

Abstract
Ca2+-induced Ca2+ release (CICR) plays an important role in the generation of cytosolic Ca2+ signals in many cell types. However, it is inherently difficult to distinguish experimentally between the contributions of messenger-induced Ca2+ release and CICR. We have directly tested the CICR sensitivity of different regions of intact pancreatic acinar cells using local uncaging of caged Ca2+. In the apical region, local uncaging of Ca2+ was able to trigger a CICR wave, which propagated toward the base. CICR could not be triggered in the basal region, despite the known presence of ryanodine receptors. The triggering of CICR from the apical region was inhibited by a pharmacological block of ryanodine or inositol trisphosphate receptors, indicating that global signals require coordinated Ca2+ release. Subthreshold agonist stimulation increased the probability of triggering CICR by apical uncaging, and uncaging-induced CICR could activate long-lasting Ca2+ oscillations. However, with subthreshold stimulation, CICR could still not be initiated in the basal region. CICR is the major process responsible for global Ca2+ transients, and intracellular variations in sensitivity to CICR predetermine the activation pattern of Ca2+ waves.
Keywords: Ca2+-induced Ca2+ release; caged Ca2+; Ca2+ wave; pancreatic acinar; Ca2+ release channels
Introduction
Ca2+-induced Ca2+ release (CICR)* is a process whereby Ca2+ triggers release of Ca2+ from intracellular stores (Endo et al., 1970; Ford and Podolsky, 1970). CICR plays a role in the generation of intracellular Ca2+ signals in many cell types (Fabiato and Fabiato, 1975; Wakui et al., 1990; Alonso et al., 1999; Solovyova et al., 2002). The CICR process is a positive feedback mechanism, which acts to amplify small elevations in the cytosolic Ca2+ concentration caused by some “trigger” mechanism such as voltage-gated Ca2+ influx after depolarization (Solovyova et al., 2002) or inositol triphosphate (IP3)–induced Ca2+ release after hormonal stimulation (Wakui et al., 1990). Virtually all models of Ca2+ oscillations depend on CICR (Berridge and Irvine, 1989; Wakui et al., 1990; Kasai et al., 1993).
CICR occurs by a mechanism whereby a rise in cytosolic Ca2+ concentration opens Ca2+ channels in the store membrane. There are two types of well-characterized Ca2+ release channels, namely IP3 receptors (IP3Rs) and ryanodine receptors (RyRs) (Pozzan et al., 1994). CICR was discovered in muscle cells, which predominantly express RyRs (Endo et al., 1970; Lai et al., 1988). Since then, characterization of isolated RyRs has confirmed their Ca2+ sensitivity (Meissner, 1994). However, IP3Rs can also be activated by a rise in the cytosolic Ca2+ concentration (Iino, 1990; Bezprozvanny et al., 1991; Miyakawa et al., 2001). This dual action of cytoplasmic Ca2+ on RyRs and IP3Rs means that different Ca2+ release channels can be linked by communication mediated by CICR. Furthermore, the regenerative nature of CICR suggests that it could play a role in the generation of spatio-temporally complex Ca2+ signals, such as Ca2+ waves (Thomas et al., 1996). Functional studies of Ca2+ release are complicated by the inherent difficulty in distinguishing between messenger-induced release and CICR. We have directly assessed the role of CICR in the generation of global Ca2+ signals by using spatially restricted uncaging of caged Ca2+ to cause elevations of cytosolic Ca2+, which are localized to specific intracellular regions of pancreatic acinar cells.
Pancreatic acinar cells are ideal for our experimental approach because they produce clearly polarized intracellular Ca2+ transients after agonist stimulation. Both local Ca2+ spikes (confined to the apical secretory granule region) and global Ca2+ waves (originating in the secretory region and then propagating toward the basal plasma membrane) can be induced by physiological doses of secretagogues (Kasai et al., 1993; Thorn et al., 1993; Toescu et al., 1994; Ashby and Tepikin, 2002). There is also nonuniform intracellular distribution of Ca2+ release channels. IP3Rs are concentrated in the apical region (Kasai et al., 1993; Thorn et al., 1993; Nathanson et al., 1994), whereas RyRs are distributed throughout the cell, both in the basal and apical regions (Leite et al., 1999; Straub et al., 2000; Fitzsimmons et al., 2000; for review see Ashby and Tepikin, 2002). It has been proposed, on the basis of indirect evidence, that there is a CICR-mediated channel cooperativity (Straub et al., 2000), which underlies the production of global Ca2+ waves (Wakui et al., 1990; Leite et al., 1999; Straub et al., 2000).
In the experiments reported here, we have used localized uncaging of caged Ca2+ in the cytosol to reveal striking regional differences in the ability to trigger CICR. The granular region displayed high sensitivity, whereas the basal region showed considerable resistance to CICR. These important regional differences were also observed in “Ca2+ ramp” experiments, where Ca2+ was uncaged slowly and uniformly in the cytosol throughout the cells. CICR induced by localized uncaging in the granular region was blocked by pharmacological inhibition of either IP3Rs or RyRs, indicating that these two types of channels must cooperate to produce CICR. The frequency with which uncaging-induced CICR occurred was increased in the presence of subthreshold concentrations of the physiological agonist cholecystokinin (CCK). A low CCK concentration, which was alone unable to trigger Ca2+ signals, could only sensitize the CICR pathway in the apical part of the cell; the basal region remained resistant to triggering of CICR. Interestingly, localized apical uncaging of Ca2+ in the presence of a subthreshold CCK concentration was not only able to trigger CICR, but could also initiate global Ca2+ oscillations. Our experiments reveal for the first time the substantial subcellular polarity of CICR in a secretory epithelium and show that this can predetermine the unidirectional polarity of Ca2+ waves. Our demonstration that CICR in the pancreatic acinar cells depends on both functional IP3Rs and RyRs explains the high apical sensitivity to CICR and the absence of CICR in the basal part of the cell, because it is only in the apical region that both types of Ca2+ release channels coexist.
Results
Localized Ca2+ uncaging reveals that the apical region has greater sensitivity to CICR than the basal region
In this part of the study, we used unstimulated pancreatic acinar cells to assess the role of CICR in the generation of global Ca2+ signals. We simultaneously imaged cytosolic Ca2+-sensitive fluorescent dyes and uncaged Ca2+ by exposure of cytosolic nitro-phenyl–EGTA (NP-EGTA) to ultraviolet laser light. The laser scanning confocal microscope used in these experiments allowed UV exposure (and hence uncaging) to occur with rapid time course and high spatial precision within predefined regions of the scan frame. The amount of Ca2+ uncaged could be controlled by alterations in the area and duration of exposure and adjustment of UV laser power. Because agonist-induced Ca2+ signals and the distribution of intracellular Ca2+ channels are highly polarized in these cells, we used localized uncaging to individually test different regions of the cell interior. Localized uncaging was induced by a brief exposure (between 5 and 50 msec) to UV laser light of a small circular region 1.6–2.2 μm in radius (e.g., dashed orange region in fluorescence image of Fig. 1 A). This resulted in uncaging of Ca2+ from NP-EGTA selectively in the specified area. The magnitude of the localized uncaging was such that the Ca2+ elevation was restricted to the immediate vicinity of uncaging, presumably due to the action of low mobility cytosolic Ca2+ buffers and removal mechanisms. The diameter of a typical uncaging region corresponded to 15–20% of the diameter of a pancreatic acinar cell. Performing the uncaging procedure in cells loaded only with fluo-4 (no caged Ca2+) caused no change in fluo-4 fluorescence, indicating that the UV exposure alone had no effect on the dye or on cellular Ca2+ homeostasis.
Figure 1.
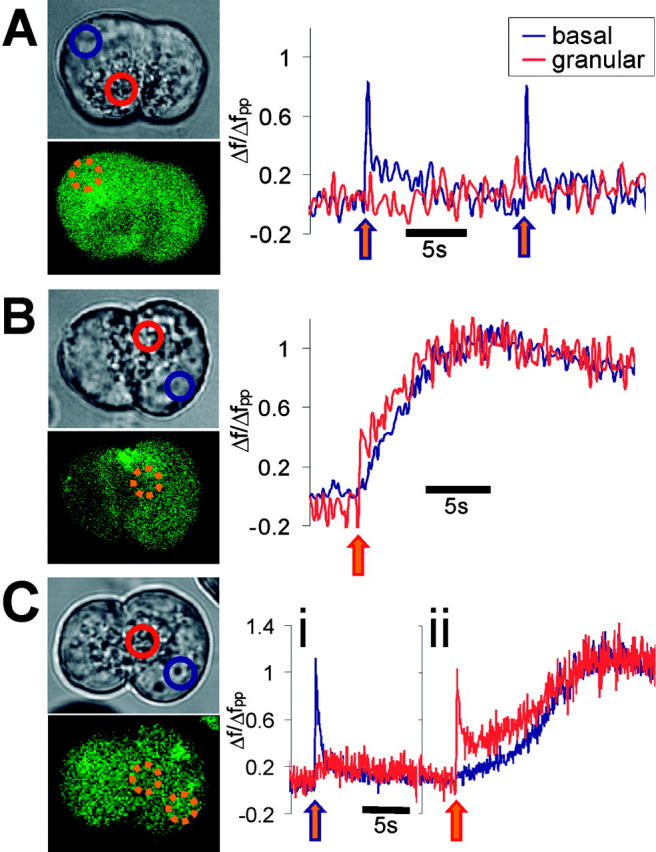
Regional variations in CICR triggered by localized uncaging. All traces represent normalized fluorescence of fluo-4 in the regions shown in accompanying transmitted image. Intracellular regions where uncaging was targeted are shown by dashed orange circles superimposed on accompanying fluorescence image. Uncaging was produced at the time points shown by arrows on the trace. The outline color of the arrow corresponds to the region of uncaging. (A) Uncaging localized to the basal region causes short-lasting, localized elevations in calcium (uncaging spikes) but fails to trigger any CICR. Fpp ratio = 1.35. (B) Uncaging of similar strength localized to the apical (secretory granule) region causes the uncaging spike that then triggers a long-lasting global CICR transient. Fpp ratio = 1.13 (C) In the same cell, (i) basal localized uncaging fails to cause CICR, but (ii) subsequent apical uncaging of identical power triggers a slowly developing global CICR transient. Fpp ratio = 1.73.
The release of Ca2+ from the uncaged NP-EGTA caused a characteristic sharp rise in the cytosolic [Ca2+] that is reported by changes in the dye fluorescence (an uncaging Ca2+ spike). Uncaging localized to the basal region caused localized Ca2+ spikes, which were entirely due to the uncaged Ca2+ (Fig. 1 A), but the basal uncaging failed to trigger any further Ca2+ release (CICR transient) (0/19 cells). In the apical region, in the absence of any other stimulation, localized Ca2+ uncaging (of similar intensity and duration as used for the basal uncaging) triggered CICR transients with relatively high frequency (25/49 cells). The CICR transient could easily be distinguished from the initial Ca2+ rise caused directly by uncaging, due to its much slower time course, which develops after the UV exposure has been terminated (Fig. 1 B). Often, the CICR transient could be seen to propagate across the cell as an apical-to-basal wave (Fig. 1 B) with similar speed to waves triggered by physiological doses of agonist.
Regional variations in CICR sensitivity were also demonstrated in the same cell using alternate basal and apical localized uncaging events (Fig. 1 C). An unstimulated cell was first exposed to localized Ca2+ uncaging in the basal region. The cytosolic Ca2+ concentration was then allowed to recover to resting levels before an apical uncaging event of identical intensity was applied. If neither of these events triggered a CICR wave, the procedure was repeated at higher UV laser power, thus uncaging more Ca2+. This cycle of localized uncaging events was repeated until a regenerative CICR wave was triggered. To account for any possible presensitization of the cell interior caused by previous uncaging spikes, the protocol was applied in reverse in other cells. In these cells, the granular region was exposed to uncaging first and then localized basal uncaging followed.
When applied first, basal uncaging caused only a localized Ca2+ spike (Fig. 1 C), as had been observed previously (Fig. 1 A). However, a Ca2+ spike caused by subsequent apical uncaging in the same cell was able to trigger a global CICR transient (five cells) (Fig. 1 C). It was never possible to initiate a CICR wave with an initial basal uncaging event. When the protocol was reversed, apical uncaging triggered a CICR wave in half of the cells (four of eight cells); in the other cells, we were not able to trigger CICR. In no cells (zero of eight cells) did the subsequent basal uncaging event trigger CICR.
Uncaging of Ca2+ could possibly activate a Ca2+-dependent phospholipase C (PLC), leading to IP3 production, which could, in turn, trigger the subsequent Ca2+ release from stores. We therefore checked whether uncaging of Ca2+ could activate PLC by measuring the real time production of IP3 in individual cells using the GFP-conjugated PH domain of PLCδ1 (GFP–PHD). This fusion construct has been used to measure cellular IP3 production in several cell types by virtue of its relative affinities for PIP2 and IP3 (Hirose et al., 1999; Nash et al., 2001; Okubo et al., 2001). In the resting cell, the GFP–PHD construct binds PIP2, which is predominantly associated with the plasma membrane, but translocates into the cytoplasm upon production of IP3 due to a higher affinity for IP3 (Hirose et al., 1999). This translocation is therefore an index of IP3 production. We expressed GFP–PH in pancreatic acinar cells by microinjection of the construct cDNA into individual cells within a cell cluster. After expression of the GFP–PH protein, cells were loaded with caged Ca2+ and the calcium-sensitive dye fura red. This approach allowed us to simultaneously monitor cellular IP3 production and cytoplasmic Ca2+ levels while performing uncaging. Photolysis of NP-EGTA caused a rapid rise in the cytosolic Ca2+ concentration, but did not cause any translocation of GFP–PHD (Fig. 2 ; n = 3). Neither localized uncaging or maximal uncaging caused any translocation. In contrast, in the same cell, application of acetylcholine (ACh) caused a rapid translocation of GFP–PHD and a rise in the cytosolic Ca2+ concentration (Fig. 2). It should be noted that the time course of translocation may not identically match the rapid kinetics of IP3 production, because it will be limited by the speed of diffusion of the relatively large protein. Also, although the relative affinity of the GFP–PHD for IP3 is high (Hirose et al., 1999), the sensitivity of the GFP–PHD for detecting IP3 is not precisely known. Nonetheless, the GFP–PHD translocation shows that although PLC is activated to produce IP3 after activation of a G-protein–coupled receptor, a rise in the cytosolic Ca2+ concentration caused by uncaging does not alone lead to production of IP3.
Figure 2.

Uncaging of Ca2 + does not stimulate PLC to produce IP3. Graph shows cytoplasmic Ca2+ and distribution of GFP–PHD in the pancreatic acinar cell shown in accompanying images (left to right: transmitted, fura red, GFP). Upper trace shows ratio of cytoplasmic to plasma membrane GFP fluorescence and lower trace shows normalized fura red fluorescence. Lower panel shows three images of GFP–PHD taken from time points indicated by the numbers on the graph. Strong uncaging at the point shown by the orange arrow causes a large rise in calcium but fails to trigger any change in the distribution of GFP–PHD, indicating that there is no production of IP3 after uncaging (compare images 1 and 2). In contrast, subsequent stimulation with 1 μM ACh causes a rapid translocation of GFP–PHD into the cytoplasm, indicated by the increase in cytoplasmic/plasma membrane fluorescence ratio and shown in image 3. This rise in IP3 level is accompanied by a sharp rise in intracellular calcium as calcium is released from stores, which is similar in peak value to that induced by uncaging.
Other intracellular messengers (cADPr and NAADP) can release Ca2+ in pancreatic acinar cells and therefore could possibly play a role during uncaging-induced CICR signals. Although there is no way presently to directly test this possibility, at this stage there is no experimental evidence of Ca2+-stimulated production of these messengers.
Differences in CICR sensitivity are not caused by regional differences in the uncaging process or by local buffering
A simultaneous uncaging protocol was used to test whether the apparent higher CICR sensitivity of the granular region was due to a larger release of Ca2+ by the uncaging process in that part of the cell. This involved simultaneous uncaging in two identical regions of the same cell; one located in the apical part and the other in the basal. This simultaneously produced Ca2+ spikes in two regions of the cell (Fig. 3 A). In this part of the study we considered only uncaging transients that were not followed by CICR. This protocol enabled us to simultaneously measure, in two distinct areas of a single cell, the change in the cytosolic free [Ca2+] due to the uncaging process.
Figure 3.
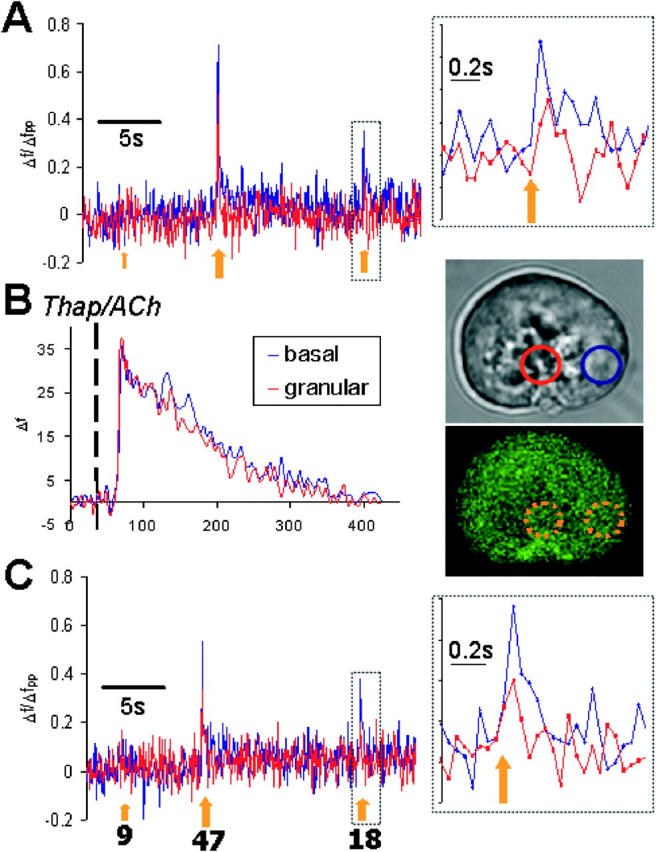
Simultaneous uncaging allows analysis of Ca2 + spikes caused by uncaging in both apical and basal regions. All traces represent normalized fluorescence levels of fluo-4 in the regions denoted in accompanying transmitted image. Traces A, B, and C were captured sequentially in time. Traces A and C followed identical scanning procedures. In these traces, images were captured at a speed of 60 msec/frame and uncaging events lasting 9, 47, and 18 msec, respectively, were triggered similarly in each trace (denoted by orange arrows). Uncaging was localized to separate granular and basal regions (indicated in fluorescence image) simultaneously. Inset shows relevant trace expanded at the 18-msec uncaging event (A and C). After the capture of trace A, the cell was perfused with 10 μM ACh and 0.5 μM thapsigargin (dashed line indicates the start of perfusion) to deplete stores and inactivate SERCA pumps. This treatment produces the characteristic transient rise in cytoplasmic Ca2+ as it is rapidly released from stores and slowly extruded from the cell. Trace C was captured after the return of the Ca2+ to a pretreatment level. Fpp ratio = 1.0.
When we compared the peak level of Ca2+ spikes caused by identical simultaneous uncagings in the basal and apical regions, significant differences were found (Fig. 3 A, inset). The mean ratio of the peak changes in Ca2+ concentration after uncaging (ΔCa2+ basal/ΔCa2+ granular) was 1.73 ± 0.09 (SEM; n = 34). Identical Ca2+ uncaging protocols clearly produce apparently larger rises in the free cytosolic Ca2+ concentration in the basal than in the apical region. It is therefore particularly remarkable that it is the Ca2+ uncaging in the apical and not in the basal part of the cell that is able to trigger a subsequent CICR transient.
There are several possible reasons for the difference in the result of uncaging in the two regions. One of these may be that there is some rapid, local CICR amplification within the uncaging spike (Lipp and Niggli, 1998). We tested this by comparing identical simultaneous uncaging protocols before and after inhibition of CICR. This was achieved by treatment with 0.5 μM of the SERCA pump inhibitor thapsigargin and 10 μM ACh to deplete the intracellular Ca2+ stores (Fig. 3 B). The relative sizes of the uncaging spikes in the apical and basal regions were similar before and after inhibition of CICR. The ratio of the peak uncaging values in thapsigargin/ACh-treated cells (ΔCa2+ basal/ΔCa2+ granular) was 1.93 ± 0.15 (SEM; n = 37), which is similar to the value reported above (1.73 ± 0.09), obtained before the treatment. This suggests that the difference in the size of the uncaging spikes in the two regions is not due to differences in local CICR amplification within the spikes.
The absolute amplitudes of uncaging spikes were reduced by ∼25% in both regions after thapsigargin/ACh treatment (Fig. 3, compare C with A). This reduction may be the result of inhibition of rapid, local CICR, but other factors, such as dye bleaching or a reduction in the NP-EGTA concentration after the first series of uncagings, may contribute to this reduction. Even if such rapid CICR does occur within the uncaging spikes, it is unlikely to underlie the differences in regional sensitivity to triggering slow global CICR waves because it is the same in both regions.
A possible explanation for the regional differences may be that the efficiency of uncaging is not equal in both regions and thus may underlie the variations observed. Although the power of the UV light is applied equally in both areas, it is possible that some scattering of the light or lower NP-EGTA concentration may reduce the uncaging in the apical region.
An intriguing possibility concerns intracellular Ca2+ buffers, which can have profound effects on the amplitude and kinetics of Ca2+ signals (Neher and Augustine, 1992). Immediately after uncaging, there is a dynamic competition for Ca2+ binding between cellular buffers fluo-4 and NP-EGTA. Therefore, the amount of Ca2+ bound by fluo-4 (and hence the increase in fluorescence) depends on this competition. The smaller amplitude of apical Ca2+ responses induced by fast, localized uncaging suggests that higher Ca2+ buffering may exist in this part of the cell than in the basal regions.
Calcium “spill-outs” from basal uncaging can trigger CICR waves originating in the apical region
It has been previously proposed that the basal region of pancreatic acinar cells undergoes CICR during agonist-induced signals (Kasai et al., 1993; Toescu et al., 1994), yet localized uncaging in this region could not trigger CICR (Fig. 1). We attempted to look for CICR in the basal region by increasing the magnitude of the basal-localized uncaging event. As the basal uncaging becomes larger, more Ca2+ can escape from the vicinity of uncaging and diffuse throughout the cytoplasm (we termed this Ca2+ “spill-outs”). These spill-outs can lead to small elevations of Ca2+ in the apical region after basal uncaging (Fig. 4) . The large Ca2+ spike in the basal region caused by the uncaging was unable to trigger a CICR wave. However, CICR could be triggered in the granular apical region by the relatively small amount of Ca2+ that diffused throughout the cell after uncaging (seven cells) (Fig. 4). The initiation site of the CICR wave was confirmed as the granular region by the early upstroke of the CICR transient in that part of the cell (Fig. 4 B). The wave then propagated from the granular to the basal pole (Fig. 4 B, images). This confirms the dramatic difference between the two regions in the ability to trigger a CICR wave. It is interesting to note, however, that after initiation, the wave propagates from the apical to the basal region in a regenerative manner.
Figure 4.

Powerful basal uncaging can trigger a regenerative CICR wave that is initiated in the granular region. Traces represent normalized fluorescence levels of fluo-4 from regions of interest shown in accompanying transmitted image. (A) The experiment is representative of those consisting of basally localized uncaging (orange region in transmitted image) followed by a cellular CICR transient. B shows an expanded section of upper trace including uncaging spike and initial phase of CICR transient. Lower panel of B shows pseudocolor confocal fluorescence images taken from time points indicated by arrows in the expanded trace. Uncaging produces a localized spike (dark blue trace and image 2) but Ca2+ also spills out of this region and throughout the cell, as shown by the slower and smaller rise in the granular part (red trace and image 3). The regenerative CICR transient is then initiated in this region, as indicated by the early rise of the granular (red) trace and images 4, 5, 6, and 7. Fpp ratio = 1.35.
Ca2+ ramp slowly elevates Ca2+ in all regions of the cell and triggers CICR first in the apical region
To verify that small regional variations in uncaging did not have unexpected effects on CICR, we explored the regional CICR sensitivity by comparing different regions at the same time under identical conditions. To achieve this, a Ca2+ ramp protocol was used to simultaneously raise Ca2+ in all parts of the cell. Throughout the scanning procedure there was a continual, low-level UV exposure in all parts of the scan frame. This caused a slow but continual uncaging of Ca2+ in all parts of the cell. As NP-EGTA was uncaged, the Ca2+ level in the cytoplasm slowly rose, causing the ramp effect. The ramp rose equally in all regions of the cell to produce a spatially homogenous cytosolic Ca2+ rise (Fig. 5) .
Figure 5.

Calcium ramp causes a steady, spatially homogenous rise in Ca2 + , which can trigger a CICR wave. Traces represent normalized fluorescence levels of fluo-4 in regions of interest indicated in accompanying transmitted image. The Ca2+ level slowly increases during the ramp phase due to continual low uncaging of Ca2+. Levels rise similarly in all regions of the cell. The ramp triggers a regenerative CICR transient, shown by the rapid change in kinetics of the rise. The CICR transient travels as a wave in an apical-to-basal direction. Fpp ratio = 0.97.
The homogenous rise in the Ca2+ concentration was able to trigger CICR, which was distinguished by a sharp increase in the rate of the cytosolic Ca2+ rise (Fig. 5). The CICR transient that was triggered then spread throughout the cell as a wave. The region where the wave was initiated displayed the earliest onset of the CICR transient. Despite the spatially homogenous nature of the ramp, the CICR wave was initiated in the apical region in 21/21 cells (Fig. 5), directly demonstrating the highly sensitive nature of this part of the cell to CICR. In the majority of the cells (18/21 cells), the wave triggered by the ramp spread from the apical region to the basal in a regenerative manner, as shown by the similar rate of rise of the CICR transient in the two areas.
Uncaging-induced CICR requires both functional IP3Rs and RyRs
Pancreatic acinar cells contain both IP3Rs and RyRs. Ca2+ release from both types of channel has been demonstrated in response to infusion of IP3 and cADPr, respectively (Thorn et al., 1993, 1994). To determine which channel(s) is involved in uncaging-induced CICR waves, we used pharmacological agents to block release from each type of channel and then assessed their effects on CICR induced by localized apical uncaging.
Caffeine is known to modify release of Ca2+ through intracellular channels (Ehrlich et al., 1994). Caffeine is generally used as an activator of CICR due to a sensitizing effect on RyRs (Fabiato, 1985; Ehrlich et al., 1994; Solovyova et al., 2002). However, caffeine (10–20 mM) also rapidly and reversibly blocks single channel currents through IP3Rs in reconstituted lipid bilayers (Bezprozvanny et al., 1994; Ehrlich et al., 1994). In pancreatic acinar cells, cytosolic Ca2+ elevations induced by IP3 are rapidly and reversibly blocked by 20 mM caffeine (Wakui et al., 1990). This is not due to depletion of intracellular Ca2+ stores, because in experiments with caffeine present from the beginning, there is no IP3-elicited Ca2+ spiking until caffeine is removed, but then Ca2+ spiking starts immediately (within seconds) (it takes several minutes to refill empty ER stores; Mogami et al., 1998). This shows that caffeine is able to block IP3Rs without triggering CICR from RyRs. Similar observations have also been made in hepatocytes (Missiaen et al., 1992), where there is evidence that both IP3 and RyRs also coexist (Sanchez-Bueno et al., 1994). Because caffeine does not inhibit RyRs (Ehrlich et al., 1994), we applied 20 mM caffeine to specifically inhibit IP3Rs. We found that fluo-4 fluorescence was considerably quenched by caffeine and so changed to an alternative Ca2+-sensitive indicator, fura red, which required the use of the whole cell patch clamp configuration to attain sufficient cytoplasmic concentrations of the dye. Under control conditions (no caffeine) in this experimental configuration, CICR was triggered by localized uncaging in the apical region in 12/19 cells tested. This proportion was similar to that exhibited by intact cells. To test the effects of caffeine, we first confirmed in each cell that ACh-induced global calcium transients were blocked and then attempted to trigger CICR by localized apical uncaging in the granular region. Uncaging-induced CICR was never observed in the presence of caffeine (zero of nine cells) (Fig. 6 A).
Figure 6.

CICR triggered by apical-localized uncaging is inhibited by pharmacological inhibition of IP3Rs and RyRs. Traces show normalized fluorescence from regions of interest, shown in accompanying transmitted images, during representative continuous experiments on cells loaded with calcium-sensitive dye and NP-EGTA. (A) The IP3 receptor antagonist caffeine blocks CICR induced by localized uncaging. (a) Application of 20 mM caffeine completely inhibits Ca2+ release triggered by 50 nM ACh, confirming the block of IP3Rs. (b) Later, still in the presence of caffeine, apically localized uncaging events (area shown by orange circle superimposed on fluorescence image) fail to trigger any CICR response. (B) The RyR antagonist ryanodine blocks CICR induced by localized uncaging. (a) 50 μM ryanodine completely blocks calcium elevations caused by 25 nM ACh. (b) In the continuous presence of ryanodine, multiple localized uncaging events (targeted at orange circle superimposed on fluorescence image) consistently fail to trigger CICR. Fpp ratio = 2.08.
We also attempted to use alternative IP3R antagonists. 2-APB and xestospongin C have been used as selective membrane-permeable IP3R antagonists (Ma et al., 2000). However, in our hands, neither 2-APB nor xestospongin C showed any suppressive effect on Ca2+ spikes triggered by infusion of 2,4,5-IP3 in pancreatic acinar cells (n = 3 and n = 4, respectively). It now appears that 2-APB is not a specific IP3 receptor antagonist, but more likely inhibits store-operated Ca2+ channels (Bakowski et al., 2001; Prakriya and Lewis, 2001). Heparin is a commonly used IP3R blocker (Ehrlich et al., 1994), but proved unsuitable for use in this experimental configuration. This is because the suppressive effect of heparin on IP3-induced spiking is somewhat variable and heparin does not consistently block ACh-induced responses. Because the CICR phenomenon under investigation is not always manifested, the number of experiments required to establish an effect of heparin proved prohibitive.
To test for involvement of RyRs in the CICR waves, we used a concentration of 50 μM ryanodine, which is usually required to block Ca2+ release from RyRs in intact cells, including pancreatic acinar cells (Sutko et al., 1997; Cancela et al., 2000; Solovyova et al., 2002). As with caffeine, we tested in each cell that this concentration blocked ACh-induced global Ca2+ responses. In those cells where the ACh effect was blocked, we then uncaged Ca2+ locally in the apical region. The presence of ryanodine completely inhibited the ability of localized apical uncaging to trigger CICR waves (six cells) (Fig. 6 B).
Subthreshold levels of the agonist CCK potentiate uncaging-induced CICR
If CICR is important in the physiological Ca2+ waves, then agonist-induced production of messengers may have an effect on CICR caused by uncaging. We therefore tested the effect of very small doses of agonist on the cell's ability to undergo CICR triggered by localized Ca2+ uncaging. We used low concentrations of CCK, which were deemed to be subthreshold because they alone did not elicit any Ca2+ elevation. Uncaging was performed in the absence and presence of these subthreshold agonist concentrations. The sensitivity to CICR was dramatically increased in the presence of CCK as shown in Fig. 7 . In this cell, multiple uncaging events were unable to cause CICR under control conditions, but when 1 pM CCK was present, a single uncaging spike of smaller size was able to trigger CICR. When pooled together, experiments of this type demonstrate the overall sensitizing effect of CCK on CICR sensitivity. In the presence of a subthreshold CCK concentration, the probability of triggering CICR by localized apical uncaging increased by ∼50%, with 28/36 cells undergoing CICR, compared with 26/49 cells when no agonist stimulation was present.
Figure 7.
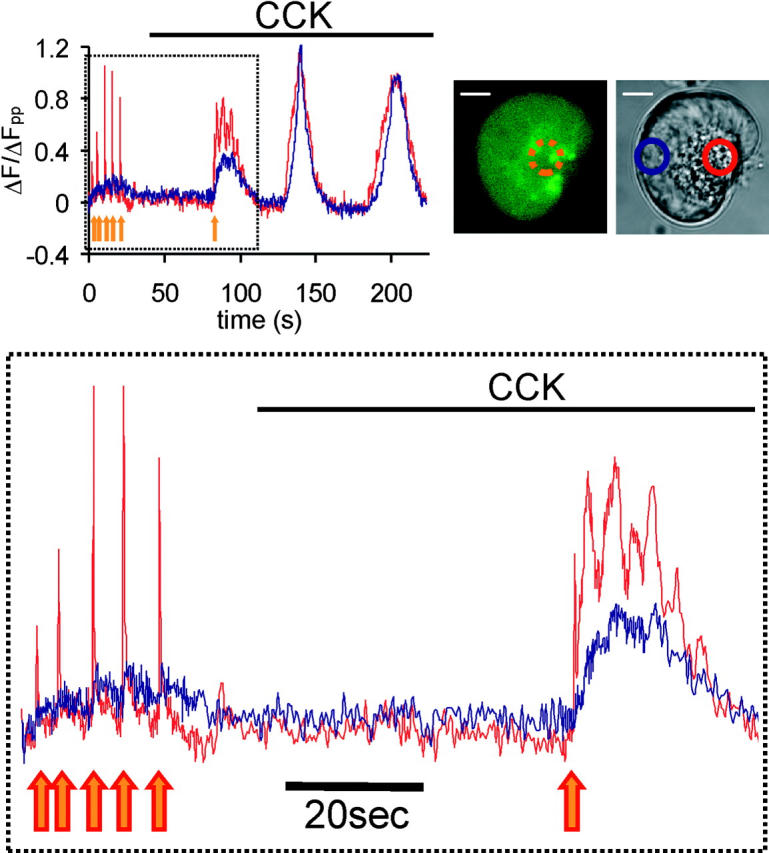
Subthreshold concentrations of agonist potentiate CICR triggered by apical uncaging. Trace shows normalized fluorescence from regions shown in transmitted image. Top trace shows full time course of experiment in which localized apical uncaging (orange circle in fluorescence image) is performed in the absence and then presence of subthreshold CCK (1 pM). Area denoted by dashed box is expanded below. In this case, multiple uncaging events fail to trigger any CICR response in the absence of any other stimulation, but single, smaller uncaging spike initiates CICR when subthreshold agonist is present. Fpp ratio = 0.84.
In light of the sensitizing effect of subthreshold CCK concentrations on the apical region, we tested the effect of similar concentrations of CCK on basal-localized uncaging. However, in the presence of subthreshold agonist stimulation, it was still impossible to trigger CICR waves by basal-localized uncaging (12 cells) (Fig. 8) . In these experiments, when the basal uncaging intensity was increased to a spill-out level, which caused the uncaged Ca2+ to diffuse throughout the cytoplasm, we did see CICR waves in 7/12 cells. All these waves were initiated in the granular region after spill-out and propagated in an apical-to-basal direction (Fig. 8).
Figure 8.

Basally localized uncaging cannot initiate CICR in the basal region even in the presence of subthreshold agonist. Trace shows normalized fluo-4 fluorescence from regions shown in accompanying transmitted image. Cells were stimulated with 1 pM CCK throughout. Basally localized uncaging (area shown by orange circle in fluorescence image) of increasing magnitude produces uncaging spikes of increasing size. None of these spikes can trigger CICR until, finally, an uncaging event of large size results in spill-out of calcium into the apical region. This calcium then triggers a CICR wave that propagates in an apical-to-basal direction. This is shown by the early rise of the apical (red) trace. Fpp ratio = 1.9.
Uncaging-induced CICR waves can trigger Ca2+ oscillations in the presence of subthreshold agonist concentrations
The most common mode of Ca2+ signaling caused by physiological levels of agonist stimulation in pancreatic acinar cells is oscillations in [Ca2+]i (Tsunoda et al., 1990; Petersen et al., 1994). In the absence of agonist stimulation, localized apical Ca2+ uncaging was able to trigger a global Ca2+ wave, but this CICR wave was never followed by any oscillatory behavior. In contrast to this, in the presence of subthreshold agonist concentrations, uncaging-induced CICR transients were often followed by further oscillations in the cytosolic Ca2+ concentration (Figs. 7 and 9) .
Figure 9.

Uncaging-induced CICR can initiate oscillations in the presence of subthreshold stimulation. Trace shows normalized fluo-4 fluorescence from regions shown in accompanying transmitted image. Cells are perfused with a low concentration of agonist (10 pM CCK) for ∼8 min to establish that this dose is subthreshold. Then, apical-localized uncaging (area shown by dashed orange circle in fluorescence image) triggers a CICR wave (this section of the time course is expanded in dashed box above). After this CICR wave, and without further uncaging, the cell produces oscillations in calcium until the agonist is removed. Fpp ratio = 1.34.
We systematically explored this phenomenon in long-lasting experiments in the presence of subthreshold agonist (Fig. 9). It was important to establish that the agonist concentration used was indeed subthreshold for the particular cell under investigation and could not have caused the oscillations alone. We did this by establishing the highest concentration of agonist that did not cause any Ca2+ signals during a long period (at least 8 min) before the uncaging. After this period, a global CICR wave was triggered by apical uncaging (Fig. 9, inset). In nine experiments, the uncaging-induced CICR wave was followed by a series of global transients, which lasted until removal of agonist (Fig. 9).
Discussion
We have shown that local uncaging of caged Ca2+ in the apical granule-containing region of pancreatic acinar cells can trigger cytosolic Ca2+ waves, which spread from the apical region toward the basal membrane (Fig. 1 B). In contrast, local Ca2+ uncaging in the basal region consistently failed to initiate Ca2+ waves (Fig. 1, A and C). The Ca2+ waves induced by Ca2+ uncaging did not involve an increase in IP3 levels (Fig. 2). The process whereby a rise in the cytosolic Ca2+ concentration triggers release of Ca2+ from intracellular stores (CICR) is generally associated with Ca2+ activation of RyRs (Meissner, 1994; Solovyova et al., 2002). RyRs of all the three known subtypes exist in pancreatic acinar cells and are distributed throughout the basolateral and apical regions (Leite et al., 1999; Fitzsimmons et al., 2000). Leite et al. (1999) even report that the density of RyRs is higher in the basal than in the apical region. In light of this, the specific localization of CICR to the apical region of the pancreatic acinar cells was therefore unexpected, although this region was shown to be particularly sensitive to Ca2+ in one other report (Kasai et al., 1993). CICR in the pancreatic acinar cells clearly depends on operational RyRs, because it was blocked by ryanodine (Fig. 6 B), but CICR was also blocked by caffeine inhibition of IP3Rs (Fig. 6 A). There is now clear evidence that the Ca2+ sensor region of the IP3R can control cytosolic Ca2+ signal generation (Miyakawa et al., 2001), and our new results indicate that in the pancreas, CICR depends on cooperation between functional IP3Rs and RyRs. The IP3Rs, in contrast to the RyRs, are specifically localized in the apical region (Kasai et al., 1993; Thorn et al., 1993; Nathanson et al., 1994; Lee et al., 1997). Our pharmacological results (Fig. 6) therefore explain why CICR initiation only occurs in the apical region, because this is the only place where IP3Rs and RyRs come into close contact. Our new data place the CICR phenomenon at the heart of the Ca2+ signal initiation process and provide a clear explanation for the original demonstration that agonist-elicited cytosolic Ca2+ waves always start in the apical region and then subsequently spread to the base (Kasai and Augustine, 1990). Once initiated, Ca2+ uncaging–induced CICR waves closely resemble agonist-induced waves in terms of initiation site (the apical region), speed, and shape of the transient. Furthermore, subthreshold agonist stimulation increased the CICR sensitivity of the apical region (Fig. 7). The similarity between Ca2+ uncaging–induced CICR transients and global waves induced by physiological levels of CCK suggests that the propagation of agonist-induced waves is driven by CICR.
Under physiological conditions, intracellular messengers (i.e., IP3, cADPr, and NAADP), which may be produced after agonist stimulation, trigger Ca2+ release primarily in the apical region (Kasai et al., 1993; Thorn et al., 1994; Cancela et al., 2002). This signal localization cannot simply be explained by exclusive presence of the relevant Ca2+ release channels in the apical region; RyRs are also present in the basal region (Leite et al., 1999). In fact, the Ca2+ releasing effects of cADPR, NAADP, and IP3 are all dependent on both functional IP3Rs and RyRs (Cancela et al., 2000). This agrees well with our present data showing that it is the coordination of IP3Rs and RyRs that enables CICR transients to be initiated and that such coordination can only occur in the apical region. These data highlight how important the process of CICR is in the generation of Ca2+ signals mediated through both IP3Rs and RyRs.
Low-level, subthreshold agonist stimulation is able to sensitize the uncaging effect and allows a single uncaging event to elicit continuing repetitive Ca2+ oscillations (Fig. 8). This suggests that CICR is involved at a very early stage immediately after the initial Ca2+ release and is crucial for the oscillatory process. Because uncaging-induced CICR empties the stores in a similar fashion whether or not agonist is present, it suggests that oscillatory behavior is not simply dependent on the loading state of intracellular stores. Thus, triggering of oscillations requires both release from intracellular stores as well as another agonist-dependent factor. Interestingly, the fact that oscillations can be triggered by CICR in what would normally be subthreshold conditions suggests that the mechanism that drives oscillations is actually more sensitive to agonist than the mechanism that triggers the initial Ca2+ release.
Our results present an intriguing paradox with regard to CICR in the basal region. Although it is impossible to initiate CICR transients in the basal region, CICR waves that originate in the apical region can travel through the basal region in a regenerative manner (indicated by the sustained rate of Ca2+ rise in the basal region, e.g., Fig.1 B and Figs. 4 and 5). This active propagation suggests that the mechanisms underlying a CICR wave must be present in the basal region. This provokes an interesting question: why is it not possible to trigger a CICR wave in the basal region even though the mechanisms exist for the propagation of the wave when it is initiated elsewhere? A possible explanation for the discrepancy between triggering and propagating CICR is that there may be close spatial localization of channels that allows very high local Ca2+ levels (larger than that generated by uncaging) to be established near receptors after release from their neighbors. These high local levels could trigger CICR from the channel and thus allow propagation of the wave. In other words, it is possible that CICR in the basal part of the cell is not completely absent but simply very insensitive to Ca2+. This may be functionally advantageous. The regional variation in CICR sensitivity ensures the polarity of the global Ca2+ signals. The apical region is where secretion takes place, and, thus, it makes sense to ensure that the Ca2+ signals are predominantly in that region. Furthermore, unwanted triggering of CICR from basal stores caused by changes in the cytosolic Ca2+ concentration (e.g., due to spontaneous opening of individual store channels or to Ca2+ influx across the basal membrane during store refilling) could have deleterious effects on normal cell function. The basal region contains the major part of the Ca2+ store in the ER, which feeds small ER extensions into the granular region (Mogami et al., 1997; Park et al., 2000). The ER store is refilled by movement of Ca2+ across the basal plasma membrane into the basal ER (Mogami et al., 1997). It is imperative for effective recharging of the stores via store-operated channels that cytosolic Ca2+ elevations near the basal membrane cannot trigger CICR. The organization of CICR within the cell, which we have described, acts to determine the polarity of calcium signals. Not only does it prevent release from occurring in the wrong part of the cell, but these differences in CICR sensitivity alone are enough to determine the activation pattern of physiological Ca2+ signals.
Materials and methods
Cell preparation and chemicals
Freshly isolated mouse pancreatic acinar cells (individual cells or small clusters) were prepared as described previously (Thorn et al., 1993) and used within 4 h. Cell clusters used for microinjection were obtained by following a similar procedure with shorter collagenase digestion (8 min). All experiments were performed at room temperature (22–24°C) in the presence of continuously perfused extracellular solutions. Fluo-4 (acetoxymethyl ester [AM] and salt forms), NP-EGTA (AM and salt), and fura red (AM and salt) were purchased from Molecular Probes. 2-APB, xestospongin C, ryanodine mixture, and various forms of heparin were from Calbiochem. Collagenase was obtained from Worthington and all other chemicals were purchased from Sigma-Aldrich.
Solutions and dye loading procedures
We used two types of extracellular solution, one containing Ca2+ and the other Ca2+ free. Both types contained 140 mM NaCl, 4.7 mM KCl, 1.13 mM MgCl2, 10 mM Hepes, and 10 mM glucose; pH 7.2 (adjusted by NaOH). In addition to this, 1 mM CaCl2 was added to Ca2+-containing solutions and 50 μM EGTA to Ca2+-free solution.
Most experiments were performed on intact cells. In those instances, the cell cytoplasm was loaded with dye/caged Ca2+ by incubation at room temperature in darkness for 25 min in solutions containing 2.5–5 μM fluo-4 AM and 5–10 μM NP-EGTA AM.
To obtain the necessary intracellular concentration of fura red, whole cell configuration was required because loading with fura red AM was not sufficient for this experimental protocol. In these experiments, the intracellular (pipette) solution contained 135 mM KCl, 10 mM NaCl, 1.5 mM MgCl2, 10 mM Hepes, and 2 mM Na2ATP; pH 7.2 (adjusted by KOH). This solution also contained 150 μM fura red and 1 mM NP-EGTA (with 0.8 mM Ca2+ preadded).
Electrophysiology
Standard patch clamp technique (Hamill et al., 1981) was employed using the EPC-8 amplifier and Pulse software (HEKA). We used pipettes having resistance of 2–4 MΩ (series resistance, 5–30 MΩ). The details of the procedure have been described previously (Thorn et al., 1993).
Confocal microscopy, Ca2+ imaging, and Ca2+ uncaging
Fluorescence images were obtained using either a ZEISS LSM 510 confocal microscope or a Leica SP2 confocal microscope with 63× water immersion objective (NA = 1.2). Cells adhered to poly-l-lysine–coated slides were imaged on the stage of an inverted microscope (Axiovert 100M; ZEISS/DMIRBE Leica). Fluo-4 and GFP were excited by a 488-nm laser line and emission was collected through a band pass filter of 505–550 nm. Fura red was excited at 488 nm and emitted light was collected through a long pass 560-nm or band pass 580–680-nm filter. During the scanning procedure, release of Ca2+ from the photolabile chelator (NP-EGTA) was achieved by exposure of predefined cytoplasmic regions to UV laser light at 364 nm and 351 nm. The systems allow rapid switching of excitation, controlled by an acousto-optical tunable filter. This means that UV exposure can be activated and deactivated on a pixel-to-pixel basis as the laser beam scans across the image frame. This allows very accurate targeting and rapidly controlled initiation and termination of calcium uncaging. Furthermore, the uncaging can be combined with confocal scanning. On occasions when maximal uncaging was required, cells were exposed to UV-filtered light from a xenon lamp.
GFP–PLCδ 1(PH) construct expression and detection
The PH domain of PLCδ1 cloned into eGFP-C1 (CLONTECH Laboratories, Inc.) cDNA plasmid was microinjected into the cytoplasm of individual pancreatic acinar cells within larger clusters (∼50–100 cells) that were adhered to poly-l-lysine–coated coverslips. The cells were incubated for 12–15 h at 37°C in Waymouth's media (GIBCO BRL) supplemented with 1% PenStrep mixture. Cells that were expressing the construct were then incubated with 10 μM fura red AM and 5 μM NP-EGTA AM for 45 min at room temperature.
Calibration of fluorescence signals
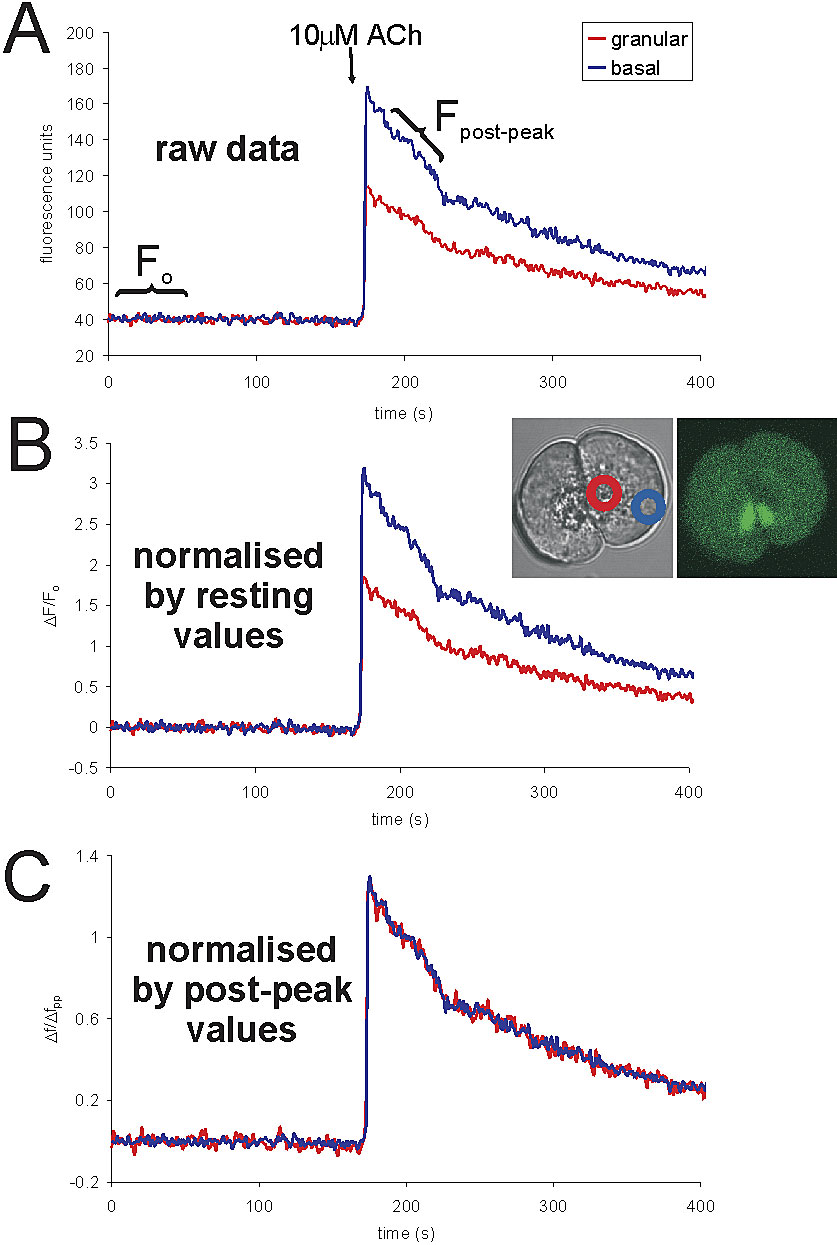
Fluo-4.
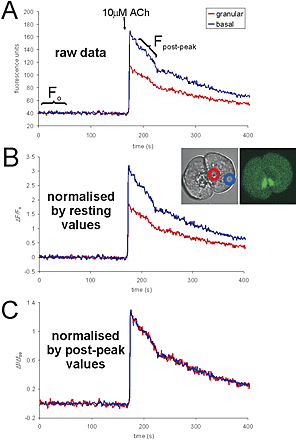
Fluo-4 is a single wavelength dye with a large dynamic range of fluorescence changes and very weak fluorescence at low resting Ca2+ levels. These properties mean that traditional normalization of fluorescence values by F0 (resting fluorescence) is prone to substantial error. At low resting calcium levels, regional fluorescence differences caused by nonuniform distribution of the dye are very small because of the low resting fluorescence of the dye (in our cells, these differences under resting conditions are often indistinguishable from background noise and autofluorescence). However, because the fluo-4 fluorescence rises so sharply upon binding of Ca2+, the differences due to dye distribution, which were small at resting Ca2+ levels, also increase substantially. This means that at elevated calcium levels, there can be substantial differences in fluorescence intensity in different regions of the cell that are not due to differences in the level of calcium (see Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200112025/DC1). These differences could not be predicted by simply looking at the resting level (F0), so we devised a novel method of normalization for fluo-4.
The principle behind our normalization procedure is the same as that used in normalization by F0. We simply compare fluorescence throughout the experiment (F) to fluorescence at a time point where we know that the Ca2+ level should be homogenous in all cytoplasmic regions (traditionally F0). The time point we use for this has to be at a time when Ca2+ is elevated so that the regional fluorescence differences are manifested. Each experiment involved, at some point, a large Ca2+ transient (induced by a supramaximal dose of ACh) that returned to resting levels over a period of many tens of seconds. Soon after the peak of the agonist-induced response, the fluorescence of fluo-4 is strong and Ca2+ levels are equal in different regions of the cell as determined by ratiometric imaging, which overcomes dye distribution artifacts (Craske et al., 1999). Therefore, we used fluorescence values from between 10 and 15 s after the peak of the large transient (“post-peak” values) rather than low resting fluorescence (traditional F0 values). This procedure eliminates inaccuracies caused by regional variations in dye concentration as well as avoids the effects of autofluorescence (which could form a substantial proportion of the resting fluorescence levels). Therefore, fluo-4 fluorescence is shown as the normalized value: ΔF/ΔFpost-peak.
The relative effects of post-peak and resting normalization are shown in Fig. S1 and values of basal/apical post-peak values are included in each figure legend. These ratios varied between 0.8 and 2.1 for different cells.
Fura red.
Because fura red has high resting levels, it is possible to use resting fluorescence to normalize these values. Changes in Ca2+ are reported as a fall in fluorescence, so the normalization shown below also reverses this trend so that an increase in normalized fluorescence reflects an increase in Ca2+:F0/F.
Online supplemental material
An online supplemental figure (Fig. S1) dealing with our fluo-4 fluorescence normalization procedures can be viewed at (http://www.jcb.org/cgi/content/full/jcb.200112025/DC1). The figure demonstrates the relative accuracy of post-peak normalization compared with normalization by resting fluorescence values.
Supplemental Material
Acknowledgments
The eGFP–PLCδ1(PH) plasmid was a gift from Dr. M. Katan (Imperial Cancer Research Fund, London, UK).
This work is supported by a Medical Research Council (MRC) Programme grant. M.C. Ashby is a Wellcome Trust Prize PhD student. O.H. Petersen is an MRC research professor.
The online version of this article includes supplemental material.
M.K. Park's present address is Department of Physiology, Sungkyunkwan University of Medicine, Suwon, 440-746, Korea.
Footnotes
Abbreviations used in this paper: ACh, acetylcholine; AM, acetoxymethyl ester; CCK, cholecystokinin; CICR, Ca2+-induced Ca2+ release; IP3, inositol trisphosphate; IP3R, IP3 receptor; NP-EGTA, nitro-phenyl–EGTA; RyR, ryanodine receptor.
References
- Alonso, M.T., M.J. Barrero, P. Michelena, E. Carnicero, I. Cuchillo, A.G. Garcia, J. Garcia-Sancho, M. Montero, and J. Alvarez. 1999. Ca2+-induced Ca2+ release in chromaffin cells seen from inside the ER with targeted aequorin. J. Cell Biol. 144:241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby, M.C., and A.V. Tepikin. 2002. Polarized calcium and calmodulin signaling in secretory epithelia. Physiol. Rev. 82:701–734. [DOI] [PubMed] [Google Scholar]
- Bakowski, D., M.D. Glitsch, and A.B. Parekh. 2001. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current I-CRAC in RBL-1 cells. J. Physiol. 532:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge, M.J., and R.F. Irvine. 1989. Inositol phosphates and cell signalling. Nature. 341:197–205. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny, I., J. Watras, and B.E. Ehrlich. 1991. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 351:751–754. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny, I., S. Bezprozvannaya, and B.E. Ehrlich. 1994. Caffeine-induced inhibition of inositol(1,4,5)-trisphosphate-gated calcium channels from cerebellum. Mol. Biol. Cell. 5:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancela, J.M., O.V. Gerasimenko, J.V. Gerasimenko, A.V. Tepikin, and O.H. Petersen. 2000. Two different but converging messenger pathways to intracellular Ca(2+) release: the roles of nicotinic acid adenine dinucleotide phosphate, cyclic ADP-ribose and inositol trisphosphate. EMBO J. 19:2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancela, J.M., F. Van Copponolle, A. Galione, A.V. Tepikin, and O.H. Petersen. 2002. Transformation of local Ca2+ spikes to global Ca2+ transients: the combinatorial roles of multiple Ca2+ releasing messengers. EMBO J. 21:909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craske, M., T. Takeo, O. Gerasimenko, C. Vaillant, K. Torok, O.H. Petersen, and A.V. Tepikin. 1999. Hormone-induced secretory and nuclear translocation of calmodulin: oscillations of calmodulin concentration with the nucleus as an integrator. Proc. Natl. Acad. Sci. USA. 96:4426–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich, B.E., E. Kaftan, S. Bezprozvannaya, and I. Bezprozvanny. 1994. The pharmacology of intracellular Ca(2+)-release channels. Trends Pharmacol. Sci. 15:145–149. [DOI] [PubMed] [Google Scholar]
- Endo, M., M. Tanaka, and Y. Ogawa. 1970. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature. 228:34–36. [DOI] [PubMed] [Google Scholar]
- Fabiato, A. 1985. Effects of ryanodine in skinned cardiac cells. Fed. Proc. 44:2970–2976. [PubMed] [Google Scholar]
- Fabiato, A., and F. Fabiato. 1975. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J. Physiol. 249:469–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimmons, T.J., I. Gukovsky, J.A. McRoberts, E. Rodriguez, F.A. Lai, and S.J. Pandol. 2000. Multiple isoforms of the ryanodine receptor are expressed in rat pancreatic acinar cells. Biochem. J. 351:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, L.E., and R.J. Podolsky. 1970. Regenerative calcium release within muscle cells. Science. 167:58–59. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hirose, K., S. Kadowaki, M. Tanabe, H. Takeshima, and M. Iino. 1999. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 284:1527–1530. [DOI] [PubMed] [Google Scholar]
- Iino, M. 1990. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J. Gen. Physiol. 95:1103–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai, H., and G.J. Augustine. 1990. Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature. 348:735–738. [DOI] [PubMed] [Google Scholar]
- Kasai, H., Y.X. Li, and Y. Miyashita. 1993. Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell. 74:669–677. [DOI] [PubMed] [Google Scholar]
- Lai, F.A., H.P. Erickson, E. Rousseau, Q.Y. Liu, and G. Meissner. 1988. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 331:315–319. [DOI] [PubMed] [Google Scholar]
- Lee, M.G., X. Xu, W. Zeng, J. Diaz, R.J. Wojcikiewicz, T.H. Kuo, F. Wuytack, L. Racymaekers, and S. Muallem. 1997. Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. J. Biol. Chem. 272:15765–15770. [DOI] [PubMed] [Google Scholar]
- Leite, M.F., J.A. Dranoff, L. Gao, and M.H. Nathanson. 1999. Expression and subcellular localization of the ryanodine receptor in rat pancreatic acinar cells. Biochem. J. 337:305–309. [PMC free article] [PubMed] [Google Scholar]
- Lipp, P., and E. Niggli. 1998. Fundamental calcium release events revealed by two-photon excitation photolysis of caged calcium in Guinea-pig cardiac myocytes. J. Physiol. 508:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H.T., R.L. Patterson, D.B. van Rossum, L. Birnbaumer, K. Mikoshiba, and D.L. Gill. 2000. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 287:1647–1651. [DOI] [PubMed] [Google Scholar]
- Meissner, G. 1994. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu. Rev. Physiol. 56:485–508. [DOI] [PubMed] [Google Scholar]
- Missiaen, L., C.W. Taylor, and M.J. Berridge. 1992. Luminal Ca2+ promoting spontaneous Ca2+ release from inositol trisphosphate-sensitive stores in rat hepatocytes. J. Physiol. 455:623–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa, T., A. Mizushima, K. Hirose, T. Yamazawa, I. Bezprozvanny, T. Kurosaki, and M. Iino. 2001. Ca(2+)-sensor region of IP(3) receptor controls intracellular Ca(2+) signaling. EMBO J. 20:1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami, H., K. Nakano, A.V. Tepikin, and O.H. Petersen. 1997. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell. 88:49–55. [DOI] [PubMed] [Google Scholar]
- Mogami, H., A.V. Tepikin, and O.H. Petersen. 1998. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO J. 17:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash, M.S., K.W. Young, G.B. Willars, R.A. Challiss, and S.R. Nahorski. 2001. Single-cell imaging of graded Ins(1,4,5)P3 production following G-protein-coupled-receptor activation. Biochem. J. 356:137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson, M.H., M.B. Fallon, P.J. Padfield, and A.R. Maranto. 1994. Localization of the type 3 inositol 1,4,5-trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar cells. J. Biol. Chem. 269:4693–4696. [PubMed] [Google Scholar]
- Neher, E., and G.J. Augustine. 1992. Calcium gradients and buffers in bovine chromaffin cells. J. Physiol. 450:273–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo, Y., S. Kakizawa, K. Hirose, and M. Iino. 2001. Visualization of IP(3) dynamics reveals a novel AMPA receptor-triggered IP(3) production pathway mediated by voltage-dependent Ca(2+) influx in Purkinje cells. Neuron. 32:113–122. [DOI] [PubMed] [Google Scholar]
- Park, M.K., O.H. Petersen, and A.V. Tepikin. 2000. The endoplasmic reticulum as one continuous Ca(2+) pool: visualization of rapid Ca(2+) movements and equilibration. EMBO J. 19:5729–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, O.H., C.C. Petersen, and H. Kasai. 1994. Calcium and hormone action. Annu. Rev. Physiol. 56:297–319. [DOI] [PubMed] [Google Scholar]
- Pozzan, T., R. Rizzuto, P. Volpe, and J. Meldolesi. 1994. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 74:595–636. [DOI] [PubMed] [Google Scholar]
- Prakriya, M., and R.S. Lewis. 2001. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 536:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Bueno, A., I. Marrero, and P.H. Cobbold. 1994. Caffeine inhibits agonists-induced cytoplasmic Ca2+ oscillations in single rat hepatocytes. Biochem. Biophys. Res. Commun. 198:728–733. [DOI] [PubMed] [Google Scholar]
- Solovyova, N., N. Veselovsky, E.C. Toescu, and A. Verkhratsky. 2002. Ca(2+) dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca(2+)-induced Ca(2+) release triggered by physiological Ca(2+) entry. EMBO J. 21:622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub, S.V., D.R. Giovannucci, and D.I. Yule. 2000. Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J. Gen. Physiol. 116:547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutko, J.L., J.A. Airey, W. Welch, and L. Ruest. 1997. The pharmacology of ryanodine and related compounds. Pharmacol. Rev. 49:53–98. [PubMed] [Google Scholar]
- Thomas, A.P., G.S. Bird, G. Hajnoczky, L.D. Robb-Gaspers, and J.W. Putney, Jr. 1996. Spatial and temporal aspects of cellular calcium signaling. FASEB J. 10:1505–1517. [PubMed] [Google Scholar]
- Thorn, P., A.M. Lawrie, P.M. Smith, D.V. Gallacher, and O.H. Petersen. 1993. Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell. 74:661–668. [DOI] [PubMed] [Google Scholar]
- Thorn, P., O. Gerasimenko, and O.H. Petersen. 1994. Cyclic ADP-ribose regulation of ryanodine receptors involved in agonist evoked cytosolic Ca2+ oscillations in pancreatic acinar cells. EMBO J. 13:2038–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu, E.C., D.V. Gallacher, and O.H. Petersen. 1994. Identical regional mechanisms of intracellular free Ca2+ concentration increase during polarized agonist-evoked Ca2+ response in pancreatic acinar cells. Biochem. J. 304:313–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda, Y., E.L. Stuenkel, and J.A. Williams. 1990. Oscillatory mode of calcium signaling in rat pancreatic acinar cells. Am. J. Physiol. 258:C147–C155. [DOI] [PubMed] [Google Scholar]
- Wakui, M., Y.V. Osipchuk, and O.H. Petersen. 1990. Receptor-activated cytoplasmic Ca2+ spiking mediated by inositol trisphosphate is due to Ca2(+)-induced Ca2+ release. Cell. 63:1025–1032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}