

Abstract
One of several features acquired by chromatin of the inactive X chromosome (Xi) is enrichment for the core histone H2A variant macroH2A within a distinct nuclear structure referred to as a macrochromatin body (MCB). In addition to localizing to the MCB, macroH2A accumulates at a perinuclear structure centered at the centrosome. To better understand the association of macroH2A1 with the centrosome and the formation of an MCB, we investigated the distribution of macroH2A1 throughout the somatic cell cycle. Unlike Xi-specific RNA, which associates with the Xi throughout interphase, the appearance of an MCB is predominantly a feature of S phase. Although the MCB dissipates during late S phase and G2 before reforming in late G1, macroH2A1 remains associated during mitosis with specific regions of the Xi, including at the X inactivation center. This association yields a distinct macroH2A banding pattern that overlaps with the site of histone H3 lysine-4 methylation centered at the DXZ4 locus in Xq24. The centrosomal pool of macroH2A1 accumulates in the presence of an inhibitor of the 20S proteasome. Therefore, targeting of macroH2A1 to the centrosome is likely part of a degradation pathway, a mechanism common to a variety of other chromatin proteins.
Keywords: XIST; macroH2A; chromatin; centrosome; aggresome
Introduction
Male and female eutherian mammals achieve equivalent levels of X-linked gene expression by silencing all but one X chromosome in cells of the developing embryo (Avner and Heard, 2001; Willard, 2000). With the exception of imprinted X inactivation in extraembryonic tissue (Huynh and Lee, 2001), the choice of which X chromosome to inactivate in the soma is random and maintained throughout subsequent cell divisions. The inactive X chromosome (Xi)* shares features common to other types of heterochromatin, including hypoacetylation of histone tails (Jeppesen and Turner, 1993; Belyaev et al., 1996; Boggs et al., 1996; Gilbert and Sharp, 1999), hypermethylation of CpG islands (Mohandas et al., 1981; Pfeifer et al., 1990), late replication in S phase (Gilbert et al., 1962; Morishma et al., 1962), and a characteristic pattern of histone H3 lysine methylation (Boggs et al., 2002; Peters et al., 2002). In addition, several unique features characterize heterochromatin of the Xi. These include the association of a large untranslated RNA, the Xi-specific transcript (XIST) (Brown et al., 1991; Brockdorff et al., 1992), and a nonrandom distribution of variants of the core histone H2A (Costanzi and Pehrson, 1998; Chadwick and Willard, 2001a,b).
The macroH2A family of H2A variants was first identified through association with nucleosomes (Pehrson and Fried, 1992). The amino-terminal third of the protein is almost identical to histone H2A, with a unique nonhistone carboxy-terminal tail. Two separate genes encode macroH2A1 and macroH2A2, both of which are enriched in Xi chromatin (Costanzi and Pehrson, 1998, 2001; Chadwick and Willard, 2001a). The enrichment of macroH2A at the Xi forms a characteristic structure in the female nucleus, referred to as a macrochromatin body (MCB). In cultured differentiating mouse embryonic stem cells (ES), an MCB appears after counting and choice of which X chromosome to inactivate has occurred (Mermoud et al., 1999; Rasmussen et al., 2000). Although macroH2A has transcriptional repression activity (Perche et al., 2000), it is not essential for the maintenance of X inactivation. The formation of an MCB is dependent upon localization of XIST RNA, as disruption of XIST results in the loss of the MCB without reactivating the Xi (Csankovszki et al., 1999; Beletskii et al., 2001). Combined, the available data indicate that macroH2A may represent one of several highly redundant mechanisms of gene silencing employed by the Xi (Mohandas et al., 1981; Singer-Sam et al., 1992; Brown and Willard, 1994; Gartler and Goldman, 1994; Csankovszki et al., 1999).
Prior to the onset of X inactivation in ES cells, a cytoplasmic concentration of macroH2A1 is evident, coincident with the centrosome (Rasmussen et al., 2000). When ES cells are stimulated to differentiate, centrosomal macroH2A disappears. More recently, a centrosome-associated pool of macroH2A1 has been observed in somatic cells as well (Mermoud et al., 2001), raising questions about the relationship between nuclear and centrosomal macroH2A1. In the present study, in order to address the spatial and temporal relationship of macroH2A1 with the centrosome and the MCB, we have investigated the distribution of macroH2A1 during the maintenance phase of X inactivation throughout the somatic cell cycle.
Results
MacroH2A1 and macroH2A2 associate with centrosomes in male and female somatic cells
A centrosomal association of macroH2A1 was observed in human somatic cells (Fig. 1 a), as previously observed in mouse (Mermoud et al., 2001), indicating that the centrosomal macroH2A1 pool is not restricted to undifferentiated ES cells (Mermoud et al., 1999; Rasmussen et al., 2000). In addition, the association with centrosomes in both XY and XX somatic cells indicates that the association is independent of X inactivation. Using independent antisera specific to either the macroH2A1 or the macroH2A2 protein, we detected both forms of macroH2A at the centrosome (Fig. 1 b).
Figure 1.

Colocalization of macroH2A with centrosomes in somatic cells. (a) Colocalization of macroH2A1 (green, FITC) with γ-tubulin (red, TRITC) in 46,XX (T-3352) and 46,XY (hTERT-BJ1) cells. Nuclei are stained with DAPI (blue). The white arrowhead indicates the position of the centrosome, determined by γ-tubulin staining. The merged macroH2A1 and γ-tubulin signals indicate overlapping signals in yellow. The white arrow indicates the location of the MCB (green) in the 46,XX nucleus. (b) Colocalization of macroH2A1 and macroH2A2 with γ-tubulin in 46,XX (hTERT-RPE1) cells by indirect immunofluorescence. Cells grown directly on microscope slides were stained with either anti-macroH2A1 or anti-macroH2A2 (aa 124–180). Metaphase chromosomes are stained with DAPI. The white arrowhead indicates the position of the centrosome, determined by γ-tubulin staining. The merged macroH2A and γ-tubulin signals indicate overlapping signals in yellow.
Both forms of macroH2A could also be detected in sucrose gradient fractions enriched for centrosomes (Fig. 2 a). In addition to a signal of anticipated size for macroH2A1, a second band of smaller size was detected in centrosome preparations (Fig. 2 a, lane 1). The same band cofractionates with full-length macroH2A1 in sucrose gradient fractions of nucleosome preparations (Fig. 2 b). Additional bands of comparable size have been detected with independent anti-macroH2A1 antisera (Costanzi et al., 2000; Mermoud et al., 2001). Although a smaller macroH2A2 band is not detected in centrosome preparations (Fig. 2 a, lane 2), a considerably weaker smaller band can be detected in nucleosome fractions with antisera specific to macroH2A2 (Fig. 2 c, lane 2). Smaller bands can also be observed in nucleosome preparations from cell lines expressing epitope-tagged forms of macroH2A1 and macroH2A2 (Fig. 2 d). Although the smaller bands observed for the epitope-tagged forms of macroH2A1 and macroH2A2 do not directly correlate to the size of the smaller bands detected with the macroH2A1 and macroH2A2 primary antisera, this may reflect either a direct effect of the presence of the epitope tag on processing or the relative stability of the endogenous proteins. Whether the smaller bands represent the potential breakdown of macroH2A during various purification protocols, or the direct result of an intracellular biological process is unknown. Further, though less likely in our view because of its reproducibility with three independent antisera, the possibility remains that the smaller bands may be unrelated to macroH2A and simply represent a shared epitope between macroH2A and another protein(s) that coexists with macroH2A in both nucleosomal and centrosomal fractions.
Figure 2.

Detection of macroH2A1 and macroH2A2 in fractions from nucleosome and centrosome preparations. (a) Immunoblot analysis of the same fraction of a centrosome preparation from a 46,XX (hTERT-RPE1) cell line detecting the presence of macroH2A1 (lane 1) or macroH2A2 (aa 124–372; lane 2). The panel below shows the relative level of γ-tubulin by immunoblotting in the same fraction. (b) Immunoblot of five consecutive sucrose gradient fractions (lanes 1–5) of a nucleosome preparation from a 46,XX (hTERT-RPE1) cell line, showing the cofractionation of full-length macroH2A1 (top arrowhead) and a smaller signal (bottom arrowhead). The relative concentration of nucleosomes/histones in each fraction is shown in the Coomassie image below. (c) Immunoblot analysis of the same nucleosome-containing fraction of a nucleosome preparation from a 46,XX (hTERT-RPE1) cell line detecting the presence of macroH2A1 (lane 1) or macroH2A2 (aa 124–372; lane 2). The panel below shows an image of the Coomassie stain of the same fraction indicating the positions of the histones. (d) Immunoblot analysis using anti-Myc mAb detecting the presence of macroH2A1-CT–Myc and macroH2A2-CT–Myc in sucrose gradient fractions of nucleosome preparations from macroH2A1-CT–Myc- and macroH2A2-CT–Myc-expressing HEK-293 cell lines. The arrowheads to the right of each image indicate the presence of the upper full-length macroH2A1/2–Myc and a smaller macroH2A1/2–Myc-derived form. The relative concentration of nucleosomes/histones in each fraction is shown in the Coomassie image below.
Both macroH2A1 and macroH2A2 concentrate in distinct bands on the human and mouse Xi
Previous observations have indicated that macroH2A1 is uniformly associated with the mouse Xi at metaphase (Costanzi and Pehrson, 1998; Mermoud et al., 1999). We have investigated the relationship of macroH2A with the human and mouse Xi and consistently observed a distinct banding pattern on the Xi chromosome (Fig. 3). As previously observed, macroH2A associates with the autosomes and active X chromosome (Xa) in human metaphase spreads as well, but at a significantly lower level than that for the Xi (Fig. 3 c and Fig. 4 a). The same banding pattern is observed using antisera specific to either macroH2A1 or macroH2A2, indicating that the bands contain both isoforms (Fig. 3 a).
Figure 3.
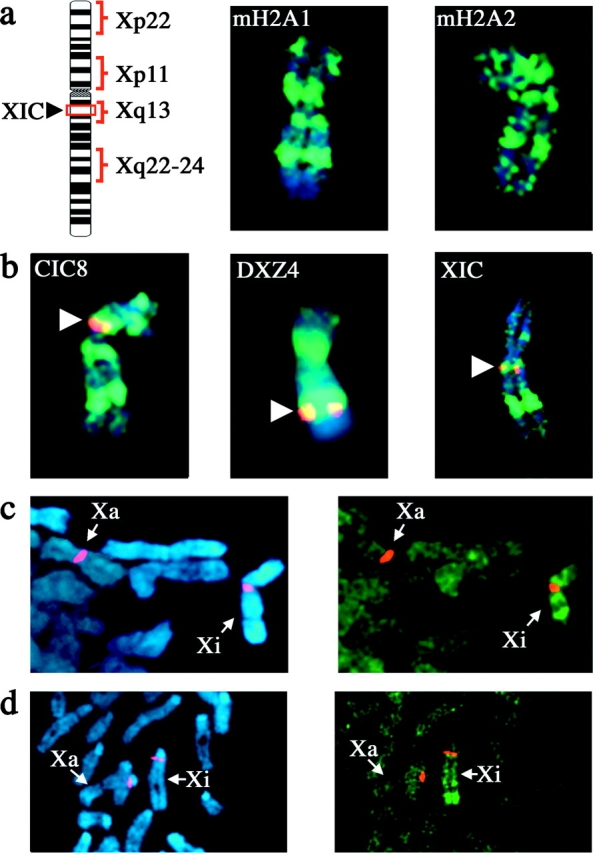
MacroH2A displays distinct banding patterns on the metaphase Xi chromosome. (a) 46,XX (hTERT-RPE1) metaphase Xi chromosome stained with macroH2A1- or macroH2A2- (aa 124–180) specific pAb by indirect immunofluorescence (green, FITC), merged with DAPI staining (blue). The ideogram represents the metaphase Xi chromosome indicating the position of the X inactivation center (XIC) and the positions of the four main bands at Xp22, Xp11, Xq13, and Xq22-24. (b) Indirect immunofluorescence of macroH2A1 banding pattern on the Xi chromosome in 46,XX (hTERT-RPE1) cells merged with FISH signals (red, TRITC) for ordered X chromosome probes, and merged with DAPI image. The white arrowheads indicate the location of the specific FISH probes. Overlapping signals are in yellow. (c) Partial metaphase spread from a 46,XX (hTERT-HME1) cell line stained with macroH2A1 by indirect immunofluorescence, merged with X-α satellite FISH signals. The Xa and Xi are indicated. (d) Partial metaphase spread from a female mouse cell line (B144) stained with macroH2A1 by indirect immunofluorescence, merged with X chromosome–specific DXwas70 FISH signals. The Xa and Xi are indicated.
Figure 4.
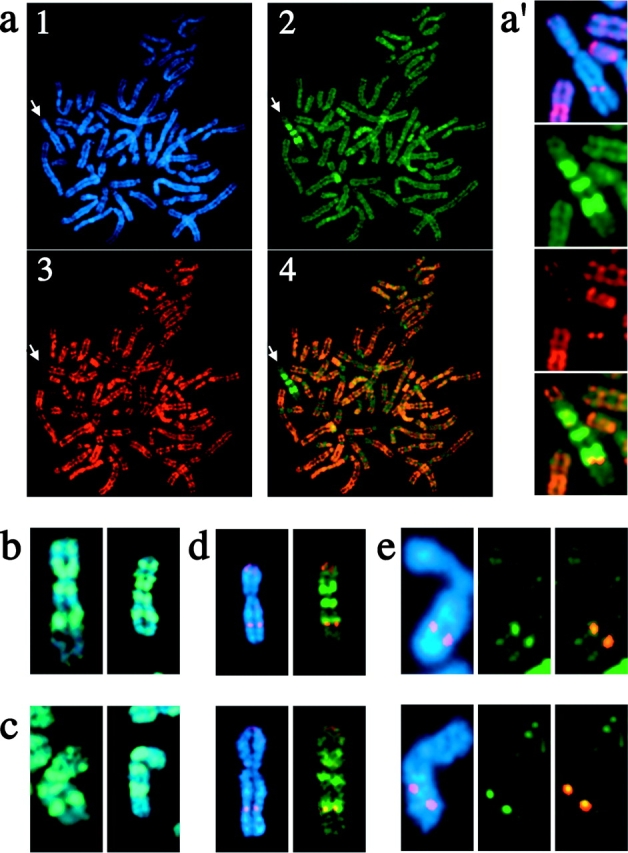
Overlap of the Xq22-24 macroH2A1 band with a band of histone H3 lysine-4 methylation centered at DXZ4. (a) Female metaphase chromosomes from a macroH2A1-CT–Myc-transfected 46,XX (hTERT-RPE1) cell line counterstained with anti-DimH3K4. Chromosomes are stained with DAPI (blue; panel 1), and the distribution of macroH2A1 (green, FITC; panel 2) and DimH3K4 (red, TRITC; panel 3) are shown. The merged distribution of macroH2A1 and DimH3K4 are shown in panel 4 with overlapping signals in orange. White arrow points to the position of the Xi. (a′) Enlarged image of the Xi indicated, showing the macroH2A1 and DimH3K4 banding patterns on the Xi. The DAPI image is merged with the DimH3K4 signal in the top panel to indicate the relative position of the DimH3K4 signal on Xq. (b) 46,XX (hTERT-RPE1) Xi chromosomes showing the distribution of macroH2A1-CT–Myc, merged with the DAPI image. The distribution is indistinguishable from that seen for the endogenous macroH2A1 protein (Fig. 2). (c) 46,XX (hTERT-RPE1) Xi chromosomes showing the distribution of macroH2A1-NT–Xpr, merged with the DAPI image. (d) 46,XX (hTERT-RPE1) Xi chromosomes showing the distribution of DimH3K4 merged with either the DAPI image or the distribution of macroH2A1-CT–Myc, with overlapping signals shown in yellow. (e) 46,XX (hTERT-RPE1) Xi chromosomes showing the DXZ4 locus, merged with the DAPI image or the DimH3K4 signal. Overlapping DXZ4 and DimH3K4 signals are shown in yellow.
Up to four macroH2A bands were observed on the Xi in a variety of human 46,XX cell lines (Fig. 3; unpublished data). To determine the precise location of each band, we stained metaphase chromosomes with macroH2A in combination with FISH using a number of ordered and previously mapped X chromosome probes. This approach placed the macroH2A bands at Xp22, Xp11, Xq13, and Xq22-24, with the most intense and consistent band at Xq22-24 (Fig. 3). Notably, the band at Xq13 was indistinguishable from a cosmid probe containing the XIST locus at the X inactivation center (Fig. 3 b). All four bands were reproduced in a 46,XX cell line overexpressing either an amino- or carboxy-terminal epitope-tagged form of macroH2A1 (Fig. 4, b and c), confirming the identity of the bands.
To extend this observation, we investigated the distribution of macroH2A1 on the mouse Xi at metaphase. As seen in humans, macroH2A1 formed a characteristic banding pattern on the mouse Xi (Fig. 3 d). Intriguingly, the location of the bands within the distal portion of the mouse Xi correlates with sequences that are syntenic with human Xp22, Xp11, Xq13, and Xq22-24 (DeBry and Seldin, 1996), suggesting a conserved role for macroH2A in these regions.
The macroH2A band at Xq22-24 overlaps with the site of histone H3 lysine-4 methylation
In addition to a reproducible banding pattern of macroH2A on the Xi, a distinct banding pattern of histone H3 lysine-4 methylation (DimH3K4) has been observed (Boggs et al., 2002). Human female metaphase chromosomes stained for macroH2A and DimH3K4 show a clear overlap of the distal boundary of the Xq22-24 macroH2A band with DimH3K4 (Fig. 4, a, a′, and d). The pattern of macroH2A and DimH3K4 at Xq22-24 is indistinguishable from that seen between macroH2A and FISH with a probe of the macrosatellite sequence DXZ4 (Fig. 3 b, middle). FISH analysis confirms that the DimH3K4 band is centered at DXZ4 on Xq24 (Fig. 4 e).
Centrosomal association of macroH2A1 alters during the cell cycle
To address a possible temporal relationship between the chromosomal and centrosomal pools of macroH2A, we examined the localization of macroH2A1 throughout the cell cycle. Female cells were synchronized chemically at either the G1–S boundary and released into S phase toward mitosis, or blocked in mitosis and released into G1 toward S phase. The appearance of an MCB was most obvious during S phase, with the loss of the MCB as cells approached mitosis (Fig. 5 a; Table I). In contrast, the proportion of cells demonstrating centrosomal localization of macroH2A1 increased as cells approached mitosis (Fig. 5 a; Table I). MCB formation after release from mitosis required ∼12 h (Fig. 5 a; Table I), whereas macroH2A1 was observed at the centrosome shortly after release. The same relationship of increased centrosome association and decreased MCB frequency as cells approach mitosis was observed in two other 46,XX cell lines (unpublished data), indicating that this is a common feature of human somatic cells.
Figure 5.
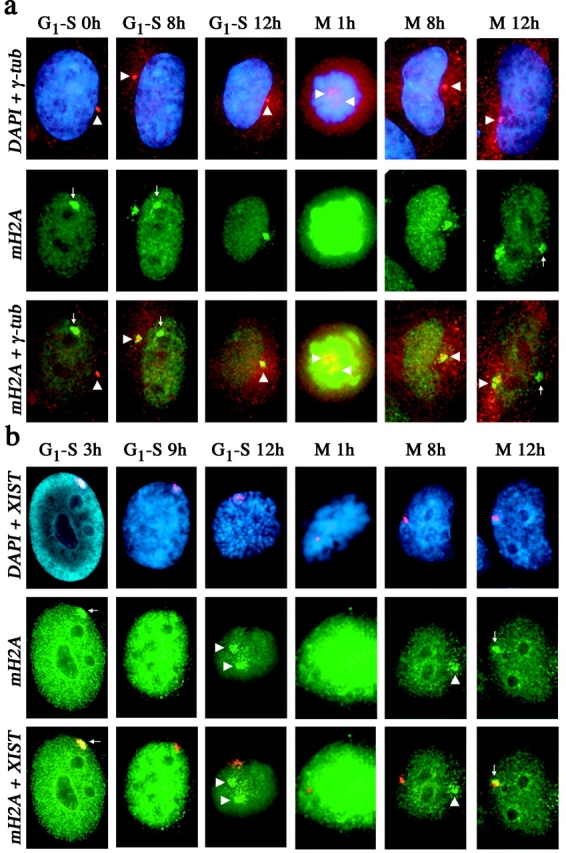
Distribution of macroH2A1 in relation to the centrosome and XIST RNA through the somatic cell cycle. (a) Distribution of macroH2A1 (green, FITC) and γ-tubulin (red, TRITC) in 46,XX (hTERT-RPE1) cells at different stages of the cell cycle as detected by indirect immunofluorescence. The nucleus is stained by DAPI (blue). Overlapping signals are shown in yellow. The positions of the centrosome (white arrowhead) and MCB (white arrow) are indicated. The stage of the cell cycle from which cells were released, G1–S or mitosis (M), are indicated along with the number of hours (h) after release. (b) Distribution of macroH2A1 and XIST RNA in 46,XX (hTERT-RPE1) cells at different stages of the cell cycle as detected by indirect immunofluorescence and RNA-FISH. The nucleus is stained by DAPI. Overlapping signals are shown in yellow. The position of the centrosome (white arrowhead) and MCB (white arrow) are indicated. The stage of the cell cycle from which cells were released, G1–S or mitosis (M), are indicated along with the number of hours (h) after release.
Table I.
Association of macroH2A1 with MCBs and γ-tubulin in 46,XX (hTERT-RPE1) cells at different stages of the cell cycle
| Cell cycle stage | Cells with MCB | Cells with centrosome association | Cells with no MCB or centrosome association |
|---|---|---|---|
| Early S phase | 95 ± 1.4 | 54 ± 2.8 | 2 ± <0.01 |
| Mid to late S phase | 74 ± 2.8 | 84 ± 2.8 | 2 ± <0.01 |
| Late S phase | 52 ± 2.8 | 93 ± 1.4 | 3 ± 1.4 |
| G2 | 22 ± 2.8 | 93 ± 1.4 | 4 ± 2.8 |
| Mitosis | 0 ± <0.01 | 5 ± 4.2 | 95 ± 4.2 |
| Early G1 | 1 ± 1.4 | 55 ± 4.2 | 45 ± 4.2 |
| Mid G1 | 3 ± 1.4 | 100 ± <0.01 | 0 ± <0.01 |
| Late G1 to early S | 39 ± 4.2 | 98 ± 2.8 | 1 ± 1.4 |
Cells were synchronized at the G1–S boundary or in mitosis and released for 0–12 h before detection of macroH2A1 and γ-tubulin by indirect immunofluorescence. Numbers of cells are given as a percentage with standard deviations (n = 100). Early S phase, cells at the G1–S boundary; mid to late S phase, G1–S + 4 h; late S phase, G1–S + 8 h; G2, G1–S + 12 h; mitosis, release from nocodazole for 1 h; early G1, mitosis + 4 h; mid G1, mitosis + 8 h; late G1 to early S, mitosis + 12 h.
Although changes in the relative level of centrosomal macroH2A1 were observed as cells passed through S phase toward mitosis, the nucleosomal concentration of macroH2A1 did not appear to change significantly (unpublished data).
Formation of an MCB follows, but does not mirror, XIST RNA accumulation
Disruption of XIST RNA results in the loss of MCB formation (Csankovszki et al., 1999; Beletskii et al., 2001), indicating dependence of macroH2A1 on XIST for Xi localization. To examine their temporal relationship in normal cells, we monitored MCB formation in relation to XIST RNA during the somatic cell cycle. XIST RNA paints the Xi during interphase (Clemson et al., 1996), but unlike mouse Xist (Duthie et al., 1999), human XIST RNA does not remain associated with the Xi during mitosis (Clemson et al., 1996). An XIST RNA domain was observed in cells throughout S phase and G2 and only dissociated from the Xi as cells entered mitosis (Fig. 5 b; Table II), consistent with earlier findings (Clemson et al., 1996). XIST RNA was expressed and formed an XIST RNA domain shortly after release from mitosis (Fig. 5 b; Table II). In contrast, MCBs dissociated from the Xi chromatin as cells approached mitosis, significantly earlier than XIST RNA, and did not rapidly reform with the XIST RNA territory shortly after mitosis (Fig. 5 b; Table II). With the exception of a very small number of cells, an MCB was present only in cells with an XIST RNA domain. This indicates that although the association of XIST RNA with the Xi is a prerequisite for an MCB, the formation of an MCB is strongly influenced by the cell cycle.
Table II. Frequency of macroH2A1 MCBs and XIST RNA association with the Xi in 46,XX (hTERT-RPE1) cells at different stages of the cell cycle.
| Cell cycle stage | Cells with MCB association with Xi | Cells XIST RNA association with Xi | Cells with no MCB XIST RNA association with Xi |
|---|---|---|---|
| Early S phase | 81 ± 9.9 | 99 ± 1.4 | 1 ± 1.4 |
| Mid to late S phase | 64 ± 5.6 | 98 ± <0.01 | 1 ± 1.4 |
| Late S phase | 48 ± 2.8 | 99 ± 1.4 | 0 ± <0.01 |
| G2 | 19 ± 4.2 | 67 ± 4.2 | 33 ± 4.2 |
| Mitosis | 0 ± <0.01 | 1 ± 1.4 | 99 ± 1.4 |
| Early G1 | 0 ± <0.01 | 68 ± 2.8 | 32 ± 2.8 |
| Mid G1 | 5 ± 1.4 | 99 ± 1.4 | 1 ± 1.4 |
| Late G1 to early S | 33 ± 4.2 | 98 ± <0.01 | 1 ± 1.4 |
Cells were synchronized at the G1–S boundary or in mitosis and released for 0–12 h before detection of macroH2A1 and XIST RNA by indirect immunofluorescence and RNA FISH. Numbers of cells are given as a percentage with standard deviations (n = 100). Early S phase, cells at the G1–S boundary; mid to late S phase, G1–S + 4 h; late S phase, G1–S + 8 h; G2, G1–S + 12 h; mitosis, release from nocodazole for 1 h; early G1, mitosis + 4 h; mid G1, mitosis + 8 h; late G1 to early S, mitosis + 12 h.
MCB formation is influenced by the cell cycle and is most prominent during S phase
To relate MCB formation directly with DNA replication during S phase, cells were synchronized and pulsed with BrdU for 1 h after different release times to detect exit and entry into S phase. Unlabeled cells after release from mitosis had not yet entered S phase (Fig. 6 a), whereas unlabeled cells after release from G1–S had exited S phase and entered G2. Chromatin of the Xi is late replicating in S phase (Gilbert et al., 1962; Morishma et al., 1962), and therefore the MCB is only labeled in late S phase cells (Fig. 6 a). This, in combination with time of release, allows accurate determination of early, middle, and late S phase. Cells released from mitosis into G1 took ∼15 h to enter S phase as detected by BrdU incorporation. Only 23% of cells in G1 had an MCB (Fig. 6 b). In contrast, MCBs were observed in 72% of cells in early S phase. MCB frequency peaked at 93% in cells 5 h after release from G1–S, before dropping sharply late in S phase and in G2. As shown in Fig. 6 b, these data clearly indicate that the appearance of an MCB is most prominent during S phase, in agreement with timing of MCB formation after mitosis and loss of MCBs as cells pass through S phase (Fig. 5; Tables I and II).
Figure 6.
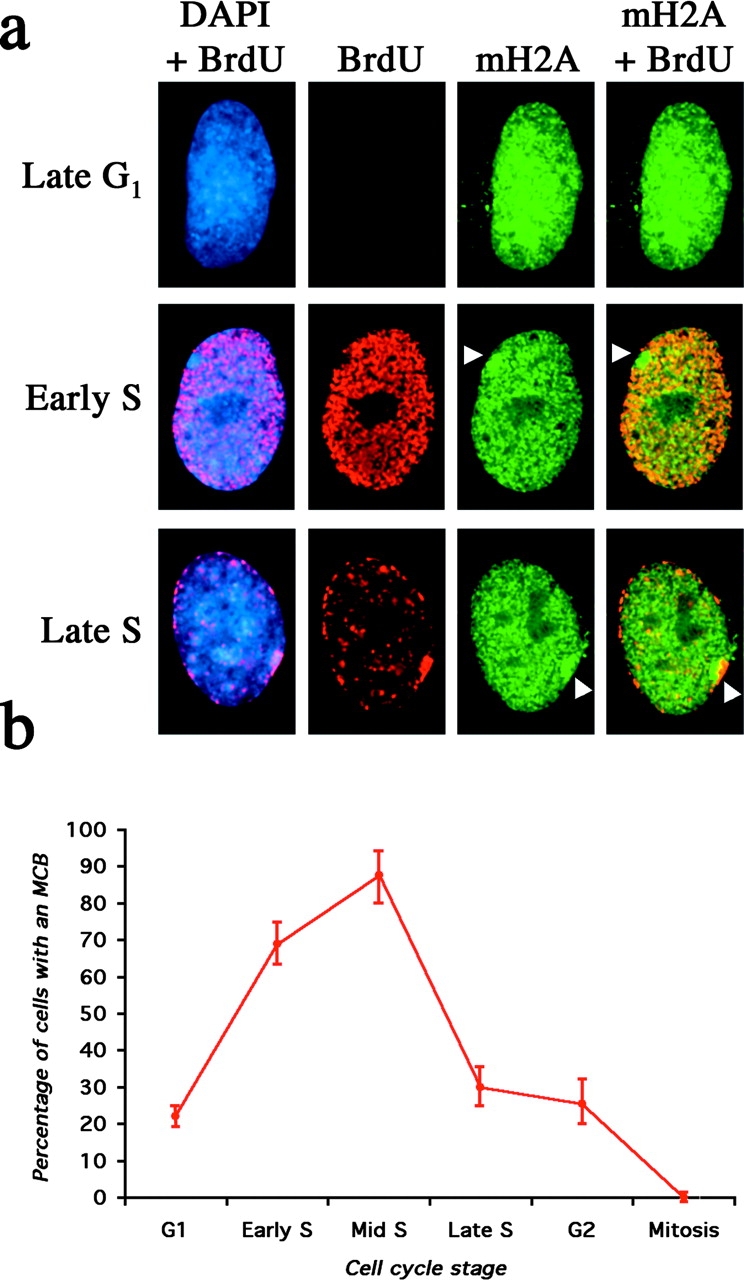
Distribution of macroH2A1 in synchronized 46,XX (hTERT-RPE1) cells in different stages of the somatic cell cycle. (a) The late G1 cell was released from mitosis for 14 h and pulsed with BrdU for 1 h before detection of macroH2A1 (green, FITC) and BrdU (red, TRITC). The nucleus is stained with DAPI (Blue). The cell has not yet entered S phase, as determined by the lack of an anti-BrdU signal. The early S phase cell was released from mitosis for 16 h and pulsed with BrdU for 1 h before detection of macroH2A and BrdU. The nucleus is stained with DAPI and merged with the anti-BrdU signal. The position of the MCB is indicated by the white arrowhead and, as indicated by the merged macroH2A1 and BrdU signals (yellow), has not yet undergone DNA replication. The late S phase cell was released from G1–S for 9 h and pulsed with BrdU for 1 h before detection of macroH2A1 and BrdU. The nucleus is stained with DAPI and merged with the anti-BrdU signal. The position of the MCB is indicated by the white arrowhead and, as indicated by the merged macroH2A1 and BrdU signals (yellow), is currently undergoing DNA replication. (b) Graph showing the frequency of MCB observation at different stages of the somatic cell cycle. Synchronized 46,XX cells (hTERT-RPE1) released for different time periods from the G1–S boundary or mitosis and pulsed for 1 h with BrdU. Cells were scored for the presence of an MCB at different cell cycle stages.
Inhibition of the 20S proteasome results in the accumulation of macroH2A1 at the centrosome
The proteasome is a large multisubunit proteolytic complex that is the major site of protein degradation (Bochtler et al., 1999). Components of the proteasome have been identified in purified centrosome fractions (Wigley et al., 1999) that are capable of degrading ubiquitinated substrates (Fabunmi et al., 2000). To evaluate the potential association of macroH2A1 with the proteasome, cells were synchronized at the G1–S boundary and in mitosis before releasing in the presence of lactacystin, an irreversible proteasome inhibitor (Fenteany et al., 1995). Centrosomal accumulation of macroH2A1 significantly increased after incubation for 12 h in lactacystin and colocalized with an enlarged ubiquitin domain (Fig. 7), whereas an accumulation of macroH2A2 was not detected (unpublished data). The dramatic accumulation of macroH2A1 at the centrosome after inhibition of the proteasome is consistent with the inability to degrade the protein, suggesting that macroH2A1 may be targeted to the centrosome for degradation. The appearance of an enlarged macroH2A1 domain at the centrosome occurs between 8–12 h after release from G1–S or mitosis. More cells acquired an enlarged centrosomal macroH2A1 domain when released from G1–S (91% of cells) than from mitosis (71%), perhaps indicating that more macroH2A1 is targeted for degradation as cells pass through S phase and G2 than in G1. This may reflect the remodeling of chromatin in preparation for mitosis and the need to remove macroH2A1 released in this process.
Figure 7.
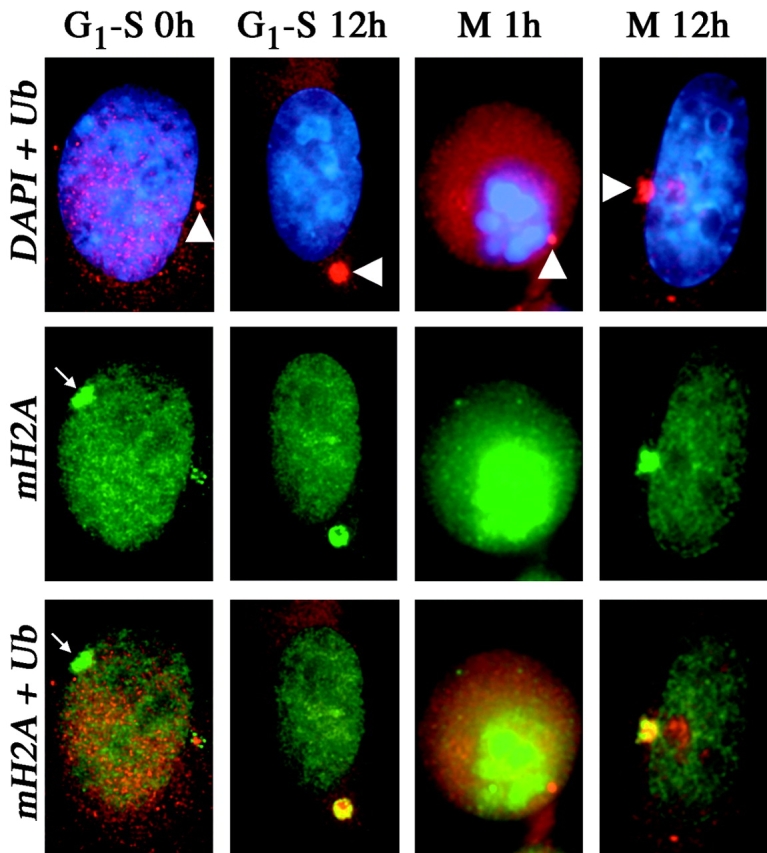
Colocalization of macroH2A1 with ubiquitin and accumulation at the centrosomal proteasome over time with inhibition of the 20S proteasome. 46,XX (hTERT-RPE1) cells were synchronized at G1–S or mitosis (M) and directly processed (0 h/1 h) or released for 12 h (12 h) in the presence of 10 μM lactacystin. The distribution of macroH2A1 (green, FITC) (mH2A) and ubiquitin (red, TRITC) (Ub) are shown by indirect immunofluorescence. The nucleus is stained with DAPI (blue). The white arrowheads indicate the position of the centrosome proteasome marked by ubiquitin, and the small white arrows indicate the position of MCBs. Overlapping macroH2A1 and ubiquitin (yellow) show colocalization and accumulation 12 h after G1–S and mitosis in the presence of the proteasome inhibitor lactacystin.
Centrosome association and accumulation is a feature of some, but not all, chromatin proteins
To determine how specific macroH2A1 association with the centrosome is, we looked at a number of other chromatin proteins for centrosome association (Fig. 8 a). Of 29 chromatin proteins tested, 8 demonstrated a clear overlap with γ-tubulin at mitosis (Fig. 8 a) and interphase (unpublished data). To address the possibility that, like macroH2A1, the centrosomal association of these chromatin proteins represents a degradation pathway, cells were treated with lactacystin and monitored for the accumulation of each chromatin protein at the centrosome (Fig. 8 b). Indeed, several of the chromatin proteins demonstrated a dramatic increase in size at the centrosome and colocalization with ubiquitin (Fig. 8 b). In contrast, DNA–methyltransferase 3a (DNMT3a) remained absent from the centrosome, but consistently accumulated at two nuclear foci (Fig. 8 b), suggesting that this protein is directed to an alternative proteasome center. Taken together, these data imply that targeting of chromatin proteins to the centrosome is a common mechanism of protein degradation.
Figure 8.
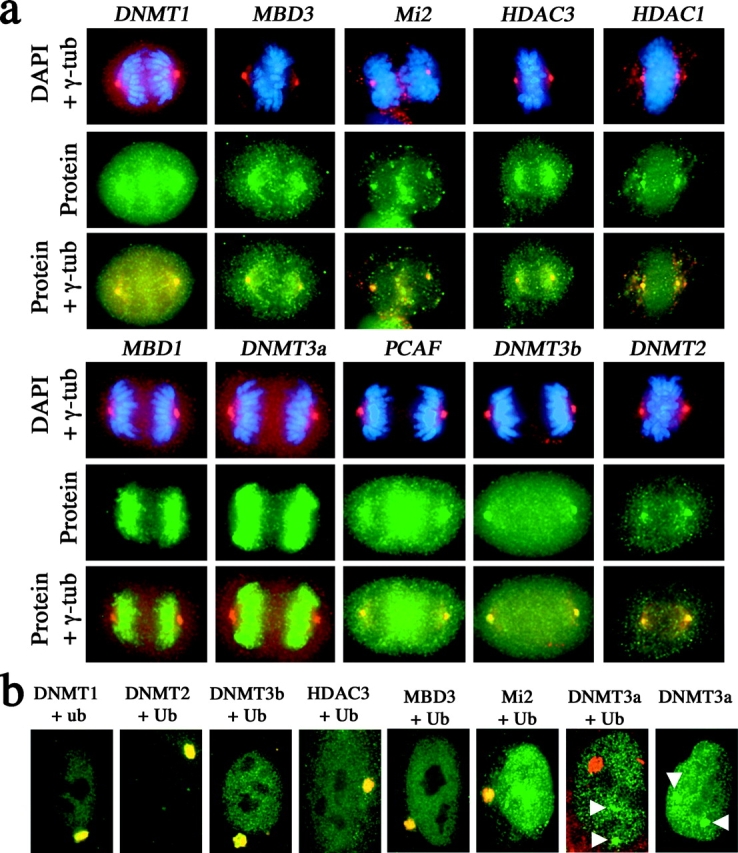
Colocalization of a number of heterochromatin proteins with centrosomes at mitosis, and accumulation at the centrosome proteasome in the presence of lactacystin. (a) Indirect immunofluorescence of heterochromatin proteins (green, FITC) with γ-tubulin (red, TRITC) in 46,XX (hTERT-RPE1) at mitosis. Chromosomes are stained with DAPI (blue). Overlapping heterochromatin protein signals and γ-tubulin signals in merged images are indicated in yellow. The different heterochromatin proteins are indicated to the left of each group of panels. (b) Extensive incubation of 46,XX (hTERT-RPE1) cells with lactacystin results in the accumulation of some, but not all, heterochromatin proteins at the centrosome proteasome. Cells were treated with 10 μM lactacystin for 14 h before processing. Ubiquitin and proteins indicated were detected by indirect immunofluorescence. Clear overlapping signals with ubiquitin can be seen (yellow). White arrowheads indicate the accumulation of Dnmt3a with two nuclear territories and not with the centrosome proteasome. Gene names are defined in the Materials and methods.
Discussion
MacroH2A1 associates with the centrosome in a manner characteristic of a degradation pathway
The association of macroH2A at the centrosome is not restricted to undifferentiated mouse ES cells (Rasmussen et al., 2000), but is a common feature of male and female mouse (Mermoud et al., 2001) and human somatic cells (Fig. 1 a). Centrosomal macroH2A is composed of both macroH2A1 and macroH2A2 (Fig. 1 b and Fig. 2 a), indicating that both proteins are spatially indistinguishable at the centrosome.
Recently, a link has been made between protein degradation pathways and the centrosome. Treatment of cells with lactacystin, a potent inhibitor of the 20S proteasome (Fenteany et al., 1995), results in the dramatic formation of perinuclear protein aggregates (Wojcik et al., 1996). In the absence of lactacystin, a variety of mutant misfolded or overexpressed proteins also accumulate in perinuclear aggregates (referred to as aggresomes) that are centered at the centrosome (Johnston et al., 1998; Garcia-Mata et al., 1999). Once aggresomes form, they appear to be highly resistant to proteolysis (Kopito and Sitia, 2000) and are thought to be a major contributor to the pathology of disease (Kopito, 2000). Most compelling is the detection and purification of active components of the proteasome at the centrosome (Wigley et al., 1999; Fabunmi et al., 2000). The accumulation of macroH2A1 at the centrosome in the presence of lactacystin (Fig. 7) suggests that macroH2A1 is targeted to the centrosome–proteasome as part of a degradation pathway. In contrast, despite the association of macroH2A2 with the centrosome (Fig. 1 b and Fig. 2 a), an accumulation was not detected in the presence of lactacystin (unpublished data). One explanation for this could be that macroH2A2 is targeted primarily to a nuclear proteasome center for degradation with centrosomal proteasome–mediated degradation being secondary. Alternatively, lactacystin may prevent the export of macroH2A2 but not macroH2A1 from the nucleus. Ultimately, this may reflect a difference in the biology of the two macroH2A proteins, or a sensitivity issue regarding the antisera. Notably, poly-ADP ribose polymerase, an activator of the 20S proteasome to degrade histones damaged by oxidation in the nucleus (Ullrich et al., 1999), is also located at the centrosome (Kanai et al., 2000) and may activate the centrosomal proteasome in a similar fashion.
Although a common feature of proteins targeted for degradation is the addition of polyubiquitin chains (Pickart, 2001), exhaustive immunoprecipitation experiments using antisera raised to ubiquitin failed to immunoprecipitate polyubiquitinated forms of macroH2A (unpublished data). Close examination of the extensive overlap in the macroH2A1 and ubiquitin signals in the aggresome indicates a hole in the macroH2A1 signal at the center of the aggresome (Fig. 7). This is also true of the macroH2A1 signal before lactacystin treatment (Fig. 7), placing macroH2A1 in the pericentriolar material, as confirmed by the absence of macroH2A1 signal at the centrioles marked by γ-tubulin (Figs. 1 and 5). Although ubiquitination is a common signal for targeting proteins to the proteasome (Bochtler et al., 1999), other proteins are targeted to the proteasome for degradation in the absence of detectable ubiquitination (Garcia-Mata et al., 1999). Therefore macroH2A1 may be targeted to the proteasome in a ubiquitin-independent fashion or else macroH2A1 is targeted through a physical association with other ubiquitinated proteins. Alternatively, the mono-ubiquitinated form of macroH2A, which is readily detectable (unpublished data), may be sufficient for targeting for degradation as previously demonstrated for histone H3 (Haas et al., 1990).
In addition to macroH2A1, a number of other chromatin proteins associate with the centrosome (Hsu and White, 1998; Barthelmes et al., 2000; Xue et al., 2000) (Fig. 8 a). Like macroH2A1, treatment of cells with lactacystin results in the association of some, but not all, chromatin proteins with aggresomes (Fig. 8 b). This indicates that targeting of chromatin proteins to the centrosomal proteasome is a fairly common mechanism. The fact that DNMT3a not associated with the centrosome (Fig. 8 a) is consistently targeted to two nuclear proteolysis centers (Fig. 8 b), and not to the centrosomal proteasome, indicates that different chromatin proteins are targeted to different proteolysis centers. In addition, export from the nucleus is not a requisite for chromatin protein degradation, and the site of degradation for each protein is specific. Why a selection of nuclear proteins is targeted to the centrosomal proteasome, instead of the proteolysis centers in the nucleus, is unclear. Given its association with gene silencing (Perche et al., 2000), it is possible that nonnucleosomal macroH2A1 may retain the ability to interact with partners in the nucleus and thus needs to be rapidly exported to prevent macroH2A1 from having detrimental effects on the cell by sequestering chromatin complexes. Alternatively, factors involved in the remodeling of chromatin, resulting in the potential release of macroH2A1, may themselves be targeted to the centrosomal proteasome, taking macroH2A1 with them.
Centrosomal concentrations of macroH2A1 alter in a cell cycle–dependent fashion. As cells proceed through S phase and G2 toward mitosis, the concentration of macroH2A1 at the centrosome increases to a level detectable by immunofluorescence (Fig. 5 a; Table I). The same trend is observed as cells proceed from mitosis through G1 toward S phase (Fig. 5 a; Table I). The accumulation of macroH2A1 at the centrosome in the presence of lactacystin is most prominent in cells as they pass through S phase toward mitosis. This is consistent with the need to target more macroH2A1 for degradation, as it is during this period that the MCB disappears, suggesting that excess quantities of macroH2A1 are required to be removed.
The association of macroH2A1 with the Xi chromatin is most prominent at S phase
A prerequisite for the formation of an MCB is the association of XIST RNA with the Xi (Csankovszki et al., 1999; Beletskii et al., 2001). Whereas XIST RNA coats the Xi through early G1 to late G2, the stable presence of XIST does not immediately direct macroH2A1 to the Xi to form an MCB (Fig. 5 b; Table II). Instead, the formation of an MCB is most common in early and middle S phase (Fig. 6 b). MacroH2A1 is unlikely to be marking chromatin for late replication, as not all sites of late replication overlap with macroH2A1 staining (Fig. 6 a), and the banding of macroH2A at metaphase (Figs. 3 and 4) does not correspond to regions of the Xi known to replicate latest in S phase (Willard, 1977).
The cell cycle–influenced appearance of an MCB suggests that macroH2A1 (and perhaps macroH2A2) may be substituting the H2A position in Xi nucleosomes at and around S phase. Although the nucleosome inner core of histones H3 and H4 is stable at interphase, H2B is more dynamic and can readily be substituted (Kimura and Cook, 2001). H2A and H2B are deposited onto chromatin as a heterodimer (Ridgway and Almouzni, 2000). Therefore it is conceivable that H2A variants, like H2B, can also dynamically exchange the H2A position, conferring alternative states to local chromatin. Preparations of nucleosomes from cells blocked at the beginning of S phase or in mitosis have comparable levels of macroH2A1 (unpublished data), despite the significant decrease in macroH2A at the Xi as the MCB disappears (Fig. 5). Put into a genomic perspective, fluctuations in the local concentrations of macroH2A1 at the Xi visualized as an MCB in females may be masked by the total concentrations of nucleosomal macroH2A1 in a cell. This may indicate that macroH2A1 at the MCB represents only a small fraction of the total concentration of macroH2A1, with the remainder functioning in autosomal chromatin (Fig. 3 c and Fig. 4 a). Indistinguishable concentrations of macroH2A in male and female nucleosome fractions and total cell extracts support this (unpublished data). More speculatively, it is conceivable that macroH2A1 at the MCB is not all nucleosomal, but functioning with other components of the dosage compensation complex at the Xi outside of the nucleosome context. Resolving this issue will require a detailed analysis of nucleosome levels of macroH2A at the Xi by chromatin immunoprecipitation analysis at different stages of the cell cycle.
One possible model to explain the functional significance of the MCB during S phase is that higher local concentrations of macroH2A may be one of many redundant mechanisms to promote the loading of the dosage compensation complex onto the daughter X and mark it as the Xi as it is synthesized.
MacroH2A is enriched at specific bands on the metaphase Xi overlapping a site of histone H3 methylation
Although the MCB is not evident before the onset of mitosis (Fig. 5; Tables I and II), macroH2A1 does remain associated with both the human and mouse Xi during mitosis as distinct bands (Figs. 3 and 4). Intriguingly, these bands appear to mimic the banding seen with Xist RNA on the mouse Xi during mitosis (Duthie et al., 1999), suggesting that macroH2A may be functioning to anchor Xist RNA in cis with the Xi. However, in humans, XIST RNA does not remain associated with the Xi during mitosis (Clemson et al., 1996). Bands enriched for macroH2A may function as reentry sites for XIST RNA and the dosage compensation complex, assisting in the rapid spread along the Xi in a manner analogous to reentry sites of the Drosophila dosage compensation complex (Meller et al., 2000). The band of macroH2A at the site of the XIST locus (Fig. 3 b) is perhaps analogous to reentry of the Drosophila dosage compensation complex at the site of the roX1 and roX2 loci (Kelley et al., 1999). Why macroH2A remains associated specifically with these regions of the chromosome is intriguing. With the exception of the proximity of macroH2A to the satellite repeat DXZ4 at Xq22-24 (Giacalone et al., 1992), there are no obvious shared features, such as gene densities or frequency of repeated elements at the chromatin of the other identified regions.
Most intriguing is the clear overlap of the macroH2A band at Xq22-24 with a band of histone H3 lysine-4 methylation (Fig. 4, a, a′, and d). The band of DimH3K4 is centered at DXZ4 (Fig. 3 e) and marks the distal edge of the macroH2A band. This demonstrates the association of a histone modification, thought primarily to associate with euchromatin and regions of transcriptional activation (Kouzarides, 2002, and references therein), with a macrosatellite repeat (Giacalone et al., 1992). Potentially, DXZ4 may act as a boundary element, delimiting the spread of macroH2A, strengthened by the H3 lysine-4 methylation. Identification of genomic sequences and chromatin modifications at the boundary of each of the macroH2A bands will provide invaluable insight into the functional significance and influence of the histone code used by the Xi.
The MCB observed in interphase and the banding of macroH2A seen at metaphase (Figs. 3 and 4) might provide similar or separate functions. Although disruption of XIST results in the loss of detectable MCB formation (Csankovszki et al., 1999; Beletskii et al., 2001), this may not effect macroH2A at the chromosome bands. Targeting of macroH2A1 and macroH2A2 in human and mouse ES cells, along with carefully directed chromatin immunoprecipitation experiments, will further our understanding of the functional significance of macroH2A in X inactivation.
Materials and methods
Cell culture and chemical treatment
Cell lines used include T-3352, a 46,XX human primary fibroblast strain (provided by Stuart Schwartz, Case Western Reserve University); hTERT-RPE1, a 46,XX telomerase-immortalized cell line derived from a human retinal pigment epithelial cell line RPE-340 (catalog no. C4000–1; CLONTECH Laboratories, Inc.); hTERT-BJ1, a 46,XY telomerase-immortalized cell line derived from a human primary foreskin fibroblast cell line (catalog no. C4001–1; CLONTECH Laboratories, Inc.); hTERT-HME1, a 46,XX telomerase-immortalized cell line derived from a human mammary epithelial cell line (catalog no. C4002–1; CLONTECH Laboratories, Inc.); and HEK-293, a female fetal kidney tumor cell line. B144 is a female mouse primary fibroblast cell line (provided by Laura Carrel, Case Western Reserve University). Cells were maintained as described previously (Chadwick and Willard, 2001a).
To establish stably transfected cell lines, hTERT-RPE1 or HEK-293 cells were transfected with 5 μg of carboxy-terminal Myc-tagged macroH2A1 (macroH2A1-CT–Myc), amino-terminal Xpress-tagged macroH2A1 (macroH2A1-NT–Xpr), or a carboxy-terminal Myc-tagged macroH2A2 (macroH2A2-CT–Myc) expression using Superfect according to the manufacturer's recommendations (QIAGEN) and grown for 48 h before selection with either 1.4 mg/ml (hTERT-RPE1) or 0.4 mg/ml (HEK-293) neomycin (GIBCO BRL).
Cell synchronization and BrdU (Sigma-Aldrich) incorporation were performed as previously described (Spector et al., 1998).
Inhibition of the 20S proteasome was achieved by washing cell lines twice with PBS and applying complete media containing 10 μM lactacystin (Calbiochem) (Fenteany et al., 1995) for the time periods indicated at 37°C in a 5% CO2 atmosphere.
Antibodies
Rabbit pAbs against the nonhistone tail region of macroH2A1 have been previously described (Chadwick and Willard, 2001a). Two glutathione-S-transferase (GST)–macroH2A2 fusion proteins covering amino acids 124–180 (aa 124–180) and amino acids 124–372 (aa 124–372) were used to raise independent rabbit pAbs to macroH2A2 (Covance Inc.). MacroH2A1 and macroH2A2 (124–180) pAbs were affinity purified. MacroH2A1 and both macroH2A2 antisera were blocked against each other using standard techniques (Harlow and Lane, 1989) to generate antisera specific to each form of the protein. Specificity was confirmed by Western analysis (unpublished data). Monoclonal anti–γ-tubulin (clone gtu-88) was obtained from Sigma-Aldrich. Mouse mAb anti-BrdU (clone BMC 9318) was obtained from Roche Diagnostics Corp. Mouse mAb antiubiquitin (clone 6C1) was obtained from Oncogene Research Products. Polyclonal antisera raised to histone deacetylases 1 (HDAC1, sc-7872) and 3 (HDAC3, sc-8138), DNA–methyltransferases 1 (Dnmt1, sc-10222), 2 (Dnmt2, sc-10227), 3a (Dnmt3a, sc-10231), and 3b (Dnmt3b, sc-10235), histone acetyltransferase p300/CBP-associated factor (PCAF, sc-8999), and methyl-DNA binding proteins 1 (MBD1, sc-9395) and 3 (MBD3, sc-9402) were obtained from Santa Cruz Biotechnology Inc. Antihistone H3 dimethyl lysine-4 (DimH3K4) was obtained from Upstate Biotechnology. Anti–Mi-2 was a gift from Paul Wade (Emory University, Atlanta, GA). Mouse mAbs anti-Myc and anti-Xpress were obtained from Invitrogen.
Immunofluorescence and FISH
Immunofluorescence and FISH was performed essentially as previously described (Chadwick and Willard, 2001b). Slides were denatured at 85°C before FISH, as opposed to 72°C, to overcome extensive sample fixation. A digoxygenin-labeled DXZ4 probe was obtained from Oncor Inc. Human X chromosome TRITC-labeled probes were generated using a nick translation kit (Vysis Inc). A mouse X chromosome–specific probe, DXwas70, was used to detect the mouse X chromosome. Human cosmid clones ICRFc100H0130 (XIC) and ICRFc100G11100 (CIC8) were obtained from the Imperial Cancer Research Fund Reference Library Database. Immuno-RNA FISH was achieved by first immunostaining followed by RNA-FISH essentially as previously described (Clemson et al., 1996).
Metaphase chromosomes were prepared as described previously (Chadwick and Willard, 2001b). Single-copy probes were blocked for repetitive sequences for 1 h at 37°C in the presence of 0.3 mg/ml human Cot-1 DNA before hybridization. Immunostaining of BrdU-pulsed cells was achieved by first fixing and staining for macroH2A as described previously (Chadwick and Willard, 2001a), before proceeding as described by Spector et al. (1998).
Images were collected with a Vysis imaging system equipped with a cooled CCD camera (Photometrics) controlled via the Quips M-FISHTM software (Vysis Inc).
Relative cell cycle stages were determined by cell cycle arrest and release in conjunction with BrdU pulse labeling. Presence or absence of BrdU incorporation was monitored relative to the number of hours after release and translated directly to subsequent surveys.
Preparation of centrosomes and nucleosomes
Centrosomes were isolated essentially as previously described (Bornens and Moudjou, 1999). Nucleosome oligomers were isolated as described previously (Chadwick and Willard, 2001b). Protein samples were separated by SDS-PAGE and transferred to a polyvinylidene diflouride membrane using standard techniques (Harlow and Lane, 1989).
Acknowledgments
The authors would like to thank Dr. Stuart Schwartz, Dr. Laura Carrel, and Jim Amos-Landgraf (Case Western Reserve University) for cell lines and constructs and Dr. Paul Wade for the anti-Mi2 antisera. The authors thank Lisa Chadwick and Cory Valley for suggestions and constructive discussion.
This work was supported by research grant GM45441 to H.F. Willard from the National Institutes of Health. B.P. Chadwick is supported by a postdoctoral fellowship from the Rett Syndrome Research Foundation.
Footnotes
Abbreviations used in this paper: DNMT, DNA-methyltransferase; ES, embryonic stem cells; MCB, macrochromatic body; Xa, active X chromosome; Xi, inactive X chromosome; XIST, Xi-specific transcript.
References
- Avner, P., and E. Heard. 2001. X-chromosome inactivation: counting, choice and initiation. Nat. Rev. Genet. 2:59–67. [DOI] [PubMed] [Google Scholar]
- Barthelmes, H.U., P. Grue, S. Feineis, T. Straub, and F. Boege. 2000. Active DNA topoisomerase IIα is a component of the salt-stable centrosome core. J. Biol. Chem. 275:38823–38830. [DOI] [PubMed] [Google Scholar]
- Beletskii, A., Y.-K. Hong, J. Pehrson, M. Egholm, and W.M. Strauss. 2001. PNA interference mapping demonstrates functional domains in the noncoding RNA Xist. Proc. Natl. Acad. Sci. USA. 98:9215–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyaev, N., A.M. Keohane, and B.M. Turner. 1996. Differential underacetylation of histones H2A, H3 and H4 on the inactive X chromosome in human female cells. Hum. Genet. 97:573–578. [DOI] [PubMed] [Google Scholar]
- Bochtler, M., L. Ditzel, M. Groll, C. Hartmann, and R. Huber. 1999. The proteasome. Annu. Rev. Biophys. Biomol. Struct. 28:295–317. [DOI] [PubMed] [Google Scholar]
- Boggs, B.A., B. Connors, R.E. Sobel, A.C. Chinault, and C.D. Allis. 1996. Reduced levels of histone H3 acetylation on the inactive X chromosome in human females. Chromosoma. 105:303–309. [DOI] [PubMed] [Google Scholar]
- Boggs, B.A., P. Cheung, E. Heard, D.L. Spector, A.C. Chinault, and C.D. Allis. 2002. Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosomes. Nat. Genet. 30:73–76. [DOI] [PubMed] [Google Scholar]
- Bornens, M., and M. Moudjou. 1999. Studying the composition and function of centrosomes in vertebrates. Mitosis and Meiosis. Vol. 61. C.L. Rieder, editor. Academic Press, Inc., Orlando, FL. 14–35. [DOI] [PubMed]
- Brockdorff, N., A. Ashworth, G.F. Kay, V.M. McCabe, D.P. Norris, P.J. Cooper, S. Swift, and S. Rastan. 1992. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 71:515–526. [DOI] [PubMed] [Google Scholar]
- Brown, C.J., and H.F. Willard. 1994. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature. 368:154–156. [DOI] [PubMed] [Google Scholar]
- Brown, C.J., A. Ballabio, J.L. Rupert, R.G. Lafreniere, M. Grompe, R. Tonlorenzi, and H.F. Willard. 1991. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 349:38–44. [DOI] [PubMed] [Google Scholar]
- Chadwick, B.P., and H.F. Willard. 2001. a. Histone H2A variants and the inactive X chromosome: identification of a second macroH2A variant. Hum. Mol. Genet. 10:1101–1113. [DOI] [PubMed] [Google Scholar]
- Chadwick, B.P., and H.F. Willard. 2001. b. A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome. J. Cell Biol. 152:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson, C.M., J.A. McNeil, H.F. Willard, and J.B. Lawrence. 1996. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol. 132:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi, C., and J.R. Pehrson. 1998. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature. 393:599–601. [DOI] [PubMed] [Google Scholar]
- Costanzi, C., and J.R. Pehrson. 2001. MacroH2A2, a new member of the MacroH2A core histone family. J. Biol. Chem. 276:21776–21784. [DOI] [PubMed] [Google Scholar]
- Costanzi, C., P. Stein, D.M. Worrad, R.M. Schultz, and J.R. Pehrson. 2000. Histone macroH2A1 is concentrated in the inactive X chromosome of female preimplantation mouse embryos. Development. 127:2283–2289. [DOI] [PubMed] [Google Scholar]
- Csankovszki, G., B. Panning, B. Bates, J.R. Pehrson, and R. Jaenisch. 1999. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 22:323–324. [DOI] [PubMed] [Google Scholar]
- DeBry, R.W., and M.F. Seldin. 1996. Human/mouse homology relationships. Genomics. 33:337–351. [DOI] [PubMed] [Google Scholar]
- Duthie, S.M., T.B. Nesterova, E.J. Formstone, A.M. Keohane, B.M. Turner, S.M. Zakian, and N. Brockdorff. 1999. Xist RNA exhibits a banded localization on the inactive X chromosome and is excluded from autosomal material in cis. Hum. Mol. Genet. 8:195–204. [DOI] [PubMed] [Google Scholar]
- Fabunmi, R.P., W.C. Wigley, P.J. Thomas, and G.N. DeMartino. 2000. Activity and regulation of the centrosome-associated proteasome. J. Biol. Chem. 275:409–413. [DOI] [PubMed] [Google Scholar]
- Fenteany, G., R.F. Standaert, W.S. Lane, S. Choi, E.J. Corey, and S.L. Schreiber. 1995. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 268:726–731. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata, R., Z. Bebok, E.J. Sorscher, and E.S. Sztul. 1999. Characterization and dynamics of aggresome formation by a cytosolic GFP chimera. J. Cell Biol. 146:1239–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartler, S.M., and M.A. Goldman. 1994. Reactivation of inactive X-linked genes. Dev. Genet. 15:504–514. [DOI] [PubMed] [Google Scholar]
- Giacalone, J., J. Friedes, and U. Francke. 1992. A novel GC-rich human macrosatellite VNTR in Xq24 is differentially methylated on active and inactive X chromosomes. Nat. Genet. 1:137–143. [DOI] [PubMed] [Google Scholar]
- Gilbert, C.W., S. Muldal, L.G. Lajthal, and J. Rowley. 1962. Time-sequence of human chromosome duplication. Nature. 195:869–873. [DOI] [PubMed] [Google Scholar]
- Gilbert, S.L., and P.A. Sharp. 1999. Promoter-specific hypoacetylation of X-inactivated genes. Proc. Natl. Acad. Sci. USA. 96:13825–13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, A., P.M. Reback, G. Pratt, and M. Rechsteiner. 1990. Ubiquitin-mediated degradation of histone H3 does not require the substrate-binding ubiquitin protein ligase, E3, or attachment of polyubiquitin chains. J. Biol. Chem. 265:21664–21669. [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1989. Antibodies: A Laboratory Manual. Cold Spring Harbour Laboratory Press, Cold Spring Harbor, NY. 726 pp.
- Hsu, L.C., and R.L. White. 1998. BRCA1 is associated with the centrosome during mitosis. Proc. Natl. Acad. Sci. USA. 95:12983–12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh, K.D., and J.T. Lee. 2001. Imprinted X inactivation in eutherians: a model of gametic execution and zygotic relaxation. Curr. Opin. Cell Biol. 13:690–697. [DOI] [PubMed] [Google Scholar]
- Jeppesen, P., and B.M. Turner. 1993. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell. 74:281–289. [DOI] [PubMed] [Google Scholar]
- Johnston, J.A., C.L. Ward, and R.R. Kopito. 1998. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai, M., M. Uchida, S. Hanai, N. Uematsu, K. Uchida, and M. Miwa. 2000. Poly(ADP-ribose) polymerase localizes to the centrosomes and chromosomes. Biochem. Biophys. Res. Commun. 278:385–389. [DOI] [PubMed] [Google Scholar]
- Kelley, R.L., V.H. Meller, P.R. Gordadze, G. Roman, R.L. Davis, and M.I. Kuroda. 1999. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell. 98:513–522. [DOI] [PubMed] [Google Scholar]
- Kimura, H., and P.R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito, R.R. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10:524–530. [DOI] [PubMed] [Google Scholar]
- Kopito, R.R., and R. Sitia. 2000. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 1:225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides, T. 2002. Histone methylation and transcriptional control. Curr. Opin. Genet. Dev. 12:198–209. [DOI] [PubMed] [Google Scholar]
- Meller, V.H., P.R. Gordadze, Y. Park, X. Chu, C. Stuckenholz, R.L. Kelley, and M.I. Kuroda. 2000. Ordered assembly of roX RNAs into MSL complexes on the dosage-compensated X chromosome in Drosophila. Curr. Biol. 10:136–143. [DOI] [PubMed] [Google Scholar]
- Mermoud, J.E., C. Costanzi, J.R. Pehrson, and N. Brockdorff. 1999. Histone macroH2A1.2 relocates to the inactive X chromosome after initiation and propagation of X-inactivation. J. Cell Biol. 147:1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermoud, J.E., A.M. Tassin, J.R. Pehrson, and N. Brockdorff. 2001. Centrosomal association of histone macroH2A1.2 in embryonic stem cells and somatic cells. Exp. Cell Res. 268:245–251. [DOI] [PubMed] [Google Scholar]
- Mohandas, T., R.S. Sparkes, and L.J. Shapiro. 1981. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science. 211:393–396. [DOI] [PubMed] [Google Scholar]
- Morishma, A., M.M. Grumbach, and J.H. Taylor. 1962. Asynchronous duplication of human chromosomes and the origin of sex chromatin. Proc. Natl. Acad. Sci. USA. 48:756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson, J.R., and V.A. Fried. 1992. MacroH2A, a core histone containing a large nonhistone region. Science. 257:1398–1400. [DOI] [PubMed] [Google Scholar]
- Perche, P., C. Vourc'h, L. Konecny, C. Souchier, M. Robert-Nicoud, S. Dimitrov, and S. Khochbin. 2000. Higher concentrations of histone macroH2A in the barr body are correlated with higher nucleosome density. Curr. Biol. 10:1531–1534. [DOI] [PubMed] [Google Scholar]
- Peters, A.H., J.E. Mermoud, D. O'Carroll, M. Pagani, D. Schweizer, N. Brockdorff, and T. Jenuwein. 2002. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 30:77–80. [DOI] [PubMed] [Google Scholar]
- Pfeifer, G.P., R.L. Tanguay, S.D. Steigerwald, and A.D. Riggs. 1990. In vivo footprint and methylation analysis by PCR-aided genomic sequencing: comparison of active and inactive X chromosomal DNA at the CpG island and promoter of human PGK-1. Genes Dev. 4:1277–1287. [DOI] [PubMed] [Google Scholar]
- Pickart, C.M. 2001. Ubiquitin enters the new millennium. Mol. Cell. 8:499–504. [DOI] [PubMed] [Google Scholar]
- Rasmussen, T.P., M.A. Mastrangelo, A. Eden, J.R. Pehrson, and R. Jaenisch. 2000. Dynamic relocalization of histone MacroH2A1 from centrosomes to inactive X chromosomes during X inactivation. J. Cell Biol. 150:1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway, P., and G. Almouzni. 2000. CAF-1 and the inheritance of chromatin states: at the crossroads of DNA replication and repair. J. Cell Sci. 113:2647–2658. [DOI] [PubMed] [Google Scholar]
- Singer-Sam, J., L. Goldstein, A. Dai, S.M. Gartler, and A.D. Riggs. 1992. A potentially critical Hpa II site of the X chromosome-linked PGK1 gene is unmethylated prior to the onset of meiosis of human oogenic cells. Proc. Natl. Acad. Sci. USA. 89:1413–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, D.L., R.D. Goldman, and L.A. Leinwand. 1998. Cells: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 2136 pp.
- Ullrich, O., T. Reinheckel, N. Sitte, R. Hass, T. Grune, and K.J. Davies. 1999. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc. Natl. Acad. Sci. USA. 96:6223–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley, W.C., R.P. Fabunmi, M.G. Lee, C.R. Marino, S. Muallem, G.N. DeMartino, and P.J. Thomas. 1999. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 145:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard, H.F. 1977. Tissue-specific heterogeneity in DNA replication patterns of human X chromosomes. Chromosoma. 61:61–73. [DOI] [PubMed] [Google Scholar]
- Willard, H.F. 2000. Sex chromosomes and X chromosome inactivation. The Molecular and Metabolic Basis of Inherited Disease. C.R. Scriver, A.L. Beaudet, W.S. Sly, and D. Valle, editors. McGraw-Hill Inc., New York. 1191–1221.
- Wojcik, C., D. Schroeter, S. Wilk, J. Lamprecht, and N. Paweletz. 1996. Ubiquitin-mediated proteolysis centers in HeLa cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur. J. Cell Biol. 71:311–318. [PubMed] [Google Scholar]
- Xue, Y., J.C. Canman, C.S. Lee, Z. Nie, D. Yang, G.T. Moreno, M.K. Young, E.D. Salmon, and W. Wang. 2000. The human SWI/SNF-B chromatin-remodeling complex is related to yeast rsc and localizes at kinetochores of mitotic chromosomes. Proc. Natl. Acad. Sci. USA. 97:13015–13020. [DOI] [PMC free article] [PubMed] [Google Scholar]