

Abstract
The appearance of protein aggregates is a characteristic of protein misfolding disorders including familial amyotrophic lateral sclerosis, a neurodegenerative disease caused by inherited mutations in Cu/Zn superoxide dismutase 1 (SOD1). Here, we use live cell imaging of neuronal and nonneuronal cells to show that SOD1 mutants (G85R and G93A) form an aggregate structure consisting of immobile scaffolds, through which noninteracting cellular proteins can diffuse. Hsp70 transiently interacts, in a chaperone activity-dependent manner, with these mutant SOD1 aggregate structures. In contrast, the proteasome is sequestered within the aggregate structure, an event associated with decreased degradation of a proteasomal substrate. Through the use of time-lapse microscopy of individual cells, we show that nearly all (90%) aggregate-containing cells express higher levels of mutant SOD1 and died within 48 h, whereas 70% of cells expressing a soluble mutant SOD1 survived. Our results demonstrate that SOD1 G85R and G93A mutants form a distinct class of aggregate structures in cells destined for neuronal cell death.
Introduction
Familial amyotrophic lateral sclerosis (fALS) is a neurodegenerative disorder resulting in the selective loss of motor neurons (Cleveland and Rothstein, 2001; Bruijn et al., 2004). Although a number of genetic defects have been linked to the development of fALS, the autosomal-dominant inheritance of mutant Cu/Zn superoxide dismutase 1 (SOD1) accounts for ∼20% of cases (Rosen et al., 1993). Although numerous potential mechanisms of cytotoxicity have been proposed, including the depletion of molecular chaperones, sequestration and dysfunction of the proteasome, impairment of axonal transport, aberrant dismutase activities, and mitochondrial disruption by SOD1 mutations (Bruijn et al., 2004), the contribution of each proposed mechanism remains unclear. Although more than 100 different SOD1 mutations cause fALS, some of which retain complete enzymatic activity (Borchelt et al., 1994; Bowling et al., 1995; Gaudette et al., 2000), it has been well established that mutant SOD1-mediated toxicity is caused by a toxic gain-of-function rather than the loss of SOD1 activity (Bruijn et al., 1998, 2004; Cleveland and Rothstein, 2001). A common feature, shared by most SOD1 mutations both in mutant SOD1 transgenic mouse models (Bruijn et al., 1997; Johnston et al., 2000; Watanabe et al., 2001; Wang et al., 2002) and in tissue samples from fALS patients (Shibata et al., 1996; Kato et al., 2000), is the appearance of intracellular inclusions containing detergent-insoluble protein aggregates. Such proteinacious inclusions also occur in many other neurodegenerative disorders (Taylor et al., 2002) and their presence suggests an inability of the protein quality control machinery (including molecular chaperones and the ubiquitin–proteasome system) to efficiently recognize, fold, and degrade this class of abnormal proteins (Kopito, 2000; Goldberg, 2003). Association of the proteasome, various molecular chaperones (i.e., Hsp40 and Hsp70), and other cellular proteins including transcription factors with aggregates (Bence et al., 2001; Watanabe et al., 2001; Kim et al., 2002; Takeuchi et al., 2002; Holmberg et al., 2004; Bennett et al., 2005) has been proposed as a common mechanism for aggregate-associated cytotoxicity (Cleveland and Rothstein, 2001; Sherman and Goldberg, 2001; Taylor et al., 2002; Goldberg, 2003; Bruijn et al., 2004).
The role of aggregates in cytoprotection or cytotoxicity of neuronal cell dysgenesis is a point of some controversy. Some studies have shown no correlation between polyglutamine-containing aggregates and increased cell death (Saudou et al., 1998; Zhou et al., 2001; Arrasate et al., 2004) whereas other studies have identified a direct relationship between aggregates and cellular toxicity (Nucifora et al., 2001; Wyttenbach et al., 2001; Jiang et al., 2003; Schaffar et al., 2004). Whether this controversy is due to biologically relevant differences associated with specific features of an aggregation-prone protein or technical differences (i.e., level of protein overexpression, cell type, method of detection/characterization) is unclear. The absence of a uniform definition for an aggregate or inclusion body may further contribute to such differences. Cell biological studies have typically used visual inspection and immunolocalization using fixed cells, complemented by biochemical methods that detect insoluble proteins using PAGE or a filter-trap assay of cell extracts. Other methods using fixed cells have used immunohistochemistry analysis and electron microscopy to visualize aggregate and inclusion structures at higher resolution (Bruijn et al., 1997, 1998). From such approaches, aggregates formed by expression of either mutant SOD1 or huntingtin have been suggested to have similar morphologies and structure. However, the recent applications of dynamic live cell imaging techniques (i.e., fluorescent loss in photobleaching [FLIP], and fluorescence resonance energy transfer [FRET]) have revealed unexpected differences in the aggregate structure of the polyglutamine proteins, ataxin 1 and mutant huntingtin (Chai et al., 2002; Kim et al., 2002; Stenoien et al., 2002; Holmberg et al., 2004), leading to the suggestion that other neurodegenerative disease-associated proteins might also form distinct classes of aggregates.
In this study, we assess the composition and dynamic properties of aggregates formed by G85R and G93A, two well-characterized mutant SOD1 proteins, to gain insight into the mechanistic events associated with SOD1 aggregate formation and cytotoxicity. Using live-cell imaging of individual PC12 neuronal and HeLa cells, we find that mutant SOD1 proteins form aggregates containing two distinct populations of protein, one highly aggregated and immobile forming a scaffold, and a second population mobile and diffusing within cavities present throughout the immobile scaffold. Additionally, in vivo these aggregates recruit, sequester, and decrease activity of the proteasome. In contrast, the molecular chaperone Hsp70 interacts transiently with mutant SOD1 aggregates. Using an automated time-lapse imaging technique to follow the fate of individual PC12 cells, we find that aggregate formation is lethal to the cell. These studies reveal that the aggregate structures formed by mutant SOD1 proteins, distinct from previously observed aggregates formed by other misfolded proteins, are associated with decreased cell survival.
Results
Mutant SOD1 forms aggregates containing mobile and immobile fractions
We monitored the in vivo dynamics of wild-type (WT) and mutant SOD1 proteins, as well as their molecular interactions in differentiated PC12 neuronal cells and HeLa cells, by expressing CFP and YFP fusions with WT (WT-CFP and WT-YFP) and mutant (G85R-CFP, G85R-YFP, G93A-CFP, G93A-YFP) SOD1 proteins. WT, G85R, and G93A fused to CFP (Fig. 1 A) or YFP (unpublished data) accumulated to equivalent levels in transiently transfected PC12 cells and both mutant proteins were detected as full-length products of expected molecular sizes. A similar level of protein accumulation was also detected in HeLa cells (unpublished data). WT-CFP and G93A-CFP displayed SOD1 enzymatic activity and inhibited the reduction of nitroblue tetrazolium (NBT) by superoxide, whereas G85R-CFP did not exhibit detectable SOD1 activity (Pasinelli et al., 1998) in either PC12 (Fig. 1 B) or HeLa cells (unpublished data). The subcellular localization of WT-CFP, G85R-CFP, and G93A-CFP was visualized in differentiated PC12 (Fig. 1 C) and HeLa cells (Fig. S1 available at http://www.jcb.org/cgi/content/full/jcb.200504050/DC1). Neuronal processes, characteristic of differentiated PC12 cells, are not apparent in these images and are visualized at lower magnification (Fig. S1). WT-CFP exhibited a diffuse pattern of protein localization throughout the cell, whereas large perinuclear aggregates were detected in cells expressing G85R-CFP and G93A-CFP. As a complement to the visual identification of aggregates, we performed a cellulose acetate filter retardation assay (Wanker et al., 1999; Wang et al., 2002). Whereas WT-CFP was soluble, both mutant proteins were trapped as 1% SDS-insoluble species, in PC12 (Fig. 1 D) and HeLa cells (unpublished data), indicative of aggregate formation (Wanker et al., 1999; Wang et al., 2002). From these results, we conclude that both mutant SOD1-YFP/CFP proteins exhibit a similar aggregation phenotype, whether expressed in PC12 neuronal cells or HeLa cells.
Figure 1.
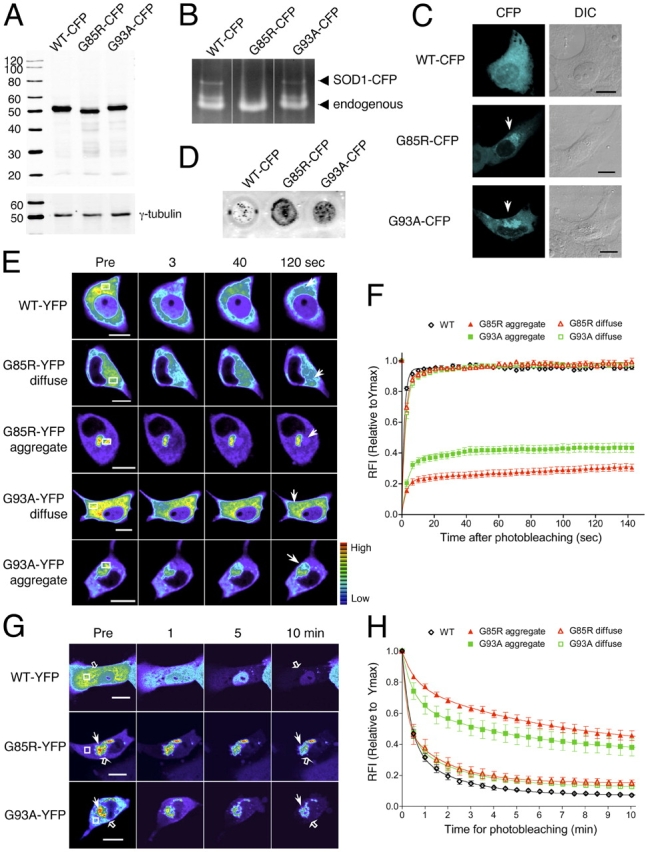
Mutant SOD1 forms aggregates containing mobile and immobile fractions. (A–H) Differentiated PC12 cells were transiently transfected for 3 d with constructs encoding WT-CFP/YFP, G85R-CFP/YFP, or G93A-CFP/YFP, as indicated. (A) Western analysis of SOD1 proteins. Whole cells extracts were harvested and analyzed by Western analysis. CFP-tagged proteins were detected with an anti-GFP antibody and γ-tubulin was used as a loading control. G85R migrates slightly faster than WT and G93A, as previously described (Bruijn et al., 1998). The anti-GFP antibody detects no obvious free CFP molecules. (B) SOD1 activity assay. Whole cell extracts were subjected to native-PAGE and SOD1 activities were detected by NBT-negative staining. The activities of both transfected and endogenous SOD1 are detected, as indicated. (C) Visualization of SOD1 proteins. CFP fusion proteins (CFP, cyan) were visualized by confocal microscopy and phase contrast microscopy (DIC). White arrows indicate aggregates. Images have been cropped to enlarge the cell body; an image showing neurites, a marker for neuronal differentiation, is found in Fig. S1. (D) Filter trap assay of SOD1 proteins. Whole cell extracts were treated with 1% SDS and subjected to filter trap analysis on a cellulose acetate membrane. Proteins that are retained on the membrane were detected with an anti-GFP antibody. (E) FRAP analysis of SOD1 proteins. Single scan images were obtained before photobleaching (Pre) of a region of interest (ROI; white box) and at the indicated times after photobleaching. Arrows indicate the photobleached area. Note that the intensity of the images is scaled differently between the samples. (F) Quantitative FRAP analysis of SOD1 proteins. The relative fluorescence intensity (RFI) was determined at each time point and is represented as the mean ± SEM (n ≥ 10 cells). (G) FLIP analysis of SOD1 proteins. Single scan images of a diffuse (open arrow) and aggregated region (closed arrow) were obtained before (Pre) and at the indicated times during continuous photobleaching of a region (white box). (H) Quantitative FLIP analysis of SOD1 proteins. The RFI was determined at each time point and is represented as the mean ± SEM (n = 5–10 cells). Bars, 10 μm.
To investigate the mobility of diffuse and aggregated SOD1 proteins, we performed qualitative and quantitative FRAP analysis in PC12 cells (Lippincott-Schwartz et al., 2001). Compared with WT-YFP and the diffusely localized G85R-YFP and G93A-YFP proteins, which were completely mobile, aggregated G85R-YFP and G93A-YFP proteins showed dramatically reduced mobility (Fig. 1, E and F, and Table I). Unexpectedly, the pattern of fluorescence recovery within either mutant SOD1 aggregate was not consistent with the presence of a single protein population, as reported with aggregates formed by the mutant huntingtin protein (Kim et al., 2002; Holmberg et al., 2004). The FRAP data suggested that both G85R and G93A mutant SOD1 aggregates are comprised of two protein populations. Mathematical analyses of FRAP data indeed supported these assertions, as both curves fit to a recovery consistent with two populations (see Materials and methods). An immobile (nondiffusing) protein population, with off-rates <0.001 s−1, predominated and accounted for 76.8 ± 1.9% (G85R-YFP) and 61.6 ± 2.2% (G93A-YFP) of total fluorescent protein within the aggregates (Fig. 1, E and F, and Table I). These fractions remained immobile even when the analysis was extended to a 10-min period (unpublished data). The presence of a slow off-rate and a persistent immobile population reveals a long residency time (τr > 1,000 s) of mutant SOD1 protein in this immobile form, corresponding to stable SOD1 aggregate structures. In contrast, the remaining 23.2 ± 1.9% (G85R-YFP) and 38.4 ± 2.2% (G93A-YFP) of protein within each aggregate was diffused (Fig. 1, E and F, and Table I).
Table I.
| Sample | Diffusing fraction | D | Nondiffusing fraction | kU |
|---|---|---|---|---|
| % | μm2/s | % | s− 1 | |
| WT-YFP | 95.0 ± 2.3 | 2.35 ± 0.60 | 5.0 ± 2.3 | N/A |
| G85R-YFP diffuse | 92.1 ± 3.1ns, a | 1.67 ± 0.29 | 7.9 ± 3.1ns, a | 0.013 ± 0.008 |
| G93A-YFP diffuse | 92.1 ± 2.9ns, a | 1.47 ± 0.23 | 7.9 ± 2.9ns, a | 0.011 ± 0.006 |
| G85R-YFP aggregate | 23.2 ± 1.9***, b | 1.27 ± 0.72 | 76.8 ± 1.9***, b | <0.001 |
| G93A-YFP aggregate | 38.4 ± 2.2***, b | 0.88 ± 0.26 | 61.6 ± 2.2***, b | <0.001 |
| LMP2-YFP (diffuse G85R-CFP) | 94.6 ± 2.8 | 2.20 ± 0.39 | 5.4 ± 2.8 | 0.012 ± 0.009 |
| LMP2-YFP (G85R-CFP aggregate) | 27.6 ± 1.2***, b | 1.78 ± 0.50 | 72.4 ± 1.2***, b | <0.001 |
| Hsp70-YFP (diffuse G85R-CFP) | 85.8 ± 1.9 | 1.76 ± 0.31 | 14.2 ± 1.8 | 0.008 ± 0.002 |
| Hsp70-YFP (G85R-CFP aggregate) | 65.2 ± 1.9***, b | 1.07 ± 0.12 | 34.8 ± 1.9***, b | 0.004 ± 0.001 |
| Hsp70-SBDx-YFP (diffuse G85R-CFP) | 92.6 ± 1.9 2***, c | 1.93 ± 0.28 | 7.4 ± 1.9 2***, c | 0.009 ± 0.006 |
| Hsp70-SBDx-YFP (G85R-CFP aggregate) | 76.4 ± 1.62***, c | 2.13 ± 0.34 | 23.6 ± 1.62***, c | 0.003 ± 0.001 |
FRAP data from Figs. 1F, 3B, and 4B were analyzed with GraphPad Prism software, as described in Supplementary Information, Methods online. Data are displayed as mean ± SEM. Two-tailed t test analysis (95% confidence) was used to compare the statistical difference between data sets: ***, P < 0.001; ns, P > 0.05.
Diffuse G85R-YFP and G93A-YFP were compared to WT-YFP.
Samples were compared to the respective diffuse sample (i.e., G85R-YFP aggregated vs. G85R-YFP diffuse).
Hsp70-SBDx-YFP mutant samples were compared to the respective Hsp70-YFP sample (i.e., Hsp70-SBDx-YFP with diffuse G85R-CFP vs. Hsp70-YFP with diffuse G85R-CFP).
The “diffuse” fractions of the mutant SOD1 proteins are not the same as the “diffuse” WT protein, as the diffusion coefficients are 1.47 μm2/s (G85R-YFP) and 1.67 μm2/s (G93A-YFP), compared with 2.35 μm2/s (WT-YFP; Table I). These results together with additional analyses (see Materials and methods) suggest that the diffuse G85R-YFP corresponds to higher-order structures of approximate molecular weights of 250 kD. This could reflect the formation of oligomers of G85R-YFP or the stable association of other cellular proteins with G85R-YFP. These results suggest that mutant SOD1 may form oligomers that are transiently diffuse, followed by higher-order oligomerization and thus populations with slower diffusion, and finally aggregation and immobilization (no diffusion). Similar to differentiated PC12 cells, we observed that both a diffuse and immobile (nondiffusing) fraction of protein exist within mutant SOD1 aggregates in undifferentiated PC12 and HeLa cells (Fig. S1). From these results, we conclude that both G85R-YFP and G93A-YFP form immobile aggregates; however, differences in the fraction of each mutant SOD1 protein present in the aggregated state reveals subtle distinctions between the two mutant proteins.
To test the hypothesis that two protein populations are indeed present within the mutant SOD1 aggregates, we used a complimentary imaging technique, FLIP. Fluorescence intensities of G85R-YFP and G93A-YFP within and outside the aggregate were measured while a region adjacent to the aggregate was continuously photobleached. We expected that a fraction of protein within the aggregate would remain intact while a second population would diffuse out of the aggregate and become photobleached. As with FRAP analyses, our FLIP data presented a better fit to a two-phase exponential decay curve, confirming the presence of two distinct populations of protein. FLIP revealed that 45% (G85R-YFP) and 38% (G93A-YFP) of the initial fluorescence within the aggregate persisted over a 10-min period (Fig. 1, G and H), supporting our hypothesis that one population of mutant SOD1 protein forms a stable aggregate structure. In contrast, the fluorescence intensities of WT-YFP, as well as the diffusely localized G85R-YFP and G93A-YFP proteins, were rapidly reduced and continued to decrease to basal fluorescence after 10 min of continuous photobleaching, indicative of a single mobile population (Fig. 1, G and H). Together, the FRAP and FLIP analyses suggest that mutant SOD1 (G85R and G93A) aggregates are composed of two protein populations, one mobile and diffusing and a second highly immobile and aggregated population.
Mutant SOD1 aggregates form a disordered “honeycomb-like” porous structure
FRAP and FLIP analyses suggested that mutant SOD1 proteins formed porous aggregate structures through which other soluble cellular proteins may diffuse. This is in contrast to aggregates formed by mutant huntingtin and polyglutamine-YFP proteins, which we and others have shown form a solid and immobile core structure (Chai et al., 2002; Kim et al., 2002; Holmberg et al., 2004). To determine whether noninteracting soluble proteins can indeed diffuse through the mutant SOD1 aggregate structures, PC12 cells were transiently cotransfected with a construct encoding an aggregation-prone protein (G85R-CFP, G93A-CFP, or httQ78-CFP) and a soluble YFP or WT-YFP. YFP (Fig. 2 A) and WT-YFP (Fig. S2 available at http://www.jcb.org/cgi/content/full/jcb.200504050/DC1) proteins displayed a diffuse and uniform pattern of localization, both within and outside the mutant SOD1 aggregates. In contrast, aggregates formed by expression of an expanded polyglutamine-containing mutant huntingtin protein (httQ78-CFP) exhibited a solid core that completely excluded YFP (Fig. 2 A). These results suggested that soluble proteins can colocalize with the aggregate structures formed by mutant SOD1.
Figure 2.
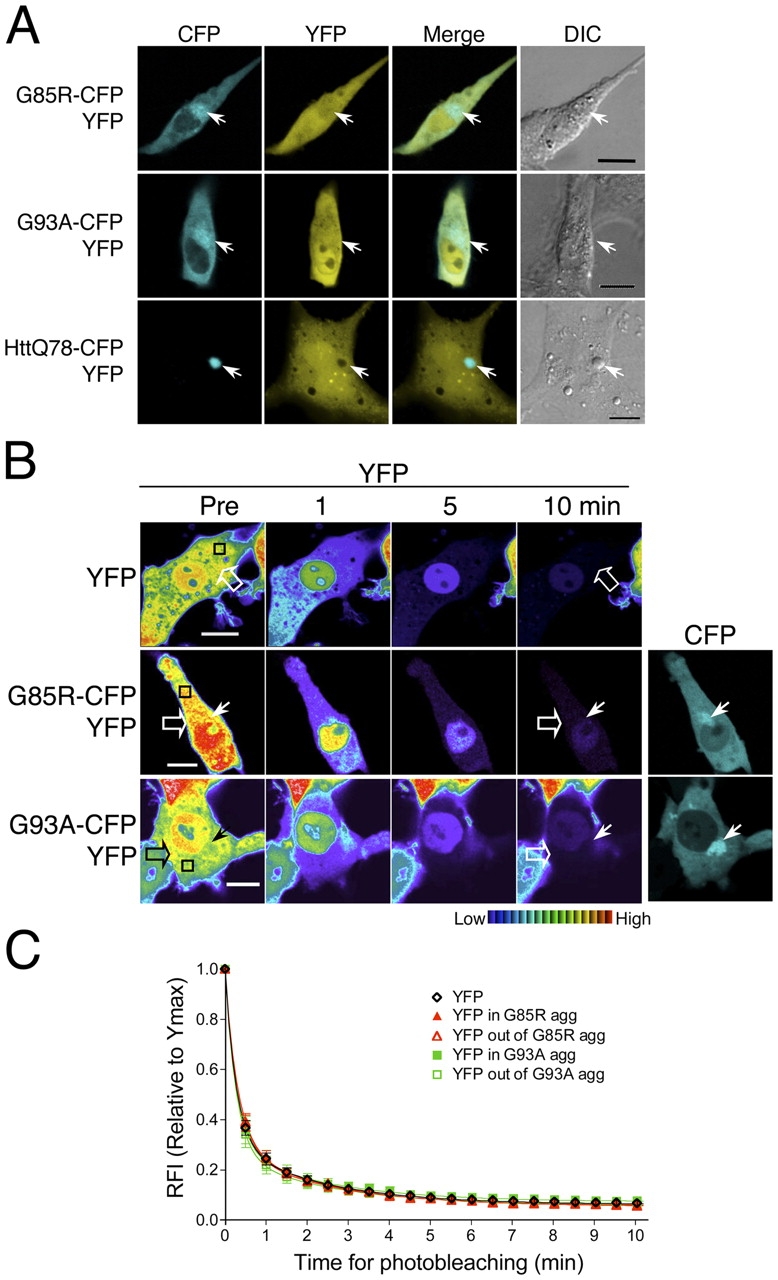
Mutant SOD1 aggregates form a disordered honeycomb-like porous structure. (A–C) Differentiated PC12 cells were transiently transfected with constructs encoding YFP together with G85R-CFP, G93A-CFP, or httQ78-CFP, as indicated. (A) Colocalization of YFP with G85R-CFP, G93A-CFP, or httQ78-CFP aggregates. YFP (YFP, yellow) and CFP fusion protein (CFP, cyan) were visualized by confocal microscopy and phase contrast microscopy (DIC). Colocalization was illustrated by merging YFP and CFP channels (Merge). Arrows indicate protein aggregates. (B and C) Differentiated PC12 cells were transiently transfected with constructs encoding YFP together with G85R-CFP or G93A-CFP, as indicated. (B) FLIP analysis of YFP. Single scan images of a diffuse (open arrow) and aggregated region (closed arrow) were obtained before (Pre) and at the indicated times during continuous photobleaching of a region (black box). (C) Quantitative FLIP analysis of YFP. The RFI was determined at each time point and is represented as the mean ± SEM (n = 5–10 cells). Bars, 10 μm.
Whereas standard colocalization methods do not establish whether associated proteins are nonspecifically trapped or diffuse within the aggregate, FLIP analysis provides a direct test, as we would expect that soluble proteins can diffuse rapidly out of the aggregate structures and become photobleached. The initial fluorescence intensities of YFP, either within or outside the mutant SOD1 (G85R-CFP or G93A-CFP) aggregates, were rapidly reduced and continued to decrease to basal fluorescence after 10 min of continuous photobleaching (Fig. 2, B and C). These results were similar to the diffusion of YFP alone, confirming that YFP molecules remain diffuse even when localized within the mutant SOD1 aggregate structures. Experiments performed in parallel revealed that WT-YFP molecules also diffuse through the mutant SOD1 aggregate structures (Fig. S2). These results support our hypothesis that mutant SOD1 proteins form porous, almost honeycomb-like aggregate structures, consisting of a highly immobile scaffold through which soluble cellular proteins can diffuse.
Hsp70 transiently interacts with mutant SOD1 aggregates
Although noninteracting proteins diffuse rapidly through the mutant SOD1 aggregate structures, we speculated that other proteins, which interact with aggregates formed by neurodegenerative disease-associated proteins, would display reduced mobility. To investigate the interaction of the molecular chaperone Hsp70 within the mutant SOD1 aggregates, PC12 cells were transiently transfected with constructs encoding G85R-CFP and a YFP-tagged Hsp70 (Hsp70-YFP) protein. This construct has been previously used in our laboratory and retains its activity as a molecular chaperone (Kim et al., 2002). FRAP analysis showed that 85.8 ± 1.9% of Hsp70-YFP was diffused in cells expressing soluble G85R-CFP (Fig. 3, A and B, and Table I). In contrast, in cells with G85R-CFP aggregates, 34.8 ± 1.9% of Hsp70-YFP was associated with the aggregates, with the remaining Hsp70-YFP in a soluble state (Fig. 3, A and B, and Table I). To address whether the association of Hsp70 reflected its chaperone function, we measured the mobility of a mutant Hsp70-YFP containing a mutation in the substrate-binding domain (Hsp70-SBDx-YFP). The mobility of Hsp70-SBDx-YFP was higher than for Hsp70-YFP with aggregated G85R-CFP (Fig. 3 B and Table I), revealing that a functional Hsp70 chaperone cycle is required for the interaction between Hsp70 and the aggregate. To further demonstrate that the Hsp70-aggregate interaction was transient, we used FLIP analysis, which showed that the fluorescence intensity of Hsp70-YFP within G85R-CFP aggregates declined rapidly to basal levels over a 10-min period of photobleaching (Fig. 3, C and D). From the results of FRAP and FLIP analysis, we conclude that Hsp70 is not sequestered, rather it is associated transiently with aggregate structures formed by mutant SOD1 proteins.
Figure 3.

Hsp70 transiently interacts with mutant SOD1 aggregates. Differentiated PC12 cells were transiently transfected with constructs encoding Hsp70-YFP or Hsp70-SBDx-YFP together with G85R-CFP, as indicated. (A) FRAP analysis of Hsp70-YFP. Single scan images were obtained before photobleaching (Pre) of a ROI (white or black box) and at the indicated times after photobleaching. Arrows indicate the photobleached area. (B) Quantitative FRAP analysis of Hsp70-YFP and Hsp70-SBDx-YFP. The RFI was determined at each time point and is represented as the mean ± SEM (n ≥ 10 cells). (Inset) The region of photobleaching recovery from 0 to 15 s is shown for clarity. (C) FLIP analysis of Hsp70-YFP. Single scan images of a diffuse (open arrow) and aggregated region (closed arrow) were obtained before (Pre) and at the indicated times during continuous photobleaching of a region (white box). (D) Quantitative FLIP analysis of Hsp70-YFP. The RFI was determined at each time point and is represented as the mean ± SEM (n = 5–10 cells). Bars, 10 μm.
Mutant SOD1 aggregates sequester the proteasome and impair its activity
Aggregates formed by many neurodegenerative disease–associated proteins, including mutant SOD1, have been shown to interact with the proteasome (Bence et al., 2001; Urushitani et al., 2002; Goldberg, 2003; Kabashi et al., 2004). To monitor interactions between mutant SOD1 and the 20S proteasome, we introduced a YFP-tagged β subunit (LMP2-YFP) into PC12 cells together with a construct encoding G85R-CFP. LMP2 is quantitatively incorporated into active proteasomes and therefore provides an excellent indicator of the functional proteasome (Reits et al., 1997; Holmberg et al., 2004). FRAP analysis indicated that 94.1 ± 3.3% of LMP2-YFP was diffuse in cells containing diffusely localized G85R-CFP (Fig. 4, A and B, and Table I). These results were similar to the previously reported mobility of LMP2-GFP in cells expressing LMP2-GFP alone (Reits et al., 1997) or in cells containing a diffusely localized WT NH2-terminal huntingtin protein (Holmberg et al., 2004). In aggregate-containing cells, the proteasome is relocalized to aggregates, with 72.1 ± 1.8% of the total LMP2-YFP immobilized within the G85R-CFP aggregate with a slow off-rate < 0.001 s−1 (Fig. 4, A and B, and Table I). The presence of a slow off-rate reveals a long residency time (τr > 1,000 s) for LMP2-YFP in this immobile state suggesting a tight interaction with the G85R-CFP aggregate.
Figure 4.

Mutant SOD1 aggregates sequester the proteasome and impair its activity. (A–D) Differentiated PC12 cells were transiently transfected with constructs encoding LMP2-YFP and G85R-CFP. (A) FRAP analysis of LMP2-YFP. Single scan images were obtained before photobleaching (Pre) of a ROI (white or black box) and at the indicated times after photobleaching. Arrows indicate the photobleached area. (B) Quantitative FRAP analysis of LMP2-YFP. The RFI was determined at each time point and is represented as the mean ± SEM (n ≥ 10 cells). (C) FLIP analysis of LMP2-YFP. Single scan images of a diffuse (open arrow) and aggregated region (closed arrow) were obtained before (Pre) and at the indicated times during continuous photobleaching of a region (white box). (D) Quantitative FLIP analysis of LMP2-YFP. The RFI was determined at each time point and is represented as the mean ± SEM (n = 5–10 cells). Note: 62.3 ± 3.7% of total LMP2-YFP fluorescence intensity is associated with the G85R-CFP aggregate (n = 7 cells; see Materials and methods). (E and F) Stable differentiated Ubi-YFP/PC12 cells were transiently transfected with constructs encoding WT-CFP, G85R-CFP, or G93A-CFP. (E) Accumulation of a proteasomal substrate. Ubi-YFP (YFP, yellow) and CFP fusion proteins (CFP, cyan) were visualized by fluorescence microscopy. Mutant SOD1 protein aggregates (closed arrows) and diffuse SOD1 (open arrows) are indicated. (F) Quantification of Ubi-YFP fluorescence. The fold increase of Ubi-YFP intensity compared with the mean intensity of nontransfected cells is represented as the mean ± SEM (n = 28–56 cells). Bars, 10 μm. Two-tailed t test analysis (95% confidence) was used to compare the statistical difference between data sets: ***, P < 0.001; *, P < 0.05; ns, P > 0.05.
To further establish the interaction of LMP2-YFP with the G85R-CFP aggregate, we used FLIP analysis. FLIP showed that 39.7 ± 10.7% of the initial fluorescence intensity of LMP2-YFP associated with G85R-CFP aggregates was retained after a 10-min period of photobleaching (Fig. 4, C and D), suggesting that the proteasome is sequestered within G85R-CFP aggregates. To verify that these results reflect the endogenous proteasome, we used indirect immunofluorescence analysis and showed that the endogenous 20S subunit of the proteasome is associated with G85R-CFP and G93A-CFP aggregates (Fig. S3 available at http://www.jcb.org/cgi/content/full/jcb.200504050/DC1). Taken together, these results demonstrate that the proteasome is sequestered into the immobile fraction of the mutant SOD1 aggregate structures.
We next examined whether sequestration of the proteasome within the mutant SOD1 aggregate has negative consequences on proteasome activity. To address this, we used an Ubi-YFP reporter construct (Holmberg et al., 2004) that was stably transfected into PC12 cells. The Ubi reporter consists of ubiquitin followed by a 40–amino acid lysine-containing linker region. The ubiquitin is rapidly cleaved off in vivo, promoting ubiquitination of the internal lysine residues and fostering degradation of the YFP-fusion protein by the proteasome. Cells in which proteasomal activity is impaired will show an accumulation of the Ubi-YFP protein. Differentiated Ubi-YFP/PC12 cells were transiently transfected with WT-CFP, G85R-CFP, or G93A-CFP. As a positive control, we transiently transfected the Ubi-YFP/PC12 cells with a construct encoding the mutant huntingtin protein (httQ78-YFP). Similar to previous reports (Bence et al., 2001; Bennett et al., 2005), we detected an increased level of Ubi-YFP accumulation (1.88 ± 0.23-fold increase; unpublished data) in cells containing httQ78-CFP aggregates. Compared with cells expressing WT-CFP or diffusely localized G93A-CFP, cells displaying a diffuse G85R-CFP localization showed an increased accumulation of Ubi-YFP (Fig. 4, E and F). A further and substantial enhancement in Ubi-YFP accumulation was observed in cells containing aggregates formed by either G85R-CFP (2.90 ± 0.19-fold increase) or G93A-CFP (2.59 ± 0.26-fold increase; Fig. 4, E and F). These findings suggest that the formation of mutant SOD1 aggregates is highly correlated with reduced in vivo proteasome activity.
Cell death caused by mutant SOD1 aggregates
To test whether aggregate formation is directly associated with neuronal cell death, differentiated PC12 cells expressing YFP, WT-YFP, G85R-YFP, or G93A-YFP were followed using live-cell time-lapse fluorescence microscopy. Individual cells were monitored, during a 48-h period (days 3 to 5 after transfection) for protein accumulation, aggregate appearance, and cell death, based on morphological change or propidium iodide (PI) staining (Fig. 5 and Video S1 available at http://www.jcb.org/cgi/content/full/jcb.200504050/DC1). Whereas WT-YFP-transfected cells did not form aggregates, the percentage of cells expressing aggregated G85R-YFP or G93A-YFP after 3 d of expression was 30 and 15%, respectively. Within the ensuing 48-h period, 15.3 ± 3.6% of nontransfected PC12 cells and 14.5 ± 3.5% of WT-YFP–transfected cells underwent cell death, whereas ∼90% (88.5 ± 1.2% for G85R-YFP and 90.3 ± 4.1% for G93A-YFP) of cells expressing a mutant SOD1 aggregate subsequently died (Fig. 5 A).
Figure 5.
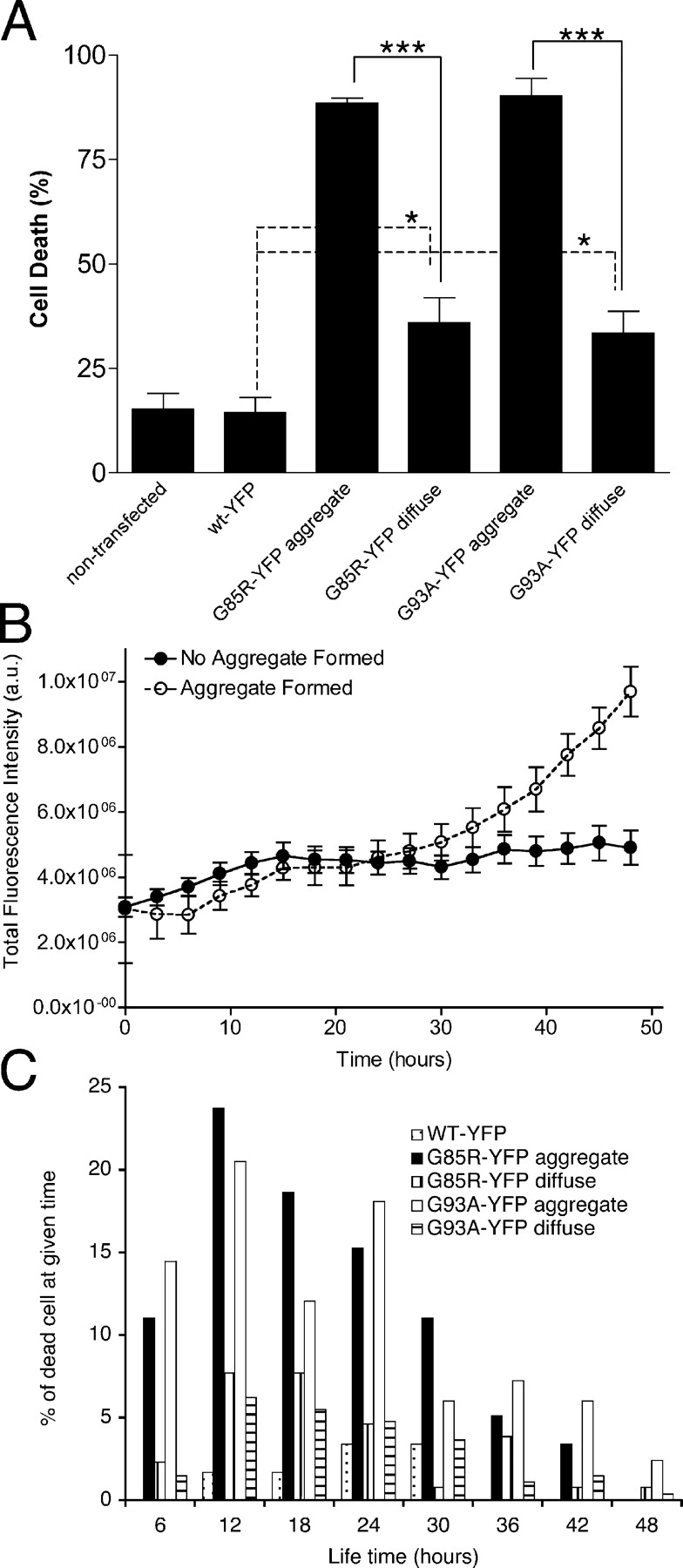
Cell death associated with mutant SOD1 aggregates. Differentiated PC12 cells were transiently transfected with constructs encoding WT-YFP, G85R-YFP, or G93A-YFP. (A) Cell death after SOD1 protein expression. Individual cells were followed for a 48-h period, from 3 to 5 d after transfection, using time-lapse fluorescence microscopy. Cell death was calculated as a percentage of dead cells at the end of the 48-h period, compared with the total number of cells. Two-tailed t test analysis (95% confidence) was used to compare the statistical difference between data sets: ***, P < 0.001; *, P < 0.05. (B) Intensity of G85R-YFP whole cell fluorescence. Whole cell fluorescence, from 3 to 5 d after transfection, was measured in PC12 cells transiently transfected with G85R-YFP. Fluorescence was measured for each time frame (every 3 h). Completed aggregate formation is set as the point of fluorescence saturation and then normalized as the 48-h time-point. n = 17 cells without aggregates and n = 21 cells with aggregates. (C) Life-time of cells after mutant SOD1 aggregate formation. Images of individual cells were taken at 2 to 3 h intervals, during the 48-h period. For cells forming visible aggregates, life-time was determined from the time when aggregates appeared (t = 0) to the time when cell death was observed. For cells that underwent cell death without the presence of visual aggregates, t = 0 was determined 3 d after transfection corresponding to a time when levels of mutant SOD1 RNA and protein levels were readily detected. Normalized life-time, t = 0 to cell death, were depicted against the percentage of dead cells at the indicated times.
An additional 30 (G85R-YFP) and 15% (G93A-YFP) of cells, in which aggregates were initially not detected, formed an aggregate during this 48-h monitoring period. We followed the level of G85R-YFP accumulation (total YFP fluorescence) before overt aggregate appearance in these cells and compared it to G85R-YFP accumulation in cells without aggregate appearance during this 48-h period (Fig. 5 B). As aggregates appeared at varying times during this 48-h period, the time-point of overt aggregate appearance was normalized as the 48-h time-point. Certain cells maintained a constant level of G85R-YFP protein reflecting a balance between synthesis and degradation without detectable aggregate formation (Fig. 5 B), whereas other cells displayed an imbalance in these processes, with a rapid increase in the level of mutant SOD1 protein, followed by the formation of large, visible aggregates (Fig. 5 B, aggregate formed). Furthermore, 70% of these aggregate-forming cells died during the subsequent 48-h monitoring period with the majority of these cells dying within 6 to 24 h after initial aggregate appearance (Fig. 5 C). In contrast, of the 30% of PC12 cells expressing diffuse mutant SOD1 proteins that exhibited cell death, we did not observe the same striking correlation with cell death (Fig. 5 C). Likewise, cells expressing soluble WT SOD1 did not undergo cell death, thus ruling out negative effects due to a flux of heterologous expression of SOD1 (Fig. 5 C). We conclude from these time-lapse studies that there is a close correlation between the appearance of mutant SOD1 aggregates, and subsequent cell death.
Discussion
In this study, we show that mutant G85R and G93A SOD1 proteins form a novel class of aggregates that correlate with cell death. Despite the difference in mutation type and in function, we observed similar biophysical properties of the aggregate species and toxicity. Using live-cell imaging, we observe that both mutant SOD1 proteins form large perinuclear aggregates that contain two populations; an immobile fraction organized into an amorphous scaffold containing mutant SOD1 and other cellular proteins such as the proteasome, and another population of mobile (diffuse) protein including mutant SOD1 in a soluble state and molecular chaperones. The structural characteristics for mutant SOD1 aggregates described here are observed in both neuronal (PC12) and nonneuronal (HeLa) cells, suggesting that these features may reflect common intrinsic properties associated with the expression of these mutant SOD1 proteins, independent of cell type.
Our live-cell analyses suggest that, even though the mutant SOD1 aggregate is structured, it is highly amorphous, with a disordered honeycomb-like scaffold through which other cellular proteins readily diffuse. This is in contrast to the aggregate structures formed by mutant huntingtin or polyglutamine-YFP that are immobile and densely packed, prohibiting the diffusion of certain soluble proteins (Chai et al., 2002; Kim et al., 2002; Holmberg et al., 2004). A third class of aggregate structures formed by expression of the polyglutamine protein, ataxin-1, is comprised of either fast or slow mobile components exchanging between the aggregate and the nucleoplasm (Stenoien et al., 2002). Taken together with our current results with mutant SOD1, we conclude that mutant huntingtin, ataxin-1, and SOD1 form aggregates that appear fundamentally distinct in their organization and structure. The molecular characteristics that distinguish these three classes were revealed only by the use of dynamic live cell imaging methods. Whether these results can be extrapolated to suggest an even more diverse family of in vivo aggregate structures, remains a question to be addressed in future studies on other neurodegenerative disease associated proteins.
Among the mechanisms proposed for aggregate-associated cytotoxicity is the association of cellular proteins with misfolded oligomers and aggregates (Bruijn et al., 1997, 2004; Cleveland and Rothstein, 2001; Pasinelli et al., 2004). Polyglutamine aggregates have been shown to recruit and sequester polyglutamine-containing transcription factors, such as TATA-binding protein (TBP) and CREB-binding protein (Steffan et al., 2000; Nucifora et al., 2001; Kim et al., 2002). The interaction between TBP and polyglutamine aggregates is dependent upon the presence of a polyglutamine stretch in TBP (Kim et al., 2002); consistent with this is the lack of colocalization of TBP with the mutant SOD1 aggregates (unpublished data). Another commonly observed class of proteins associated with various aggregates are components of the protein quality control machinery, including Hsp70 and the proteasome (Bence et al., 2001; Cleveland and Rothstein, 2001; Watanabe et al., 2001; Kim et al., 2002; Takeuchi et al., 2002; Urushitani et al., 2002; Bruijn et al., 2004; Bennett et al., 2005; Holmberg et al., 2004; Kabashi et al., 2004). Our results show that, while Hsp70 interacts transiently with mutant SOD1 aggregates in a chaperone-dependent manner, Hsp70 is not sequestered within the aggregate, similar to what was observed with mutant huntingtin (Kim et al., 2002). Likewise, both the endogenous proteasome and the transiently transfected LMP2 proteasomal subunit interact with the mutant SOD1 aggregate. However, FRAP and FLIP analyses showed that the proteasome was sequestered irreversibly. Coincident with the sequestration of the proteasome within the mutant SOD1 aggregate, we observe decreased proteasome activity; however, our results do not establish whether proteasome impairment occurred before or after the appearance of aggregates. Whereas we observed a slight increase in Ubi-YFP accumulation in cells expressing diffusely localized G85R-YFP, a greater inhibitory effect on proteasome activity was observed in cells with visible aggregates, similar to what has been observed for mutant huntingtin (Bence et al., 2001; Bennett et al., 2005). However, this contrasts with mutant ataxin-1 aggregates that do not irreversibly sequester the colocalized proteasome (Stenoien et al., 2002). We conclude that the biochemical properties of each misfolded protein determine the intrinsic features of aggregate structure, the nature of interactions with other cellular proteins, and the dynamics of these interactions.
In our studies, mutant SOD1 aggregates localize near the microtubule organizing centers and are surrounded by α-tubulin (unpublished observations), consistent with previous studies showing that mutant SOD1 forms aggresomes through a microtubule-dependent process (Johnston et al., 1998, 2000; Kopito, 2000). Although aggresome formation may be a component of the cellular response to the accumulation of misfolded proteins when the capacity of the proteasome is exceeded (Kopito, 2000), the formation of such structures will also inevitably increase the local concentration of mutant SOD1 protein and associated proteins. As the proteasome has been implicated as a crucial regulator of multiple essential cellular processes, including gene expression, apoptosis, the cell cycle, development, and DNA repair (for review see Wolf and Hilt, 2004), aberrant interactions initiated by aggregate formation may result in the dysregulation of numerous pathways critical to cell survival.
The appearance of mutant SOD1 aggregates in mouse models of fALS (Bruijn et al., 1997; Johnston et al., 2000; Watanabe et al., 2001; Wang et al., 2002) and in tissue samples from patients (Shibata et al., 1996; Kato et al., 2000) has suggested a role for aggregation in cellular toxicity. However, other observations with mutant SOD1 (A4V and V148G) have concluded that aggregate formation does not correlate with neuronal death in differentiated PC12 cells (Lee et al., 2002). In an effort to address this, we performed time-lapse experiments to follow the fate of individual differentiated PC12 cells expressing mutant SOD1 and showed that cell death correlates with an increased levels of mutant SOD1 protein and the formation of aggregates and occurs within 6 to 24 h after aggregates are visually detected. At any given time-point, only a small fraction (20–30%) of mutant SOD1-expressing cells has visible aggregates. Such variation may be due to the intrinsic heterogeneity of transient or stably expressing cell lines or perhaps due to differential levels of protein expression and cell-to-cell variation in protein homeostasis. If only these aggregate-containing cells are marked for cell death within the subsequent 24-h time period, the overall percentage of cells undergoing cell death at any single time point is much smaller, yet for the fraction of cells with aggregates, nearly all are destined for cell death. Moreover, as these aggregate-containing cells die, they are replaced by other cells that express aggregates. Using live-cell time-lapse microscopy, we are able to demonstrate the rapid cell death that ensues upon appearance of mutant SOD1 aggregates and clearly show that aggregate formation leads to cell death. Whereas aggregate-containing PC12 cells died rapidly, in contrast, 70% of cells with diffuse mutant SOD1 remained viable over the 120-h experimental period. A recent observation with cells containing huntingtin aggregates showed survival for more than 120 h and that aggregate-containing cells even exhibited a reduced risk for cell death (Arrasate et al., 2004). Even though some of these differences can be attributed to various technical differences (i.e., level of protein overexpression, cell type, method of detection/characterization), our studies on mutant SOD1 aggregates clearly indicate a direct link to neuronal cell death.
Materials and methods
Constructs
The pEYFP-N1-Hsp70 and the Hsp70 substrate binding domain deletion mutant (Hsp70-SBDx-YFP) were previously described (Kim et al., 2002). The pEYFP-N1-LMP2, pTRE2hyg-HttQ78-CFP, and pEYF-N1-Ubi-YFP constructs were previously described (Holmberg et al., 2004). The pTRE-YFP or pTRE-CFP vectors were generated by PCR amplification of YFP from pEYFP-N1 or CFP from pECFP-N1 (CLONTECH Laboratories, Inc.) and subcloning into the pTRE2hyg (CLONTECH Laboratories, Inc.). pTRE-SOD1-WT-YFP, pTRE-SOD1-WT-CFP, pTRE-SOD1-G85R-YFP, and pTRE-SOD1-G85R-CFP were generated by PCR amplification of WT SOD1 and G85R mutant SOD1 from plQL01 or plQL03 (gift from Dr. Q. Liu, Harvard Medical School, Boston, MA), respectively, and inserting into pTRE-YFP or pTRE-CFP. pTRE-SOD1-G93A-YFP and pTRE-SOD1-G93A-CFP were generated by PCR-based site-directed mutagenesis at G93A of pTRE-SOD1-WT-YFP or pTRE-SOD1-WT-CFP. All constructs were verified by sequencing.
Cell culture and transfection
PC12 Tet-Off cells (CLONTECH Laboratories, Inc.) were maintained in DME supplemented with 10% heat-inactivated horse serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μg/ml G418 at 37°C in a 5% CO2-humidified atmosphere. PC12 Tet-Off cells were differentiated with 100 ng/ml nerve growth factor (Sigma-Aldrich) for 7 d and were continuously maintained in medium containing nerve growth factor for the remainder of all studies, as previously described (Vignali et al., 1996). For colocalization studies, cells were grown in Matrigel-coated (BD Biosciences) two-well glass slide chambers (Lab-Tek). For live cell analysis, cells were grown in Matrigel-coated 35-mm glass-bottom dishes (MatTek Corp.). Transient transfections were performed using LipofectAmine-PLUS reagent using twice the manufacturer-recommended volume and 3 μg of SOD1 DNA constructs. Cotransfection of pEYFP-N1-Hsp70 with the SOD1 constructs was performed at a ratio of 1:10 (Hsp70/SOD1) and of pEYFP-N1-LMP2 at 1:3 (LMP2/SOD1). Ubi-YFP/PC12 cells were generated by cotransfecting PC12 Tet-Off cells with the pEYFP-N1-Ubi-YFP construct and a puromycin-resistance pBSpacΔp construct (gift from Dr. Y. Minami, Osaka University, Osaka, Japan), using LipofectAmine-PLUS (Invitrogen) and selecting with 2.5 μg/ml puromycin.
Western analysis
Transfected PC12 cells were harvested, resuspended in 20 mM Hepes, pH 7.9, with 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 1 mM PMSF and lysed using freeze–thaw. Total protein amount was quantitated using a Bradford protein assay (Bio-Rad Laboratories). Protein lysates (10 μg) were resuspended in SDS sample buffer and loaded onto a 12.5% SDS-PAGE gel. The gel was transferred to a nitrocellulose membrane and immunoblotted using an IRDye 800-fused anti-GFP antibody (600–132-215; Rockland Immunochemicals, Inc.) to detect CFP- or YFP-fused proteins. As a loading control, γ-tubulin was detected using an anti–γ-tubulin antibody (GTU-88; Sigma-Aldrich). Primary antibodies were detected with an anti–mouse Alexa Fluor 680-conjugated secondary antibody (Invitrogen). Blots were analyzed using an Odyssey infrared imaging system (LI-COR Bioscience).
SOD1 activity assay
The same protein lysates (15 μg) used for Western analysis, were loaded onto a 10% native PAGE gel. The gel was incubated in a riboflavin-NBT (Sigma-Aldrich) solution at room temperature for 15 min (in the dark, as riboflavin is light sensitive). After removal of riboflavin-NBT, the gel was incubated with 0.1% TEMED for 15 min and exposed to light, to induce superoxide synthesis.
Filter-trap assay
The same protein lysates (100 μg) used for Western analysis, were resuspended in a 1% SDS solution in PBS, and loaded onto a cellulose acetate membrane (0.2 μm; Advantec MFS, Inc.). The membrane was washed with 1% SDS in PBS and protein was detected as per Western blotting protocol.
Visualization of fluorescently tagged proteins
Transfected PC12 cells were fixed in 4% formaldehyde in PBS for 10 min, quenched in 0.1 M Tris-HCl, pH 8.0, for 5 min, washed in PBS at room temperature, and mounted in Vectashield antifading solution (Vector Laboratories). Fixed samples were examined using a Leica TCS SP2/Leica DM-IRE2 inverted confocal microscope equipped with 63× oil objective lens (Leica). Approximately 300 different transfected cells were scored, in three independent experiments, for aggregate formation. For measurement of LMP2 relocalization, a whole-cell Z stack (each slice = 0.5 μm) was acquired, a maximum projection created, and whole-cell intensity of LMP2-YFP measured against the intensity localized to the aggregate (n = 7 different cells). For live-cell imaging, transfected PC12 cells were maintained at 37°C for the duration of the experiment. Photobleaching for FRAP and FLIP analyses was performed as described previously (Kim et al., 2002; Holmberg et al., 2004), using a Zeiss LSM 510 Meta Axiovert confocal microscope (Carl Zeiss MicroImaging Inc.) and a 40× oil objective lens (FRAP) and a Leica TCS SP2/Leica DM-IRE2 inverted confocal microscope and a 63× oil objective lens (FLIP). The 514-nm laser wavelength was used at 100% laser power in photobleaching of the YFP fusion proteins. For FRAP analysis, single-scan images were taken at 5× zoom power and an area of 12.5 μm2 was bleached for 3 s (20 iterations), after which an image was collected every 3 s. For FLIP analysis, a single image was taken at 4× zoom power and an area of 8.4 μm2 was bleached away from the aggregate. Subsequently, images were collected every 30 s during continuous photobleaching. Average fluorescence intensities for FRAP and FLIP were determined using Metamorph software (Universal Imaging Corp.). Relative fluorescence intensities (RFI) for FRAP and FLIP were calculated using RFI = ([Net/N1t]/[Ne0/N10]) × 100, as previously described (Kim et al., 2002; Holmberg et al., 2004). FRAP data were fit as described in Online supplemental materials. FLIP data were fit with GraphPad Prism (GraphPad Software), using a two-phase exponential decay method, which yielded a better curve fit than a one-phase exponential decay method. All images were processed by Adobe Photoshop software (Adobe Systems Inc.).
Ubi-YFP reporter assay
Differentiated Ubi-YFP/PC12 cells were transfected with indicated constructs and 72 h later, the cells were fixed and examined using a Zeiss Axiovert 200 inverted microscope equipped with a 20× objective lens, CFP (ex 436/20, em 480/40), and a YFP (ex 500/20, em 535/30) filter set (Carl Zeiss MicroImaging, Inc.). YFP intensities in nuclei of individual transfected or nontransfected cells were measured using Metamorph software.
Time-lapse survival assay
PC12 cells were transfected with constructs and 72 h later, analyzed using time-lapse microscopy. Cells were maintained at 37°C, with a 5% CO2 live-cell incubation chamber, for the duration of the experiment. Imaging was performed on a Zeiss Axiovert 200 inverted microscope, equipped with a 20× objective lens, an objective heater, and an electromechanical stage for automated x, y, and z positioning. Images at 15–20 different sites were acquired at 2–3 h intervals, during a 48-h period. For each site, seven different Z planes (2 μm) were acquired to account for cellular movement in the Z plane. Images were acquired of the FITC channel (YFP-fused proteins), Texas red channel (PI; Sigma-Aldrich), and DIC. After the 48-h period, images were aligned into a time-lapse animation using Zeiss AxioVision imaging software. Individual cells were followed over the 48-h period and scored for time of aggregate appearance and cell death, according to morphological change or PI staining. Over 200 individual cells, for each construct, were scored over three independent experiments. All images were processed by Adobe Photoshop software.
Statistical analysis
GraphPad Prism software was used to fit all curves and determine statistical values. All data are depicted as mean ± SEM. Two-tailed t test analysis (95% confidence) was used to compare the statistical difference between data sets, where applicable.
FRAP curve fitting and mathematical modeling
Two empirical formulas, one described by Ellenberg et al. (1997) and Lippincott-Schwartz et al. (2001),
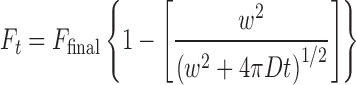 |
(1) |
and a second described by Axelrod et al. (1976),
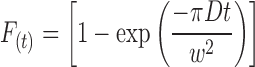 |
(2) |
have been commonly used for fitting FRAP data and the determination of diffusion coefficients. However, neither of these equations yielded a completely satisfactory fit to our experimental data, suggesting that these proteins exhibit a behavior that cannot be explained solely by diffusion, rather also involves either the formation of higher-order complexes or binding interactions with one or more cytoplasmic macromolecules or structures. To incorporate such interactions into an optimal, biologically significant mathematical model of the mobility of WT and mutant SOD1-YFP proteins, we fit FRAP data using a recently described biphasic recovery equation by Carrero et al. (2004)(see equation 53 therein),
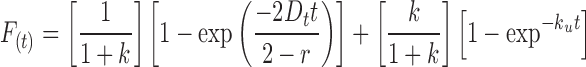 |
(3) |
characterized by a fast recovery phase produced by a fraction 1/(1 + k) of the protein population diffusing rapidly with a diffusional transfer coefficient Dt, and a slow recovery phase produced by the “turnover” of a fraction k/(1 + k) of the protein population bound with an unbinding (off) rate ku. All FRAP curves were fit with equation 3, using GraphPad Prism software (GraphPad Software), yielding satisfactory fits with residual mean squares s2 < 0.0006 s−1. We then used the relationship by Carrero et al. (2004),
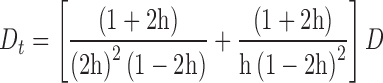 |
(4) |
to calculate the diffusion coefficient D from the experimentally derived diffusional transfer coefficient Dt. From the diffusion coefficients, we have applied the Stokes-Einstein formula,
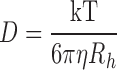 |
(5) |
where the Boltzmann constant k, the viscosity of the solution η, and temperature T all remained constant throughout our studies, to yield a relationship between the diffusion coefficient and the Stokes' radius of the molecule, D ∼ Rh −1. Assuming that the molecule in question is a sphere with a Stokes' radius Rh and a volume proportional to its molecular weight, we can relate the diffusion coefficient to molecular weight by D ∼ M−1/3. As WT SOD1 forms a highly stable homodimer, we expect WT-SOD1 fused to YFP to form a complex with a molecular weight of ∼90 kD. Using D ∼ M−1/3 or where C is a constant to be calculated, and the experimentally derived diffusion coefficients (Table I), we approximated the average molecular weights resulting in the reduced diffusion for each protein population.
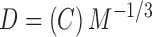 |
(6) |
Immunofluorescence analysis
The protocol for visualization of the 20-s endogenous proteasome was performed as previously described (Holmberg et al., 2004).
Online supplemental material
Fig. S1 shows mutant SOD1 aggregates retain similar properties in nonneuronal cells. Fig. S2 demonstrates WT-YFP also diffuses through mutant SOD1 aggregates. Fig. S3 shows that mutant SOD1 aggregates affect the localization of endogenous proteasome. Video S1 depicts how PC12 cells forming mutant SOD1 aggregates die rapidly. Online supplemental materials are available at http://www.jcb.org/cgi/content/full/jcb.200504050/DC1.
Acknowledgments
We thank Dr. Q. Liu for SOD1 constructs, Dr. R.A. Lamb and Dr. G.P. Leser for sharing their time-lapse microscope, and Dr. W. Russin, the Bioimaging Facility at Northwestern University (Evanston, IL), the Cell Imaging Facility at Northwestern Medical School (Chicago, IL), and S. Fox and K.E. Staniszewski for technical support.
This study was supported by grants from the National Institutes of Health (NIH), the Amyotrophic Lateral Sclerosis Association, and the Daniel F. and Ada L. Rice Foundation. G. Matsumoto and C.I. Holmberg were supported by Human Frontier Science Program Organization long-term fellowships, A. Stojanovic was supported by a training grant from the NIH (T32 AG00260; Drug Discovery Training in Age-related Disorders), and S. Kim was supported by the Mechanisms in Aging and Dementia Training Program from the National Institutes of Aging (T32 AG20506).
Abbreviations used in this paper: fALS, familial amyotrophic lateral sclerosis; FLIP, fluorescent loss in photobleaching; NBT, nitroblue tetrazolium; PI, propidium iodide; RFI, relative fluorescence intensity; ROI, region of interest; SOD1, Cu/Zn superoxide dismutase 1; TBP, TATA-binding protein; WT, wild type.
References
- Arrasate, M., S. Mitra, E.S. Schweitzer, M.R. Segal, and S. Finkbeiner. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431:805–810. [DOI] [PubMed] [Google Scholar]
- Axelrod, D., D.E. Koppel, J. Schlessinger, E. Elson, and W.W. Webb. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 16:1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence, N.F., R.M. Sampat, and R.R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292:1552–1555. [DOI] [PubMed] [Google Scholar]
- Bennett, E.J., N.F. Bence, R. Jayakumar, and R.R. Kopito. 2005. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell. 17:351–365. [DOI] [PubMed] [Google Scholar]
- Borchelt, D.R., M.K. Lee, H.S. Slunt, M. Guarnieri, Z.S. Xu, P.C. Wong, R.H. Brown Jr., D.L. Price, S.S. Sisodia, and D.W. Cleveland. 1994. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA. 91:8292–8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling, A.C., E.E. Barkowski, D. McKenna-Yasek, P. Sapp, H.R. Horvitz, M.F. Beal, and R.H. Brown Jr. 1995. Superoxide dismutase concentration and activity in familial amyotrophic lateral sclerosis. J. Neurochem. 64:2366–2369. [DOI] [PubMed] [Google Scholar]
- Bruijn, L.I., M.W. Becher, M.K. Lee, K.L. Anderson, N.A. Jenkins, N.G. Copeland, S.S. Sisodia, J.D. Rothstein, D.R. Borchelt, D.L. Price, and D.W. Cleveland. 1997. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 18:327–338. [DOI] [PubMed] [Google Scholar]
- Bruijn, L.I., M.K. Houseweart, S. Kato, K.L. Anderson, S.D. Anderson, E. Ohama, A.G. Reaume, R.W. Scott, and D.W. Cleveland. 1998. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 281:1851–1854. [DOI] [PubMed] [Google Scholar]
- Bruijn, L.I., T.M. Miller, and D.W. Cleveland. 2004. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 27:723–749. [DOI] [PubMed] [Google Scholar]
- Carrero, G., E. Crawford, M.J. Hendzel, and G. de Vries. 2004. Characterizing fluorescence recovery curves for nuclear proteins undergoing binding events. Bull. Math Biol. 66:1515–1545. [DOI] [PubMed] [Google Scholar]
- Chai, Y., J. Shao, V.M. Miller, A. Williams, and H.L. Paulson. 2002. Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc. Natl. Acad. Sci. USA. 99:9310–9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland, D.W., and J.D. Rothstein. 2001. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2:806–819. [DOI] [PubMed] [Google Scholar]
- Ellenberg, J., E.D. Siggia, J.E. Moreira, C.L. Smith, J.F. Presley, H.J. Worman, and J. Lippincott-Schwartz. 1997. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 138:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudette, M., M. Hirano, and T. Siddique. 2000. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1:83–89. [DOI] [PubMed] [Google Scholar]
- Goldberg, A.L. 2003. Protein degradation and protection against misfolded or damaged proteins. Nature. 426:895–899. [DOI] [PubMed] [Google Scholar]
- Holmberg, C.I., K.E. Staniszewski, K.N. Mensah, A. Matouschek, and R.I. Morimoto. 2004. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23:4307–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H., F.C. Nucifora Jr., C.A. Ross, and D.B. DeFranco. 2003. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 12:1–12. [DOI] [PubMed] [Google Scholar]
- Johnston, J.A., C.L. Ward, and R.R. Kopito. 1998. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J.A., M.J. Dalton, M.E. Gurney, and R.R. Kopito. 2000. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 97:12571–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi, E., J.N. Agar, D.M. Taylor, S. Minotti, and H.D. Durham. 2004. Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 89:1325–1335. [DOI] [PubMed] [Google Scholar]
- Kato, S., M. Takikawa, K. Nakashima, A. Hirano, D.W. Cleveland, H. Kusaka, N. Shibata, M. Kato, I. Nakano, and E. Ohama. 2000. New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1:163–184. [DOI] [PubMed] [Google Scholar]
- Kim, S., E.A. Nollen, K. Kitagawa, V.P. Bindokas, and R.I. Morimoto. 2002. Polyglutamine protein aggregates are dynamic. Nat. Cell Biol. 4:826–831. [DOI] [PubMed] [Google Scholar]
- Kopito, R.R. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10:524–530. [DOI] [PubMed] [Google Scholar]
- Lee, J.P., C. Gerin, V.P. Bindokas, R. Miller, G. Ghadge, and R.P. Roos. 2002. No correlation between aggregates of Cu/Zn superoxide dismutase and cell death in familial amyotrophic lateral sclerosis. J. Neurochem. 82:1229–1238. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2:444–456. [DOI] [PubMed] [Google Scholar]
- Nucifora, F.C., Jr., M. Sasaki, M.F. Peters, H. Huang, J.K. Cooper, M. Yamada, H. Takahashi, S. Tsuji, J. Troncoso, V.L. Dawson, et al. 2001. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 291:2423–2428. [DOI] [PubMed] [Google Scholar]
- Pasinelli, P., D.R. Borchelt, M.K. Houseweart, D.W. Cleveland, and R.H. Brown Jr. 1998. Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc. Natl. Acad. Sci. USA. 95:15763–15768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli, P., M.E. Belford, N. Lennon, B.J. Bacskai, B.T. Hyman, D. Trotti, and R.H. Brown Jr. 2004. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 43:19–30. [DOI] [PubMed] [Google Scholar]
- Reits, E.A., A.M. Benham, B. Plougastel, J. Neefjes, and J. Trowsdale. 1997. Dynamics of proteasome distribution in living cells. EMBO J. 16:6087–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen, D.R., T. Siddique, D. Patterson, D.A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J.P. O'Regan, H.X. Deng, et al. 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 362:59–62. [DOI] [PubMed] [Google Scholar]
- Saudou, F., S. Finkbeiner, D. Devys, and M.E. Greenberg. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 95:55–66. [DOI] [PubMed] [Google Scholar]
- Schaffar, G., P. Breuer, R. Boteva, C. Behrends, N. Tzvetkov, N. Strippel, H. Sakahira, K. Siegers, M. Hayer-Hartl, and F.U. Hartl. 2004. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol. Cell. 15:95–105. [DOI] [PubMed] [Google Scholar]
- Sherman, M.Y., and A.L. Goldberg. 2001. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 29:15–32. [DOI] [PubMed] [Google Scholar]
- Shibata, N., A. Hirano, M. Kobayashi, T. Siddique, H.X. Deng, W.Y. Hung, T. Kato, and K. Asayama. 1996. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J. Neuropathol. Exp. Neurol. 55:481–490. [DOI] [PubMed] [Google Scholar]
- Steffan, J.S., A. Kazantsev, O. Spasic-Boskovic, M. Greenwald, Y.Z. Zhu, H. Gohler, E.E. Wanker, G.P. Bates, D.E. Housman, and L.M. Thompson. 2000. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA. 97:6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien, D.L., M. Mielke, and M.A. Mancini. 2002. Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat. Cell Biol. 4:806–810. [DOI] [PubMed] [Google Scholar]
- Takeuchi, H., Y. Kobayashi, T. Yoshihara, J. Niwa, M. Doyu, K. Ohtsuka, and G. Sobue. 2002. Hsp70 and Hsp40 improve neurite outgrowth and suppress intracytoplasmic aggregate formation in cultured neuronal cells expressing mutant SOD1. Brain Res. 949:11–22. [DOI] [PubMed] [Google Scholar]
- Taylor, J.P., J. Hardy, and K.H. Fischbeck. 2002. Toxic proteins in neurodegenerative disease. Science. 296:1991–1995. [DOI] [PubMed] [Google Scholar]
- Urushitani, M., J. Kurisu, K. Tsukita, and R. Takahashi. 2002. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 83:1030–1042. [DOI] [PubMed] [Google Scholar]
- Vignali, G., J. Niclas, M.T. Sprocati, R.D. Vale, C. Sirtori, and F. Navone. 1996. Differential expression of ubiquitous and neuronal kinesin heavy chains during differentiation of human neuroblastoma and PC12 cells. Eur. J. Neurosci. 8:536–544. [DOI] [PubMed] [Google Scholar]
- Wang, J., G. Xu, and D.R. Borchelt. 2002. High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol. Dis. 9:139–148. [DOI] [PubMed] [Google Scholar]
- Wanker, E.E., E. Scherzinger, V. Heiser, A. Sittler, H. Eickhoff, and H. Lehrach. 1999. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 309:375–386. [DOI] [PubMed] [Google Scholar]
- Watanabe, M., M. Dykes-Hoberg, V.C. Culotta, D.L. Price, P.C. Wong, and J.D. Rothstein. 2001. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 8:933–941. [DOI] [PubMed] [Google Scholar]
- Wolf, D.H., and W. Hilt. 2004. The proteasome: a proteolytic nanomachine of cell regulation and waste disposal. Biochim. Biophys. Acta. 1695:19–31. [DOI] [PubMed] [Google Scholar]
- Wyttenbach, A., J. Swartz, H. Kita, T. Thykjaer, J. Carmichael, J. Bradley, R. Brown, M. Maxwell, A. Schapira, T.F. Orntoft, et al. 2001. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum. Mol. Genet. 10:1829–1845. [DOI] [PubMed] [Google Scholar]
- Zhou, H., S.H. Li, and X.J. Li. 2001. Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J. Biol. Chem. 276:48417–48424. [DOI] [PubMed] [Google Scholar]