

Abstract
Human immunodeficiency virus (HIV)-1 infection depends on multiple lateral interactions between the viral envelope and host cell receptors. Previous studies have suggested that these interactions are possible because HIV-1 receptors CD4, CXCR4, and CCR5 partition in cholesterol-enriched membrane raft domains. We generated CD4 partitioning mutants by substituting or deleting CD4 transmembrane and cytoplasmic domains and the CD4 ectodomain was unaltered. We report that all CD4 mutants that retain raft partitioning mediate HIV-1 entry and CD4-induced Lck activation independently of their transmembrane and cytoplasmic domains. Conversely, CD4 ectodomain targeting to a nonraft membrane fraction results in a CD4 receptor with severely diminished capacity to mediate Lck activation or HIV-1 entry, although this mutant binds gp120 as well as CD4wt. In addition, the nonraft CD4 mutant inhibits HIV-1 X4 and R5 entry in a CD4+ cell line. These results not only indicate that HIV-1 exploits host membrane raft domains as cell entry sites, but also suggest new strategies for preventing HIV-1 infection.
Keywords: CD4, lipid rafts, HIV-1, Lck, infection inhibition
Introduction
The plasma membrane is a specialized structure that channels and integrates the information that flows continuously between a cell and its environment. The cell plasma membrane also constitutes the initial barrier against infection by intracellular pathogens. Contrary to the view of the plasma membrane as a homogenous phospholipid backbone loaded with proteins, it is now established that it is a highly sophisticated structure assembled of distinct lipid domains that functionally organize the proteins embedded in the bilayer. One of these microdomain types, termed rafts, is formed by glycosphingolipids (GSL),* sphingomyelin, and cholesterol packaged in the external leaflet of the plasma membrane (1). Due to the high melting point of lipids, membrane rafts are in a rigid, ordered state (2). Rafts nonetheless retain substantial lateral and rotational mobility. They are viewed as moving platforms of ordered membrane-bearing proteins with a specific preference for this lipid environment, such as glycosylphosphatidylinositol (GPI)-anchored and double-acylated cytoplasmic proteins.
Rafts may function as devices to control membrane protein–protein interactions and several mechanisms can be envisioned. First, it is reported that interactions occur preferentially between proteins sharing identical lipid environments (3). Accordingly, the interaction of raft-associated proteins with membrane nonraft proteins is very restricted (4). Second, proteins initially segregated in distinct elementary raft units might be brought together by raft coalescence, which could generate supramolecular complexes of raft-associated proteins in a single clustering event. For instance, lateral cross-linking of GPI-anchored raft proteins triggers raft-associated tyrosine kinase activation (2) even though these two types of proteins cannot directly interact. A third possibility is that a protein that initially partitions in a nonraft membrane region might be recruited into a preexisting raft, enabling raft-dependent protein–protein interactions. Therefore, raft domains may organize protein interactions in time and space by regulating raft coalescence and/or controlling the recruitment of nonraft proteins to these domains. Nevertheless, this scenario might be even more complex as membrane protein segregation not only between raft and nonraft domains, but also between distinct raft subtypes, influences lateral organization of the plasma membrane (5).
The temporal and spatial control of protein interactions at the plasma membrane regulates cell signaling integration and pathogen infection of cells (6). CD4 takes part in these two processes, as it participates in the integration of TCR signaling by recruiting p56lck and is also the primary HIV-1 cell surface receptor (7, 8). To perform these functions, CD4 must interact laterally with the TCR for T cell activation and with members of the chemokine receptor family, such as CXCR4 or CCR5, to mediate HIV-1 entry (9–11). Chemokine receptors act as necessary coreceptors for HIV-1 infection, as gp120 binding to CD4 is insufficient to promote virus entry (12).
Although the role of rafts in CD4-mediated signaling is well established (8), the importance of CD4 association with rafts for HIV-1 entry remains a subject of debate (13). HIV-1 entry is impeded after raft disruption by membrane cholesterol sequestration (14–16). Favoring a role for rafts in HIV-1 infection, the inhibition of GSL synthesis blocks HIV-1 entry (17, 18). Cholesterol depletion experiments have suggested that large CD4–gp120 coreceptor complexes are the consequence of raft clustering initially induced by HIV-1 binding to CD4 (14). Even assuming that CD4 and CCR5 might be constitutively associated at the cell surface (19), HIV–host membrane fusion is a cooperative process requiring multiple CD4–gp120–CCR5 complexes (20), which may be formed by lateral raft rearrangements. Viral entry would thus be enabled if CD4 and chemokine receptor partition into raft domains. Accordingly, it has been reported that the HIV-1 coreceptors CCR5 and CXCR4 partition and signal in rafts after ligand- or gp120-induced clustering (5, 14, 21–23). A recent report suggests, however, that raft domains may not have a determinant role in HIV-1 infection (24). According to this latter interpretation, CD4 partitioning in nonraft membranes would not affect HIV-1 entry. Conversely, if HIV-1 uses host raft membranes as entry sites, CD4 partitioning into nonraft membranes would impede viral infection.
Here, we analyzed these possibilities by generating CD4 mutants in the cytoplasmic and transmembrane domains. These mutants shared the extracellular domain of the WT CD4 form. We generated a CD4 mutant that partitions in the nonraft plasma membrane fraction by replacing CD4 transmembrane and cytoplasmic domains with the transmembrane and a very short cytoplasmic tail from the low density lipoprotein (LDL)-R. This CD4 mutant (CD4–LDL) neither activates Lck after CD4 cross-linking nor mediates HIV-1 entry, which are two independent CD4 functions. The failure of the CD4–LDL mutant to mediate these processes is the sole consequence of CD4 extracellular domain localization in a nonraft membrane fraction, because a raft-associated CD4–GPI mutant that lacks transmembrane and cytoplasmic CD4 domains activates Lck and mediates HIV-1 entry. Moreover, the nonraft CD4 mutant prevents X4 and R5 HIV-1 strain infection of CD4+ T cells. These results highlight the fundamental role of membrane rafts in regulating the protein–protein interactions required for HIV-1 entry and suggest new strategies for preventing HIV-1 infection.
Materials and Methods
Generation of CD4 Mutants.
Selected transmembrane amino acids of WT CD4 were replaced with alanine by site-directed mutagenesis (Stratagene). The synthetic oligonucleotide primer pairs 5′-GCAGCCAATGGCCGCGGCTGCGGCGGGGGGCGTCGCCG-3′ and 5′-CGGCGACGCCCCCCGCCGCAGCCGCGGCCATTGGCTGC-3′ (CD4-4A374); 5′-CCCTGATTGTGGCGGCGGCCGCCGCCGGCCTCCTG-3′ and 5′-CAGGAGGCCGGCGGCGGCCGCCGCCACAATCAGGG-3′ (CD4-4A377); 5′-GGGGGCGTCGCCGGCGCCGCGGCTTTCATTGGGCTAGGC-3′ and 5′-GCCTAGCCCAATGAAAGCCGCGGCGCCGGCGACGCCCCC-3′ (CD4-3A383); 5′-CGCCGGCCTCCTGCTTGCCGCTGGGCTAGGCATC-3′ and 5′-GATGCCTAGCCCAGCGGCAAGCAGGAGGCCGGCG-3′ (CD4-2A386); 5′-CATTGGGCTAGGCGCCGCCGCCTGTGTCAGGTGCCGGC-3′ and 5′-GCCGGCACCTGACACAGGCGGCGGCGCCTAGCCCAATG-3′ (CD4-3A391); 5′-GGGCTAGGCATCGCCGCCGCTGTCAGGTGCCGGCACCG-3′ and 5′-CGGTGCCGGCACCTGACAGCGGCGGCGATGCCTAGCCC-3′ (CD4-3A392); 5′-TCTGTGTCAGGGCCCGGCACCGAAGGCG-3′ and 5′-CGCCTTCGGTGCCGGGCCCTGACACAGA-3′ (CD4-C397A); and 5′-GGCTAGGCATCGCCGCCGCTGTCAGGGCCCGGCACCGAAGGCGC-3′ and 5′-GCGCCTTCGGTGCCGGGCCCTGACAGCGGCGGCGATGCCTAGCC-3′ (CD4-C394/7A) were used to extend CD4 cDNA cloned in pCDNA-3.1 with PfuTurbo DNA polymerase. The product was DpnI-treated to specifically digest the parental template. The resulting plasmids were transformed in Epicurian Coli XL1-Blue supercompetent cells and clones (Stratagene) containing the mutated CD4 cDNA selected by sequencing.
To construct CD4 chimeras, the luminal CD4 domain was cloned in HindIII/KpnI-digested pcDNA3.1A/myc/his (Invitrogen) by PCR using 5′-GCCAAGCTTATGAACCGGGG AGTC-3′ and 5′-AGAGGTACCCATTGGCTGCACCGG-3′ to produce pCD4ext. A GPI consensus sequence was rescued from the GFP-GPI using 5′-AAAGGTACCTATGATCCGC GCCCA-3′ and 5′-CCCGCGAATTCTTAAAGAACATT-3′ and was cloned in KpnI/EcoRI-digested pCD4ext. To construct CD4–LDL, transmembrane and juxtamembrane LDL-R were rescued from LGFP-GT46 (both plasmids were provided by P. Keller, Max Planck Institute of Cell Biology and Genetics, Dresden, Germany; reference 25) using 5′-GCAACGGTACCGCTCTGTCCATTG-3′ and 5′-CTACTCGAGGTTCTTAAGCCGCCA-3′ and were cloned in KpnI/EcoRI-opened pCD4ext. For CD4–LDL–CD4, the LDL-R transmembrane domain was amplified using 5′-GAGGAATTCCCATAGAAGGAAGAC-3′ as reverse oligonucleotide and the fragment was cloned in pCD4ext. The CD4 intracellular domain, maintaining the double acylation sequence and the stop codon, was PCR amplified with 5′-GAGGAATTCTGTGTCAGGTGCCGG-3′ and 5′-GACCTCGAGTCAAATGGGGCTACA-3′, and then cloned in EcoRI/XhoI-digested pCD4ext LDL-R transmembrane vector.
HEK-293 cells were transfected using a standard calcium phosphate method. Transfection efficiency was 74–85%, as determined with an enhanced green fluorescent protein vector. CD4 levels were maximal 24–96 h after transfection as analyzed by FACS® (EPICS; Coulter) using an FITC–anti-CD4 (Immunotech) or an FITC-labeled control antibody. The percentage of CD4+ cells observed after subtracting the control was multiplied by the average fluorescence intensity to calculate surface CD4 expression for each mutant (26).
Flotation Experiments.
HEK-293 cells expressing the CD4 mutants were lysed at 4°C in TXNE buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% Triton X-100) plus protease inhibitors. Detergent-resistant membranes (DRMs) were isolated by ultracentrifugation (170,000 g) for 4 h at 4°C in a 30–35% OptiPrep™ gradient (Nycomed; reference 21). Normalized protein amounts were analyzed by Western blot with anti-CD4 (Leu3A; Becton Dickinson), anti–transferrin receptor (TfR; Zymed Laboratories), and anti–VIP-21 (Santa Cruz Biotechnology, Inc.) antibodies.
Immunofluorescence and gp120-induced Patching.
For colocalization of CD4 with GM1, HEK-293 transfected with CD4 mutants were incubated for 30 min at 12°C with anti-CD4 (Leu3A) followed by a Cy3-anti–mouse antibody (Jackson ImmunoResearch Laboratories). Finally, FITC-cholera toxin was added for 5 min at 12°C.
For gp120 copatching, HEK-293 cells expressing CD4 mutants were incubated with recombinant gp120 (T cell line–adapted X4 virus, isolate IIIB; Intracel) in PBS/0.2% BSA. Cells were incubated for 30 min at 12°C with rabbit polyclonal anti-gp120, FITC–anti-CD4 (OKT4; Ortho Diagnostics), and biotinylated anti-CXCR4 (FAB172; R&D Systems). Finally, anti–rabbit Cy3 antibody and streptavidin-Cy5 (Jackson ImmunoResearch Laboratories) were added.
In all cases, cells were fixed with 3.7% paraformaldehyde in PBS for 5 min on ice, mounted in Vectashield medium (Vector Laboratories), and visualized by confocal laser microscopy (Leica).
CD4–Lck Association and In Vitro Kinase (IVK) Assays.
HEK-293 cells expressing CD4 and Lck were serum starved for 4 h, lysed in 50 mM Hepes, pH 8.0, 150 mM NaCl, 1% Triton X-100, plus phosphatases and proteases inhibitors, and then precipitated with equal amounts of anti-CD4 (HP2/6; provided by F. Sánchez-Madrid, Hospital de la Princesa, Madrid, Spain; reference 27) and agarose-coupled anti–mouse antibody (Sigma-Aldrich). IVK assays were performed with the pellets using enolase (Sigma-Aldrich) as a substrate (28).
CD4-induced Lck Activation.
Cells were cotransfected with CD4 mutants, Lck and CD8ζ (provided by E.L. Reinherz, Dana-Farber Cancer Institute, Boston, MA; reference 28). Cells were serum starved and incubated for 20 min at 4°C with anti-CD4 and anti–mouse antibodies (27). Plates were then incubated at 37°C for the times indicated and lysed as described above. Equal amounts (200 μg) were precipitated with anti-CD8 antibody (B9.4.2; reference 29) and SDS-PAGE–resolved pellets blotted sequentially with peroxidase-coupled antiphosphotyrosine, anti-Lck COOH-terminal (Upstate Biotechnology), and anti-CD3ζ (386) antibodies. IVK assays were performed by the immunoprecipitation of cell lysates with anti-Lck COOH-terminal antibody plus protein A–Sepharose (Amersham Biosciences) as described above.
gp120 Binding Analysis.
Mock-transfected or CD4 construct–transfected HEK-293 cells were incubated sequentially with 10 μg/ml gp120IIIB and an anti-gp120 antibody, and then with FITC–anti-CD4 and phycoerythrin-goat anti–rabbit (Caltag) antibodies (all for 30 min at 4°C) to monitor CD4 expression and gp120 binding, respectively. gp120 binding was analyzed in CD4+ gated cells. Control samples were processed similarly, omitting gp120.
HIVenv-induced Cell–Cell Fusion.
HEK-293 cells were cotransfected with the CD4 constructs, the pSCluc plasmid harboring the firefly luciferase gene under the control of the vaccinia virus 7.5 promoter (provided by D. Rodríguez, Centro Nacional de Biotecnología, Madrid, Spain), and the promoterless renilla luciferase plasmid (30). CD4 expression was analyzed 24 h after transfection by flow cytometry. HIV-1env IIIB was introduced into effector HEK-293 cells by infection for 1 h at 37°C with recombinant vaccinia virus. 12 h after infection, 105 effector cells cultured in 100 μg/ml rifampicin were mixed with HEK-293-CD4–expressing cells for 6 h at 37°C, and cell–cell fusion was analyzed by luciferase activity measurement in cell lysates (25 mM Tris-phosphate, pH 7.8, 1% Triton X-100, 1 mM EDTA, 1 mM DTT, 8 mM MgCl2, 15% glycerol). Luciferase activity was calculated as the quotient between firefly and renilla activity values.
Generation of Recombinant Replication-defective HIV Pseudotypes.
For single-round infections, pNL4-3.Luc.R-E- (provided by N. Landau, AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD) was pseudotyped with HIV-1NL4-3 env, HIV-1Ada, and vesicular stomatitis virus (VSV)-G env as previously described (14). HEK-293-CCR5 or MT-2-CCR5 cells (provided by J. Alcamí, Instituto Salud Carlos III, Madrid, Spain) expressing selected CD4 mutants were transduced with viral supernatants (1 and 0.1 multiplicity of infection) for 2 h at 37°C, and infectivity was determined after 24 h.
Biotinylation of Cells.
Mock, CD4wt, or CD4–LDL cells were biotinylated using EZ-Link Sulfo-NHS-Biotin (Pierce Chemical Co.) according to the manufacturer's instructions. Cells were lysed with RIPA (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% deoxycholate) and equal amounts (100 μg) of lysates were precipitated with agarose-avidin for 1 h at 4°C. Pellets were washed and resolved in SDS-PAGE. Western blots were probed sequentially with anti-6xHis (Sigma-Aldrich), anti-CD4 (Leu3A), and peroxidase-streptavidin (Sigma-Aldrich).
HIV-1 Infection of MT-2-CCR5 Cells.
Mock-, CD4wt-, or CD4–LDL-transfected MT-2-CCR5 cells were incubated with NL4-3 or BaL viral stocks (1 or 10 ng p24 antigen/106 cells) for 2 h at 37°C. 0.5 × 106/ml cells were cultured in complete RPMI medium. Cell-free supernatants were collected daily and tested for p24 antigen (Coulter).
Results
Generation of CD4 Mutants with Differential Raft Partitioning.
Double acylation and GPI modification are major signals for protein partitioning in rafts by anchoring proteins to the inner or outer leaflet of the membrane raft, respectively. Nonetheless, integral membrane proteins have no clear consensus signal that indicates preferential raft association. The best studied raft-associated transmembrane protein is the influenza hemagglutinin, whose raft targeting is determined by three acylation acceptor cysteines and specific amino acids in its transmembrane domain (31, 32). CD4 has a 26–amino acid transmembrane region with two putative palmitoylation acceptor cysteines in the juxtamembrane domain (33). We generated a panel of CD4 chimeras and mutants that affect both transmembrane and cytoplasmic domains (Fig. 1 A). The CD4 extracellular domain was fused to the LDL-R transmembrane and juxtamembrane region (CD4–LDL). As a control for this construct, a CD4 mutant was generated by replacing the CD4 transmembrane domain with that of the LDL-R (CD4–LDL–CD4). This mutant retains the palmitoylated cysteines. The CD4 ectodomain was also fused to a GPI consensus sequence (CD4–GPI) to target CD4 luminal domain to rafts. Finally, we generated CD4 mutants, including three in which palmitoylated Cys394 and/or Cys397 are eliminated by alanine scanning of the transmembrane and juxtamembrane CD4 domains.
Figure 1.

Partitioning of CD4 mutants into distinct membrane domains. (A) The scheme shows the amino acid sequence of the CD4 mutants generated. Mutations or foreign sequences added to the CD4 extracellular domain are indicated in bold. (B) HEK-293 cells expressing CD4 mutants were fractionated in flotation gradients and CD4 partitioning was analyzed by Western blot. Fraction 1 represents the top and fraction 5 represents the bottom of the gradient. Filters were hybridized with anti-TfR and anti-VIP21 (caveolin-1) as controls for nonraft- and raft-associated proteins, respectively. (C–G) Confocal microscopy of CD4 mutant–expressing cells stained with cholera toxin β subunit (green) and anti-CD4 antibody (red). Yellow staining indicates colocalization of the molecules. The two-color overlay shows the representative cells for (C) CD4wt, (D) CD4–GPI, (E) CD4–LDL, (F) CD4–LDL–CD4, and (G) CD4–C394/397A (n = 50/mutant). Bar, 5 μm.
We analyzed raft partitioning of the CD4 mutants by isolating a DRM fraction enriched in raft-associated proteins (6). HEK-293 cells expressing the CD4 mutants were extracted with Triton X-100 and the DRM fraction was isolated in density gradients. A large proportion of CD4wt, CD4–GPI, and CD4–LDL–CD4 proteins copurify with caveolin in the DRM fraction, whereas CD4–LDL copurifies with the TfR in the nonraft compartment (Fig. 1 B). Because CD4–LDL and CD4–LDL–CD4 have identical transmembrane domains, the results suggest that the transmembrane sequence does not contain the main determinants for CD4 partitioning in rafts. Supporting this idea, all CD4 transmembrane mutants showed DRM partitioning comparable to that of CD4wt (unpublished data). Single CD4–3A392, CD4–C397A (unpublished data), or double CD4–C394/397A (Fig. 1 B) palmitoylation mutants also partition in the DRM fraction, suggesting that CD4 acylation is not a major determinant for CD4 association to DRM.
DRM analysis proved useful in identifying parameters affecting raft partitioning of proteins, although weak raft associations might be lost after detergent extraction (6). These misinterpretations can be avoided in copatching experiments, as antibody-induced lateral clustering stabilizes protein–lipid interactions. We analyzed raft partitioning of the CD4 constructs in live HEK-293 cells by copatching CD4 and the cholera toxin β subunit, which binds to the raft-associated ganglioside GM1. Confocal analysis indicated that CD4wt (Fig. 1 C), CD4–GPI (Fig. 1 D), CD4–LDL–CD4 (Fig. 1 F), the palmitoylation CD4–C394/397A mutant (Fig. 1 G), as well as the transmembrane CD4 mutants (unpublished data), colocalize extensively with GM1. Conversely, CD4–LDL and GM1 show segregated staining patterns (Fig. 1 E), confirming that this chimera does not partition in rafts.
Partitioning of the CD4 Ectodomain in Rafts Is Sufficient to Activate LCK.
We studied CD4 chimera and mutant association to, and activation of, raft-p56lck. Lck association with the palmitoylation-deficient CD4 mutants was reported (33), which suggests that neither the CD4 transmembrane nor juxtamembrane domain plays a role in Lck activation. Accordingly, chimeras with the CD4 cytoplasmic domain bind to Lck in cotransfected HEK-293 cells independently of the transmembrane domain (Fig. 2 A). Because CD4–GPI and CD4–LDL do not associate with Lck directly (Fig. 2 A), they were used to analyze CD4 partitioning effects on Lck activation. HEK-293 cells were cotransfected with the CD4 constructs, p56lck, and a chimeric protein composed of the CD8 extramembrane and transmembrane domains fused to the CD3ζ chain (CD8ζ; reference 28), an Lck substrate (34). Cross-linking of CD4–GPI induces Lck association with CD8ζ in a time-dependent manner and also induces an increase in CD8ζ tyrosine phosphorylation (Fig. 2 B). This is not observed in cells expressing the nonraft CD4–LDL chimera. Furthermore, CD4–GPI cross-linking increases Lck activity in anti-Lck immunoprecipitates, as measured by IVK assays (Fig. 2 C). Again, CD4–LDL cross-linking produces no detectable increase in Lck activity (Fig. 2 C). These results suggest that if the CD4 extracellular domain is located in rafts, the cytoplasmic CD4 domain is not necessary to activate Lck. Both Lck recruitment to CD8ζ and Lck activation are more robust in CD4wt– (Fig. 2, B and C) or CD4–LDL–CD4–cross-linked cells (unpublished data) than in CD4–GPI cells, which suggests that prior Lck interaction with the CD4 cytoplasmic tail enhances subsequent binding to targets and kinase activation.
Figure 2.

Raft partitioning, but not association, is a requisite for CD4-induced Lck activation. (A) HEK-293 cells coexpressing the indicated mutants and Lck were anti-CD4 precipitated. CD4-associated tyrosine kinase activity was determined in an IVK assay using enolase as a substrate. The migration of both Lck and enolase is indicated by arrows. (B) HEK-293 cells coexpressing Lck, CD8ζ and CD4wt, and CD4–GPI or CD4–LDL were incubated with anti-CD4 for the times indicated. Additional cross-linking was induced with a secondary anti–mouse antibody. After lysis, anti-CD8 immunoprecipitates were blotted sequentially with antiphosphotyrosine (PY), anti-Lck, and anti-CD3ζ antibodies as indicated. CD4, Lck, and CD8ζ expression levels for each condition were determined by Western blot. (C) IVK assay of anti-Lck immunoprecipitates from lysates in B. Results represent two independent experiments.
Exclusion of CD4 Extracellular Domain from Raft Domains Impedes HIV-1 Entry.
We analyzed whether raft partitioning of CD4 affects its HIV-1 receptor function. We confirmed that gp120 binding was comparable among the distinct CD4 mutants (Fig. 3 A), indicating that chimeric domains do not affect correct CD4 ectodomain folding. No significant gp120 binding was detected in nontransfected cells (unpublished data). The CD4 constructs were tested in a luciferase-based, HIV-1env IIIB–mediated cell–cell fusion assay. Because HIV-1–induced fusion is receptor density dependent (35), a DNA concentration range was used to confirm similar mutant CD4 cell surface expression (Fig. 3 B). HEK-293Env–HEK-293CD4 fusion increases luciferase activity at comparable levels in raft-associated CD4wt, CD4–GPI, CD4–LDL–CD4 (Fig. 3 C), and the transmembrane and juxtamembrane mutant-expressing cells (unpublished data). Nonetheless, fusion is completely abrogated in cells expressing nonraft CD4–LDL (Fig. 3 C). CD4–LDL and CD4–LDL–CD4 share an identical transmembrane domain, indicating that the inserted LDL fragment does not influence Env-mediated fusion. As efficient HIV-1–mediated fusion occurs in cells expressing CD4–GPI, the lack of HIV-1 fusion in CD4–LDL cells is probably due to partitioning of this mutant in a nonraft membrane fraction.
Figure 3.
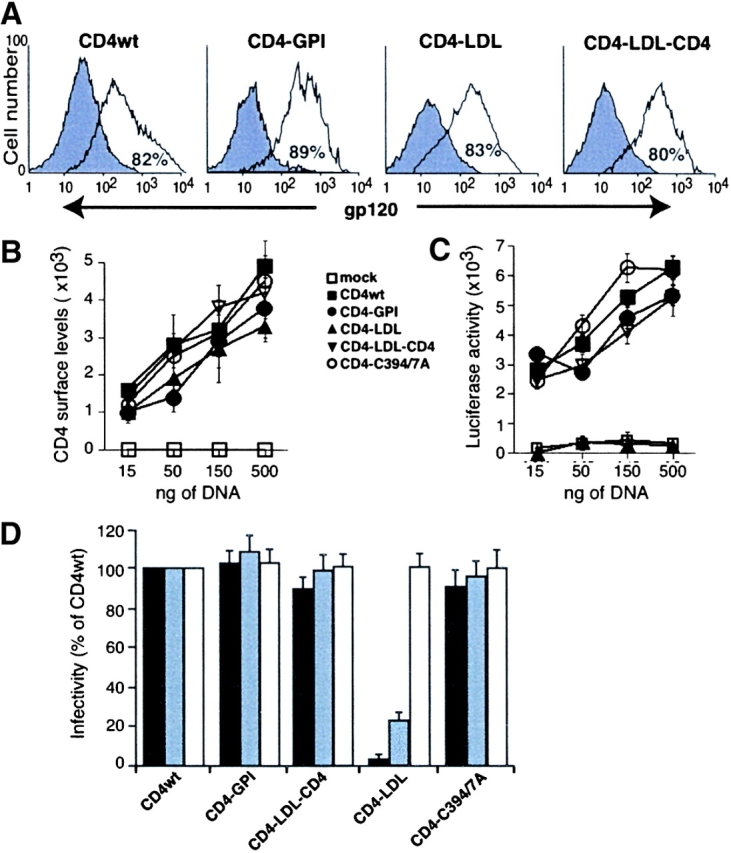
The nonraft CD4–LDL mutant does not allow HIV-1 entry. (A) HEK-293 cells expressing CD4 chimeras were incubated alone (filled) or with recombinant gp120 (open). Anti-gp120 antibody fluorescence intensity was then recorded in the CD4+ gated cells by flow cytometry. The percentage of gp120-binding CD4+ cells is indicated in each panel. A representative experiment is shown (n = 3). (B) HEK-293 cells were transfected with different amounts of cDNA as indicated, and CD4 immunoreactivity on the cell surface was analyzed by flow cytometry. Cell surface levels of CD4 were calculated by multiplying the percentage of CD4+ cells and the mean fluorescence intensity for each condition. Data represent the mean ± SD of duplicate points (n = 4). (C) The cells in B were mixed with HIV-1IIIB env–expressing HEK-293 cells and cell–cell fusion events were measured. Luciferase activity values were normalized using a promoterless renilla plasmid. (D) Single-round infection experiments were performed in HEK-293-CCR5 cells expressing the indicated CD4 mutants using a replication-defective NL4-3 virus pseudotyped with HIVNL4–3 (solid bar), HIVAda (gray bar), or VSV-G (open bar) envelopes. Cell infection, detected as an increase in luciferase activity, was normalized considering CD4wt as 100%. Data represent mean ± SD of duplicate points (n = 4).
To analyze the effect of CD4 partitioning on viral entry, we performed single-round infections with a replication-defective HIV-1 NL4-3 variant pseudotyped with HIV-X4 (NL4-3), HIV-R5 (Ada), or VSV-G envelopes. HEK-293–CCR5 cells, an established cell line expressing CCR5 at high levels, were used as targets. Efficient viral entry was observed in all raft-associated CD4 mutants, whereas cells expressing the nonraft CD4–LDL mutant were refractory to infection by either X4- or R5-pseudotyped HIV-1 variants (Fig. 3 D). CD4–LDL cells are nonetheless infected by VSV envelope-pseudotyped NL4-3 variants (Fig. 3 D), which indicates that raft partitioning of CD4 is a specific requisite for HIV-1 entry. Although CD4–LDL drastically affects X4 and R5 virus entry, we found that the inhibitory effect of the nonraft CD4–LDL mutant is more dramatic for X4 enveloped viruses than for the R5 pseudotypes. This difference may be the consequence of the high, nonphysiological CCR5 levels expressed by this cell line. Indeed, coreceptor density has been shown to influence both HIV-1 infectivity and entry inhibitor sensitivity (36, 37).
CD4 Partitioning in Rafts Is Required for gp120-induced Receptor Clustering.
According to the raft hypothesis, CD4–LDL may not support HIV-1 entry, as the CD4–gp120 coreceptor fusion complex cannot be formed. Using confocal microscopy, we analyzed the ability of selected CD4 mutants to form high order molecular complexes of gp120, CD4, and CXCR4. We observed gp120-induced patches in cells expressing raft-associated CD4wt (Fig. 4 A), CD4–GPI (Fig. 4 B), and CD4–LDL–CD4 (Fig. 4 C), in which gp120 (red), CXCR4 (blue), and CD4 (green) colocalize (white staining). Conversely, in CD4–LDL-expressing cells, anti–gp120-induced patching promotes small aggregates of gp120 and CD4 (yellow) that do not colocalize with CXCR4 (Fig. 4 D). Thus, when gp120–CD4 complexes are formed in rafts, anti–gp120-induced clustering may trigger lateral coalescence of CD4–gp120- and CXCR4-bearing rafts, resulting in the assembly of gp120, CD4, and CXCR4 complexes. However, CD4–LDL–gp120 complexes formed in nonraft membranes will not trigger coalescence of CXCR4-containing rafts, resulting in the inability of this mutant to form CD4–gp120–CXCR4 complexes. These results suggest that CD4 partitioning in membrane rafts is necessary for the CD4–gp120–CXCR4 complex formation.
Figure 4.
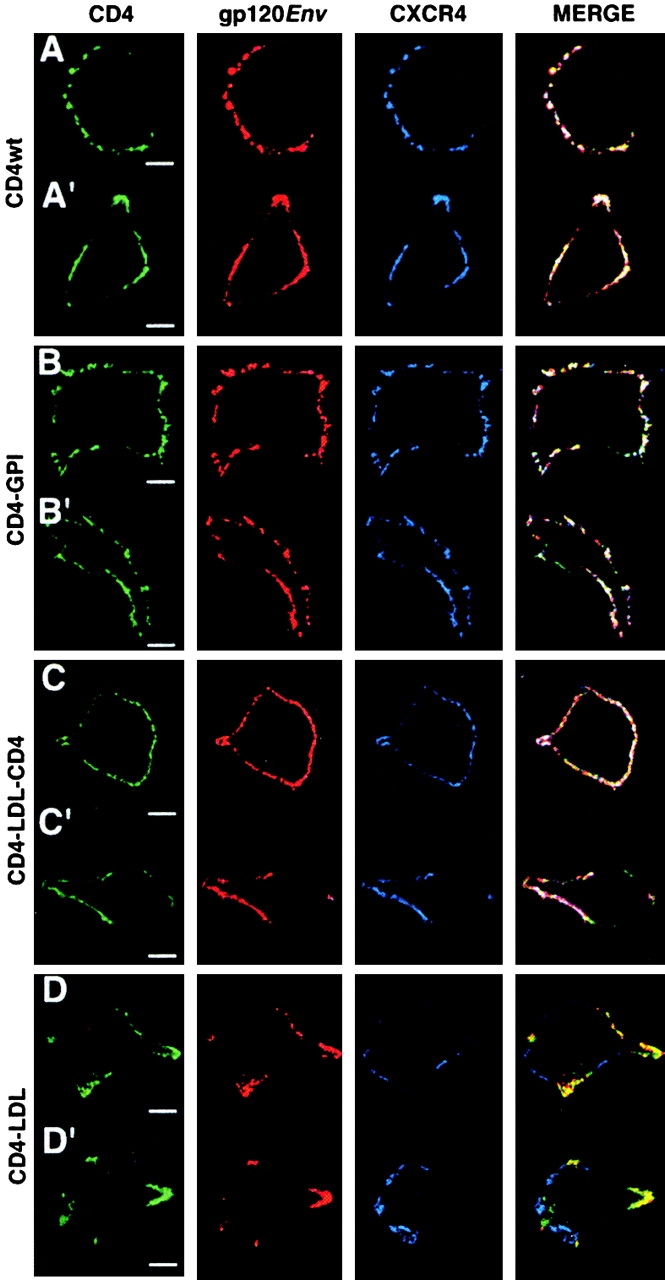
Raft partitioning of the CD4 extracellular domain is required for gp120-induced lateral association of CD4 and CXCR4. HEK-293 cells expressing (A and A′) CD4wt, (B and B′) CD4–GPI, (C and C′) CD4–LDL–CD4, and (D and D′) CD4–LDL were incubated with recombinant gp120IIIB and copatched with anti-gp120 (red), anti-CD4 (green), and anti-CXCR4 (blue) as indicated, and then analyzed by confocal microscopy. The three-color overlay is shown in the merge panel. Representative cells are shown (n = 50–60/mutant). Bar, 5 μm.
Nonraft CD4–LDL Prevents HIV-1 Infection in CD4+ T Cells.
The immunofluorescence results provide a molecular basis to explain why the nonraft CD4–LDL mutant does not support HIV-1 entry. As this mutant binds to HIV-1Env, we examined whether CD4–LDL prevents HIV-1 infection of CD4+ cells. The MT-2-CCR5 cell line expresses CD4 as well as CXCR4 and CCR5 coreceptors, rendering it susceptible to infection by X4 and R5 viral strains (14). Therefore, we transfected MT-2-CCR5 cells with the nonraft CD4–LDL mutant. A transfection efficiency of 44 ± 7% was estimated using a green fluorescent protein reporter plasmid. We analyzed ectopic cell surface CD4 expression by the biotinylation of mock-, CD4wt- and CD4–LDL-transfected MT-2-CCR5 cells. CD4–LDL has a 6xHis tag. Anti-6xHis antibody detects a specific band in avidin-precipitated cell lysates (Fig. 5 A), indicating that CD4–LDL is biotinylated and therefore expressed on the surface of these cells. The total CD4 levels in these blots confirm ectopic CD4wt expression (Fig. 5 A).
Figure 5.

Expression of the nonraft CD4–LDL mutant prevents HIV-1 entry in CD4+ cells. (A) Mock-, CD4wt-, or CD4–LDL-transfected MT-2-CCR5 cells were biotinylated to analyze cell surface CD4 expression. Biotin-labeled proteins were precipitated with agarose-coupled avidin and sequentially blotted with anti-6xHis and anti-CD4 antibodies. Total biotinylated proteins in the same immunoprecipitates were developed with avidin. (B) The cells in A were exposed to R5 (BaL) or X4 (NL4-3) HIV-1 viral strains at the indicated doses and productive infection followed by measurement of p24 antigen levels. Data shown are mean ± SD of triplicate points (n = 3). ▪, mock; •, CD4wt; ▴, CD4–LDL. (C) Mock-, CD4wt-, and CD4–LDL-expressing MT-2-CCR5 cells were infected with a HIVNL4–3 env-pseudotyped replication-defective NL4-3 virus. Infected cells were determined 24 h later by measuring luciferase activity. Data are mean ± SD (n = 4).
Productive HIV-1 infection was examined daily by recording p24 levels in mock-, CD4wt-, or CD4–LDL-expressing MT-2-CCR5 cells infected with NL4-3 (X4) or BaL (R5) strains. CD4–LDL expression delays p24 kinetics compared with mock- or CD4wt-expressing cells (Fig. 5 B) independently of the viral strain tested. This suggests that the nonraft CD4–LDL mutant inhibits HIV-1 infection of CD4+ cells. We explored this inhibitory effect in single-round infection experiments. Mock-, CD4wt-, or CD4–LDL-expressing MT-2-CCR5 cells were exposed to a replication-defective NL4-3 HIV-1–pseudotyped virus. The infection ratio of MT-2–CD4–LDL cells with this HIV-1 variant was 60% lower than that observed in mock or CD4wt cells, which again indicates that the CD4–LDL mutant impairs HIV-1 entry in MT-2-CCR5 cells.
Discussion
HIV-1 entry is a multistep process that requires virus attachment to the cell surface via CD4, an essential interaction with a coreceptor to trigger conformational changes in the viral fusion protein, and the viral–cell membrane fusion reaction itself. The use of drugs to sequester membrane cholesterol or inhibit GSL synthesis provided indirect evidence that these entry steps are unlikely to occur at random on the cell surface, but are confined to specific membrane regions termed rafts (14–18). The primary finding in this study is that CD4 partitioning into raft domains is critical for its function as an HIV receptor, as well as for CD4-mediated signaling, two independent CD4 processes. By engineering the transmembrane and cytoplasmic CD4 domains, we generated the nonraft CD4–LDL mutant that retains HIV-1–binding capacity, but does not allow viral infection by blocking viral entry. These results provide direct evidence that HIV-1 exploits raft domains on the host plasma membrane as entry sites into the cell.
Earlier studies indicated that the cytoplasmic, transmembrane, or juxtamembrane regions are not needed for CD4 function as an effective HIV-1 receptor (38, 39). Concurring with this, we found that the CD4–GPI mutant mediates HIV-1 entry as efficiently as CD4wt. Even though this mutant cannot directly interact with Lck, CD4–GPI cross-linking induces Lck activation, further suggesting that CD4 cytoplasmic and transmembrane domains are unnecessary for CD4-mediated signal transduction. It is not known how GPI-anchored proteins communicate with the intracellular milieu, although GPI-induced signaling is thought to depend on the raft structure in membranes (2). Because rafts normally contain few molecules, the clustering of GPI-anchored proteins would cause small rafts to coalesce, bringing raft-associated transmembrane proteins into proximity. Alternatively, the clustering of GPI-anchored proteins may induce transmembrane protein recruitment to rafts, which may function as adaptors between GPI proteins and intracellular kinases (2). In contrast to CD4–GPI, the nonraft CD4–LDL mutant, which also lacks CD4 transmembrane and cytoplasmic domains, does not activate Lck and cannot mediate HIV-1 infection. These results suggest that CD4 extracellular domain location in rafts is the major determinant of CD4 function in mediating signaling and viral infection.
Nevertheless, the potential role of cytoplasmic and/or transmembrane domains in determining CD4 association to rafts remains an open question. The affinity of a membrane protein for rafts can be increased by oligomerization, acylation, coupling to a raft-associated molecule, or conformational changes in a transmembrane region (6). The mutation of specific amino acids in the transmembrane domain, replacement of the membrane-spanning region with that of the LDL-R, or the elimination of the acylation acceptor cysteines is insufficient to force CD4 partitioning to nonraft regions. Comparison of CD4–LDL–CD4 and the CD4–LDL mutants suggests that the CD4 cytoplasmic domain exercises considerable influence on CD4 association to rafts, for instance, by enabling interaction with p56lck. This is nonetheless unlikely, as CD4–LDL–CD4 is also raft associated, even in the absence of Lck expression. Oligomerization is an important determinant of transmembrane protein partitioning in rafts. Proteins such as LDL-R, which does not partition in rafts by DRM criteria, are partially raft associated after oligomerization (3). Cross-linking with anti-CD4 does not increase raft partitioning of the CD4–LDL mutant, however, which lacks the cytoplasmic LDL-R region. This differential raft partitioning by LDL-R and CD4–LDL may explain the HIV-1 infection described in cells expressing a CD4–LDL-R chimera (38), whereas our CD4–LDL mutant fails to support viral infection. A final, intriguing possibility is that multiple determinants in the cytoplasmic, transmembrane, or even in the extracellular domain of CD4 are responsible for drawing CD4 into membrane rafts. Decoding the molecular signals that trigger CD4 association to rafts warrants additional research.
The most striking finding in this study is that the nonraft CD4–LDL mutant hinders infection by X4 and R5 viruses in a CD4+ MT-2 cell line. This inhibition probably occurs at initial phases of infection, as CD4–LDL also prevents MT-2 infection by a replication-deficient virus variant. The true inhibitory ability of the CD4–LDL mutant on HIV-1 infection is nonetheless difficult to assess based on the experiments presented here. First, only 40% of the MT-2-CCR5 cells are transfected by the nonraft mutant. Second, CD4–LDL expression would decrease throughout the spreading infection, as cell expression of this mutant is not selected during the experimental period. These two features may cause underestimation of the inhibitory capacity of CD4–LDL on spreading and single-round HIV-1 infection of CD4+ cells. We are currently generating stable cell lines expressing distinct CD4wt/CD4–LDL ratios.
A mechanism by which CD4–LDL inhibits HIV-1 infection may be postulated from the observation that the formation of high order complexes between gp120 and cell receptors is impaired in cells expressing the nonraft CD4–LDL mutant. The plasma membrane is probably in dynamic equilibrium between domains in raft and nonraft phases, which may preclude or predispose to protein–protein interactions. CD4–LDL may therefore be unable to mediate HIV-1 entry, as the CD4–Env complexes formed in a nonraft membrane fraction cannot further interact with coreceptors. This mechanism may also operate in CD4+ MT-2 cells, suggesting that CD4–LDL acts as a competitive inhibitor by preventing HIVEnv interaction with “fusion-competent” CD4 molecules on the cell surface. Alternatively, gp120 binding to CD4–LDL in a nonraft environment may prevent viral interaction with other host cofactors involved in HIV-1 entry. The GSL may act as alternative HIV-1 entry cofactors, due to their direct, CD4-dependent association with the viral envelope (17, 18, 40). Because rafts are GSL-enriched, such an interaction would not occur in cells expressing nonraft CD4–LDL. Finally, it is also possible that CD4–LDL binding to gp120 outside rafts cannot promote the conformational changes in the viral Env protein necessary for coreceptor binding. If this is the case, CD4–LDL partitioning in nonraft membranes would again be the major cause of this failure, as the raft-associated CD4–LDL–CD4 mutant, which also has the LDL-R transmembrane domain, efficiently mediates the formation of ternary CD4–gp120 coreceptor complexes.
In light of this study and reports showing the importance of lipid components in HIV budding (41, 42), it is tempting to speculate that the manipulation of specific host membrane raft components would be useful in designing new strategies to prevent and/or block HIV-1 infection. These therapeutic approaches would be suitable for both X4 and R5 viral strains and would obviate the problem of resistance mutants observed using current treatments.
Acknowledgments
We thank C. Hernández for technical assistance, M. Muñoz and K. Harshman for DNA sequencing, and C. Mark for editorial assistance.
S. Jiménez-Baranda is supported by a predoctoral fellowship from the Pharmacia Corporation. The Department of Immunology and Oncology was founded and is supported by the CSIC and the Pharmacia Corporation.
G. del Real and S. Jiménez-Baranda contributed equally to this work.
Footnotes
Abbreviations used in this paper: DRM, detergent-resistant membrane; GPI, glycosylphosphatidylinositol; GSL, glycosphingolipids; IVK, in vitro kinase; LDL, low density lipoprotein; TfR, transferrin receptor; VSV, vesicular stomatitis virus.
References
- 1.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- 2.Brown, D., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- 3.Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodgers, W., and J. Rose. 1996. Exclusion of CD45 inhibits activity of p56lck associated with glycolipid–enriched membrane domains. J. Cell Biol. 135:1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez-Mouton, C., J. Abad, E. Mira, R. Lacalle, E. Gallardo, S. Jimenez-Baranda, I. Illa, A. Bernad, S. Mañes, and C. Martinez-A. 2001. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. USA. 98:9642–9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Goot, F., and T. Harder. 2001. Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin. Immunol. 13:89–97. [DOI] [PubMed] [Google Scholar]
- 7.Maddon, P., A. Dalgleish, J. McDougal, P. Clapham, R. Weiss, and R. Axel. 1986. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 47:333–348. [DOI] [PubMed] [Google Scholar]
- 8.Xavier, R., T. Brennan, Q. Li, C. McCormack, and B. Seed. 1998. Membrane compartimentalization is required for efficient T cell activation. Immunity. 8:723–732. [DOI] [PubMed] [Google Scholar]
- 9.Moore, J., A. Trkola, and T. Dragic. 1997. Co-receptors for HIV-1 entry. Curr. Opin. Immunol. 9:551–562. [DOI] [PubMed] [Google Scholar]
- 10.Berger, E., R. Doms, E. Fenyo, B. Korber, D. Littman, J. Moore, Q. Sattentau, H. Schuitemaker, and J. Sodroski. 1998. A new classification for HIV-1. Nature. 391:240. [DOI] [PubMed] [Google Scholar]
- 11.Littman, D. 1998. Chemokine receptors: keys to AIDS pathogenesis? Cell. 93:677–680. [DOI] [PubMed] [Google Scholar]
- 12.Mellado, M., J. Rodriguez-Frade, S. Mañes, and C. Martinez-A. 2001. Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu. Rev. Immunol. 19:397–421. [DOI] [PubMed] [Google Scholar]
- 13.Mañes, S., R. Lacalle, C. Gomez-Mouton, G. del Real, E. Mira, and C. Martinez-A. 2001. Membrane raft microdomains in chemokine receptor function. Semin. Immunol. 13:147–157. [DOI] [PubMed] [Google Scholar]
- 14.Mañes, S., G. del Real, R. Lacalle, P. Lucas, C. Gomez-Mouton, S. Sanchez-Palomino, R. Delgado, J. Alcami, E. Mira, and C. Martinez-A. 2000. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 1:190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao, Z., L. Cimakasky, R. Hampton, D. Nguyen, and J. Hildreth. 2001. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retroviruses. 17:1009–1019. [DOI] [PubMed] [Google Scholar]
- 16.Khanna, K., K. Whaley, L. Zeitlin, T. Moench, K. Mehrazar, R. Cone, Z. Liao, J. Hildreth, T. Hoen, L. Shultz, et al. 2002. Vaginal transmission of cell-associated HIV-1 in the mouse is blocked by a topical, membrane-modifying agent. J. Clin. Invest. 109:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammache, D., N. Yahi, M. Maresca, G. Pieroni, and J. Fantini. 1999. Human erythrocyte glycosphingolipids as alternative cofactors for human immunodeficiency virus type 1 (HIV-1) entry: evidence for CD4-induced interactions between HIV-1 gp120 and reconstituted membrane microdomains of glycosphingolipids (Gb3 and GM3). J. Virol. 73:5244–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hug, P., H. Lin, T. Korte, X. Xiao, D. Dimitrov, J. Wang, A. Puri, and R. Blumenthal. 2000. Glycosphingolipids promote entry of a broad range of human immunodeficiency virus type 1 isolates into cell lines expressing CD4, CXCR4, and/or CCR5. J. Virol. 74:6377–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao, X., L. Wu, T. Stantchev, Y. Feng, S. Ugolini, H. Chen, Z. Shen, J. Riley, C. Broder, Q. Sattentau, et al. 1999. Constitutive cell surface association between CD4 and CCR5. Proc. Natl. Acad. Sci. USA. 96:7496–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhmann, S., E. Platt, S. Kozak, and D. Kabat. 2000. Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J. Virol. 74:7005–7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mañes, S., E. Mira, C. Gómez-Moutón, R. Lacalle, P. Keller, J. Labrador, and C. Martínez-A. 1999. Membrane raft microdomains mediate front-rear polarity in migrating cells. EMBO J. 18:6211–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorice, M., T. Garofalo, R. Misasi, A. Longo, V. Mattei, P. Sale, V. Dolo, R. Gradini, and A. Pavan. 2001. Evidence for cell surface association between CXCR4 and ganglioside GM3 after gp120 binding in SupT1 lymphoblastoid cells. FEBS Lett. 506:55–60. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen, D., and D. Taub. 2002. CXCR4 function requires membrane cholesterol: implications for HIV infection. J. Immunol. 168:4121–4126. [DOI] [PubMed] [Google Scholar]
- 24.Kozak, S., J. Heard, and D. Kabat. 2002. Segregation of CD4 and CXCR4 into distinct lipid microdomains in T lymphocytes suggests a mechanism for membrane destabilization by human immunodeficiency virus. J. Virol. 76:1802–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pralle, A., P. Keller, E. Florin, K. Simons, and J. Hörber. 2000. Sphingolipid–cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J. Cell Biol. 148:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lama, J., A. Mangasarian, and D. Trono. 1999. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 9:622–631. [DOI] [PubMed] [Google Scholar]
- 27.Carrera, A., F. Sánchez-Madrid, M. López-Botet, C. Bernabeu, and M. De Landazuri. 1987. Involvement of the CD4 molecule in a post-activation event on T cell proliferation. Eur. J. Immunol. 17:179–186. [DOI] [PubMed] [Google Scholar]
- 28.Carrera, A., H. Paradis, L. Borlado, T. Roberts, and C. Martínez-A. 1995. Lck unique domain influences Lck specificity and biological function. J. Biol. Chem. 270:3385–3391. [DOI] [PubMed] [Google Scholar]
- 29.Malissen, B., N. Rebai, A. Liabeuf, and C. Mawas. 1982. Human cytotoxic T cell structures associated with expression of cytolysis. I. Analysis at the clonal cell level of the cytolysis-inhibiting effect of 7 monoclonal antibodies. Eur. J. Immunol. 12:739–747. [DOI] [PubMed] [Google Scholar]
- 30.Mira, E., R. Lacalle, M. González, C. Gómez-Moutón, J. Abad, A. Bernad, C. Martínez-A., and S. Mañes. 2001. A role for chemokine receptor transactivation in growth factor signaling. EMBO Rep. 2:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheiffele, P., M. Roth, and K. Simons. 1997. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 16:5501–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melkonian, K., A. Ostermeyer, J. Chen, M. Roth, and D. Brown. 1999. Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J. Biol. Chem. 274:3910–3917. [DOI] [PubMed] [Google Scholar]
- 33.Crise, B., and J. Rose. 1992. Identification of palmitoylation sites on CD4, the human immunodeficiency virus receptor. J. Biol. Chem. 267:13593–13597. [PubMed] [Google Scholar]
- 34.Chu, K., and D. Littman. 1994. Requirement for kinase activity of CD4-associated p56lck in antibody-triggered T cell signal transduction. J. Biol. Chem. 269:24095–24101. [PubMed] [Google Scholar]
- 35.Bannert, N., D. Schenten, S. Craig, and J. Sodroski. 2000. The level of CD4 expression limits infection of primary rhesus monkey macrophages by a T-tropic simian immunodeficiency virus and macrophagetropic human immunodeficiency viruses. J. Virol. 74:10984–10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nokta, M., X. Li, J. Nichols, M. Mallen, A. Pou, D. Asmuth, and R. Pollard. 2001. Chemokine/CD4 receptor density ratios correlate with HIV replication in lymph node and peripheral blood of HIV-infected individuals. AIDS. 15:161–169. [DOI] [PubMed] [Google Scholar]
- 37.Derdeyn, C., J. Decker, J. Sfakianos, Z. Zhang, W. O'Brien, L. Ratner, G. Shaw, and E. Hunter. 2001. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J. Virol. 75:8605–8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bedinger, P., A. Moriarty, R. von Borstel, N. Donovan, K. Steimer, and D. Littman. 1988. Internalization of the human immunodeficiency virus does not require the cytoplasmic domain of CD4. Nature. 334:162–165. [DOI] [PubMed] [Google Scholar]
- 39.Diamond, D., R. Finberg, S. Chaudhuri, B. Sleckman, and S. Burakoff. 1990. Human immunodeficiency virus infection is efficiently mediated by a glycolipid-anchored form of CD4. Proc. Natl. Acad. Sci. USA. 87:5001–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puri, A., P. Hug, K. Jernigan, J. Barchi, H. Kim, J. Hamilton, J. Wiels, G. Murray, R. Brady, and R. Blumenthal. 1998. The neutral glycophingolipid globotriaosylceramide promotes fusion mediated by a CD4-dependent CXCR4-utilizing HIV type 1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA. 95:14435–14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng, Y.-H., A. Plemenitas, T. Linnemann, O. Fackler, and B. Peterlin. 2001. Nef increases infectivity of HIV via lipid rafts. Curr. Biol. 11:875–879. [DOI] [PubMed] [Google Scholar]
- 42.Ono, A., and E. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA. 98:13925–13930. [DOI] [PMC free article] [PubMed] [Google Scholar]