 2003) |
| RGS18 | Osteoclasts
Hematopoietic stem cells Platelets |
↑ RANKL-induced osteoclastogenesis* | (Iwai, et al., 2007)
(Park, et al., 2001; Yowe, et al., 2001) (Gagnon, et al., 2002) |
| RGS21 | Taste bud cells | N/A | (von Buchholtz, et al., 2004) |
Loss-of-function phenotypes in RGS-deficient cells. Studies of knockout mice or cells there from unless otherwise noted (
SiRNA;
Ribozyme;
Blocking antibody). N/A: not assessed; LN: lymph node
Table II.
Interacting Partners of R4-RGS Proteins within GPCR pathways
Table III.
Interacting Partners of R4-RGS Proteins outside of GPCR pathways
Abbreviations
- AC
adenylyl cyclase
- AR
adrenergic receptor
- AMF
aluminum magnesium fluoride
- EGFR
epidermal growth factor receptor
- GAP
GTPase accelerating protein
- PIP3
phosphatidylinositol (1,4,5) trisphosphate
- GIRK channel
G-protein regulated inward-rectifier potassium channels
- GPCR
G protein-coupled receptor
- 3iL
3rd intracellular loop
- NRB
neurabin
- mAch
muscarinic acetycholine
- PKA
protein kinase A
- PKG
protein kinase G
- PLC
phospholipase C
- RTK
receptor tyrosine kinases
- RGS
regulators of G protein signaling
- SPL
spinophilin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramow-Newerly M, Ming H, Chidiac P. Modulation of subfamily B/R4 RGS protein function by 14-3-3 proteins. Cell Signal. 2006a;18(12):2209–2222. doi: 10.1016/j.cellsig.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Abramow-Newerly M, Roy AA, Nunn C, Chidiac P. RGS proteins have a signalling complex: interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell Signal. 2006b;18(5):579–591. doi: 10.1016/j.cellsig.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Adams LD, Geary RL, Li J, Rossini A, Schwartz SM. Expression profiling identifies smooth muscle cell diversity within human intima and plaque fibrous cap: loss of RGS5 distinguishes the cap. Arterioscler Thromb Vasc Biol. 2006;26(2):319–325. doi: 10.1161/01.ATV.0000196647.45718.d6. [DOI] [PubMed] [Google Scholar]
- Agenes F, Bosco N, Mascarell L, Fritah S, Ceredig R. Differential expression of regulator of G-protein signalling transcripts and in vivo migration of CD4+ naive and regulatory T cells. Immunology. 2005;115(2):179–188. doi: 10.1111/j.1365-2567.2005.02146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5(9):943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- Anger T, Zhang W, Mende U. Differential contribution of GTPase activation and effector antagonism to the inhibitory effect of RGS proteins on Gq-mediated signaling in vivo. J Biol Chem. 2004;279(6):3906–3915. doi: 10.1074/jbc.M309496200. [DOI] [PubMed] [Google Scholar]
- Bakker SC, Hoogendoorn ML, Hendriks J, Verzijlbergen K, Caron S, Verduijn W, Selten JP, Pearson PL, Kahn RS, Sinke RJ. The PIP5K2A and RGS4 genes are differentially associated with deficit and non-deficit schizophrenia. Genes Brain Behav. 2007;6(2):113–119. doi: 10.1111/j.1601-183x.2006.00234.x. [DOI] [PubMed] [Google Scholar]
- Beadling C, Druey KM, Richter G, Kehrl JH, Smith KA. Regulators of G protein signaling exhibit distinct patterns of gene expression and target G protein specificity in human lymphocytes. J Immunol. 1999;162(5):2677–2682. [PubMed] [Google Scholar]
- Benians A, Leaney JL, Tinker A. Agonist unbinding from receptor dictates the nature of deactivation kinetics of G protein-gated K+ channels. Proc Natl Acad Sci U S A. 2003;100(10):6239–6244. doi: 10.1073/pnas.1037595100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benians A, Nobles M, Hosny S, Tinker A. Regulators of G-protein signaling form a quaternary complex with the agonist, receptor, and G-protein. A novel explanation for the acceleration of signaling activation kinetics. J Biol Chem. 2005;280(14):13383–13394. doi: 10.1074/jbc.M410163200. [DOI] [PubMed] [Google Scholar]
- Benzing T, Yaffe MB, Arnould T, Sellin L, Schermer B, Schilling B, Schreiber R, Kunzelmann K, Leparc GG, Kim E, Walz G. 14-3-3 interacts with regulator of G protein signaling proteins and modulates their activity. J Biol Chem. 2000;275(36):28167–28172. doi: 10.1074/jbc.M002905200. [DOI] [PubMed] [Google Scholar]
- Berger M, Bergers G, Arnold B, Hammerling GJ, Ganss R. Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood. 2005;105(3):1094–1101. doi: 10.1182/blood-2004-06-2315. [DOI] [PubMed] [Google Scholar]
- Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell. 1996;86(3):445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- Bernstein LS, Grillo AA, Loranger SS, Linder ME. RGS4 binds to membranes through an amphipathic alpha -helix. J Biol Chem. 2000;275(24):18520–18526. doi: 10.1074/jbc.M000618200. [DOI] [PubMed] [Google Scholar]
- Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI, Hepler JR. RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem. 2004;279(20):21248–21256. doi: 10.1074/jbc.M312407200. [DOI] [PubMed] [Google Scholar]
- Berthebaud M, Riviere C, Jarrier P, Foudi A, Zhang Y, Compagno D, Galy A, Vainchenker W, Louache F. RGS16 is a negative regulator of SDF-1-CXCR4 signaling in megakaryocytes. Blood. 2005;106(9):2962–2968. doi: 10.1182/blood-2005-02-0526. [DOI] [PubMed] [Google Scholar]
- Blumer KJ. Vision: the need for speed. Nature. 2004;427(6969):20–21. doi: 10.1038/427020a. [DOI] [PubMed] [Google Scholar]
- Bodenstein J, Sunahara RK, Neubig RR. N-terminal residues control proteasomal degradation of RGS2, RGS4, and RGS5 in human embryonic kidney 293 cells. Mol Pharmacol. 2007;71(4):1040–1050. doi: 10.1124/mol.106.029397. [DOI] [PubMed] [Google Scholar]
- Bondjers C, Kalen M, Hellstrom M, Scheidl SJ, Abramsson A, Renner O, Lindahl P, Cho H, Kehrl J, Betsholtz C. Transcription profiling of platelet-derived growth factor-B-deficient mouse embryos identifies RGS5 as a novel marker for pericytes and vascular smooth muscle cells. Am J Pathol. 2003;162(3):721–729. doi: 10.1016/S0002-9440(10)63868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden NA, Scott RJ, Tooney PA. Altered expression of regulator of G-protein signalling 4 (RGS4) mRNA in the superior temporal gyrus in schizophrenia. Schizophr Res. 2007;89(1–3):165–168. doi: 10.1016/j.schres.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science. 1997;275(5306):1640–1643. doi: 10.1126/science.275.5306.1640. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Meyer-Lindenberg A, Honea RA, Straub RE, Pezawas L, Egan MF, Vakkalanka R, Kolachana B, Verchinski BA, Sust S, Mattay VS, Weinberger DR, Callicott JH. Allelic variation in RGS4 impacts functional and structural connectivity in the human brain. J Neurosci. 2007;27(7):1584–1593. doi: 10.1523/JNEUROSCI.5112-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunemann M, Frank M, Lohse MJ. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci U S A. 2003;100(26):16077–16082. doi: 10.1073/pnas.2536719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo LA, Pagnin E, Davis PA, Sartori M, Ceolotto G, Pessina AC, Semplicini A. Increased expression of regulator of G protein signaling-2 (RGS-2) in Bartter’s/Gitelman’s syndrome. A role in the control of vascular tone and implication for hypertension. J Clin Endocrinol Metab. 2004;89(8):4153–4157. doi: 10.1210/jc.2004-0498. [DOI] [PubMed] [Google Scholar]
- Chen C, Zheng B, Han J, Lin SC. Characterization of a novel mammalian RGS protein that binds to Galpha proteins and inhibits pheromone signaling in yeast. J Biol Chem. 1997;272(13):8679–8685. doi: 10.1074/jbc.272.13.8679. [DOI] [PubMed] [Google Scholar]
- Chen CK, Wieland T, Simon MI. RGS-r, a retinal specific RGS protein, binds an intermediate conformation of transducin and enhances recycling. Proc Natl Acad Sci U S A. 1996;93(23):12885–12889. doi: 10.1073/pnas.93.23.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Kozasa T, Bondjers C, Betsholtz C, Kehrl JH. Pericyte-specific expression of Rgs5: implications for PDGF and EDG receptor signaling during vascular maturation. Faseb J. 2003;17(3):440–442. doi: 10.1096/fj.02-0340fje. [DOI] [PubMed] [Google Scholar]
- Chowdari KV, Bamne M, Joel W, Talkowski ME, Mirnics K, Levitt P, Lewis DA, Nimgaonkar VL. Linkage Disequilibrium Patterns and Functional Analysis of RGS4 Polymorphisms in Relation to Schizophrenia. Schizophr Bull. 2007 doi: 10.1093/schbul/sbm042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MA, Sethi PR, Lambert NA. Active Galpha(q) subunits and M3 acetylcholine receptors promote distinct modes of association of RGS2 with the plasma membrane. FEBS Lett. 2007;581(4):764–770. doi: 10.1016/j.febslet.2007.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dekker E, Hoenderop JG, Nilius B, Bindels RJ. The epithelial calcium channels, TRPV5 & TRPV6: from identification towards regulation. Cell Calcium. 2003;33(5–6):497–507. doi: 10.1016/s0143-4160(03)00065-4. [DOI] [PubMed] [Google Scholar]
- Denecke B, Meyerdierks A, Bottger EC. RGS1 is expressed in monocytes and acts as a GTPase-activating protein for G-protein-coupled chemoattractant receptors. J Biol Chem. 1999;274(38):26860–26868. doi: 10.1074/jbc.274.38.26860. [DOI] [PubMed] [Google Scholar]
- Denker SP, McCaffery JM, Palade GE, Insel PA, Farquhar MG. Differential distribution of alpha subunits and beta gamma subunits of heterotrimeric G proteins on Golgi membranes of the exocrine pancreas. J Cell Biol. 1996;133(5):1027–1040. doi: 10.1083/jcb.133.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien A, Druey KM. RGS16 function is regulated by epidermal growth factor receptor-mediated tyrosine phosphorylation. J Biol Chem. 2001;276(51):48532–48538. doi: 10.1074/jbc.M108862200. [DOI] [PubMed] [Google Scholar]
- Digby GJ, Lober RM, Sethi PR, Lambert NA. Some G protein heterotrimers physically dissociate in living cells. Proc Natl Acad Sci U S A. 2006;103(47):17789–17794. doi: 10.1073/pnas.0607116103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Guzman JN, Tkatch T, Chen S, Goldberg JA, Ebert PJ, Levitt P, Wilson CJ, Hamm HE, Surmeier DJ. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci. 2006;9(6):832–842. doi: 10.1038/nn1700. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Song J, Apanovitch DM, DiBello PR, Gillen KM. Regulation of G protein signalling in yeast. Semin Cell Dev Biol. 1998;9(2):135–141. doi: 10.1006/scdb.1998.0218. [DOI] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A. 1997;94(19):10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowal L, Elliott J, Popov S, Wilkie TM, Scarlata S. Determination of the contact energies between a regulator of G protein signaling and G protein subunits and phospholipase C beta 1. Biochemistry. 2001;40(2):414–421. doi: 10.1021/bi001923+. [DOI] [PubMed] [Google Scholar]
- Druey KM, Blumer KJ, Kang VH, Kehrl JH. Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature. 1996;379(6567):742–746. doi: 10.1038/379742a0. [DOI] [PubMed] [Google Scholar]
- Dulin NO, Pratt P, Tiruppathi C, Niu J, Voyno-Yasenetskaya T, Dunn MJ. Regulator of G protein signaling RGS3T is localized to the nucleus and induces apoptosis. J Biol Chem. 2000;275(28):21317–21323. doi: 10.1074/jbc.M910079199. [DOI] [PubMed] [Google Scholar]
- Ebert PJ, Campbell DB, Levitt P. Bacterial artificial chromosome transgenic analysis of dynamic expression patterns of regulator of G-protein signaling 4 during development. I. Cerebral cortex. Neuroscience. 2006;142(4):1145–1161. doi: 10.1016/j.neuroscience.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdely HA, Lahti RA, Lopez MB, Myers CS, Roberts RC, Tamminga CA, Vogel MW. Regional expression of RGS4 mRNA in human brain. Eur J Neurosci. 2004;19(11):3125–3128. doi: 10.1111/j.0953-816X.2004.03364.x. [DOI] [PubMed] [Google Scholar]
- Estes JD, Thacker TC, Hampton DL, Kell SA, Keele BF, Palenske EA, Druey KM, Burton GF. Follicular dendritic cell regulation of CXCR4-mediated germinal center CD4 T cell migration. J Immunol. 2004;173(10):6169–6178. doi: 10.4049/jimmunol.173.10.6169. [DOI] [PubMed] [Google Scholar]
- Fowler CE, Aryal P, Suen KF, Slesinger PA. Evidence for association of GABA(B) receptors with Kir3 channels and regulators of G protein signalling (RGS4) proteins. J Physiol. 2007;580(Pt 1):51–65. doi: 10.1113/jphysiol.2006.123216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Thumer L, Lohse MJ, Bunemann M. G Protein activation without subunit dissociation depends on a G{alpha}(i)-specific region. J Biol Chem. 2005;280(26):24584–24590. doi: 10.1074/jbc.M414630200. [DOI] [PubMed] [Google Scholar]
- Furuya M, Nishiyama M, Kimura S, Suyama T, Naya Y, Ito H, Nikaido T, Ishikura H. Expression of regulator of G protein signalling protein 5 (RGS5) in the tumour vasculature of human renal cell carcinoma. J Pathol. 2004;203(1):551–558. doi: 10.1002/path.1543. [DOI] [PubMed] [Google Scholar]
- Gagnon AW, Murray DL, Leadley RJ. Cloning and characterization of a novel regulator of G protein signalling in human platelets. Cell Signal. 2002;14(7):595–606. doi: 10.1016/s0898-6568(02)00012-8. [DOI] [PubMed] [Google Scholar]
- Garnier M, Zaratin PF, Ficalora G, Valente M, Fontanella L, Rhee MH, Blumer KJ, Scheideler MA. Up-regulation of regulator of G protein signaling 4 expression in a model of neuropathic pain and insensitivity to morphine. J Pharmacol Exp Ther. 2003;304(3):1299–1306. doi: 10.1124/jpet.102.043471. [DOI] [PubMed] [Google Scholar]
- Georgoussi Z, Leontiadis L, Mazarakou G, Merkouris M, Hyde K, Hamm H. Selective interactions between G protein subunits and RGS4 with the C-terminal domains of the mu- and delta-opioid receptors regulate opioid receptor signaling. Cell Signal. 2006;18(6):771–782. doi: 10.1016/j.cellsig.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Geurts M, Maloteaux JM, Hermans E. Altered expression of regulators of G-protein signaling (RGS) mRNAs in the striatum of rats undergoing dopamine depletion. Biochem Pharmacol. 2003;66(7):1163–1170. doi: 10.1016/s0006-2952(03)00447-7. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Gold SJ, Han MH, Herman AE, Ni YG, Pudiak CM, Aghajanian GK, Liu RJ, Potts BW, Mumby SM, Nestler EJ. Regulation of RGS proteins by chronic morphine in rat locus coeruleus. Eur J Neurosci. 2003;17(5):971–980. doi: 10.1046/j.1460-9568.2003.02529.x. [DOI] [PubMed] [Google Scholar]
- Gold SJ, Heifets BD, Pudiak CM, Potts BW, Nestler EJ. Regulation of regulators of G protein signaling mRNA expression in rat brain by acute and chronic electroconvulsive seizures. J Neurochem. 2002;82(4):828–838. doi: 10.1046/j.1471-4159.2002.01002.x. [DOI] [PubMed] [Google Scholar]
- Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26(22):3122–3142. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- Grafstein-Dunn E, Young KH, Cockett MI, Khawaja XZ. Regional distribution of regulators of G-protein signaling (RGS) 1, 2, 13, 14, 16, and GAIP messenger ribonucleic acids by in situ hybridization in rat brain. Brain Res Mol Brain Res. 2001;88(1–2):113–123. doi: 10.1016/s0169-328x(01)00038-9. [DOI] [PubMed] [Google Scholar]
- Grillet N, Dubreuil V, Dufour HD, Brunet JF. Dynamic expression of RGS4 in the developing nervous system and regulation by the neural type-specific transcription factor Phox2b. J Neurosci. 2003;23(33):10613–10621. doi: 10.1523/JNEUROSCI.23-33-10613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillet N, Pattyn A, Contet C, Kieffer BL, Goridis C, Brunet JF. Generation and characterization of Rgs4 mutant mice. Mol Cell Biol. 2005;25(10):4221–4228. doi: 10.1128/MCB.25.10.4221-4228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross V, Tank J, Obst M, Plehm R, Blumer KJ, Diedrich A, Jordan J, Luft FC. Autonomic nervous system and blood pressure regulation in RGS2-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1134–1142. doi: 10.1152/ajpregu.00246.2004. [DOI] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Yan Z. RGS4 modulates serotonin signaling in prefrontal cortex and links to serotonin dysfunction in a rat model of schizophrenia. Mol Pharmacol. 2007;71(4):1030–1039. doi: 10.1124/mol.106.032490. [DOI] [PubMed] [Google Scholar]
- Hague C, Bernstein LS, Ramineni S, Chen Z, Minneman KP, Hepler JR. Selective inhibition of alpha1A-adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J Biol Chem. 2005;280(29):27289–27295. doi: 10.1074/jbc.M502365200. [DOI] [PubMed] [Google Scholar]
- Han J, Mark MD, Li X, Xie M, Waka S, Rettig J, Herlitze S. RGS2 determines short-term synaptic plasticity in hippocampal neurons by regulating Gi/o-mediated inhibition of presynaptic Ca2+ channels. Neuron. 2006a;51(5):575–586. doi: 10.1016/j.neuron.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Han JI, Huang NN, Kim DU, Kehrl JH. RGS1 and RGS13 mRNA silencing in a human B lymphoma line enhances responsiveness to chemoattractants and impairs desensitization. J Leukoc Biol. 2006b;79(6):1357–1368. doi: 10.1189/jlb.1105693. [DOI] [PubMed] [Google Scholar]
- Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, Schwartz O, Kehrl JH. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity. 2005;22(3):343–354. doi: 10.1016/j.immuni.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Hao J, Michalek C, Zhang W, Zhu M, Xu X, Mende U. Regulation of cardiomyocyte signaling by RGS proteins: differential selectivity towards G proteins and susceptibility to regulation. J Mol Cell Cardiol. 2006;41(1):51–61. doi: 10.1016/j.yjmcc.2006.04.003. [DOI] [PubMed] [Google Scholar]
- He W, Cowan CW, Wensel TG. RGS9, a GTPase accelerator for phototransduction. Neuron. 1998;20(1):95–102. doi: 10.1016/s0896-6273(00)80437-7. [DOI] [PubMed] [Google Scholar]
- Hein P, Frank M, Hoffmann C, Lohse MJ, Bunemann M. Dynamics of receptor/G protein coupling in living cells. Embo J. 2005;24(23):4106–4114. doi: 10.1038/sj.emboj.7600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo K, Ha SH, Chae YC, Lee S, Oh YS, Kim YH, Kim SH, Kim JH, Mizoguchi A, Itoh TJ, Kwon HM, Ryu SH, Suh PG. RGS2 promotes formation of neurites by stimulating microtubule polymerization. Cell Signal. 2006;18(12):2182–2192. doi: 10.1016/j.cellsig.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Hepler JR, Berman DM, Gilman AG, Kozasa T. RGS4 and GAIP are GTPase-activating proteins for Gq alpha and block activation of phospholipase C beta by gamma-thio-GTP-Gq alpha. Proc Natl Acad Sci U S A. 1997;94(2):428–432. doi: 10.1073/pnas.94.2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Ruppersberg JP, Mark MD. New roles for RGS2, 5 and 8 on the ratio-dependent modulation of recombinant GIRK channels expressed in Xenopus oocytes. J Physiol. 1999;517(Pt 2):341–352. doi: 10.1111/j.1469-7793.1999.0341t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heximer SP, Cristillo AD, Forsdyke DR. Comparison of mRNA expression of two regulators of G-protein signaling, RGS1/BL34/1R20 and RGS2/G0S8, in cultured human blood mononuclear cells. DNA Cell Biol. 1997a;16(5):589–598. doi: 10.1089/dna.1997.16.589. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N, Oliveira-dos-Santos A, Penninger JM, Muslin AJ, Steinberg TH, Wyss JM, Mecham RP, Blumer KJ. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J Clin Invest. 2003;111(4):445–452. doi: 10.1172/JCI15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heximer SP, Srinivasa SP, Bernstein LS, Bernard JL, Linder ME, Hepler JR, Blumer KJ. G protein selectivity is a determinant of RGS2 function. J Biol Chem. 1999;274(48):34253–34259. doi: 10.1074/jbc.274.48.34253. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc Natl Acad Sci U S A. 1997b;94(26):14389–14393. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Ward RJ, Cavalli A, Carr IC, Milligan G. Differential capacities of the RGS1, RGS16 and RGS-GAIP regulators of G protein signaling to enhance alpha2A-adrenoreceptor agonist-stimulated GTPase activity of G(o1)alpha. J Neurochem. 2001;78(4):797–806. doi: 10.1046/j.1471-4159.2001.00479.x. [DOI] [PubMed] [Google Scholar]
- Holland SJ, Gale NW, Mbamalu G, Yancopoulos GD, Henkemeyer M, Pawson T. Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature. 1996;383(6602):722–725. doi: 10.1038/383722a0. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54(3):527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- Hong JX, Wilson GL, Fox CH, Kehrl JH. Isolation and characterization of a novel B cell activation gene. J Immunol. 1993;150(9):3895–3904. [PubMed] [Google Scholar]
- Huang C, Hepler JR, Gilman AG, Mumby SM. Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc Natl Acad Sci U S A. 1997;94(12):6159–6163. doi: 10.1073/pnas.94.12.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15(5):1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Huang J, Pashkov V, Kurrasch DM, Yu K, Gold SJ, Wilkie TM. Feeding and fasting controls liver expression of a regulator of G protein signaling (Rgs16) in periportal hepatocytes. Comp Hepatol. 2006;5:8. doi: 10.1186/1476-5926-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhou H, Mahavadi S, Sriwai W, Murthy KS. Inhibition of Galphaq-dependent PLC-beta1 activity by PKG and PKA is mediated by phosphorylation of RGS4 and GRK2. Am J Physiol Cell Physiol. 2007;292(1):C200–208. doi: 10.1152/ajpcell.00103.2006. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Hirokawa M, Satani N, Kinoshita T, Watanabe Y, Inoue H, Tone S, Ishikawa T, Minatogawa Y. Molecular cloning and characterization of a steroid receptor-binding regulator of G-protein signaling protein cDNA. Gene. 2001;273(2):207–214. doi: 10.1016/s0378-1119(01)00589-3. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Inoue S, Muramatsu M, Minatogawa Y. Characterization and identification of a steroid receptor-binding protein, SRB-RGS. Biol Pharm Bull. 2007;30(6):1056–1064. doi: 10.1248/bpb.30.1056. [DOI] [PubMed] [Google Scholar]
- Ingi T, Krumins AM, Chidiac P, Brothers GM, Chung S, Snow BE, Barnes CA, Lanahan AA, Siderovski DP, Ross EM, Gilman AG, Worley PF. Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. J Neurosci. 1998;18(18):7178–7188. doi: 10.1523/JNEUROSCI.18-18-07178.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Fujita S, Yamada M, Hosaka Y, Kurachi Y. Phosphatidylinositol 3,4,5-trisphosphate and Ca2+/calmodulin competitively bind to the regulators of G-protein-signalling (RGS) domain of RGS4 and reciprocally regulate its action. Biochem J. 2005a;385(Pt 1):65–73. doi: 10.1042/BJ20040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Ikushima M, Kurachi Y. In vivo interaction between RGS4 and calmodulin visualized with FRET techniques: possible involvement of lipid raft. Biochem Biophys Res Commun. 2005b;338(2):839–846. doi: 10.1016/j.bbrc.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Ishii M, Inanobe A, Kurachi Y. PIP3 inhibition of RGS protein and its reversal by Ca2+/calmodulin mediate voltage-dependent control of the G protein cycle in a cardiac K+ channel. Proc Natl Acad Sci U S A. 2002;99(7):4325–4330. doi: 10.1073/pnas.072073399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam TC, Asplund AC, Lindvall JM, Nygren L, Liden J, Kimby E, Christensson B, Smith CI, Sander B. High level of cannabinoid receptor 1, absence of regulator of G protein signalling 13 and differential expression of Cyclin D1 in mantle cell lymphoma. Leukemia. 2003;17(9):1880–1890. doi: 10.1038/sj.leu.2403057. [DOI] [PubMed] [Google Scholar]
- Itoh M, Nagatomo K, Kubo Y, Saitoh O. Alternative splicing of RGS8 gene changes the binding property to the M1 muscarinic receptor to confer receptor type-specific Gq regulation. J Neurochem. 2006;99(6):1505–1516. doi: 10.1111/j.1471-4159.2006.04220.x. [DOI] [PubMed] [Google Scholar]
- Iwai K, Koike M, Ohshima S, Miyatake K, Uchiyama Y, Saeki Y, Ishii M. RGS18 Acts as a Negative Regulator of Osteoclastogenesis by Modulating Acid-Sensing OGR1/NFAT Signaling Pathway. J Bone Miner Res. 2007 doi: 10.1359/jbmr.070612. [DOI] [PubMed] [Google Scholar]
- Jaen C, Doupnik CA. Neuronal Kir3.1/Kir3.2a channels coupled to serotonin 1A and muscarinic m2 receptors are differentially modulated by the “short” RGS3 isoform. Neuropharmacology. 2005;49(4):465–476. doi: 10.1016/j.neuropharm.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Jaen C, Doupnik CA. RGS3 and RGS4 differentially associate with G protein-coupled receptor-Kir3 channel signaling complexes revealing two modes of RGS modulation. Precoupling and collision coupling. J Biol Chem. 2006;281(45):34549–34560. doi: 10.1074/jbc.M603177200. [DOI] [PubMed] [Google Scholar]
- Jean-Baptiste G, Li X, Yang Z, Heubach J, Gaudio S, Khoury C, Ravens U, Greenwood MT. Beta adrenergic receptor-mediated atrial specific up-regulation of RGS5. Life Sci. 2005;76(13):1533–1545. doi: 10.1016/j.lfs.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. Differential regulation of G protein-gated inwardly rectifying K(+) channel kinetics by distinct domains of RGS8. J Physiol. 2001;535(Pt 2):335–347. doi: 10.1111/j.1469-7793.2001.00335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EN, Druey KM. Functional characterization of the G protein regulator RGS13. J Biol Chem. 2002;277(19):16768–16774. doi: 10.1074/jbc.M200751200. [DOI] [PubMed] [Google Scholar]
- Johnson EN, Seasholtz TM, Waheed AA, Kreutz B, Suzuki N, Kozasa T, Jones TL, Brown JH, Druey KM. RGS16 inhibits signalling through the G alpha 13-Rho axis. Nat Cell Biol. 2003;5(12):1095–1103. doi: 10.1038/ncb1065. [DOI] [PubMed] [Google Scholar]
- Johnson KD, Boyer ME, Kang JA, Wickrema A, Cantor AB, Bresnick EH. Friend of GATA-1-independent transcriptional repression: a novel mode of GATA-1 function. Blood. 2007;109(12):5230–5233. doi: 10.1182/blood-2007-02-072983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrl JH, Sinnarajah S. RGS2: a multifunctional regulator of G-protein signaling. Int J Biochem Cell Biol. 2002;34(5):432–438. doi: 10.1016/s1357-2725(01)00141-8. [DOI] [PubMed] [Google Scholar]
- Kehrl JH, Srikumar D, Harrison K, Wilson GL, Shi CS. Additional 5′ exons in the RGS3 locus generate multiple mRNA transcripts, one of which accounts for the origin of human PDZ-RGS3. Genomics. 2002;79(6):860–868. doi: 10.1006/geno.2002.6773. [DOI] [PubMed] [Google Scholar]
- Kim SD, Sung HJ, Park SK, Kim TW, Park SC, Kim SK, Cho JY, Rhee MH. The expression patterns of RGS transcripts in platelets. Platelets. 2006;17(7):493–497. doi: 10.1080/09537100600758123. [DOI] [PubMed] [Google Scholar]
- Kimple AJ, Willard FS, Giguere PM, Johnston CA, Mocanu V, Siderovski DP. The RGS protein inhibitor CCG-4986 is a covalent modifier of the RGS4 Galpha-interaction face. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbapap.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3(7):475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- Kurrasch DM, Huang J, Wilkie TM, Repa JJ. Quantitative real-time polymerase chain reaction measurement of regulators of G-protein signaling mRNA levels in mouse tissues. Methods Enzymol. 2004;389:3–15. doi: 10.1016/S0076-6879(04)89001-3. [DOI] [PubMed] [Google Scholar]
- Kveberg L, Ryan JC, Rolstad B, Inngjerdingen M. Expression of regulator of G protein signalling proteins in natural killer cells, and their modulation by Ly49A and Ly49D. Immunology. 2005;115(3):358–365. doi: 10.1111/j.1365-2567.2005.02174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladds G, Goddard A, Hill C, Thornton S, Davey J. Differential effects of RGS proteins on G alpha(q) and G alpha(11) activity. Cell Signal. 2007;19(1):103–113. doi: 10.1016/j.cellsig.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Lan KL, Sarvazyan NA, Taussig R, Mackenzie RG, DiBello PR, Dohlman HG, Neubig RR. A point mutation in Galphao and Galphai1 blocks interaction with regulator of G protein signaling proteins. J Biol Chem. 1998;273(21):12794–12797. doi: 10.1074/jbc.273.21.12794. [DOI] [PubMed] [Google Scholar]
- Larminie C, Murdock P, Walhin JP, Duckworth M, Blumer KJ, Scheideler MA, Garnier M. Selective expression of regulators of G-protein signaling (RGS) in the human central nervous system. Brain Res Mol Brain Res. 2004;122(1):24–34. doi: 10.1016/j.molbrainres.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Leyte A, Barr FA, Kehlenbach RH, Huttner WB. Multiple trimeric G-proteins on the trans-Golgi network exert stimulatory and inhibitory effects on secretory vesicle formation. Embo J. 1992;11(13):4795–4804. doi: 10.1002/j.1460-2075.1992.tb05585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Adams LD, Wang X, Pabon L, Schwartz SM, Sane DC, Geary RL. Regulator of G protein signaling 5 marks peripheral arterial smooth muscle cells and is downregulated in atherosclerotic plaque. J Vasc Surg. 2004;40(3):519–528. doi: 10.1016/j.jvs.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Li X, Chen L, Ji C, Liu B, Gu J, Xu J, Zou X, Gu S, Mao Y. Isolation and expression pattern of RGS21 gene, a novel RGS member. Acta Biochim Pol. 2005;52(4):943–946. [PubMed] [Google Scholar]
- Liang Y, Li C, Guzman VM, Chang WW, Evinger AJ, 3rd, Sao D, Woodward DF. Identification of a novel alternative splicing variant of RGS5 mRNA in human ocular tissues. Febs J. 2005;272(3):791–799. doi: 10.1111/j.1742-4658.2004.04516.x. [DOI] [PubMed] [Google Scholar]
- Liu W, Yuen EY, Allen PB, Feng J, Greengard P, Yan Z. Adrenergic modulation of NMDA receptors in prefrontal cortex is differentially regulated by RGS proteins and spinophilin. Proc Natl Acad Sci U S A. 2006;103(48):18338–18343. doi: 10.1073/pnas.0604560103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Sun EE, Klein RS, Flanagan JG. Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell. 2001;105(1):69–79. doi: 10.1016/s0092-8674(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Maier O, Ehmsen E, Westermann P. Trimeric G protein alpha subunits of the Gs and Gi families localized at the Golgi membrane. Biochem Biophys Res Commun. 1995;208(1):135–143. doi: 10.1006/bbrc.1995.1315. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci. 2001;22(7):368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- Mark MD, Herlitze S. G-protein mediated gating of inward-rectifier K+ channels. Eur J Biochem. 2000;267(19):5830–5836. doi: 10.1046/j.1432-1327.2000.01670.x. [DOI] [PubMed] [Google Scholar]
- Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology. 2004;47(5):724–733. doi: 10.1016/j.neuropharm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Mittmann C, Chung CH, Hoppner G, Michalek C, Nose M, Schuler C, Schuh A, Eschenhagen T, Weil J, Pieske B, Hirt S, Wieland T. Expression of ten RGS proteins in human myocardium: functional characterization of an upregulation of RGS4 in heart failure. Cardiovasc Res. 2002;55(4):778–786. doi: 10.1016/s0008-6363(02)00459-5. [DOI] [PubMed] [Google Scholar]
- Moratz C, Hayman JR, Gu H, Kehrl JH. Abnormal B-cell responses to chemokines, disturbed plasma cell localization, and distorted immune tissue architecture in Rgs1−/− mice. Mol Cell Biol. 2004;24(13):5767–5775. doi: 10.1128/MCB.24.13.5767-5775.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moratz C, Kang VH, Druey KM, Shi CS, Scheschonka A, Murphy PM, Kozasa T, Kehrl JH. Regulator of G protein signaling 1 (RGS1) markedly impairs Gi alpha signaling responses of B lymphocytes. J Immunol. 2000;164(4):1829–1838. doi: 10.4049/jimmunol.164.4.1829. [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84(6):889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Mutneja M, Berton F, Suen KF, Luscher C, Slesinger PA. Endogenous RGS proteins enhance acute desensitization of GABA(B) receptor-activated GIRK currents in HEK-293T cells. Pflugers Arch. 2005;450(1):61–73. doi: 10.1007/s00424-004-1367-1. [DOI] [PubMed] [Google Scholar]
- Nagata Y, Oda M, Nakata H, Shozaki Y, Kozasa T, Todokoro K. A novel regulator of G-protein signaling bearing GAP activity for Galphai and Galphaq in megakaryocytes. Blood. 2001;97(10):3051–3060. doi: 10.1182/blood.v97.10.3051. [DOI] [PubMed] [Google Scholar]
- Neer EJ. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80(2):249–257. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- Neitzel KL, Hepler JR. Cellular mechanisms that determine selective RGS protein regulation of G protein-coupled receptor signaling. Semin Cell Dev Biol. 2006;17(3):383–389. doi: 10.1016/j.semcdb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Niu J, Scheschonka A, Druey KM, Davis A, Reed E, Kolenko V, Bodnar R, Voyno-Yasenetskaya T, Du X, Kehrl J, Dulin NO. RGS3 interacts with 14-3-3 via the N-terminal region distinct from the RGS (regulator of G-protein signalling) domain. Biochem J. 2002;365(Pt 3):677–684. doi: 10.1042/BJ20020390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nlend MC, Bookman RJ, Conner GE, Salathe M. Regulator of G-protein signaling protein 2 modulates purinergic calcium and ciliary beat frequency responses in airway epithelia. Am J Respir Cell Mol Biol. 2002;27(4):436–445. doi: 10.1165/rcmb.2002-0012OC. [DOI] [PubMed] [Google Scholar]
- Oliveira-Dos-Santos AJ, Matsumoto G, Snow BE, Bai D, Houston FP, Whishaw IQ, Mariathasan S, Sasaki T, Wakeham A, Ohashi PS, Roder JC, Barnes CA, Siderovski DP, Penninger JM. Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc Natl Acad Sci U S A. 2000;97(22):12272–12277. doi: 10.1073/pnas.220414397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooe A, Kato K, Noguchi S. Possible involvement of CCT5, RGS3, and YKT6 genes up-regulated in p53-mutated tumors in resistance to docetaxel in human breast cancers. Breast Cancer Res Treat. 2007;101(3):305–315. doi: 10.1007/s10549-006-9293-x. [DOI] [PubMed] [Google Scholar]
- Ou-Yang YS, Tu Y, Yang F. The mutation in the N-terminal domain of RGS4 disrupts PA-conferred inhibitory effect on GAP activity. Biosci Rep. 2003;23(4):213–224. doi: 10.1023/b:bire.0000007694.71158.02. [DOI] [PubMed] [Google Scholar]
- Ouyang YS, Tu Y, Barker SA, Yang F. Regulators of G-protein signaling (RGS) 4, insertion into model membranes and inhibition of activity by phosphatidic acid. J Biol Chem. 2003;278(13):11115–11122. doi: 10.1074/jbc.M212606200. [DOI] [PubMed] [Google Scholar]
- Owen VJ, Burton PB, Mullen AJ, Birks EJ, Barton P, Yacoub MH. Expression of RGS3, RGS4 and Gi alpha 2 in acutely failing donor hearts and end-stage heart failure. Eur Heart J. 2001;22(12):1015–1020. doi: 10.1053/euhj.2000.2578. [DOI] [PubMed] [Google Scholar]
- Park ES, Echetebu CO, Soloff S, Soloff MS. Oxytocin stimulation of RGS2 mRNA expression in cultured human myometrial cells. Am J Physiol Endocrinol Metab. 2002;282(3):E580–584. doi: 10.1152/ajpendo.00437.2001. [DOI] [PubMed] [Google Scholar]
- Park IK, Klug CA, Li K, Jerabek L, Li L, Nanamori M, Neubig RR, Hood L, Weissman IL, Clarke MF. Molecular cloning and characterization of a novel regulator of G-protein signaling from mouse hematopoietic stem cells. J Biol Chem. 2001;276(2):915–923. doi: 10.1074/jbc.M005947200. [DOI] [PubMed] [Google Scholar]
- Patten M, Bunemann J, Thoma B, Kramer E, Thoenes M, Stube S, Mittmann C, Wieland T. Endotoxin induces desensitization of cardiac endothelin-1 receptor signaling by increased expression of RGS4 and RGS16. Cardiovasc Res. 2002;53(1):156–164. doi: 10.1016/s0008-6363(01)00443-6. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Popov S, Yu K, Kozasa T, Wilkie TM. The regulators of G protein signaling (RGS) domains of RGS4, RGS10, and GAIP retain GTPase activating protein activity in vitro. Proc Natl Acad Sci U S A. 1997;94(14):7216–7220. doi: 10.1073/pnas.94.14.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov SG, Krishna UM, Falck JR, Wilkie TM. Ca2+/Calmodulin reverses phosphatidylinositol 3,4, 5-trisphosphate-dependent inhibition of regulators of G protein-signaling GTPase-activating protein activity. J Biol Chem. 2000;275(25):18962–18968. doi: 10.1074/jbc.M001128200. [DOI] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature. 1994;370(6485):143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- Riddle EL, Rana BK, Murthy KK, Rao F, Eskin E, O’Connor DT, Insel PA. Polymorphisms and haplotypes of the regulator of G protein signaling-2 gene in normotensives and hypertensives. Hypertension. 2006;47(3):415–420. doi: 10.1161/01.HYP.0000200714.81990.61. [DOI] [PubMed] [Google Scholar]
- Rogers JH, Tamirisa P, Kovacs A, Weinheimer C, Courtois M, Blumer KJ, Kelly DP, Muslin AJ. RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J Clin Invest. 1999;104(5):567–576. doi: 10.1172/JCI6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig RR. Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol Pharmacol. 2007;71(1):169–175. doi: 10.1124/mol.106.028670. [DOI] [PubMed] [Google Scholar]
- Romero DG, Plonczynski MW, Gomez-Sanchez EP, Yanes LL, Gomez-Sanchez CE. RGS2 is regulated by angiotensin II and functions as a negative feedback of aldosterone production in H295R human adrenocortical cells. Endocrinology. 2006;147(8):3889–3897. doi: 10.1210/en.2005-1532. [DOI] [PubMed] [Google Scholar]
- Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- Roy AA, Baragli A, Bernstein LS, Hepler JR, Hebert TE, Chidiac P. RGS2 interacts with Gs and adenylyl cyclase in living cells. Cell Signal. 2006a;18(3):336–348. doi: 10.1016/j.cellsig.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Roy AA, Lemberg KE, Chidiac P. Recruitment of RGS2 and RGS4 to the plasma membrane by G proteins and receptors reflects functional interactions. Mol Pharmacol. 2003;64(3):587–593. doi: 10.1124/mol.64.3.587. [DOI] [PubMed] [Google Scholar]
- Roy AA, Nunn C, Ming H, Zou MX, Penninger J, Kirshenbaum LA, Dixon SJ, Chidiac P. Up-regulation of endogenous RGS2 mediates cross-desensitization between Gs and Gq signaling in osteoblasts. J Biol Chem. 2006b;281(43):32684–32693. doi: 10.1074/jbc.M604416200. [DOI] [PubMed] [Google Scholar]
- Saitoh O, Kubo Y, Miyatani Y, Asano T, Nakata H. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 1997;390(6659):525–529. doi: 10.1038/37385. [DOI] [PubMed] [Google Scholar]
- Saitoh O, Kubo Y, Odagiri M, Ichikawa M, Yamagata K, Sekine T. RGS7 and RGS8 differentially accelerate G protein-mediated modulation of K+ currents. J Biol Chem. 1999;274(14):9899–9904. doi: 10.1074/jbc.274.14.9899. [DOI] [PubMed] [Google Scholar]
- Saitoh O, Masuho I, Itoh M, Abe H, Komori K, Odagiri M. Distribution of regulator of G protein signaling 8 (RGS8) protein in the cerebellum. Cerebellum. 2003;2(2):154–160. doi: 10.1080/14734220309409. [DOI] [PubMed] [Google Scholar]
- Saitoh O, Masuho I, Terakawa I, Nomoto S, Asano T, Kubo Y. Regulator of G protein signaling 8 (RGS8) requires its NH2 terminus for subcellular localization and acute desensitization of G protein-gated K+ channels. J Biol Chem. 2001;276(7):5052–5058. doi: 10.1074/jbc.M006917200. [DOI] [PubMed] [Google Scholar]
- Saitoh O, Murata Y, Odagiri M, Itoh M, Itoh H, Misaka T, Kubo Y. Alternative splicing of RGS8 gene determines inhibitory function of receptor type-specific Gq signaling. Proc Natl Acad Sci U S A. 2002;99(15):10138–10143. doi: 10.1073/pnas.152085999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh O, Odagiri M. RGS8 expression in developing cerebellar Purkinje cells. Biochem Biophys Res Commun. 2003;309(4):836–842. doi: 10.1016/j.bbrc.2003.08.083. [DOI] [PubMed] [Google Scholar]
- Salim S, Sinnarajah S, Kehrl JH, Dessauer CW. Identification of RGS2 and type V adenylyl cyclase interaction sites. J Biol Chem. 2003;278(18):15842–15849. doi: 10.1074/jbc.M210663200. [DOI] [PubMed] [Google Scholar]
- Sarrouilhe D, di Tommaso A, Metaye T, Ladeveze V. Spinophilin: from partners to functions. Biochimie. 2006;88(9):1099–1113. doi: 10.1016/j.biochi.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Schoeber JP, Topala CN, Wang X, Diepens RJ, Lambers TT, Hoenderop JG, Bindels RJ. RGS2 inhibits the epithelial Ca2+ channel TRPV6. J Biol Chem. 2006;281(40):29669–29674. doi: 10.1074/jbc.M606233200. [DOI] [PubMed] [Google Scholar]
- Schwable J, Choudhary C, Thiede C, Tickenbrock L, Sargin B, Steur C, Rehage M, Rudat A, Brandts C, Berdel WE, Muller-Tidow C, Serve H. RGS2 is an important target gene of Flt3-ITD mutations in AML and functions in myeloid differentiation and leukemic transformation. Blood. 2005;105(5):2107–2114. doi: 10.1182/blood-2004-03-0940. [DOI] [PubMed] [Google Scholar]
- Semplicini A, Lenzini L, Sartori M, Papparella I, Calo LA, Pagnin E, Strapazzon G, Benna C, Costa R, Avogaro A, Ceolotto G, Pessina AC. Reduced expression of regulator of G-protein signaling 2 (RGS2) in hypertensive patients increases calcium mobilization and ERK1/2 phosphorylation induced by angiotensin II. J Hypertens. 2006;24(6):1115–1124. doi: 10.1097/01.hjh.0000226202.80689.8f. [DOI] [PubMed] [Google Scholar]
- Shi CS, Lee SB, Sinnarajah S, Dessauer CW, Rhee SG, Kehrl JH. Regulator of G-protein signaling 3 (RGS3) inhibits Gbeta1gamma 2-induced inositol phosphate production, mitogen-activated protein kinase activation, and Akt activation. J Biol Chem. 2001;276(26):24293–24300. doi: 10.1074/jbc.M100089200. [DOI] [PubMed] [Google Scholar]
- Shi GX, Harrison K, Han SB, Moratz C, Kehrl JH. Toll-like receptor signaling alters the expression of regulator of G protein signaling proteins in dendritic cells: implications for G protein-coupled receptor signaling. J Immunol. 2004;172(9):5175–5184. doi: 10.4049/jimmunol.172.9.5175. [DOI] [PubMed] [Google Scholar]
- Shi GX, Harrison K, Wilson GL, Moratz C, Kehrl JH. RGS13 regulates germinal center B lymphocytes responsiveness to CXC chemokine ligand (CXCL)12 and CXCL13. J Immunol. 2002;169(5):2507–2515. doi: 10.4049/jimmunol.169.5.2507. [DOI] [PubMed] [Google Scholar]
- Shin DM, Dehoff M, Luo X, Kang SH, Tu J, Nayak SK, Ross EM, Worley PF, Muallem S. Homer 2 tunes G protein-coupled receptors stimulus intensity by regulating RGS proteins and PLCbeta GAP activities. J Cell Biol. 2003;162(2):293–303. doi: 10.1083/jcb.200210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnarajah S, Dessauer CW, Srikumar D, Chen J, Yuen J, Yilma S, Dennis JC, Morrison EE, Vodyanoy V, Kehrl JH. RGS2 regulates signal transduction in olfactory neurons by attenuating activation of adenylyl cyclase III. Nature. 2001;409(6823):1051–1055. doi: 10.1038/35059104. [DOI] [PubMed] [Google Scholar]
- Slessareva JE, Routt SM, Temple B, Bankaitis VA, Dohlman HG. Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein alpha subunit at the endosome. Cell. 2006;126(1):191–203. doi: 10.1016/j.cell.2006.04.045. [DOI] [PubMed] [Google Scholar]
- Smith FD, Oxford GS, Milgram SL. Association of the D2 dopamine receptor third cytoplasmic loop with spinophilin, a protein phosphatase-1-interacting protein. J Biol Chem. 1999;274(28):19894–19900. doi: 10.1074/jbc.274.28.19894. [DOI] [PubMed] [Google Scholar]
- Stauss HM. Regulator of G protein signaling (RGS2)-deficient mice: a novel model to study autonomic nervous system function. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1091–1092. doi: 10.1152/ajpregu.00057.2005. [DOI] [PubMed] [Google Scholar]
- Su Z, Xu P, Ni F. Single phosphorylation of Tyr304 in the cytoplasmic tail of ephrin B2 confers high-affinity and bifunctional binding to both the SH2 domain of Grb4 and the PDZ domain of the PDZ-RGS3 protein. Eur J Biochem. 2004;271(9):1725–1736. doi: 10.1111/j.1432-1033.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- Sullivan BM, Harrison-Lavoie KJ, Marshansky V, Lin HY, Kehrl JH, Ausiello DA, Brown D, Druey KM. RGS4 and RGS2 bind coatomer and inhibit COPI association with Golgi membranes and intracellular transport. Mol Biol Cell. 2000;11(9):3155–3168. doi: 10.1091/mbc.11.9.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Kaltenbronn KM, Steinberg TH, Blumer KJ. RGS2 is a mediator of nitric oxide action on blood pressure and vasoconstrictor signaling. Mol Pharmacol. 2005;67(3):631–639. doi: 10.1124/mol.104.007724. [DOI] [PubMed] [Google Scholar]
- Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, Kaltenbronn KM, Blumer KJ, Siderovski DP, Zhu Y, Mendelsohn ME. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med. 2003;9(12):1506–1512. doi: 10.1038/nm958. [DOI] [PubMed] [Google Scholar]
- Tank J, Obst M, Diedrich A, Brychta RJ, Blumer KJ, Heusser K, Jordan J, Luft FC, Gross V. Sympathetic nerve traffic and circulating norepinephrine levels in RGS2-deficient mice. Auton Neurosci. 2007 doi: 10.1016/j.autneu.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89(2):251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Thaminy S, Auerbach D, Arnoldo A, Stagljar I. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res. 2003;13(7):1744–1753. doi: 10.1101/gr.1276503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinker A. The selective interactions and functions of regulators of G-protein signalling. Semin Cell Dev Biol. 2006;17(3):377–382. doi: 10.1016/j.semcdb.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Tosetti P, Pathak N, Jacob MH, Dunlap K. RGS3 mediates a calcium-dependent termination of G protein signaling in sensory neurons. Proc Natl Acad Sci U S A. 2003;100(12):7337–7342. doi: 10.1073/pnas.1231837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH, Burns ME, Calvert PD, Gouras P, Baylor DA, Goff SP, Arshavsky VY. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282(5386):117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- Tseng CC, Zhang XY. Role of regulator of G protein signaling in desensitization of the glucose-dependent insulinotropic peptide receptor. Endocrinology. 1998;139(11):4470–4475. doi: 10.1210/endo.139.11.6282. [DOI] [PubMed] [Google Scholar]
- Tu Y, Woodson J, Ross EM. Binding of regulator of G protein signaling (RGS) proteins to phospholipid bilayers. Contribution of location and/or orientation to Gtpase-activating protein activity. J Biol Chem. 2001;276(23):20160–20166. doi: 10.1074/jbc.M101599200. [DOI] [PubMed] [Google Scholar]
- von Buchholtz L, Elischer A, Tareilus E, Gouka R, Kaiser C, Breer H, Conzelmann S. RGS21 is a novel regulator of G protein signalling selectively expressed in subpopulations of taste bud cells. Eur J Neurosci. 2004;19(6):1535–1544. doi: 10.1111/j.1460-9568.2004.03257.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu M, Mullah B, Siderovski DP, Neubig RR. Receptor-selective effects of endogenous RGS3 and RGS5 to regulate mitogen-activated protein kinase activation in rat vascular smooth muscle cells. J Biol Chem. 2002;277(28):24949–24958. doi: 10.1074/jbc.M203802200. [DOI] [PubMed] [Google Scholar]
- Wang X, Zeng W, Kim MS, Allen PB, Greengard P, Muallem S. Spinophilin/neurabin reciprocally regulate signaling intensity by G protein-coupled receptors. Embo J. 2007;26(11):2768–2776. doi: 10.1038/sj.emboj.7601701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zeng W, Soyombo AA, Tang W, Ross EM, Barnes AP, Milgram SL, Penninger JM, Allen PB, Greengard P, Muallem S. Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat Cell Biol. 2005;7(4):405–411. doi: 10.1038/ncb1237. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Milligan G. A key serine for the GTPase-activating protein function of regulator of G protein signaling proteins is not a general target for 14-3-3 interactions. Mol Pharmacol. 2005;68(6):1821–1830. doi: 10.1124/mol.105.015073. [DOI] [PubMed] [Google Scholar]
- Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature. 1996;383(6596):172–175. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- Wess J. G-protein-coupled receptors: molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. Faseb J. 1997;11(5):346–354. [PubMed] [Google Scholar]
- Wickman KD, Iniguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE. Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature. 1994;368(6468):255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- Willard FS, Kimple AJ, Johnston CA, Siderovski DP. A direct fluorescence-based assay for RGS domain GTPase accelerating activity. Anal Biochem. 2005;340(2):341–351. doi: 10.1016/j.ab.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Willars GB. Mammalian RGS proteins: multifunctional regulators of cellular signalling. Semin Cell Dev Biol. 2006;17(3):363–376. doi: 10.1016/j.semcdb.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Xie Z, Gong MC, Su W, Turk J, Guo Z. Group VIA Phospholipase A2 (iPLA2beta) Participates in Angiotensin II-induced Transcriptional Up-regulation of Regulator of G-protein Signaling-2 in Vascular Smooth Muscle Cells. J Biol Chem. 2007;282(35):25278–25289. doi: 10.1074/jbc.M611206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zeng W, Popov S, Berman DM, Davignon I, Yu K, Yowe D, Offermanns S, Muallem S, Wilkie TM. RGS proteins determine signaling specificity of Gq-coupled receptors. J Biol Chem. 1999;274(6):3549–3556. doi: 10.1074/jbc.274.6.3549. [DOI] [PubMed] [Google Scholar]
- Yalcin B, Willis-Owen SA, Fullerton J, Meesaq A, Deacon RM, Rawlins JN, Copley RR, Morris AP, Flint J, Mott R. Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice. Nat Genet. 2004;36(11):1197–1202. doi: 10.1038/ng1450. [DOI] [PubMed] [Google Scholar]
- Yamada M, Inanobe A, Kurachi Y. G protein regulation of potassium ion channels. Pharmacol Rev. 1998;50(4):723–760. [PubMed] [Google Scholar]
- Yan Y, Chi PP, Bourne HR. RGS4 inhibits Gq-mediated activation of mitogen-activated protein kinase and phosphoinositide synthesis. J Biol Chem. 1997;272(18):11924–11927. doi: 10.1074/jbc.272.18.11924. [DOI] [PubMed] [Google Scholar]
- Yang J, Kamide K, Kokubo Y, Takiuchi S, Tanaka C, Banno M, Miwa Y, Yoshii M, Horio T, Okayama A, Tomoike H, Kawano Y, Miyata T. Genetic variations of regulator of G-protein signaling 2 in hypertensive patients and in the general population. J Hypertens. 2005;23(8):1497–1505. doi: 10.1097/01.hjh.0000174606.41651.ae. [DOI] [PubMed] [Google Scholar]
- Yowe D, Weich N, Prabhudas M, Poisson L, Errada P, Kapeller R, Yu K, Faron L, Shen M, Cleary J, Wilkie TM, Gutierrez-Ramos C, Hodge MR. RGS18 is a myeloerythroid lineage-specific regulator of G-protein-signalling molecule highly expressed in megakaryocytes. Biochem J. 2001;359(Pt 1):109–118. doi: 10.1042/0264-6021:3590109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Xu X, Popov S, Mukhopadhyay S, Chidiac P, Swistok J, Danho W, Yagaloff KA, Fisher SL, Ross EM, Muallem S, Wilkie TM. The N-terminal domain of RGS4 confers receptor-selective inhibition of G protein signaling. J Biol Chem. 1998;273(52):34687–34690. doi: 10.1074/jbc.273.52.34687. [DOI] [PubMed] [Google Scholar]
- Zhang S, Watson N, Zahner J, Rottman JN, Blumer KJ, Muslin AJ. RGS3 and RGS4 are GTPase activating proteins in the heart. J Mol Cell Cardiol. 1998;30(2):269–276. doi: 10.1006/jmcc.1997.0591. [DOI] [PubMed] [Google Scholar]
- Zhang W, Anger T, Su J, Hao J, Xu X, Zhu M, Gach A, Cui L, Liao R, Mende U. Selective loss of fine tuning of Gq/11 signaling by RGS2 protein exacerbates cardiomyocyte hypertrophy. J Biol Chem. 2006;281(9):5811–5820. doi: 10.1074/jbc.M507871200. [DOI] [PubMed] [Google Scholar]
- Zhou J, Moroi K, Nishiyama M, Usui H, Seki N, Ishida J, Fukamizu A, Kimura S. Characterization of RGS5 in regulation of G protein-coupled receptor signaling. Life Sci. 2001;68(13):1457–1469. doi: 10.1016/s0024-3205(01)00939-0. [DOI] [PubMed] [Google Scholar]
- Zuber AM, Singer D, Penninger JM, Rossier BC, Firsov D. Increased Renal Responsiveness to Vasopressin and Enhanced V2 Receptor Signaling in RGS2−/− Mice. J Am Soc Nephrol. 2007;18(6):1672–1678. doi: 10.1681/ASN.2007010032. [DOI] [PubMed] [Google Scholar]