

Abstract
Extracellular amyloid β peptides (Aβs) have long been thought to be a primary cause of Alzheimer's disease (AD). Now, detection of intracellular neuronal Aβ1–42 accumulation before extracellular Aβ deposits questions the relevance of intracellular peptides in AD. In the present study, we directly address whether intracellular Aβ is toxic to human neurons. Microinjections of Aβ1–42 peptide or a cDNA-expressing cytosolic Aβ1–42 rapidly induces cell death of primary human neurons. In contrast, Aβ1–40, Aβ40–1, or Aβ42–1 peptides, and cDNAs expressing cytosolic Aβ1–40 or secreted Aβ1–42 and Aβ1–40, are not toxic. As little as a 1-pM concentration or 1500 molecules/cell of Aβ1–42 peptides is neurotoxic. The nonfibrillized and fibrillized Aβ1–42 peptides are equally toxic. In contrast, Aβ1–42 peptides are not toxic to human primary astrocytes, neuronal, and nonneuronal cell lines. Inhibition of de novo protein synthesis protects against Aβ1–42 toxicity, indicating that programmed cell death is involved. Bcl-2, Bax-neutralizing antibodies, cDNA expression of a p53R273H dominant negative mutant, and caspase inhibitors prevent Aβ1–42-mediated human neuronal cell death. Taken together, our data directly demonstrate that intracellular Aβ1–42 is selectively cytotoxic to human neurons through the p53–Bax cell death pathway.
Keywords: Alzheimer's disease; amylois β peptide; p53; Bax; neurotoxicity
Introduction
Alzheimer's disease (AD)* is a progressive neurodegenerative disease of the central nervous system characterized by the presence of extracellular senile plaques, intraneuronal neurofibrillary tangles (NFTs), and the loss of synapses and neurons. The amyloid β peptide (Aβ) is a major component of the extracellular senile plaque. Aβ naturally arises from the metabolic processing of the amyloid precursor protein (APP) in the ER, the Golgi apparatus, or the endosomal–lysosomal pathway, and most is normally secreted as a 40– (Aβ1–40) or 42– (Aβ1–42) amino acid peptide (Martin et al., 1995; Chyung et al., 1997; Tienari et al., 1997; Lee et al., 1998; Morishima-Kawashima and Ihara, 1998; Greenfield et al., 1999; Perez et al., 1999). However, an NH2-terminally truncated form of Aβ1–42 (Aβ42) accumulates in the ER in aging cell cultures (Greenfield et al., 1999; Yang et al., 1999). Recently, the presence of intracellular Aβ42 has been detected in the brains of individuals with AD or Down's syndrome, and in APP transgenic mice and aging monkeys. In AD brains, the intracellular Aβ42 accumulates as aggregates or granules in the cytoplasm of neurons (Gouras et al., 2000; D'Andrea et al., 2001). The accumulation of Aβ precedes the appearance of NFTs and senile plaques and is observed in regions affected early in AD: the hippocampus and entorhinal cortex. The intracellular Aβ does not appear to be fibrillar, as it is not stained by Bielchowsky silver stain, Thioflavin S, or Congo red, nor does it require formic acid treatment for immunostaining (Gouras et al., 2000). In Down's syndrome brains, accumulation of intracellular Aβ1–28 and Aβ40 precedes Aβ42, and these intracellular Aβ accumulations precede the appearance of diffuse plaques, senile core plaques, and NFT formation (Gyure et al., 2001). In aging monkeys, nonfibrillar neuronal and nonneuronal intracellular Aβ precede the deposition of extracellular and fibrillar Aβ (Martin et al., 1994). Furthermore, the accumulation of intracellular Aβ also precedes plaque formation in mutant APP/presenilin (PS)-1 transgenic mice (Wirths et al., 2001), accumulates in the APPV717F mutation where synaptic loss precedes extracellular Aβ deposition (Masliah et al., 1996; Hsia et al., 1999; Li et al., 1999), and is associated with neuronal loss in PS-1 transgenic mice in the absence of extracellular Aβ deposition. Although extensive research has been conducted on the relevance of extracellular Aβ deposits in AD (Selkoe, 1998), the biological significance of the accumulation of intracellular Aβ is not known. The early appearance of the intracellular Aβ42 suggests that it could be the initial cause of neuronal dysfunction and neuronal loss in AD. To determine if intracellular Aβ may be detrimental to neurons, we microinjected Aβ1–40, Aβ1–42, and control reverse peptides Aβ40–1 and Aβ42–1 or cDNAs expressing cytosolic or secreted Aβ1–40 and Aβ1–42 in primary cultures of human neurons, neuronal, and nonneuronal cell lines. Aβ1–42 was selectively toxic to human neurons. Microinjections of the Aβ1–42 peptide in the presence of transcriptional and translational inhibitors showed that the cytotoxicity of Aβ1–42 required de novo protein synthesis. Because of the known role of the p53 transcriptionally regulated proapoptotic Bax protein in neuronal cell death (Xiang et al., 1998), we assessed the effect of the antiapoptotic Bcl-2 protein and Bax neutralizing antibodies on Aβ1–42-mediated neurotoxicity. Both eliminated toxicity. Furthermore, the p53R273H dominant negative mutant, which prevents transactivation of Bax expression (Aurelio et al., 2000), eliminated Aβ1–42 toxicity. Lastly, caspase-6–, but not caspase-3–type inhibitors, prevented Aβ1–42 toxicity. We conclude that Aβ1–42 is selectively toxic to human neurons through activation of the p53 and Bax proapoptotic pathway.
Results
Intracellular Aβ1–42, but not Aβ1–40, Aβ42–1, or Aβ40–1, is neurotoxic to human neurons
To directly determine whether intracellular Aβ is toxic to human neurons, we microinjected aged Aβ1–40, Aβ1–42, or the reverse control peptide Aβ40–1 into the cytoplasm of primary cultured human neurons. Aβ1–42 induces significant cell death in 60% of microinjected neurons within 24 h of injection (Fig. 1, A and B). Cell death further increases to 78 and 90% at 48 and 96 h after injection. However, Aβ1–40, the control reverse peptides, Aβ40–1, Aβ42–1, or the fluorescent marker dye Dextran Texas red (DTR), do not affect cell viability for up to 4 d (Fig. 1 B). After 16 d of injection, only Aβ1–40 induces 50% cell death. Because we inject 25 pl/shot of a 10-nM concentration of peptide, the above results indicate that 0.25 × 10−18 moles of Aβ1–42 delivered into the neuron cytosol is sufficient to induce rapid neuronal death. A 100-fold dilution in the amount injected still induces 50% cell death in these neurons (Fig. 1 C). Using confocal microscopy, we estimated the volume of these human neurons at 4.97 ± 0.8 nl (n = 3). The nuclei occupy >50% of the cell, thus the cytosolic area is ∼2.5 nl. Therefore, the actual toxic concentration of injected Aβ1–42 is 0.25 × 10−18 to 0.25 × 10−20 moles/2.5 nl, which equals 10−10 to 10−12 M, or 1 to 100 pM. These neurons do not undergo cell death even with 10 μM of extracellular Aβ1–42, Aβ1–40, or Aβ40–1, a concentration known to induce cell death in a variety of neuronal cell lines (Paradis et al., 1996; Klein et al., 2001). To test if this particular batch of peptide might be neurotoxic, we treated the neurons with 10 μM of these peptides for 24 h. Neither extracellular aged Aβ1–40, Aβ1–42, or Aβ40–1 are toxic to these neurons after 24 h of treatment (Fig. 1 D). Therefore, the toxicity of intracellular Aβ1–42 is at least 100,000 times greater than extracellular Aβ. These results indicate that an infinitesimal amount of intracellular Aβ1–42 is detrimental to human neurons. Calculation of the number of molecules of Aβ1–42 injected in neurons based on the Avogadro number shows maximal toxicity with 150,055 molecules and 50% toxicity with 1505.5 molecules/neuron. The level of toxic Aβ1–42 is probably at least 10,000-fold lower than the amount of immunologically detectable intracellular Aβ1–42 in AD neurons. However, because neurons in the brain are bathed in extracellular milieu that promotes their survival, the in vivo neurons may resist higher concentrations of intracellular Aβ1–42 than the neurons in culture. Finally, to confirm the toxicity of naturally produced intracellular Aβ peptides, neurons were microinjected with cDNA constructs expressing cytosolic or secreted Aβ1–40 and Aβ1–42. As observed with the synthetic Aβ1–42 peptide, only the cytosolically expressed Aβ1–42 was toxic, whereas secreted Aβ1–42 or cytosolic or secreted Aβ1–40 did not induce cell death in neurons (Fig. 1 E).
Figure 1.
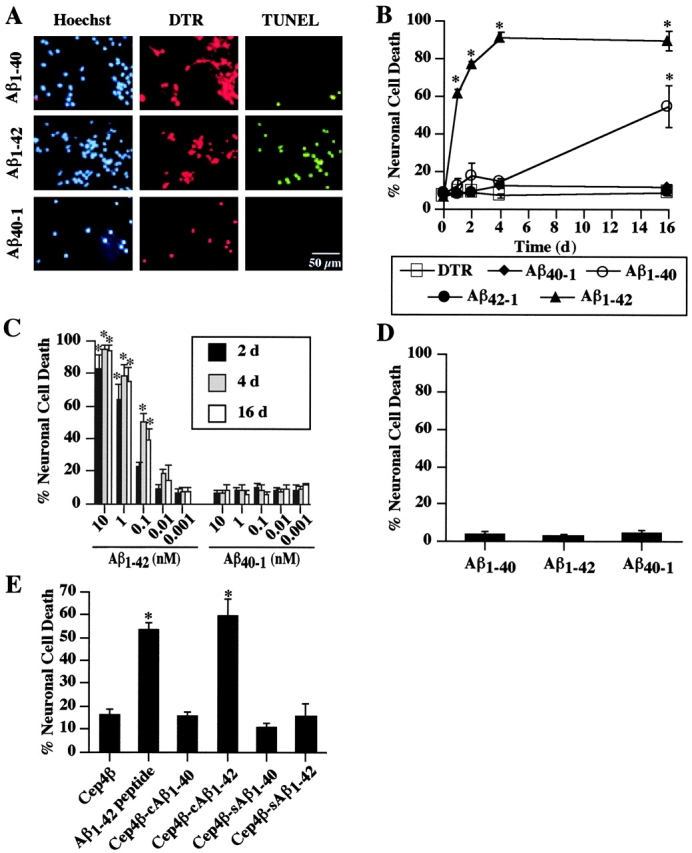
Intracellular Aβ neurotoxicity in primary human neurons. (A). Fluorescent photomicrographs of microinjected neurons. Neurons were microinjected with the peptides in DTR and incubated 24 h before staining with TUNEL for cell death or Hoechst for nuclear stain. (B) Aged Aβ1–40, Aβ1–42, Aβ42–1, and Aβ40–1 peptides (10 nM) were microinjected into the cytosol of human neurons and cell death was measured by TUNEL at 1, 2, 4, and 16 d after injection. Two-way ANOVAs (dftime = 4; dftreatment = 3) followed by Sheffé's test were performed to determine the statistical significance between Aβ-injected and control DTR-injected neurons. *, P<0.01. (C) Various doses of Aβ1–42 and Aβ40–1 were injected into human neurons and cell death was determined by TUNEL staining at 2, 4, or 16 d after injection. Two-way ANOVAs (dftime = 2; dftreatment = 29) followed by Sheffé's test were performed to determine the statistical significance. *, P < 0.01. (D) Human neurons were exposed to 10 μM extracellular Aβ1–40, Aβ1–42, and Aβ40–1 for 24 h and stained with propidium iodide to reveal cellular nuclei and TUNEL to reveal cell death. (E) Cell death in neurons 24 h after microinjection with pCep4β episomal cDNA constructs expressing cytosolic Aβ1–40 and Aβ1–42 (cAβ) or secreted Aβ1–40 and Aβ1–42 (sAβ). One-way ANOVA (df = 5) followed by Sheffé's test determined a statistically significant difference between the Cep4β construct alone and Aβ1–42 peptide or Cep4β-cAβ1–42 expression construct. *, P < 0.01. For B–E, the data represent the mean ± SEM of three independent experiments.
Nonfibrillized Aβ1–42 is neurotoxic
Because the fibrillar form of Aβ is commonly seen in the senile plaques in AD brains and has long been proposed to be more toxic than soluble Aβ (Pike et al., 1993), we examined the toxicity of both fibrillized and nonfibrillized Aβ peptides. Transmission electron microscopy on the preparation of Aβ confirms the fibrillar and nonfibrillar nature of the Aβ preparation (Fig. 2 A). The nonfibrillized Aβ1–42 peptides show well defined globular as well as diffuse aggregate morphology. Similar well-defined globular structures are also seen in the nonfibrillized Aβ1–40 preparation, but are much less abundant than in the Aβ1–42 preparation. The fibrillized Aβ1–42 peptide shows a heterogeneous mixture of fibrils of various sizes, protofibrils, and globular structures. The fibrils in the Aβ1–40 preparation were less heterogeneous and consisted of thick fibrillar aggregates and thin aligned fibrils. The large Aβ1–40 fibrils appear not to pass through the micropipette, and thus were unlikely to have been injected in neurons (unpublished data). However, some of the smaller fibrils from the Aβ1–42 and Aβ1–40 preparation did pass through the pipette. Both fibrillized and nonfibrillized preparations of Aβ1–42 induce 50–90% neuronal cell death between 24–96 h after injection (Fig. 2 B). In contrast, neither fibrillized nor nonfibrillized Aβ1–40 cause significant cell death. Previously, it was shown that dimers/oligomers of Aβ are toxic to neurons when applied in the extracellular milieu (Walsh et al., 1999). Western blot analysis of the nonfibrillized and fibrillized Aβ peptides shows that the Aβ1–42 peptide forms aggregates with the expected size of Aβ dimers, trimers, and oligomers. In contrast, the nonfibrillized and fibrillized Aβ1–40 peptides show a 6.5-kD aggregate and with longer exposure, a smear of higher MW oligomers (unpublished data). Taken together, these data show that the fibrils are not required for toxicity and suggest that either the monomeric Aβ1–42 is significantly more toxic than the monomeric Aβ1–40 or the dimers, trimers, and oligomers of the nonfibrillized and fibrillized Aβ1–42 peptide may be the toxic forms of the Aβ peptides.
Figure 2.
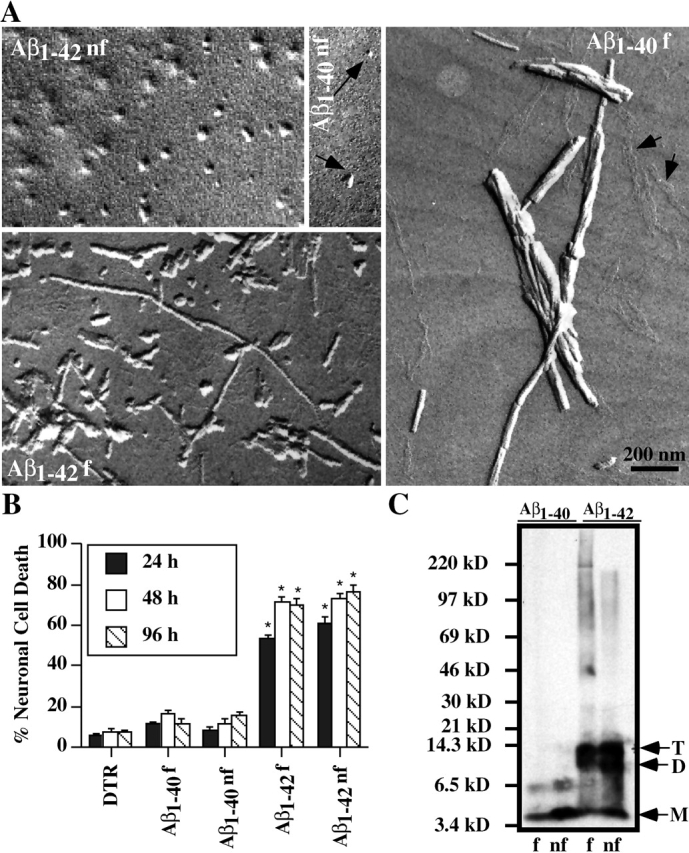
Soluble and fibrillar Aβ 1–42 are toxic to human neurons. (A) Electron micrographs of nonfibrillized (nf) and fibrillized (f) Aβ1–40 and Aβ1–42. In the Aβ1–40 nf, the arrows point to the rare globular structures while in the Aβ1–40 f, the arrows point to the small aligned fibrils. (B) Nonfibrillized or fibrillized Aβ1–40 or Aβ1–42 (10 nM) were injected into human neurons and neuronal cell death assessed by TUNEL staining at 24, 48, and 96 h after injection. The data represent the mean ± SEM of three independent experiments. One-way ANOVA (df = 14) followed by Sheffé's test were performed to determine the statistical significance between Aβ-injected and control DTR-injected neurons. *, P < 0.01. (C) Western blot analysis of fibrillized or nonfibrillized Aβ1–40 and Aβ1–42 with 6E10. M, D, and T represent the monomeric, dimeric and trimeric forms, respectively. A longer exposure revealed a smear also in the fibrillized Aβ1–40 (unpublished data).
Intracellular Aβ1–42 toxicity appears selective to human neurons
Because neurons specifically undergo cell death in AD, we explored the specificity of intracellular Aβ toxicity in primary cultured human astrocytes. The volume of these cells is 10 times that of human neurons, therefore, we injected 100 nM of Aβ1–42 into the cytosol of astrocytes to keep the same concentration of injected Aβ. In human neurons, cell death is easily observed in the DTR-microinjected cells by TUNEL and condensed nuclear DNA (Fig. 3 A). In contrast, microinjected astrocytes do not show any sign of condensed chromatin by Hoechst staining, nor positive nuclear TUNEL staining (Fig. 3 A). The faint green fluorescence detected in these cells is the result of bleed-through from the red DTR fluorescence on our microscope. Similarly, microinjections of the Aβ1–42 peptide into human neuroblastoma, La-N-1 and M17 cell lines, teratocarcinoma NT2 cells, mice NIH 3T3 fibroblasts, and BHK cells fail to induce cell death. Quantitative analysis on 600 microinjected cells from three independent experiments confirms that among the cell lines tested, only human primary neurons are susceptible to the intracellular Aβ1–42 toxicity (Fig. 3 B). Whether this toxicity occurs in vivo in adult human neurons remains to be determined. However, the in vivo intracellular toxicity of Aβ1–42 occurs in transgenic animals (LaFerla et al., 1995). Taken together, our data demonstrates that intracellular Aβ1–42 is selectively toxic to these human neurons.
Figure 3.
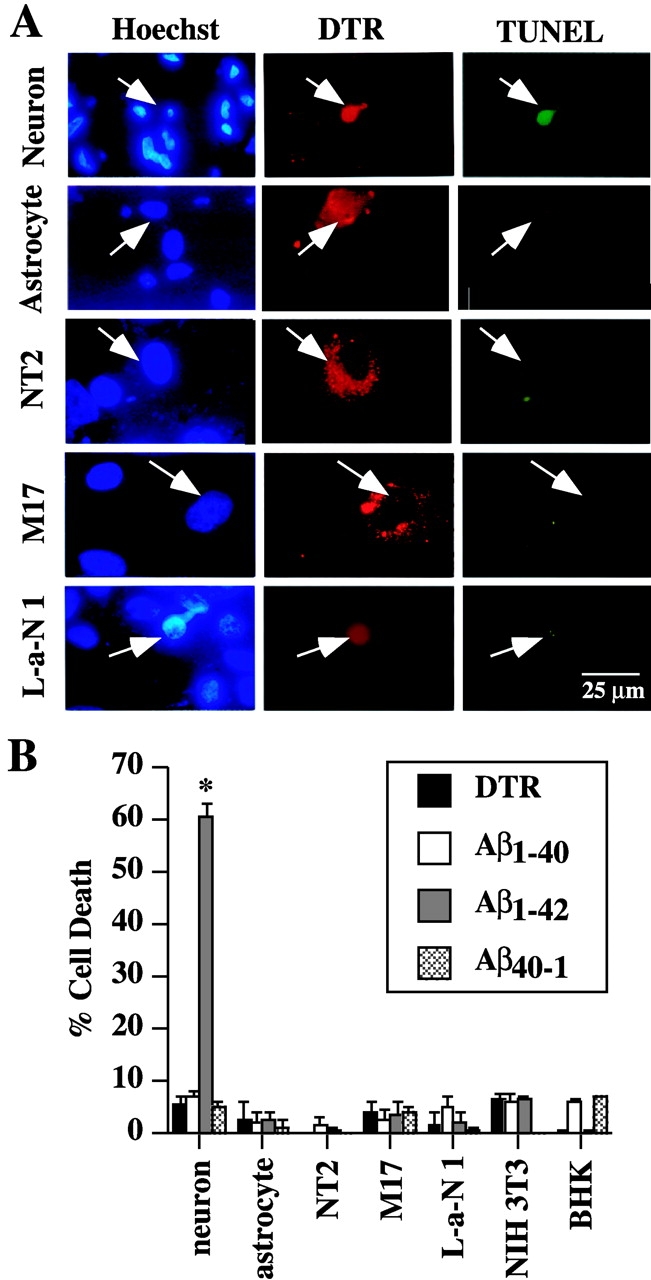
Intracellular Aβ1–42 is not toxic to primary human astrocytes, neuroblastoma, teratocarcinoma, fibroblast, and kidney cell lines. (A) Cells were microinjected with 100 nM Aβ1–42 and DTR and incubated for 24 h. TUNEL staining identified cell death and Hoechst staining detected nuclei. (B) Cell survival quantitation in several cell lines injected with DTR and Aβ1–40, Aβ1–42, or Aβ40–1 and incubated 24 h. The data represent the mean ± SEM of three independent experiments. One-way ANOVA (df = 27) followed by Sheffé's test determined the statistical significance between Aβ-injected and control DTR-injected cells. *, P < 0.01.
Aβ1–42 neurotoxicity requires do novo protein synthesis
To address the underlying molecular mechanism of intracellular Aβ toxicity, we microinjected Aβ1–42 into neurons and incubated the cells in the presence or absence of the transcriptional inhibitor, actinomycin D, and the translational inhibitor, cycloheximide, for 24 h. Both cycloheximide and actinomycin D efficiently block Aβ1–42-induced neuronal death (Fig. 4). These results indicate that de novo protein synthesis is necessary for intracellular Aβ toxicity.
Figure 4.

Intracellular Aβ 1–42 toxicity requires de novo protein synthesis. Neuronal cell death in non or Aβ1–42 -injected neurons incubated in the absence (−) or presence of 5 μg/ml cycloheximide (CHX) or 5 μM actinomycin D (ACTD) for 48 h. Neurons were preincubated for 1 h in CHX and ACTD before microinjections. The data represent the mean ± SEM of three independent experiments. One-way ANOVA (df = 5) followed by Sheffé's test determined a statistical significant difference between Aβ1–42 in absence and presence of CHX and ACTD but not between CHX and ACTD treatment of non and Aβ1–42-injected neurons. *, P < 0.01.
Bax is responsible for intracellular Aβ toxicity
Because the proapoptotic Bax protein can induce rapid human neuronal cell death through overexpression (Bounhar et al., 2001), we suspected that Bax could be involved in Aβ1–42-mediated cell death. Therefore, we microinjected a human cDNA expression construct of Bcl-2, the major anti-Bax protein, with the Aβ1–42 peptide (Fig. 5 A). The microinjection of the Bcl-2 construct completely eliminates Aβ1–42-mediated neurotoxicity as observed before against Bax (Bounhar et al., 2001). In contrast, coinjection of an APP cDNA construct does not alter Aβ1–42-mediated cell death.
Figure 5.
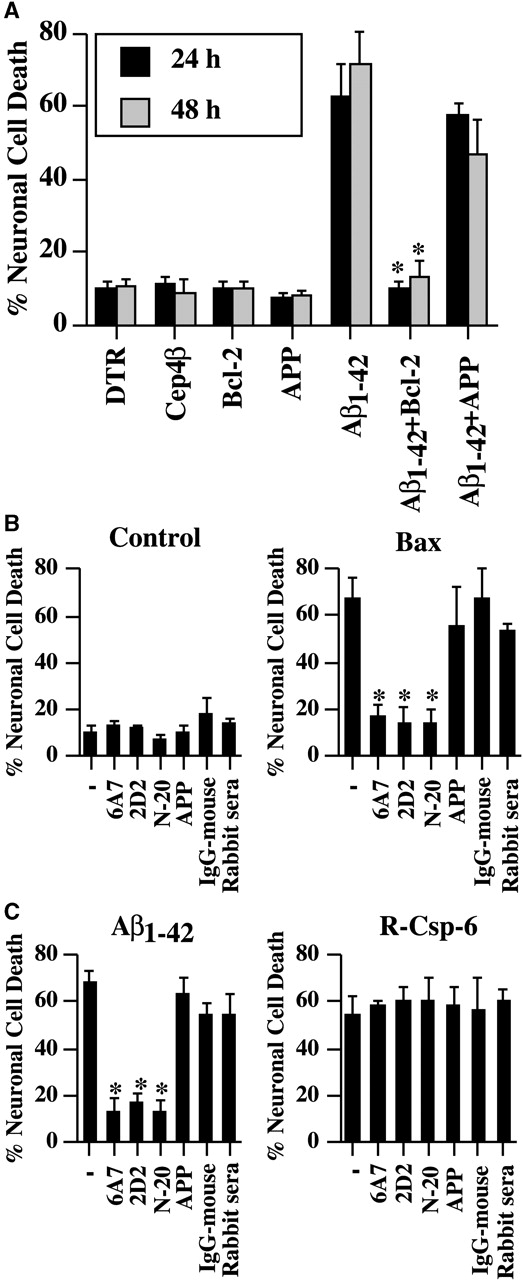
Inhibition of Aβ 1–42 -mediated neuronal cell death with Bcl-2 and Bax-neutralizing antibodies. (A) Cell death in neurons coinjected with Aβ1–42 and Bcl-2 or APP pCep4β eukaryotic cDNA expression episomal construct. *P < 0.01. (B) Neuronal cell death in DTR alone (control), Bax cDNA, Aβ1–42 peptide, or recombinant active caspase-6 (R-Csp-6)-microinjected neurons in the absence (−) or presence of monoclonal Bax antibodies, 6A7 or 2D2, Bax polyclonal antisera, N-20, APP monoclonal antibody 22C11, mouse IgG, or rabbit nonimmune sera. Cells were incubated 24 h after microinjections. The data represent the mean ± SEM of three independent experiments. One-way ANOVA (df = 6) followed by Sheffé's test determined the significance of the difference between the insult in absence and presence of the antibodies. *, P < 0.01.
To confirm that Bax is involved, we tested whether antibodies against human Bax could neutralize Bax-mediated killing. We found that two monoclonal antibodies, 6A7 and 2D2, and the polyclonal antisera N-20, but not an anti-APP antibody, rabbit serum nor a mouse IgG, could neutralize the pro-apoptotic properties of Bax (Fig. 5 B). Similarly, these anti-Bax antibodies completely neutralized the Aβ1–42-mediated neurotoxicity. In contrast, none of these antibodies had any effect on recombinant active caspase-6–mediated cell death. Therefore, these experiments show that anti-Bax antibodies specifically inhibit Bax-mediated or Aβ1–42-mediated cell death and indicate that Aβ1–42 induces cell death through Bax activation. Together, these results implicate Bax in Aβ1–42-mediated neuronal cell death.
p53 is involved in intracellular Aβ1–42-mediated neurotoxicity
Bax is transcriptionally regulated by p53; therefore, we tested whether p53 activation was involved in intracellular Aβ1–42-mediated neurotoxicity. We chose to use the p53R273H dominant negative (p53DN) mutant because it effectively inhibits p53 transcriptional activation of Bax (Aurelio et al., 2000). Whereas the expression of p53 wild-type (WT) or p53DN does not induce neuronal apoptosis in absence or presence of Aβ1–40, the p53DN, but not the p53WT, effectively inhibits Aβ1–42-mediated neurotoxicity (Fig. 6). The inability of the p53DN to inhibit the toxicity of Bax when expressed from the CMV promoter of a cDNA construct or to inhibit cell death by recombinant active caspase-6 attests to the specificity of the p53DN against Aβ1–42-mediated cell death. Together with the inhibition of Aβ1–42-mediated neurotoxicity using Bax antibodies, these results suggest that Aβ1–42 activates p53 which regulates Bax expression to induce cell death.
Figure 6.

Inhibition of Aβ1–42-mediated neuronal cell death with p53 dominant negative R273H mutant but not p53 wild type. Neuronal cell death in DTR (Ctl), Aβ1–40 peptide, Aβ1–42 peptide, R-Csp-6, or Bax cDNA alone (−) or comicroinjected with cDNA expressing wild-type (WT) or dominant negative (DN) p53 in neurons. Microinjected cells were incubated 48 h. The data represent the mean ± SEM of three independent experiments. One-way ANOVA (df = 14) followed by Sheffé's test showed a statistically significant difference between Aβ1–42 without and with p53DN-injected neurons. *, P < 0.01.
Caspase inhibitors prevent Aβ1–42-mediated cell death
Previously, we have shown that serum deprivation induces caspase-6–, but not caspase-3–mediated cell death in human neurons (LeBlanc et al., 1999). To assess the role of caspases in Aβ1–42-mediated neurotoxicity, we incubated Aβ1–42-injected neurons in the presence or absence of various caspase inhibitors. The results show that the pan caspase inhibitor, Z-VAD-fmk, the caspase-6–like inhibitor, Z-VEID-fmk, and the caspase-8–like inhibitor, Z-IETD-fmk, completely prevent Aβ1–42-induced neuronal cell death (Fig. 7). In contrast, the caspase-1–like inhibitor, Z-YVAD-fmk and the caspase-3–like inhibitor, Z-DEVD-fmk, do not inhibit Aβ1–42-induced cell death. Therefore, the data show that caspase-6– or caspase-8–like activities regulate Aβ1–42-mediated neuronal apoptosis.
Figure 7.
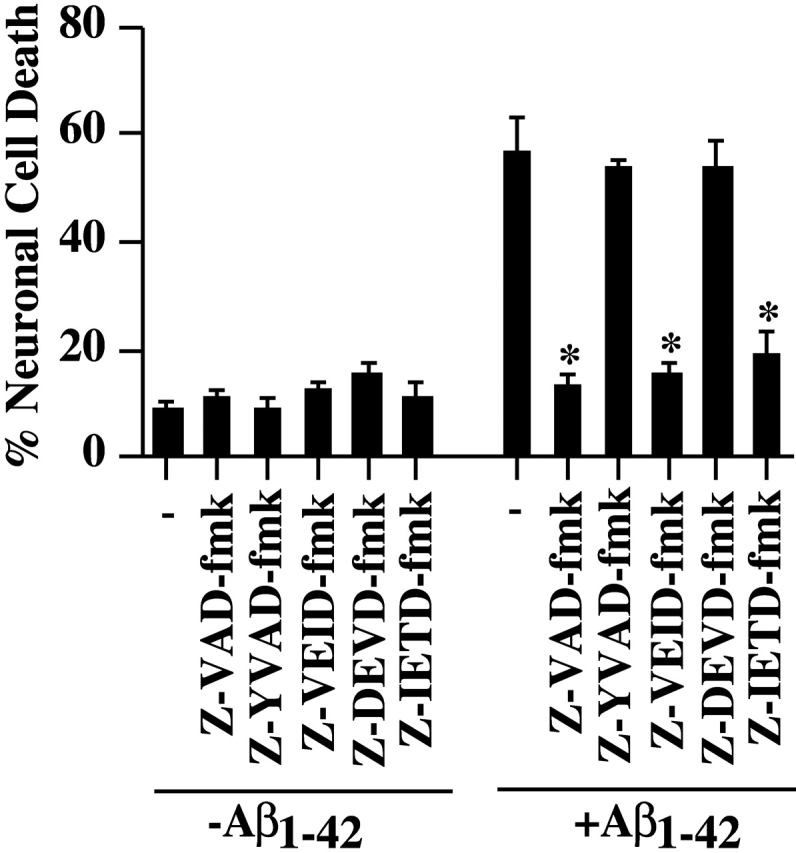
Inhibition of Aβ1–42-mediated neuronal cell death with caspase inhibitors. Neurons were preincubated for 1 h in the presence of 5 μM of each inhibitor, microinjected with Aβ1–42 peptide and incubated for 24 h in the presence of the inhibitors before revealing cell death of injected cells with TUNEL. The data represent the mean ± SEM of three independent experiments. One-way ANOVA (df = 11) followed by Sheffé's test showed a statistically significant difference between Aβ1–42 and Aβ1–42 + Z-VAD-fmk, Z-VEID-fmk, and Z-IETD-fmk. *, P < 0.01.
Discussion
The accumulation of intraneuronal Aβ42 before the appearance of senile plaques and NFT in AD questions the relevance of intraneuronal Aβ42 in neurons (Gouras et al., 2000; D'Andrea et al., 2001). In the present manuscript, we show that (a) very low levels of intracellular Aβ1–42, but not Aβ1–40, Aβ42–1, or Aβ40–1, are selectively cytotoxic to human neurons; (b) nonfibrillized Aβ1–42 peptide is as toxic as the fibrillized peptide and toxicity may be attributable to the formation of Aβ1–42 oligomers; and (c) Aβ1–42 toxicity is mediated through the p53 and Bax cell death pathway. Therefore, our findings support the hypothesis that intracellular accumulation of Aβ1–42 may be an initial cause of neuronal dysfunction and loss in AD. Intracellular accumulation of Aβ1–42 preceding other pathological lesions is observed not only in AD (Gouras et al., 2000; D'Andrea et al., 2001), but also in cells or transgenic mice expressing AD-associated PS-1 mutations, APP mutations, or double APP/PS-1 mutations (Masliah et al., 1996; Sudoh et al., 1998; Xia et al., 1998; Chui et al., 1999; Petanceska et al., 2000; Wirths et al., 2001) and in aging monkeys (Martin et al., 1994). Some of these animal models have been shown to present behavioral abnormalities or neurodegeneration before the appearance of extracellular plaques (Chui et al., 1999; Hsia et al., 1999). In addition, despite most studies showing that extracellular Aβ deposition in senile plaques does not correlate well with the severity of AD (Terry, 1999), analysis of total Aβ levels in protein extracts from brains of well-ascertained cognitively impaired patients reveals an elevation of both Aβ40 and Aβ42 levels with increasing cognitive deficits (Naslund et al., 2000). In the frontal cortex, the increased Aβ levels precede significant NFT pathology. Therefore, these data suggest that intracellular Aβ may be involved in cognitive decline. Because a positive correlation exists between cognitive decline and loss of synaptophysin or neurons, our results indicate that the intracellular Aβ could be the initial insult leading to neuronal dysfunction or death in AD (Masliah et al., 1989, 2001; Gomez-Isla et al., 1996).
The in vivo cytotoxicity of intracellular Aβ1–42 is supported by the results of LaFerla and colleagues (1995) who showed that neurofilament L promoter-directed expression of cytosolic Aβ1–42 results in neuronal cell death in mice (LaFerla et al., 1996). Additionally, our experiments show that levels of Aβ1–42 likely to be more physiological can rapidly induce cell death and that this toxicity is restricted to a nonfibrillar form of Aβ1–42 peptide.
The extreme toxicity of intracellular Aβ1–42 raises some important questions. First, can Aβ1–42, which is made through the secretory pathway, access the cytosol? Normally, it should not. However, there are three possible modes of entry of Aβ1–42 into the cytosol. Insoluble Aβ42 in the ER could access the cytosol through the ER quality control system where misfolded proteins are reverse translocated to the cytosol, ubiquitinated, and degraded through the proteosome system (Lippincott-Schwartz et al., 1988; Bonifacino et al., 1990; Bonifacino and Lippincott-Schwartz, 1991; Greenfield et al., 1999). In aging, the degradation of the Aβ1–42 could be compromised by a reduction of proteosomal activity thus resulting in cytosolic Aβ1–42 neurotoxicity (Merker et al., 2001). Second, newly synthesized Aβ1–42 could be directed to endosomes/lysosomes either from the trans-Golgi or by endocytosis of secreted peptide. In this situation, the Aβ1–42 would have to be released in the cytosol through breakdown of membrane or passive diffusion of the peptide from endosomes/lysosomes into the cytosol. Interestingly, it has been shown that Aβ1–42, but not Aβ1–40, increases lysosomal membrane permeability possibly resulting in leakage of the Aβ1–42 in the cytosol (Yang et al., 1998). Third, the secreted Aβ1–42 could passively diffuse back into the cytosol through the plasma membrane. Many have reported the neuronal toxicity of the extracellular Aβ peptides on primary and cell line systems (for review see Klein et al., 2001). In contrast to these findings, we were unable to find toxicity of the extracellular Aβ1–42 or Aβ1–40 in our cultures of human neurons despite high concentrations of the peptides. However, we have previously shown that the amyloid peptides render neurons vulnerable to a secondary insult (Paradis et al., 1996). Others have also observed this effect in human neuronal cultures (Mattson et al., 1992). Possibly, the secondary stress increases Aβ receptors or endocytosis or alters the plasma membrane permeability resulting in the diffusion of the extracellular Aβ1–42 peptide inside the cell. However, this latter possibility is unlikely to be a primary cause of AD, as intracellular Aβ1–42 accumulation precedes senile plaques but nevertheless could contribute to a secondary round of cell death after the deposition of extracellular amyloid.
The selective toxicity of Aβ1–42 compared with Aβ1–40 poses another interesting problem: the extracellular Aβ peptide toxicity has been attributed to the fibrillar, protofibrillar, or aggregating properties of Aβ (for review see Klein et al., 2001). For intracellular Aβ1–42 toxicity, there exists three possibilities. First, it could be that the extra two amino acids on Aβ1–42 are responsible for inducing toxicity. Second, as has previously been suggested, amyloid fibrils could be the toxic molecules. However, both less toxic Aβ1–40 and extremely toxic Aβ1–42 form significant amounts of fibrils and nonfibrillized Aβ1–42 is still very toxic. Third, the oligomers observed by Western blot analysis in the nonfibrillized and fibrillized Aβ1–42 could be responsible for cell death (Walsh et al., 1999). Furthermore, it remains a possibility that the forms observed by electron microscopy and by Western blot analysis change when microinjected in neurons due to association with certain cytosolic factors (Walsh et al., 1999; Nilsberth et al., 2001). Therefore, much work will be required to elucidate the toxic structure of the Aβ1–42 peptides.
We further show that Aβ1–42 mediates neurotoxicity through the known p53 and Bax cell death pathway. p53 expression increases in the transgenic neurons of cytosolically expressed Aβ1–42 and in AD neurons (LaFerla et al., 1996; de la Monte et al., 1997, 1998; Kitamura et al., 1997; Seidl et al., 1999). In addition, synthetic p53 inhibitors can prevent extracellular Aβ mediated toxicity of hippocampal neuron cultures (Culmsee et al., 2001). Extracellular Aβ can upregulate Bax expression or require Bax to mediate cytotoxicity (Paradis et al., 1996; Selznick et al., 2000; Culmsee et al., 2001). Furthermore, Bax protein levels increase in AD (MacGibbon et al., 1997; Nagy and Esiri, 1997; Su et al., 1997; Tortosa et al., 1998; Giannakopoulos et al., 1999), although this was not confirmed by all studies (Engidawork et al., 2001). Therefore, the identification of the role of the p53–Bax cell death pathway in intracellular Aβ1–42-mediated neurotoxicity reveals additional therapeutic targets that could be used against AD. Because p53 is activated through phosphorylation, the results suggest that Aβ1–42 induces a kinase or inhibits a phosphatase responsible for this phosphorylation. Given the known importance of tau hyperphosphorylation in AD, one wonders whether the activation of p53 could be linked to the phosphorylation of tau through intracellular Aβ1–42 induction of kinase activity.
The mechanism by which p53 is activated by intracellular Aβ1–42 remains to be elucidated. It is interesting to note that Aβ1–42 interacts with ERAB, an intracellular ER and mitochondria-localized member of the alcohol dehydrogenase family (Yan et al., 1997). In transfected Cos cells, ERAB facilitates the toxicity of extracellular Aβ1–42 and APPV717G mutant (Yan et al., 1999). Whether ERAB is involved in cytosolic Aβ1–42 toxicity remains to be determined but since ERAB is localized to mitochondria and ER, it is unlikely to have access to the injected Aβ1–42 unless mitochondrial ERAB is released into the cytosol during apoptosis as are many other mitochondrial factors.
It is well known that Bax activates caspases by promoting the release of mitochondrial cytochrome c, which forms an apoptosome with a number of other factors including caspases (Adrain and Martin, 2001). The caspase-9 has specifically been shown to be activated by cytochrome c and further activate caspase-3. Our result showing that the caspase-6 and -8, but not the caspase-3 inhibitors, prevent Aβ1–42 toxicity is surprising. The lack of involvement of caspase-3 is likely due to the fact that these neurons contain high levels of inhibitor of apoptosis proteins known to inhibit caspase-3 (Zhang and LeBlanc; unpublished data). These results are consistent with our previous observation that caspase-6, but not caspase-3, is activated in serum-deprived primary human neurons and that only recombinant active caspase-6 induces apoptosis of human primary neurons (LeBlanc et al., 1999; Zhang et al., 2000). Because little is known of caspase-6 regulation, much more work will be required to understand how caspase-6 is activated in the presence of intracellular Aβ1–42.
Uncovering the acute human neuronal toxicity of intracellular Aβ1–42 questions the validity of the currently developed therapies against extracellular amyloid in AD. If intracellular Aβ1–42 precedes extracellular amyloid deposits, then anti-amyloid therapies need to be cell permeable. Otherwise, they may have little effect on the prevention or early treatment of AD but could be beneficial to prevent further damage incurred by the extracellular amyloid. However, if the intracellular Aβ1–42 is a consequence of extracellular amyloid deposits, then these therapies would presumably be advantageous to AD patients. Attempts directed toward the inhibition of the secretases responsible for the production of Aβ1–42 should also have a favorable impact on the disease assuming that it is only Aβ1–42 that is the problem in AD and not an underlying general problem with the secretory pathway resulting in the misfolding and cytosolic accumulation of other insoluble proteins. Our study indicates that in addition to the antiamyloid strategies, antineuronal cell death therapies against p53, Bax, or caspases could be extremely valuable in preventing neuronal loss in AD.
Materials and methods
cDNA clones
Human Bcl-2 cDNA was obtained from Dr. Walter Nishioka (Vical, Inc.). Bax cDNA was amplified from a human neuron cDNA library (Bounhar et al., 2001). APP695 cDNA in pCep4β was obtained from Steve Younkin (Mayo Clinic, Jacksonville, FL). The cDNAs were cloned into pCep4β (Invitrogen). The p53 wild-type and p53R273H dominant negative cDNAs were produced by Arnold Levine, and were cloned in the pCMV-NEO vector (Hinds et al., 1990). These cDNAs were purified through GlassMAXTM (GIBCO BRL) and diluted at 30 ng/μl in PBS before the microinjections.
Primers were designed to amplify secreted and cytosolic Aβ1–40 and Aβ1–42 from APP695. These primers amplify the entire Aβ sequence with an additional methionine ATG codon at the 5′ end and a stop codon at the 3′ end to ensure translation. Cytosolic Aβ was amplified with the Aβ1–40/42 forward primer 5′-TCA CTC GAG AAT GGA TGC AGA ATT CCG ACA T-3′ (contains a built-in 5′ XhoI site) and Aβ1–42 reverse primer 5′-ATG GAT CCT TAC GCT ATG ACA ACA CCG AA-3′ (has a 3′ BamH1 site) or Aβ1–40 reverse primer 5′-TCG ATC CTT AGA CAA CAC CGC CCA CCA TG-3′ (has a 3′ BamH1 site). Secreted Aβ was made by linking the APP signal peptide (SP) to the Aβ sequence. The SP was amplified with APP-SP1 forward primer, 5′-TTA CTC GAG ATG CTG CCC GGT TTG GCA-3′ (contains a 5′ XhoI site) and APP-SP2 reverse primer, 5′-GGA ATT CTG CAT CCA TCG CCC GAG CCG TCC AGG C-3′ (contains a 3′ EcoR1 site). A ligation between EcoR1- cleaved PCR-amplified Aβ coding sequence and the EcoR1-cleaved SP sequence was reamplified with the Aβ1–40/42 forward primer and Aβ1–40 or Aβ1–42 reverse primers. The PCR-amplified SP and Aβ sequence were cloned into the prokaryotic pBSKII and eukaryotic episomal pCep4β vectors through the XhoI/BamHI restriction sites. All clones were restriction mapped and sequenced before use.
Neutralizing Bax antibodies
Monoclonal anti-Bax 6A7 (amino acids 12–24; PharMingen) and 2D2 (amino acids 3–16; Trevigen), polyclonal anti-Bax N-20 (Santa Cruz Biotechnology, Inc.), monoclonal anti-APP 22C11 (Roche), mouse IgG, or rabbit sera were diluted at 50 μg/ml (for 6A7, 2D2, mouse IgG, and 22C11) or 25 μg/ml (for polyclonal anti-Bax and rabbit sera) in PBS before use. A toxicity curve was done to determine these concentrations as the highest nontoxic concentrations that can be injected in neurons.
Cell cultures
Primary cultures of human neurons and astrocytes were prepared from 11–17-wk-old fetal brains as described previously (LeBlanc, 1995). The McGill University Institutional Review Board (Montréal, Québec, Canada) has approved this procedure following Canadian Institutes of Health and National Institutes of Health ethical guidelines. Brain tissues were dissociated with 0.25% trypsin (GIBCO BRL) in PBS at 37°C for 15 min. The trypsin was inactivated with 10% decomplemented FBS (HyClone). The dissociated cells were triturated in 0.1 mg/ml DNaseI (GIBCO BRL), filtered successively through 130 μm nylon mesh (Sefar Canada, Inc.) and Falcon 70-μm cell strainers (Becton Dickinson), and centrifuged at 5,000 g for 10 min at 10°C to pellet the cells. The cell pellet was washed once with PBS and once with MEM (GIBCO BRL) in Earle's balanced salt solution containing 0.225% sodium bicarbonate, 1 mM sodium pyruvate, 2 mM l-glutamine, 0.1% dextrose, antibiotic Pen-Strep (all from GIBCO BRL), and 5% decomplemented FBS. The cells were plated at a density of 3 × 106 cells/ml on poly-l-lysine–coated ACLAR™ (Cat. No., 33C; thickness, 0.5 mm; Allied Chemical) coverslips. Neuron cultures were treated successively three times with 1 mM fluorodeoxyuridine (GIBCO BRL) at feeding, and subsequently every week to prevent proliferation of dividing cells. In general, the neurons attach to the coverslips within 24 h and develop dense neuritic networks within 3 d. The cultures contain 90–95% neurons and 5–10% astrocytes (LeBlanc, 1995). Microinjections or treatments were performed 10 d after plating for neurons and astrocytes.
Human neuroblastoma M17 cells were obtained from Dr. J. Biedler (Cellular Biochemistry and Genetics, New York, NY) and cultured on ACLAR™ coverslips at 106 cells/ml in OPTI-MEM (GIBCO BRL) containing 5% FBS. Human teratocarcinoma NT2 (Stratagene) and neuroblastoma La-N-1 cells, a gift from Dr. L. Culp (Case Western Reserve University, Cleveland, OH), were cultured on ACLAR™ coverslips at 106 cells/ml in DME (GIBCO BRL) containing 10% FBS. BHK cells, a gift from William Bowers (University of Rochester, Rochester, NY), and the mice NIH3T3 fibroblasts, a gift from Dr. Stephane Richard (McGill University, Montréal, Québec, Canada) were grown in DME and 10% FBS.
Aβ peptides
Initially (for Fig. 1), Aβ peptides (Bachem) were dissolved in sterile distilled water at 25 μM and incubated at 37°C for 5 d. The peptides' stock solutions were frozen and diluted in PBS immediately before microinjection. Thereafter, nonfibrillar Aβ peptides (American Peptide Co.) were disaggregated at 25 μM in 5 mM Tris buffer pH 7.4, an aliquot diluted to 0.25 μM and immediately frozen at −20°C in aliquots of 50 μl. The remaining 25-μM solution was incubated at 37°C in Eppendorf tubes with continuous mixing by inversion to fibrillize the peptides. After incubation, the samples were removed, vortexed, sonicated twice for 1 min in a bath type sonicator (ELMA GmbH & Co. KG), and frozen at −20°C in 50 μl aliquots. Each aliquot was used once to avoid possible effects of freeze and thaw cycles.
Electron microscopy
A 3-μl aliquot of Aβ peptide was placed on freshly cleaved mica plates (BioForce Laboratory, Inc.). The specimens were air dried and subsequently transferred to a Balzers High-Vacuum Freeze-Etch Unit (model 301) under a 1.3 × 10−4 Pa vacuum. The specimens were shadowed with platinum (BAL-TEC EM-Technology and Application, NH) at a 30° angle and coated with a carbon film platinum (BAL-TEC EM-Technology and Application). The replicas were detached from the mica by flotation in deionized water and transferred onto a 300-mesh grid (Canemco, Inc.). The grids were examined with a Joel 200FX transmission electron microscope (Joel) at 21,000× magnification.
Western blot analysis of Aβ peptides
Nonfibrillar and fibrillar forms of Aβ1–40 and Aβ1–42 (5 μg) were added to sample buffer and electrophoresed on a triple layer (4%, 10%, 16.5%) Tris-Tricine gel at 50 V for 1 h followed by 70 V for 16 h (Schagger and Von Jagow, 1987). The proteins were transferred to Immobilon-P PVDF Membrane (Millipore) at 200 milliamps for 2 h. The membrane was blocked by 5% nonfat milk in Tris buffered saline with 0.1% Tween 20 (TBST) at room temperature for 1 h, incubated with a 1/100 dilution of the anti-Aβ1–17 antibody 6E10 (Signet) and detected by chemiluminescence.
Microinjection
Thin-walled Borosilicate glass capillaries (OD 1.0 mm, ID 0.5 mm) with microfilament (MTW100F-4; World Precision Instrument) were pulled with a Flaming/Brown Micropipette Puller (P-87; Sutter) to obtain injection needles with a tip diameter of ∼0.5 μm. Microinjections were performed in the cytosol of each cell using the Eppendorf Microinjector 5246 and Burleigh Micromanipulator MIS-5000. Human neurons were injected with 25 pl/shot at an injection pressure of 100 hPa, a compensation pressure of 50 hPa, and an injection time of 0.1 s. Human astrocytes, M17, NT2, La-N-1, BHK, and NIH 3T3 cells were injected with 8 pl/shot at an injection pressure of 50 hPa, a compensation pressure of 30 hPa and an injection time of 0.1 s. The diluted peptides were injected at the indicated concentrations with 100 μg/ml DTR (MW: 3000; Molecular Probes) as a fluorescent marker to recognize the injected cells. Approximately 90% neurons and NT2 cells, and 50% astrocytes, M17, La-N-1, BHK, and NIH 3T3 cells survive the injections for at least 16 d.
Measurement of neuronal apoptosis
Cells were fixed in freshly prepared 4% paraformaldehyde/4% sucrose for 20 min at room temperature and permeabilized in 0.1% Triton X-100, 0.1% sodium citrate on ice for 2 min. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) was performed using the In situ Cell Death Detection Kit I as described by the manufacturer (Roche). The percentage of cell death was determined as the ratio of the number of DTR-TUNEL–double positive cells over the total number of DTR-positive cells.
Hoechst staining was used to recognize cell nuclei and detect apoptotic nuclear condensation and fragmentation. Hoechst dye (Intergen) was dissolved in sterile distilled water at 200 μg/ml and diluted 500 times in PBS immediately before staining. After the incubation for TUNEL staining, cells were washed three times for 10 min each in PBS, treated with the diluted Hoechst dye for 15 min at room temperature (in the dark), washed three times for 10 min each in PBS, washed once in water for 5 min, and mounted with Immunon™ mounting medium (Shandon) onto glass slides to be observed under the fluorescence microscope.
Treatment with caspase inhibitors, cycloheximide, and actinomycin D
Caspase pan inhibitor, Z-Valine-Alanine-Aspartic acid-fluoromethylketone (Z-VAD-fmk) (Biomol), caspase-1 inhibitor, Z-Tyrosine-Valine-Alanine-Aspartic acid-fmk (Z-YVAD-fmk), caspase-6 inhibitor, Z-Valine-Glutamic acid-Isoleucine-Aspartic acid-fmk (Z-VEID-fmk), caspase-3 inhibitor, Z-Aspartic acid-Glutamic acid-Valine-Aspartic acid-fmk (Z-DEVD-fmk), and caspase-8 inhibitor, Z-Isoleucine-Glutamic acid-Threomine-Aspartic acid-fmk (Z-IETD-fmk) (Sigma-Aldrich), were dissolved in 100% DMSO (Sigma-Aldrich) at 20 mM and were diluted at 5 μM into culture media immediately before use. Stock solutions of 5 mg/ml cycloheximide (Sigma-Aldrich) and 200 μM actinomycin D (Sigma-Aldrich) were made in sterile distilled water and diluted to 5 μg/ml for cycloheximide and 5 μM for actinomycin D in the culture media immediately before use. The media was changed every 48 h.
Statistical evaluation
One-way or two-way analyses of variance (ANOVAs) with post hoc tests (Statview 5.01) determined the statistical significance of the difference between treatments. The Dunnett's test was used when comparing several groups with one certain group (control), for example, comparison between different treatment groups vs. untreated group. The Sheffé's test was applied when comparing between every other group, for example, comparison between each treatment group. A P value of <0.05 was taken as the criteria for statistical significance.
Acknowledgments
We acknowledge the contribution of Neurochem, Inc. (A. LeBlanc), Fonds de Recherche en Santé du Québec (A. LeBlanc), and the Alzheimer Society of Canada (Y. Zhang) for this work. We also gratefully thank Dr. Robert J. Chalifour (Neurochem Inc.) for the preparation of the amyloid peptides, Jennifer Hammond for the technical preparation of the human neuron cultures, Megan Blacker and Dr. Emily Vereker for the construction of the Aβ vectors, Dr. H. Vali for help with the electron microscopy, and Drs. Patrick Tremblay and Francine Gervais for useful suggestions pertaining to this work.
Footnotes
Abbreviations used in this paper: AD, Alzheimer's disease; Aβ, amyloid β peptide; APP, amyloid precursor protein; DTR, Dextran Texas red; NFT, neurofibrillary tangle; PS, presenilin; SP, signal peptide; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
References
- Adrain, C., and S.J. Martin. 2001. The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Trends Biochem. Sci. 26:390–397. [DOI] [PubMed] [Google Scholar]
- Aurelio, O.N., X.T. Kong, S. Gupta, and E.J. Stanbridge. 2000. p53 mutants have selective dominant-negative effects on apoptosis but not growth arrest in human cancer cell lines. Mol. Cell. Biol. 20:770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino, J., and J. Lippincott-Schwartz. 1991. Degredation of proteins within the endoplasmic reticulum. Curr. Opin. Biol. 3:592–600. [DOI] [PubMed] [Google Scholar]
- Bonifacino, J.S., C.K. Suzuki, and R.D. Klausner. 1990. A peptide sequence confers retention and rapid degradation in the endoplasmic reticulum. Science. 247:79–82. [DOI] [PubMed] [Google Scholar]
- Bounhar, Y., Y. Zhang, C. Goodyer, and A. LeBlanc. 2001. Prion protein protects against Bax-mediated cell death. J. Biol. Chem. 276:39145–39149. [DOI] [PubMed] [Google Scholar]
- Chui, D.H., H. Tanahashi, K. Ozawa, S. Ikeda, F. Checler, O. Ueda, H. Suzuki, W. Araki, H. Inoue, K. Shirotani, et al. 1999. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat. Med. 5:560–564. [DOI] [PubMed] [Google Scholar]
- Chyung, A., B. Greenberg, D. Cook, R. Doms, and V. Lee. 1997. Novel β-secretase cleavage of β-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J. Cell Biol. 138:671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee, C., X. Zhu, Q.S. Yu, S.L. Chan, S. Camandola, Z. Guo, N.H. Greig, and M.P. Mattson. 2001. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 77:220–228. [DOI] [PubMed] [Google Scholar]
- D'Andrea, M.R., R.G. Nagele, H.Y. Wang, P.A. Peterson, and D.H. Lee. 2001. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 38:120–134. [DOI] [PubMed] [Google Scholar]
- de la Monte, S.M., Y.K. Sohn, and J.R. Wands. 1997. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J. Neurol. Sci. 152:73–83. [DOI] [PubMed] [Google Scholar]
- de la Monte, S.M., Y.K. Sohn, N. Ganju, and J.R. Wands. 1998. P53- and CD95-associated apoptosis in neurodegenerative diseases. Lab. Invest. 78:401–411. [PubMed] [Google Scholar]
- Engidawork, E., T. Gulesserian, R. Seidl, N. Cairns, and G. Lubec. 2001. Expression of apoptosis related proteins in brains of patients with Alzheimer's disease. Neurosci. Lett. 303:79–82. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos, P., E. Kovari, A. Savioz, F. de Bilbao, M. Dubois-Dauphin, P.R. Hof, and C. Bouras. 1999. Differential distribution of presenilin-1, Bax, and Bcl-X(L) in Alzheimer's disease and frontotemporal dementia. Acta Neuropathol. 98:141–149. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla, T., R. Hollister, H. West, S. Mui, J. Growdon, R. Petersen, J. Parisi, and B. Hyman. 1996. Neuronal loss correlated with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41:17–24. [DOI] [PubMed] [Google Scholar]
- Gouras, G.K., J. Tsai, J. Naslund, B. Vincent, M. Edgar, F. Checler, J.P. Greenfield, V. Haroutunian, J.D. Buxbaum, H. Xu, et al. 2000. Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 156:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield, J.P., J. Tsai, G.K. Gouras, B. Hai, G. Thinakaran, F. Checler, S.S. Sisodia, P. Greengard, and H. Xu. 1999. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. USA. 96:742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyure, K.A., R. Durham, W.F. Stewart, J.E. Smialek, and J.C. Troncoso. 2001. Intraneuronal Aβ-amyloid precedes development of amyloid plaques in Down syndrome. Arch. Pathol. Lab. Med. 125:489–492. [DOI] [PubMed] [Google Scholar]
- Hinds, P.W., C.A. Finlay, R.S. Quartin, S.J. Baker, E.R. Fearon, B. Vogelstein, and A.J. Levine. 1990. Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: a comparison of the “hot spot” mutant phenotypes. Cell Growth Differ. 1:571–580. [PubMed] [Google Scholar]
- Hsia, A.Y., E. Masliah, L. McConlogue, G.Q. Yu, G. Tatsuno, K. Hu, D. Kholodenko, R.C. Malenka, R.A. Nicoll, and L. Mucke. 1999. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc. Natl. Acad. Sci. USA. 96:3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura, Y., S. Shimohama, W. Kamoshima, Y. Matsuoka, Y. Nomura, and T. Taniguchi. 1997. Changes of p53 in the brains of patients with Alzheimer's disease. Biochem. Biophys. Res. Commun. 232:418–421. [DOI] [PubMed] [Google Scholar]
- Klein, W.L., G.A. Krafft, and C.E. Finch. 2001. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 24:219–224. [DOI] [PubMed] [Google Scholar]
- LaFerla, F.M., B.T. Tinkle, C.J. Bieberich, C.C. Haudenschild, and G. Jay. 1995. The Alzheimer's Aβ peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat. Genet. 9:21–30. [DOI] [PubMed] [Google Scholar]
- LaFerla, F.M., C.K. Hall, L. Ngo, and G. Jay. 1996. Extracellular deposition of beta-amyloid upon p53-dependent neuronal cell death in transgenic mice. J. Clin. Invest. 98:1626–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc, A.C. 1995. Increased production of 4 kDa amyloid β peptide in serum deprived human primary neuron cultures: possible involvement of apoptosis. J. Neurosci. 15:7837–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc, A.C., H. Liu, C. Goodyer, C. Bergeron, and J. Hammond. 1999. Caspase-6 role in apoptosis of human neurons, amyloidogenesis and Alzheimer's disease. J. Biol. Chem. 274:23426–23436. [DOI] [PubMed] [Google Scholar]
- Lee, S.J., U. Liyanage, P.E. Bickel, W. Xia, P.T. Lansbury, and K.S. Kosik. 1998. A detergent-insoluble membrane compartment contains A beta in vivo. Nat. Med. 4:730–734. [DOI] [PubMed] [Google Scholar]
- Li, Q.X., C. Maynard, R. Cappai, C.A. McLean, R.A. Cherny, T. Lynch, J.G. Culvenor, J. Trevaskis, J.E. Tanner, K.A. Bailey, et al. 1999. Intracellular accumulation of detergent-soluble amyloidogenic A beta fragment of Alzheimer's disease precursor protein in the hippocampus of aged transgenic mice. J. Neurochem. 72:2479–2487. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., J.S. Bonifacino, L.C. Yuan, and R.D. Klausner. 1988. Degradation from the endoplasmic reticulum: Disposing of newly synthesized proteins. Cell. 54:209–220. [DOI] [PubMed] [Google Scholar]
- MacGibbon, G.A., P.A. Lawlor, E.S. Sirimanne, M.R. Walton, B. Connor, D. Young, C. Williams, P. Gluckman, R.L. Faull, P. Hughes, and M. Dragunow. 1997. Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimer's disease hippocampus. Brain Res. 750:223–234. [DOI] [PubMed] [Google Scholar]
- Martin, L.J., C.A. Pardo, L.C. Cork, and D.L. Price. 1994. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am. J. Pathol. 145:1358–1381. [PMC free article] [PubMed] [Google Scholar]
- Martin, B.L., G. Schrader-Fisher, J. Busciglio, M. Duke, P. Paganetti, and B.A. Yankner. 1995. Intracellular accumulation of β-amyloid in cells expressing the Swedish mutant amyloid precursor protein. J. Biol. Chem. 270:26727–26730. [DOI] [PubMed] [Google Scholar]
- Masliah, E., R.D. Terry, R.M. DeTeresa, and L.A. Hansen. 1989. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett. 103:234–239. [DOI] [PubMed] [Google Scholar]
- Masliah, E., A. Sisk, M. Mallory, L. Mucke, D. Schenk, and D. Games. 1996. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J. Neurosci. 16:5795–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah, E., M. Mallory, M. Alford, R. DeTeresa, L.A. Hansen, D.W. McKeel, Jr., and J.C. Morris. 2001. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 56:127–129. [DOI] [PubMed] [Google Scholar]
- Mattson, M.P., B. Cheng, D. Davis, K. Bryant, I. Lieberburg, and R.E. Rydel. 1992. β-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12:376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker, K., A. Stolzing, and T. Grune. 2001. Proteolysis, caloric restriction and aging. Mech. Aging Dev. 122:595–615. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima, M., and Y. Ihara. 1998. The presence of amyloid beta-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochemistry. 37:15247–15253. [DOI] [PubMed] [Google Scholar]
- Nagy, Z.S., and M.M. Esiri. 1997. Apoptosis-related protein expression in the hippocampus in Alzheimer's disease. Neurobiol. Aging. 18:565–571. [DOI] [PubMed] [Google Scholar]
- Naslund, J., V. Haroutunian, R. Mohs, K.L. Davis, P. Davies, P. Greengard, and J.D. Buxbaum. 2000. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 283:1571–1577. [DOI] [PubMed] [Google Scholar]
- Nilsberth, C., A. Westlind-Danielsson, C.B. Eckman, M.M. Condron, K. Axelman, C. Forsell, C. Stenh, J. Luthman, D.B. Teplow, S.G. Younkin, et al. 2001. The “Arctic” APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat. Neurosci. 4:887–893. [DOI] [PubMed] [Google Scholar]
- Paradis, E., H. Douillard, M. Koutroumanis, C. Goodyer, and A. LeBlanc. 1996. Amyloid β peptide of Alzheimer's disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J. Neurosci. 16:7533–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, R.G., S. Soriano, J.D. Hayes, B. Ostaszewski, W. Xia, D.J. Selkoe, X. Chen, G.B. Stokin, and E.H. Koo. 1999. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 274:18851–18856. [DOI] [PubMed] [Google Scholar]
- Petanceska, S.S., M. Seeger, F. Checler, and S. Gandy. 2000. Mutant presenilin 1 increases the levels of Alzheimer amyloid beta-peptide Abeta42 in late compartments of the constitutive secretory pathway. J. Neurochem. 74:1878–1884. [DOI] [PubMed] [Google Scholar]
- Pike, C.J., D. Burdick, A.J. Walencewicz, C.G. Glabe, and C.W. Cotman. 1993. Neurodegeneration induced by β-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 13:1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger, H., and G. Von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 KDa. Anal. Biochem. 166:368–379. [DOI] [PubMed] [Google Scholar]
- Seidl, R., S. Fang-Kircher, B. Bidmon, N. Cairns, and G. Lubec. 1999. Apoptosis-associated proteins p53 and APO-1/Fas (CD95) in brains of adult patients with Down syndrome. Neurosci. Lett. 260:9–12. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J. 1998. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 8:447–453. [DOI] [PubMed] [Google Scholar]
- Selznick, L., T.S. Zheng, R.A. Flavell, P. Rakic, and K. Roth. 2000. Amyloid beta induced neuronal death is bax-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 59:271–279. [DOI] [PubMed] [Google Scholar]
- Su, J.H., G. Deng, and C. Cotman. 1997. Bax protein expression is increased in Alzheimer's disease brain: correlations with DNA damage, Bcl-2 expression and brain pathology. J. Neuropathol. Exp. Neurol. 56:86–93. [DOI] [PubMed] [Google Scholar]
- Sudoh, S., Y. Kawamura, S. Sato, R. Wang, T.C. Saido, F. Oyama, Y. Sakaki, H. Komano, and K. Yanagisawa. 1998. Presenilin 1 mutations linked to familial Alzheimer's disease increase the intracellular levels of amyloid beta-protein 1-42 and its N-terminally truncated variant(s) which are generated at distinct sites. J. Neurochem. 71:1535–1543. [DOI] [PubMed] [Google Scholar]
- Terry, R.D. 1999. The neuropathology of Alzheimer disease and the structural basis of its cognitive alterations. Alzheimer Disease. R.D. Terry, editor. Lippincott, Williams and Wilkins, Philadelphia. 187–206.
- Tienari, P., N. Ida, E. Ikonen, M. Simons, A. Weidemann, G. Multhaup, C. Masters, C. Dotti, and K. Beyreuther. 1997. Intracellular and secreted Alzheimer β-amyloid species are generated by distinct mechanisms in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA. 94:4125–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortosa, A., E. Lopez, and I. Ferrer. 1998. Bcl-2 and Bax protein expression in Alzheimer's disease. Acta Neuropathol. 95:407–412. [DOI] [PubMed] [Google Scholar]
- Walsh, D.M., D.M. Hartley, Y. Kusumoto, Y. Fezoui, M.M. Condron, A. Lomakin, G.B. Benedek, D.J. Selkoe, and D.B. Teplow. 1999. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 274:25945–25952. [DOI] [PubMed] [Google Scholar]
- Wirths, O., G. Multhaup, C. Czech, V. Blanchard, S. Moussaoui, G. Tremp, L. Pradier, K. Beyreuther, and T.A. Bayer. 2001. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 306:116–120. [DOI] [PubMed] [Google Scholar]
- Xia, W., J. Zhang, B.L. Ostaszewski, W.T. Kimberly, P. Seubert, E.H. Koo, J. Shen, and D.J. Selkoe. 1998. Presenilin 1 regulates the processing of beta-amyloid precursor protein C-terminal fragments and the generation of amyloid beta-protein in endoplasmic reticulum and Golgi. Biochemistry. 37:16465–16471. [DOI] [PubMed] [Google Scholar]
- Xiang, H., Y. Kinoshita, M. Knudson, S. Korsmeyer, P. Schwartzkroin, and R. Morrisson. 1998. Bax involvement in p53-mediated neuronal cell death. J. Neurosci. 18:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, S.D., J. Fu, C. Soto, X. Chen, H. Zhu, F. Al-Mohanna, K. Collison, A. Zhu, E. Stern, T. Saido, et al. 1997. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 389:689–695. [DOI] [PubMed] [Google Scholar]
- Yan, S.D., Y. Shi, A. Zhu, J. Fu, H. Zhu, Y. Zhu, L. Gibson, E. Stern, K. Collison, F. Al-Mohanna, et al. 1999. Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Abeta-induced cytotoxicity. J. Biol. Chem. 274:2145–2156. [DOI] [PubMed] [Google Scholar]
- Yang, A.J., D. Chandswangbhuvana, L. Margol, and C.G. Glabe. 1998. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J. Neurosci. Res. 52:691–698. [DOI] [PubMed] [Google Scholar]
- Yang, A.J., D. Chandswangbhuvana, T. Shu, A. Henschen, and C.G. Glabe. 1999. Intracellular accumulation of insoluble, newly synthesized abetan-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1-42. J. Biol. Chem. 274:20650–20656. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., C. Goodyer, and A. LeBlanc. 2000. Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, 6, 7 and 8. J. Neuroscience. 20:8384–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]