

Abstract
To identify the physiological role of Hck, a functionally redundant member of the Src family of tyrosine kinases expressed in myelomonocytic cells, we generated HckF/F “knock-in” mice which carry a targeted tyrosine (Y) to phenylalanine (F) substitution of the COOH-terminal, negative regulatory Y499-residue in the Hck protein. Unlike their Hck−/− “loss-of-function” counterparts, HckF/F “gain-of-function” mice spontaneously acquired a lung pathology characterized by extensive eosinophilic and mononuclear cell infiltration within the lung parenchyma, alveolar airspaces, and around blood vessels, as well as marked epithelial mucus metaplasia in conducting airways. Lungs from HckF/F mice showed areas of mild emphysema and pulmonary fibrosis, which together with inflammation resulted in altered lung function and respiratory distress in aging mice. When challenged transnasally with lipopolysaccharide (LPS), HckF/F mice displayed an exaggerated pulmonary innate immune response, characterized by excessive release of matrix metalloproteinases and tumor necrosis factor (TNF)α. Similarly, HckF/F mice were highly sensitive to endotoxemia after systemic administration of LPS, and macrophages and neutrophils derived from HckF/F mice exhibited enhanced effector functions in vitro (e.g., nitric oxide and TNFα production, chemotaxis, and degranulation). Based on the demonstrated functional association of Hck with leukocyte integrins, we propose that constitutive activation of Hck may mimic adhesion-dependent priming of leukocytes. Thus, our observations collectively suggest an enhanced innate immune response in HckF/F mice thereby skewing innate immunity from a reversible physiological host defense response to one causing irreversible tissue damage.
Keywords: knock-in mutation, LPS, endotoxemia, macrophage, neutrophil
Introduction
Hck is a member of the highly conserved Src-family of cytoplasmic protein tyrosine kinases that transduce a variety of extracellular signals, which ultimately affect cellular processes including proliferation, differentiation and migration. Src family kinases (SFKs)* can be divided into two groups, those that exhibit a relatively ubiquitous pattern of expression (i.e., Src, Yes, Fyn) and those whose expression is restricted to the hemopoietic system (e.g., Lck in T lymphocytes and Hck in myelomonocytic cell lineages; reference 1). The well-defined modular structure of SFKs comprises a relatively divergent, NH2-terminal “unique” domain, which is subject to posttranslational lipid modifications thereby targeting SFKs to the plasma membrane. Src homology 3 (SH3) and 2 (SH2) domains, and a tyrosine kinase catalytic domain (2) follow the “unique” domain. The catalytic activity of SFKs is regulated, both positively and negatively, by tyrosine phosphorylation of highly conserved tyrosine (Y) residues. Phosphorylation of a single conserved Y residue in the COOH terminus of SFKs (e.g., Y499 in murine Hck or Y527 in chicken c-Src) by the protein kinase Csk renders SFKs inactive as a result of an intramolecular interaction between the phosphorylated tyrosine (pY) residue and their own SH2 domain. Disruption of this interaction, either as a result of dephosphorylation, or substitution of the COOH-terminal regulatory Y residue with phenylalanine (F; e.g., HckY499F), or COOH-terminal truncation mutations as observed in the virally transduced v-Src oncoprotein, results in constitutive activation of SFKs (3–8). In contrast to phosphorylation of the COOH-terminal regulatory tyrosine residue, autophosphorylation of a tyrosine residue within the kinase domain of SFKs (e.g., HckY388) acts to positively regulate their catalytic activity. Thus, activation of SFKs requires both disruption of the COOH-terminal regulatory tyrosine-SH2 domain interaction and autophosphorylation of the regulatory tyrosine residue within the kinase domain.
Unique functions for individual SFKs have been identified from studies of “loss-of-function” mouse models engineered by the genetic disruption of specific family members. However, the phenotypes of the resulting mice were often subtle due to functional redundancy between individual family members, as revealed by the subsequent analysis of compound knockout mutant mice. In particular, the full extent of Hck function remains elusive, as the phenotype of Hck−/− mice was confined to a mild defect in Fc receptor-independent phagocytic activity of macrophages (9). Further analysis of Hck deficiency, in the context of Hck−/−Fgr−/− and Hck−/−Fgr−/−Lyn−/− compound mutant mice, suggested a much broader, albeit redundant, role for Hck in neutrophil and macrophage mediated host defense and inflammatory responses (9–10).
Here, we translate into the laboratory mouse a strategy previously established by us in cultured cells (11) to identify the physiological functions of an individual member within a functionally redundant protein family (such as SFKs) by introducing a constitutively activating “gain-of-function” mutation into the corresponding endogenous gene. This strategy ensures, without prior characterization of the cis-acting regulatory gene sequences, faithful tempo-spatial recapitulation of the expression pattern of the wild-type version of the protein. Introduction of a “gain-of-function” mutation into the hck gene (i.e., HckY499F) resulted in mice that exhibited an enhanced innate immune response, characterized by constitutive priming of leukocytes and culminating in acquired leukocyte consolidation in the lung. Additionally, the mice homozygous for this mutation, referred to as HckF/F mice, were highly susceptible to LPS-induced pulmonary and systemic inflammatory responses.
Materials and Methods
Targeting Construct and Generation of Mice
The “replacement-type” targeting vector pHck 499F-IRESneo used in the present study represents a modified version of the previously described “insertion-type” targeting vector pHCK499F (11). In brief, the SalI-XhoI fragment from pHCK499F, encompassing 0.8 and 6.6 kb of homologous sequence flanking the last coding exon of the mouse hck gene, was used to introduce the tyrosine-499 to phenylalanine substitution (TAT → TTC), as well as a novel BglII restriction site over the translational stop codon at amino acid position 504. A BamHI-NheI-BamHI adaptor was then introduced into the BglII site and a Neor-IRES cassette inserted into the NheI site as a XbaI/NheI fragment.
Twenty-five million W9.5 embryonic stem (ES) cells (129/Sv) were electroporated with the SalI linearized pHck 499F-IRESneo targeting construct. 5 out of 92 neomycin-resistant colonies were found to be correctly targeted, as judged by Southern blotting of genomic DNA with a fragment mapping upstream of the 5′-end of the targeting vector. Allele-specific RT-PCR was used to confirm the nucleotide sequence of the mutated exon in the targeted allele. Two recombinant ES cell lines were injected into C57BL/6 blastocysts and the resulting germline chimeras mated with C57BL/6 females to establish a HckF/F breeding colony.
Assays on Live Mice
LPS-triggered Innate Immunity in Mice.
An innate cellular inflammatory response was induced by instillation of 10 μg of the gram-negative bacterial cell-wall component LPS (Escherichia coli Serotype 026:B6 from Sigma-Aldrich; dissolved in 50 μl PBS) into the lungs of anesthetized mice using a transnasal challenge method (12). For necropsy, mice were anesthetized with ketamine/xylazine (15 mg/kg and 30 mg/kg, intraperitoneally, respectively) and a blood sample withdrawn from the abdominal aorta. Both total and viable cells counts (using ethidium bromide/acridine orange; Molecular Probes) were performed on bronchoalveloar lavage fluids (BAL-F) from the lungs of the mice. Cell types and differential counts were determined according to standard morphological criteria on Diff-Quick (Dade Baxter) stained cytospins of undiluted BAL-F. To assess the response of mice to systemic challenge with LPS, a telemetric temperature sensor (ELAMS temperature transponder system) was implanted subcutaneously in the backs of age-, weight-, and sex-matched mice. 3 d later, mice received a single intraperitoneal injection of the indicated dose of LPS. The body weight, core body temperature, and survival of the mice were monitored for up to 7 d after injection.
Measurement of Mouse Lung Function.
Each mouse was anesthetized with 0.1 ml/10 g of a mixture containing xylazine (2 mg/ml; Bayer) and ketamine (40 mg/ml; Parnell Laboratories). Two-thirds of the dose was given to induce anesthesia, with the remainder given when the animal was attached to the ventilator. Top-up doses were given approximately each 40–60 min, as required. Once surgical anesthesia had been established, a tracheostomy was performed with a polyethylene cannula (10-mm long and 0.813-mm inner diameter, respectively) inserted. Mice were ventilated with a tidal volume of 8 ml/kg at a rate of 450 breaths per minute, with a positive end-expiratory pressure of 0.2 kPa, using a custom-designed ventilator (flexiVent; Scireq). The special features of this ventilator include a precision computer-controlled piston that is capable of accurately producing any desired wave-form and measuring of the delivered volume (and thus flow) by tracking the piston movement (with appropriate corrections for gas compression). The animal handling and study protocol conformed with the guidelines of the Australian National Health and Medical Research Council and were approved by the Animal Ethics Committee of the Institute for Child Health Research.
Lung function was measured during brief pauses (t = 6 s) of ventilation, during which the animal was switched from the ventilation circuit to the measurement circuit. A forcing function, with 25 frequency components between 1 and 25 Hz was generated by the loudspeaker and delivered to the animal via a wave tube (length = 100 cm, inner diameter = 0.116 cm). The phase content of the forcing function was optimized to limit the peak-to-peak amplitude to 2 cm H2O. Measurements were made at trans-respiratory pressures of 0, 2, 5, 10, 15, and 20 cm H2O. The measurement circuit was pressurized to the desired pressure before the animal being switched into the circuit. Approximately 3 s were allowed for the animal to equilibrate with the measurement circuit before the forcing function was introduced. At each pressure four discrete data epochs were collected, separated by at least 30 s of normal ventilation, with a positive end expiratory pressure (PEEP) of 2 cm H20. Individual spectra were averaged and the constant phase model (CPM; reference 13) fitted with the impedance of the respiratory system (Zrs; Zrs = R + jωI + (G − jH)/ωα, where R is the Newtonian resistance (primarily located in the airways but containing a contribution from the chest wall), I is the inertance, G is the coefficient of tissue damping (which reflects the resistive properties of the lung tissues), H is the coefficient of tissue elastance (which reflects tissue stiffness), ω is the angular frequency, and α represents the reciprocal frequency-dependent behavior of G + H.
Measurement of Erythrocyte Half-Life.
Erythrocytes from C57/Bl6 mice were labeled with 200 μCi of Na2[51Cr]O3 (300 mCi/mg chromium; Amersham Biosciences) as described (14). The labeled erythrocytes were diluted with isotonic saline and injected into the tail vein of 8-wk-old wild-type (i.e., HckY/Y) and HckF/F mice. Approximately 70 μl of blood was subsequently collected (via saphenous tail vein bleeding) from each recipient mouse 1, 5, 8, 12, 15, and 18 d later. Radioactivity associated with erythrocytes was quantitated with a gamma counter.
Cell Culture
Bone marrow–derived macrophages (BMDMs) were derived from bone marrow cells aspirated from the femurs of 8- to 12-wk-old mice. Bone marrow cells were flushed into BM-DMEM (DMEM supplemented with 10% FCS and 20% L cell-conditioned medium) and cultured in BM-DMEM at 37°C in a humidified atmosphere of 10% CO2. Nonadherent cells were collected 24 h later, resuspended in fresh BM-DMEM and cultured for an additional 4 to 6 d. PMNs from mouse femurs were aspirated into Ca2+/Mg2+-free HBSS and then purified through a discontinuous Percoll gradient as described previously (15). Greater than 95% of the purified cells were positive for the granulocyte marker Gr-1. Eosinophils were purified, to greater than 95% purity, from BAL-F by lectin affinity selection as described previously (16).
In experiments where pervanadate was used to inhibit in vivo protein tyrosine phosphatase activity, sodium orthovanadate (20 mM in water) was mixed 1:4 with H2O2 (30%), incubated for 5 min at room temperature (17) and then immediately added to BMDMs to a final concentration of 1.5 μM.
Cultured BMDMs were stimulated with the indicated concentrations of LPS (1 mg/ml stock solution in PBS containing 1% FCS) preceded in some cases by priming for 2 h with recombinant murine INF-γ (15 U/ml; Roche Molecular Diagnostics).
Cellular Assays
In vitro migration of PMNs was assessed using modified Boyden chambers (18, 19). In brief, 105 PMNs were seeded in the upper chamber of a 24-well dish containing a tissue culture insert with a porous polycarbonate filter membrane (8 μM; Becton Dickinson) that had been precoated with 10 μg/ml fibrinogen for 1 h at 37°C. The chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine (fMLP; 200 nM) was then added to the upper and/or lower chambers in the indicated combinations. At the specified time, the filter inserts were removed, washed extensively, and the cells attached to the inserts fixed in 4% paraformaldehyde and stained with Giemsa. The chemotactic capacity of the PMNs was quantitated by counting in five randomly chosen fields the number of cells on the underside of the filters.
Priming, activation, and degranulation of PMNs, evident from a progressive gain in “forward scatter” and loss of “side scatter,” was analyzed by FACS® (20). In brief, PMNs were resuspended in PBS containing 0.2% BSA and 5 mM glucose, or in some cases adhered to fibrinogen-coated 24-well plates for 30 min at 37°C, and then stimulated at 37°C with TNFα (20 ng/ml) or PMA (1 μg/ml). After fixation with ice-cold 8% formaldehyde in PBS, the cells were stained with a Gr-1 antibody and analyzed by FACS®. Gr-1hi cells were gated and both “forward” and “side” scatter profiles analyzed.
Release of nitric oxide by LPS-stimulated BMDMs and reactive oxygen species (i.e., O2 −) by TNFα (20 ng/ml) or fMLP (200 nM) stimulated PMNs was as described previously by Stuehr et al. (21) and Berton et al. (22), respectively. The values were corrected for the number of live cells in each well as assessed by MTT stain.
The capacity of BMDMs to phagocytose either opsonized or nonopsonized sheep red blood cells that had been labeled with Na2[51Cr]O3 was performed according to a standard procedure (23).
Immunoblotting, In Vitro Kinase Assays and Zymography
Cells were lysed directly in lysis buffer (1% Triton X-100, 0.1% SDS, 10% glycerol, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1 mM Na2VO3, 50 mM NaF, and complete protease inhibitors) for 30 min on ice. Lysates were clarified by centrifugation at 13,000 g for 10 min at 4°C, and protein concentrations measured with a BCA protein assay kit (Pierce Chemical Co.). Immunoblotting and immunoprecipitation of cell lysates was performed by standard techniques. The anti-phosphotyrosine (4G10; UBI), anti-Hck rabbit polyclonal (from C. Lowell, University of California at San Francisco, San Francisco, CA) and rat monoclonal (24), anti-Lyn (Santa Cruz Biotochnology, Inc.), anti-iNOS (Santa Cruz Biotochnology, Inc.), Gr-1 (BD Biosciences), anti-cbl (W. Langdon, University of Western Australia, Perth, Australia), anti-paxillin (Zymed Laboratories), and anti-SHP-1 (from H.-C. Cheng, University of Melbourne, Melbourne, Australia) antibodies were from the indicated sources.
The in vitro kinase activity of Hck was assessed in immunoprecipitates of whole cell lysates and equalized for Hck protein. The immunoprecipitates were washed three times with lysis buffer, then once with kinase wash buffer (20 mM Hepes, pH 7.4, 0.1% NP-40, and 0.1 mM sodium orthovanadate). The immunoprecipitates were then incubated at room temperature for 10 min in 30 μl of kinase buffer (20 mM Hepes, pH 7.4, 10 mM MnCl2, 0.1% NP-40, and 0.1 mM sodium orthovanadate) containing 10 μCi [γ-32P] ATP (25). Reactions were terminated by the addition of an equal volume of 2× SDS-PAGE sample buffer and heating for 5 min at 95°C. Phosphorylated proteins were resolved by SDS-PAGE and subsequently analyzed using a PhosphorImager screen (Molecular Dynamics).
Zymography was used to assess total protease activity in BAL-F from LPS-treated mice. Specifically, 100 μl of BAL-F was concentrated by precipitation with trichloroacetic acid and then resuspended in nonreducing SDS-PAGE sample buffer. The samples were then applied to 10% SDS-PAGE gel slabs containing either α-casein (2 mg/ml; Sigma-Aldrich) or gelatin (2 mg/ml; Labchem). After electrophoresis, gels were removed and washed twice in 2.5% Triton X-100 and then incubated overnight at 37°C in zymography buffer (50 mM Tris, pH 7.5, 5 mM CaCl2, 1 mM ZnCl2, 0.01% NaN3). The gels were then stained for 45 min with Coomassie Brilliant Blue 250-R (Sigma-Aldrich), followed by extensive destaining. Protease activity is indicated by regions of negative staining.
Northern Blot Analysis
Northern blotting of poly(A)+ RNA isolated from BMDM cells and mouse spleens was performed by standard techniques.
Histology
To ensure consistent morphological preservation of lungs, mice were anesthetized and then perfusion fixed via a tracheal cannula with 4% formaldehyde at exactly 200 mm H2O pressure for 4 h at room temperature. Fixed tissues were embedded in paraffin and sections stained either with hematoxylin and eosin, Alcian Blue/periodic acid-Schiff to stain mucus, or trichromic acid to detect extracellular matrix. All histological assessments were performed blind.
ELISA
Mouse TNFα, IL-5, and eotaxin levels were determined by interpolation of standard curves using standard sandwich ELISA (R&D Systems Quantikine for TNFα and BD Biosciences antibody pairs for IL-5 and eotaxin). All limits of detection were 10 ng/ml.
Results
Generation of HckF/F Mice.
To generate mice that express a constitutively active mutant of Hck (i.e., HckY499F), we used gene targeting in ES cells to introduce a tyrosine (Y499) to phenylalanine (F) substitution in exon 13 of the endogenous hck gene. The promoterless, bicistronic targeting vector pHCK499F-IRESneo contains 0.8 kb of intron 12 and 6.6 kb of 3′ homologous sequences flanking the mutant exon 13 with the Y499F substitution (TAT → TTC), and an IRES-neomycin resistance cassette (Fig. 1 a). This strategy was adopted to take advantage of the fact that the hck promoter is active in ES cells (11) and to ensure that the bicistronic mutant hck F-IRESneo RNA species contained all the 3′-UTR sequence information that might normally regulate the stability of endogenous wild-type hck Y mRNA transcripts. Targeted heterozygous HckY/F ES cells, recovered at a frequency of 1 in 19 antibiotic-resistant ES cell colonies, gave rise to germline-transmitting chimeric mice upon injection into host blastocysts. Interbreeding of heterozygous offspring yielded fertile homozygous mutant (HckF/F) mice at the expected Mendelian ratio (unpublished data). The mice were of normal appearance and had an average life span of 22 ± 4 mo when housed in a conventional animal facility.
Figure 1.

Generation of HckF/F mice and biochemical analysis of HckF/F BMDMs. (a) Gene targeting strategy for introducing the Y499F mutation into the endogenous hck gene in ES cells. A region of the murine hck gene is schematically depicted with numbers of the coding exons (boxes) and the 3-UTR. The targeting vector pHCK499F-IRESneo contains the TAT to TTC substitution in the nucleotide triplet encoding amino acid 499, a stop codon, and an IRES-neomycin resistance (IRESneo) cassette. A diagnostic digest with BamHI (Ba) yields a fragment of 7.5 kb from the wild-type allele and a 2.4-kb fragment from the targeted allele when hybridized with a probe external to the targeting vector (marked by *). (b) Reduced levels of HckF protein in BMDM. Equal amounts of total cell lysates derived from mice of the indicated genotypes were subjected to Western blotting with anti-Hck antibody. The blot was stripped and re-probed with an anti-Lyn antibody. Arrows indicate the positions of the p59/p56 Hck and p56/p53 Lyn isoforms, respectively. (c) Northern blot analysis of BMDM RNA. Poly(A)+RNA was hybridized with a full-length cDNA clone encoding murine Hck. Dicistronic hck F-IRESneo mRNA transcripts are ∼1.4 kb larger than the wild-type hck mRNA transcripts. A gapdh probe was subsequently used to assess RNA loading. (d) Elevated levels of tyrosine-phosphorylated proteins in HckF/F mutant cells. Total cell lysates from BMDMs derived from mice of the indicated genotypes were subjected to Western blotting with anti-phosphotyrosine (pY) antibodies. The blot was then stripped and reprobed with an anti-Hck antibody. (e) HckF exhibits elevated activity in vitro. Equalized amounts of Hck were immunoprecipitated from lysates of BMDMs (bottom panel) and subjected to either Western blotting with anti-phosphotyrosine (pY) antibody (top panel) or an auto-phosphorylation assay in the presence of γ-[32P]-ATP (middle panel). (f) Enhanced tyrosine-phosphorylation in BMDMs in the presence of pervanadate. Total cell lysates prepared from BMDMs that had been cultured in the presence or absence of 1.5 μM pervanadate (VO4) were subjected to Western blotting with an anti-pY antibody. (g) The lysates used in f were also used to analyze tyrosine-phosphorylation of the Hck substrates c-Cbl and paxillin. 1 mg of total cell lysate was immunoprecipitated with the indicated antibody and then subjected to Western blotting with an anti-pY antibody. Stripped blots were subsequently reprobed with anti-Cbl and anti-paxillin antibodies to confirm equal protein loading.
Elevated Hck Kinase Activity in Cells Derived from HckF/F Mice.
To assess the impact of this “gain-of-function” mutation into the hck gene on tyrosine phosphorylation of proteins in myelomonocytic cells, whole cell lysates of BMDMs derived from both mutant HckF/F and wild-type (HckY/Y) mice were subjected to Western blotting with anti-Hck and anti-phosphotyrosine antibodies. Significantly, we reproducibly detected reduced (approximately fourfold) levels of both isoforms of Hck in BMDMs and other primary myelomonocytic cell types derived from HckF/F mutant mice when compared with those from HckY/Y mice (Fig. 1 b). By contrast, the protein levels of the related Src-family kinase Lyn were not affected in BMDMs derived from HckF/F mutant mice. The specific reduction of HckF protein was correlated with reduced steady-state RNA level of hck F-IRESneo transcripts in BMDMs (Fig. 1 c) and in the spleen (unpublished data) when compared with wild-type hck Y RNA. However, we determined similar RNA stability of hck F-IRESneo and hck Y transcripts in the presence of the RNA polymerase II inhibitor 5,6-dichloro-1β-D-ribofuranosylbenzimidazole (unpublished data). Despite reduced levels of HckF protein in whole cell lysates of the BMDMs, we observed modestly increased levels of overall tyrosine phosphorylation in a manner dependent on the dosage of the mutant gene (Fig. 1 d). Furthermore, in vitro kinase assays performed on equalized amounts of Hck protein, revealed a three- to fivefold increase in the specific activity of the HckF protein (Fig. 1 e). By contrast, Hck immunoprecipitated from HckY/Y BMDMs did not exhibit any detectable tyrosine phosphorylation when using the phosphotyrosine-specific antibody 4G10 (Fig. 1 e, top panel). This finding suggests endogenous protein tyrosine phosphatases (PTPs) act to keep HckF in an un-(auto)phosphorylated state (26), thereby suppressing to some extent its catalytic activity. To test this notion, BMDMs from HckF/F mice were treated with the pan-PTP inhibitor pervanadate. As shown in Fig. 1 f, pervanadate treatment dramatically enhanced the levels of tyrosine phosphorylation of cellular proteins, including that of the bone fide Hck-substrates c-Cbl and paxillin (27) in BMDMs from HckF/F mice (Fig. 1 g). Collectively, these results confirm that the Y499F mutation introduced into the endogenous hck gene results in the expression of a constitutively active form of Hck. However, as a result of the counteracting mechanisms (reduced steady-state level of HckY protein, elevated activity of endogenous PTPs), the kinase activity of Hck is only moderately elevated in HckF/F “gain-of-function” mice (Fig. 1 e).
Pulmonary Pathology in HckF/F Mice.
HckF/F mice appeared superficially healthy throughout their adult life. Upon histological examination, however, the lungs of all adult HckF/F mice (3–4 mo and older) showed areas of atelectasis, inflammatory infiltration into the interstitium and alveoli, and some areas of enlarged airspaces. These lungs were consistently characterized by increased cellularity within the parenchyma, accumulation of mononuclear inflammatory cells in areas around airways, alveolar airspaces, and blood vessels, as well as by the presence of large macrophages despite specific pathogen-free (SPF)-housing (Fig. 2 , a and b). Eosinophils represented the most prominent cell type within the infiltrates (30 ± 9% of total BAL-F cells in HckF/F mice versus 2 ± 1% in HckY/Y mice). Additionally, spiculate crystalline forms, likely to represent degranulated eosinophilic protein, often accompanied the eosinophilic matter in the alveolar spaces as well as engulfed crystalline eosinophilic material was frequently observed in macrophages (see below). The epithelium lining conducting airways was frequently hypertrophic and many epithelial cells were enlarged due to excessive production and/or accumulation of mucus material (Fig. 2, c and d). Furthermore, lungs from HckF/F mice showed areas of mild emphysema, as histologically evident from alveolar enlargement with the loss of the orderly appearance of the acinus, and signs of fibrosis characterized by deposition of extracellular matrix in the alveolar septa (Fig. 2, e and f). Concomitantly, BAL-F recovered from the lungs of mutant mice revealed elevated levels of IL-5 (70 ± 27 pg/ml in HckY/Y versus 17 ± 5 pg/ml in HckY/Y) and eotaxin (37 ± 8 pg/ml in HckY/Y versus 23 ± 4 pg/ml in HckY/Y), features associated with eosinophilia (28, 29). At weaning, lungs of HckF/F mice were indistinguishable from those of HckY/Y mice (Fig. 3 , a and b). Irrespective of whether mice were housed in a conventional or a SPF-environment, areas of localized pulmonary consolidations became evident around ∼5-6 wk of age in HckF/F mice and progressed thereafter (Fig. 3, c and d). In rare cases, pulmonary adenocarcinomas were detected in HckF/F mice approaching 2 yr of age (Fig. 3, e and f).
Figure 2.
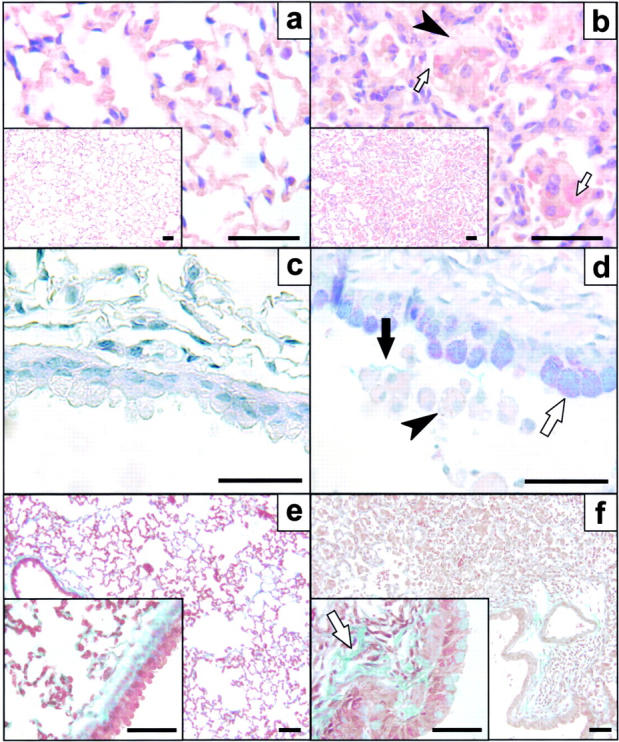
Histological analysis of lungs from HckF/F mice. Cross sections of lungs collected from HckY/Y mice (a, c, and e) and HckF/F mice (b, d, and f) at 3 mo of age. Mutant lungs consistently showed accumulation of monocytes and macrophages in the alveolar airspaces in lungs of HckF/F mice (b and f). Alveolar macrophages in mutant lungs were frequently vacuolated (b and d; arrowheads) with eosinophilic cytoplasm (panel b inset; arrows). Epithelial cells lining the conducting airways of mutant mice displayed extensive mucus cell metaplasia (blue/purple stain in d; white arrow) resulting in mucus aggregates in the airway lumen (d, black arrow) when compared with wild-type mice (c). Mutant lungs were characterized by excessive fibrotic deposits of extracellular matrix in the alveolar septa (pale green stain in f, arrow). Histological stains were hematoxylin and eosin (a and b), alcian blue/periodic acid-Schiff (c and d), or Masson's trichrome (e and f). Bar = 40 μm.
Figure 3.
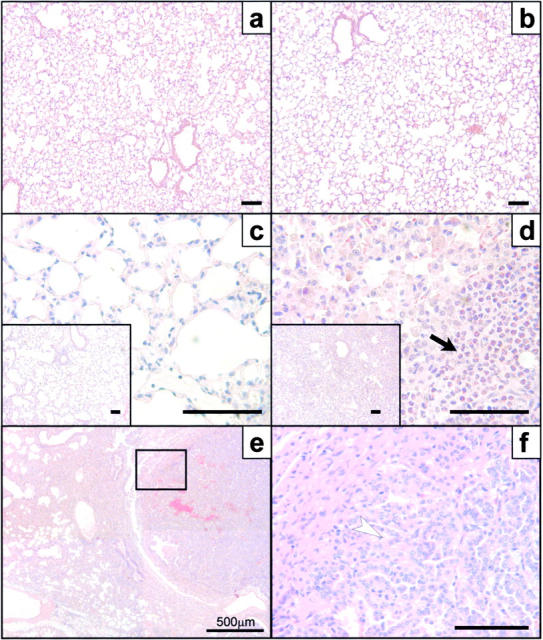
Acquired pulmonary changes in HckF/F mice. Cross sections of lungs collected from HckY/Y mice (a and c) and HckF/F mice (b, d, e, and f). The lungs of mutant mice look histologically indistinguishable at weaning age (a and b), but show the characteristic accumulation of macrophages and eosinophils (arrow) by 6 wk of age (c and d). Occasionally, old mutant mice (∼2 yr of age) develop pulmonary adenocarcinoma (e and f). Panel f represents a magnification of the boxed area in e and shows well-differentiated adenocarcinoma composed of irregular glands lined by neoplastic epithelial cells (arrowhead). Note the extensive consolidation in the nonmalignant sections of the lung (e). Histological stains were hematoxylin and eosin. Bar = 100 μm.
Assessment of Lung Function in HckF/F Mice.
To assess the functional consequences of the extensive lung parenchymal inflammation, we analyzed pulmonary mechanics by the low-frequency forced oscillation technique (13, 30). Consistent with the major histological changes affecting the parenchyma, no differences in baseline resistance (Raw) or the coefficient of tissue damping (G) were observed between mice of the two genotypes (Fig. 4) . However, HckF/F mutant mice showed decreased tissue elastance of the lung tissue (H) at baseline (PEEP = 2 cm H2O) compared with HckY/Y wild-type mice. It is highly likely this pathological stiffening of the lungs at low to medium lung volume directly results from the inflammatory infiltrates, thickening of alveolar septa and areas of atelectasis. These differences disappeared at PEEP lung volumes approaching total lung capacity, as atelectatic areas become reinflated. As the shape of the volume-pressure curve and tissue stiffness for lungs approaching their total capacity more readily reflects the tension skeleton (which spreads throughout the lung in an organized fashion and consists of collagen and elastin fibers), our data suggest that there were no major abnormalities in the axial skeleton of the lungs in HckF/F mutant mice. These clinical features were consistently observed in aging HckF/F mice, which frequently showed signs of respiratory distress as evident by their use of accessory muscles. Notably, the pulmonary changes observed in HckF/F mice are reminiscent of some of the features associated with asthma and chronic obstructive pulmonary disease in humans.
Figure 4.
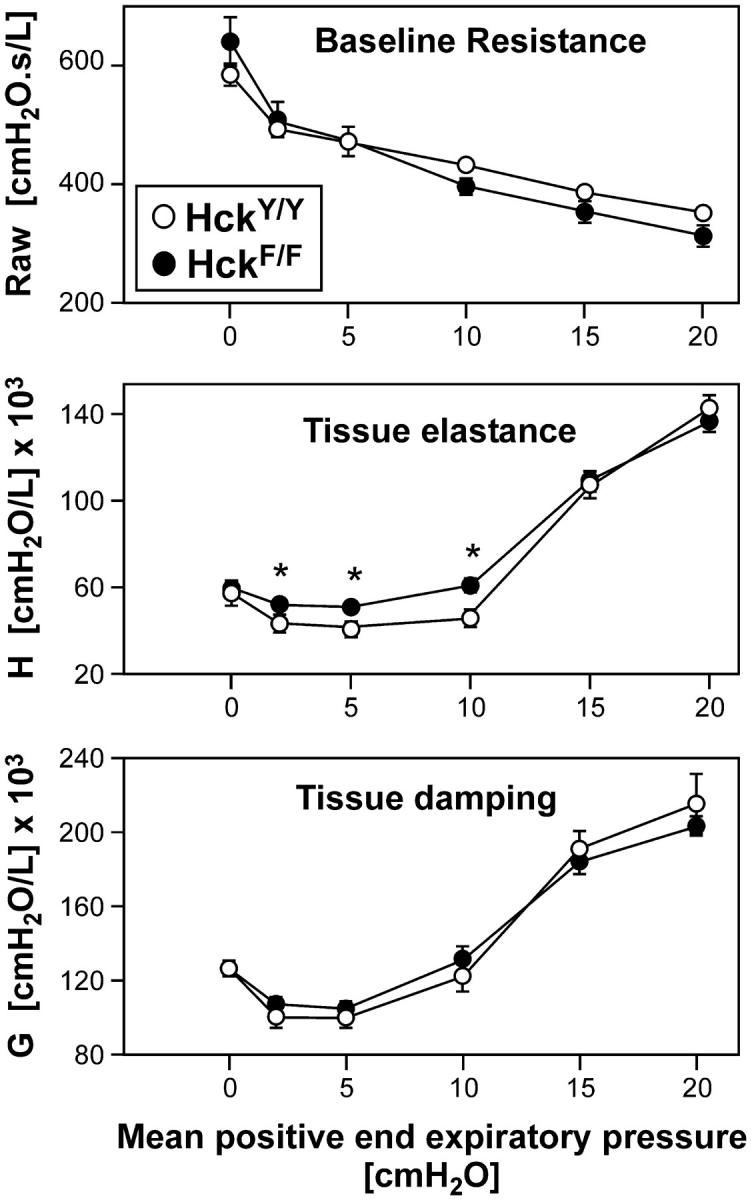
Impaired lung function in HckF/F mice. Lung function analysis by oscillatory mechanics indicated that despite intense inflammation and incipient emphysema, HckF/F mice did not have increased baseline resistance (Raw) or marked altered tissue damping (G) when compared with wild-type mice. However, mutant lungs were stiffer (increased tissue elastance, H), particularly at low lung volumes, possibly resulting from extensive pulmonary consolidation. Mean ± SD (n = 6); *P < 0.05.
Enhanced Pulmonary Innate Immune Response in HckF/F Mice.
The acquired pulmonary pathology in HckF/F mice and the proposed involvement of Src family kinases in inflammatory reactions (10, 19), suggested the underlying cause of the lung pathology in HckF/F mice is an enhanced innate immune response that results in a mild but chronic pulmonary inflammation process. To test this hypothesis, we elicited an acute and local inflammatory response in the lungs of mice by transnasally instilling LPS. Such a pulmonary challenge with LPS elicits a rapid and transient neutrophilic inflammatory response, followed by macrophage invasion into the alveolar spaces (31). Although no difference between the basal number of neutrophils in the BAL-F of unchallenged HckF/F and HckY/Y mice was observed, the extent of neutrophilic accumulation in BAL-F of LPS-challenged HckY/Y mice, which peaked ∼24 h after challenge, was threefold higher than that seen in BAL-F from identically treated HckF/F mice (Fig. 5 a). By contrast, the number of macrophages and eosinophils in BAL-F from unchallenged HckF/F mice were profoundly elevated in comparison to those detected in BAL-F from unchallenged HckY/Y mice, with macrophages accounting for about one-third of all cells recovered in BAL-F from HckF/F mice. Challenging HckF/F or HckY/Y mice with LPS led to a comparable relative increase (approximately twofold) in the number of macrophages in BAL-F, while the LPS challenge had little effect on the number of eosinophils in BAL-F of mice of either genotype. Interestingly, we observed a transient decrease in the number of macrophages, and to a lesser extent eosinophils, in the BAL-F from mice of either genotype 2 h after LPS challenge, possibly due to their homotypic interactions and increased leukocyte adhesion to the lung epithelium. As macrophages and neutrophils, rather than eosinophils, are traditionally thought to be the major hemopoietic cell types expressing Hck, we compared Hck activity associated with equal numbers of fractionated BAL-F cells of unchallenged HckF/F mice. This analysis revealed approximately threefold higher kinase activity associated with the eosinophil fraction when compared with the lectin-extracted mixture of neutrophils and macrophages (Fig. 5 b), thereby possibly suggesting a cell-autonomous effect of Hck in eosinophils. Furthermore, analysis of BAL-F from LPS-challenged HckF/F mice, but not from similarly challenged HckY/Y mice, revealed a rapid and transient increase in levels of the proinflammatory cytokine TNFα (Fig. 5 c), a finding consistent with the enhanced leukocyte accumulation in the lungs of HckF/F mice.
Figure 5.
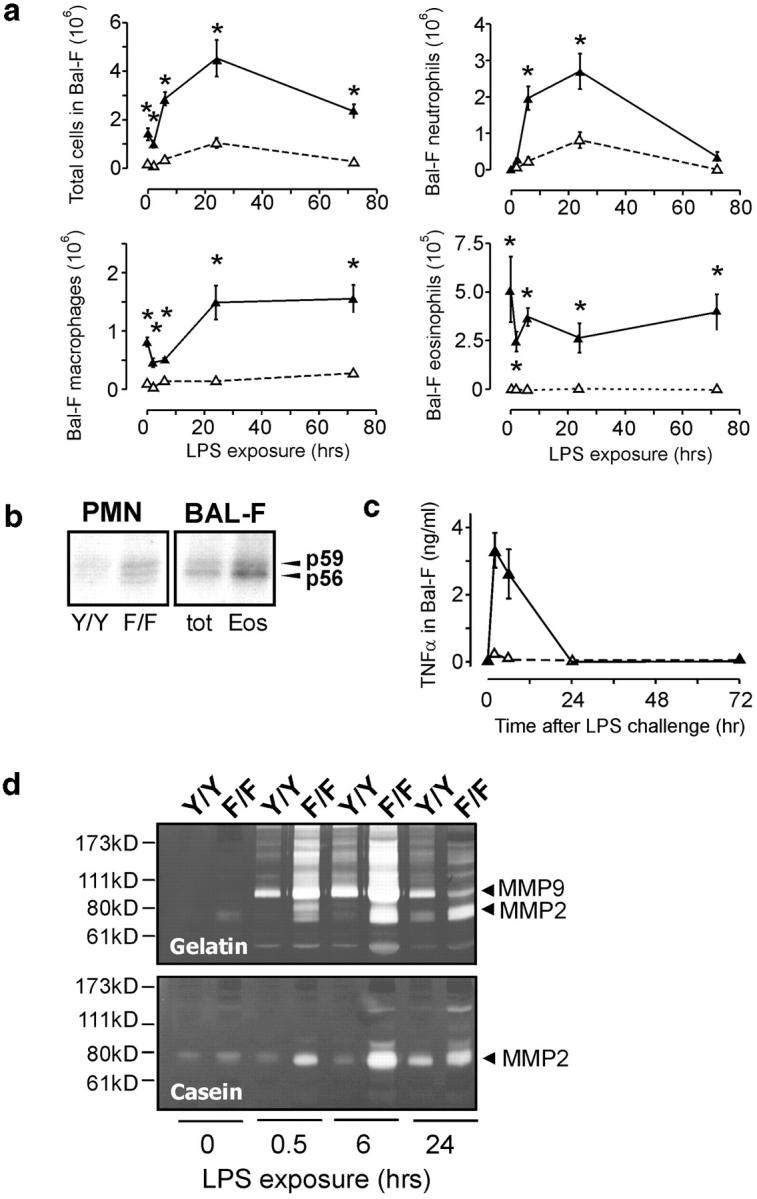
Enhanced pulmonary innate immune response in HckF/F mice. (a) Cellular composition of BAL-F cells from mutant HckF/F (filled symbols) and wild-type HckY/Y mice (open symbols) of 10 wk of age after transnasal challenge with LPS (10 μg/mouse). BAL-F was collected as described in Materials and Methods and cells from eight mice were counted and classified by their morphological appearance in cytospins. Mean ± SD, *P < 0.01. Data were analyzed by oneway analysis of variance (ANOVA) followed by Newman-Kreuls multiple comparison tests. (b) In vitro Hck-kinase activity of leukocytes. PMNs were collected from the bone-marrow of adult HckF/F and HckY/Y mice, whereas total BAL-F cells came only from lungs of unchallenged adult HckF/F mice, as BAL-F of HckY/Y contains negligible numbers of eosinophils. The eosinophil-enriched (Eos) population represented the lectin-bound proportion of total BAL-F cells (tot) as described in Materials and Methods. 500 μg of cell lysate was then immunoprecipitated with an anti-Hck antibody and in vitro autokinase assays were performed in the presence of γ-[32P]-ATP. (c) TNFα levels in BAL-F from LPS-challenged mice. After a single transnasal challenge with LPS (10 μg/mouse), levels of TNFα in BAL-F from HckF/F and HckY/Y mice (three mice of each genotype per experiment) were measured. Filled symbols: HckF/F mice, open symbols: HckY/Y mice. (d) MMP-9 and MMP-2 activity in BAL-F from LPS-challenged mice. After a single transnasal challenge with LPS (10 μg/mouse), BAL-F was recovered from the mice at the indicated time points and MMP-9 and MMP-2 activity was assayed by zymography using gelatin and casein as the respective substrates. Zones of MMP activity appear as unstained bands corresponding to the apparent molecular weights of MMP-2 and MMP-9, respectively.
The sustained presence of inflammatory cells in the lungs of HckF/F mice is likely to trigger enhanced turn-over of pulmonary tissue, possibly culminating in emphysema. As metalloproteinases (MMPs) are critical components in the continual remodeling of alveolar tissues and are implicated in emphysema and leukocyte diapedesis, we used zymography to investigate MMP-2 and MMP-9 activity in BAL-F from LPS-challenged mice. Analysis of BAL-F from LPS-challenged HckF/F mice revealed exaggerated induction of MMP-2 and MMP-9 activity when compared with that detected in BAL-F from LPS-challenged HckY/Y mice (Fig. 5 d). Similarly, serine protease activity, which largely reflects neutrophil elastase activity, was elevated in BAL-F from HckF/F mutant mice when compared with HckY/Y mice (unpublished data).
Increased Sensitivity of HckF/F Mice to Systemic Endotoxemia.
Although there has been conflicting evidence for the redundant involvement of Hck, Lyn, and Fgr in the endotoxic response (10, 19), our data clearly indicated an enhanced sensitivity to LPS in the lungs of mice expressing a constitutively active form of Hck. To extend this observation, we monitored the sensitivity of HckF/F mice to endotoxemia after systemic administration of a single dose of LPS. A dose–response survival study following age-, weight-, and genetic background-matched adult mice over 5 d revealed survival of more than 50% of HckY/Y mice treated with 24 mg/kg of LPS. By contrast, HckF/F mice showed 80% mortality at this dose of LPS, while administration of 12 mg/kg of LPS resulted in ∼50% mortality in these mice (Fig. 6 a). To characterize altered susceptibility of HckF/F mice to sublethal doses of LPS, we attempted to objectively monitor LPS-associated illness in terms of changes in the body weight and core body temperature registered with an implanted telemetric thermometer device. After a single challenge with LPS, surviving HckF/F mice showed exacerbated weight loss and hypothermia (which is a rodent-specific response to LPS; Fig. 6 b), as well as profoundly elevated levels of circulating TNFα 6 h post LPS (8 mg/kg) administration (30.8 ng/ml ± 13.2 ng/ml in HckF/F mice versus 1.5 ng/ml ± 1.3 ng/ml in HckY/Y mice).
Figure 6.

Enhanced susceptibility of HckF/F mice to systemic LPS-challenges. (a) Reduced survival of HckF/F mutant mice compared with HckY/Y wild-type mice after a single intraperitoneal injection of LPS at 6 mg/kg (▾); 12 mg/kg (▪); 16 mg/kg (▴) or 24 mg/kg (•) and a 5 d observation period for survival. Five mice were used per treatment group. (b) Enhanced morbidity of HckF/F mice in response to administration of sublethal amounts of LPS. 3 d before LPS administration, a telemetric temperature-measuring device was implanted subcutaneously in the back of mice and temperature and weight recorded at the indicated time points after a single intraperitoneal injection of LPS at 2 mg/kg (U); 4 mg/kg (Δ); 8 mg/kg (○) or vehicle (♦). Each curve represents the median of five mice per group, except for the 8 mg/kg HckF/F group which only depicts the results from the three surviving mice.
Elevated LPS-responsiveness of HckF/F Macrophages In Vitro.
The above in vivo data suggested that the enhanced susceptibility of HckF/F mice to LPS might be a consequence of the hyperactive nature of their leukocytes, in particular macrophages and neutrophils. We therefore analyzed biological markers that correlate with the activation of BMDMs by LPS, including the release of the anti-microbial agent nitric oxide (NO) and the pro-inflammatory cytokine TNFα. We observed that BMDMs derived from HckF/F mice generally showed enhanced sensitivity to low concentrations of LPS, irrespective of whether the cells had been primed with IFN-γ, and that the response of mutant BMDMs to higher concentrations of LPS was enhanced by two to threefold (Fig. 7 , a and b). The increased release of NO into the culture medium of LPS-stimulated BMDMs from HckF/F mice is likely to be explained by the increased expression of inducible nitric oxide synthetase (iNOS; Fig. 7 c). Comparison of these activation parameters between naive, unprimed BMDMs from HckF/F mice and IFN-γ–primed BMDMs from HckY/Y mice suggested that the expression of constitutively active HckF might result in cell-autonomous priming of BMDMs. Consistent with this hypothesis, the spontaneous release of NO or TNFα by BMDMs from HckF/F mice was not detected (unpublished data).
Figure 7.
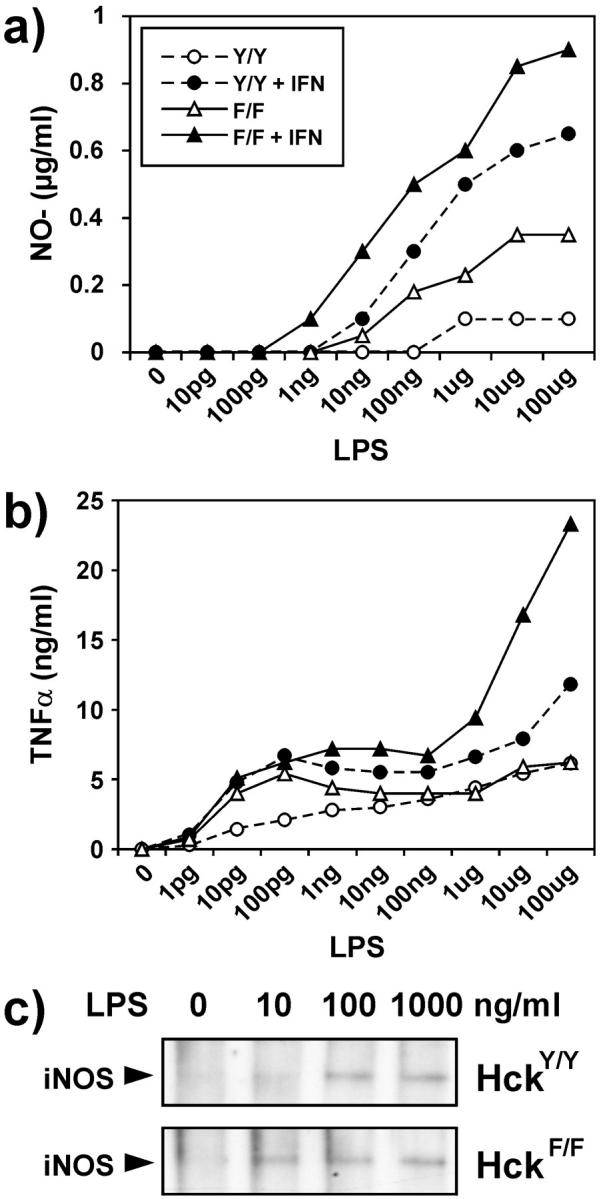
Exaggerated in vitro response of BMDMs to LPS. (a and b) Release of NO2 − (a) and TNFα (b) by BMDMs, derived from 8-wk-old HckF/F and HckY/Y mice and exposed to the indicated amount of LPS for 48 h. Where indicated, the cultures were primed with IFN-γ (15 U/ml) 2 h before LPS treatment. Values are the median of quadruplicate cultures. (c) Induction of nitric oxide synthetase protein (iNOS) in LPS-treated BMDMs of HckF/F and HckY/Y mice was assessed by Western blotting of 50 μg of total cell lysates after a 24-h stimulation with LPS at the indicated concentration.
Enhanced Phagocytosis by HckF/F Macrophages.
The phagocytic activity of macrophages is crucial for the clearance of leukocytes at sites of inflammation and tissue injury. Electron microscopic analysis of alveolar macrophages in HckF/F mice revealed the abundant presence of spiculate crystalline matter highly reminiscent of Charcot-Leyden crystals (32) within the cells (Fig. 8 , a and b). Similarly, histological signs of erythro-phagocytosis were also abundantly apparent in alveolar macrophages (Fig. 8 c) and circulating peripheral blood monocytes from HckF/F mice, which coincided with mild reticulocytosis in these mice (unpublished data). Thus, we assessed the in vivo half-life of 51Cr-labeled erythrocytes in both HckF/F and HckY/Y control mice. As shown in Fig. 8 d, the extrapolated half-life of erythrocytes in HckY/Y mice was ∼31 d, whereas the half-life of erythrocytes in HckF/F mice was ∼22 d. Collectively, these observations suggested that monocytes and macrophages in HckF/F mice have enhanced phagocytic activity. Indeed, when compared with BMDMs derived from HckY/Y mice, BMDMs from HckF/F mice exhibited an enhanced ability to phagocytose nonopsonized sheep red blood cells (SRBCs; Fig. 8 e). By contrast, no difference in the phagocytic activity of BMDMs from wild-type and HckF/F mice toward opsonized SRBCs was detected, a finding consistent with an earlier report that Fc-receptor-mediated uptake of particles is Hck-independent mice (9).
Figure 8.


Enhanced phagocytic activity of HckF/F macrophages. (a–c) Electron micrographs of alveolar macrophages from HckF/F mice showing the presence of eosinophilic crystals (a, original magnification ×25,000), which are often associated with secondary lysosomes (b; arrowhead, original magnification ×50,500), and frequently contain ingested erythrocytes (c; arrowhead, original magnification ×25,000). (d) Half-life of erythrocytes in HckF/F and HckY/Y mice. Levels of 51Cr-labeled erythrocytes in the peripheral blood of HckF/F and HckY/Y mice were determined at the indicated time points after transfusion. The amount of radioactivity associated with erythrocyte 30 min after transfusion was taken as 100%. The data represent means ± SD (n = 10). (e) Phagocytosis of SRBCs by BMDMs. Nonopsonized or anti-SRBC antiserum opsonized 51Cr-labeled SRBC were added to cultures of BMDMs derived from HckF/F or HckY/Y mice. After a 2-h incubation, the cultures were washed extensively and radioactivity released after lysis of the BMDMs was expressed as a proportion of total radioactivity added to each well. Data represent mean ± SD of quadruplicate cultures from a representative experiment.
Enhanced Activity of Neutrophils from HckF/F Mice Is Dependent on Cell Adhesion.
The enhanced innate immune response of leukocytes observed after LPS administration to HckF/F mice, and the functional association of Hck with leukocyte integrins prompted us to investigate the adhesive and migratory properties of isolated polymorphic neutrophils (PMNs) on the β2-integrin ligand fibrinogen. When assayed in Boyden chambers, PMNs derived from HckF/F mice had a modestly enhanced migratory capacity when compared with PMNs from wild-type mice (unpublished data). However, this difference became more pronounced when the capacity of the PMNs to migrate along a gradient of the chemotactic peptide fMLP was assessed (Fig. 9 a). Similarly, BMDMs derived from HckF/F mice exhibited an enhanced capacity to migrate when tested in in vitro “wound-healing” assays (unpublished data).
Figure 9.
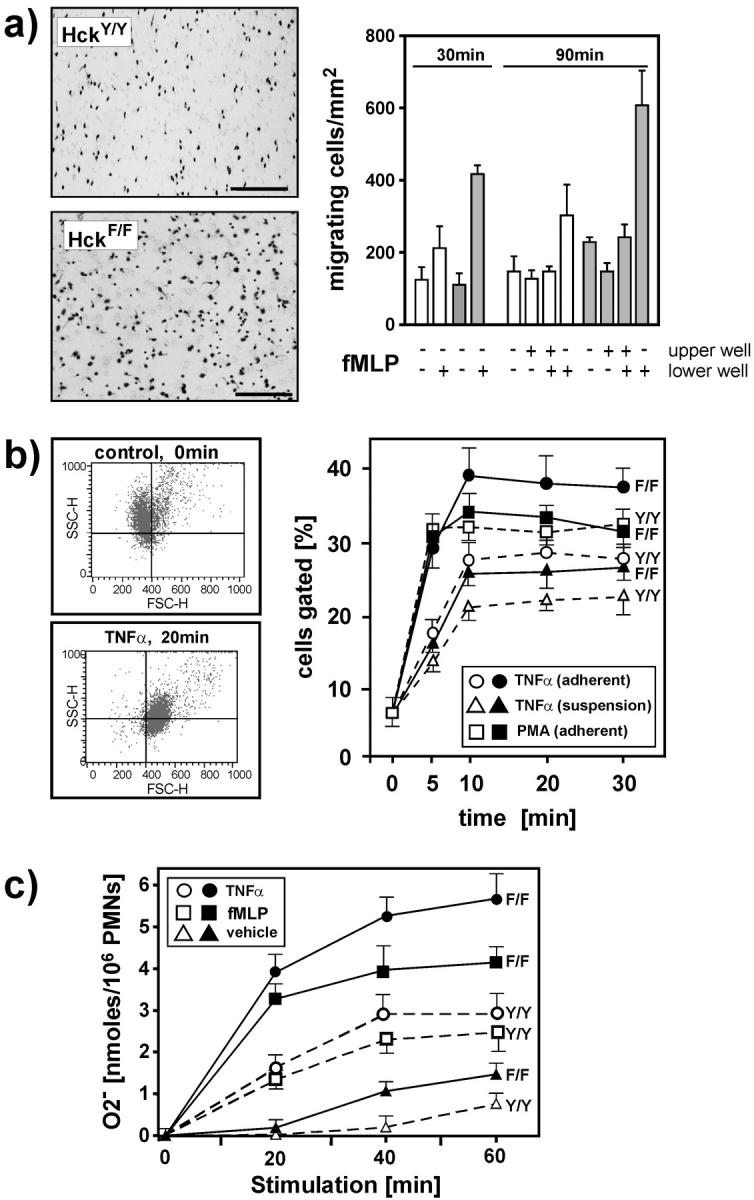
Enhanced adhesion-dependent activity of HckF/F PMNs in vitro. (a) PMNs from HckF/F (black bars) and HckY/Y (white bars) mice were seeded in the top well of a modified Boyden chamber containing a fibronectin-coated insert studded with 8-μM pores. The chemoattractant fMLP (200 nM) as added to lower and/or upper well as indicated. After incubation at 37°C for the indicated periods of time, the inserts were removed, stained with Giemsa and the number of PMNs that had migrated to the underside of the inserts (left panels) were quantified by counting the number of cells in five separate fields on two inserts per time point (right panel). Mean ± SD. (b) Activation and degranulation response of Gr-1–positive PMNs as determined by FACS® analysis. HckY/Y (white symbols) and HckF/F PMNs (black symbols) were either preincubated for 30 min at 37°C in fibrinogen-coated 24-well plates (“adherent”) or directly stimulated (“suspension”) with TNFα (20 ng/ml) or PMA (1 mg/ml). Cells were fixed at the indicated time points, stained for Gr-1, and changes in forward scatter (FSC) and side scatter (SSC) profiles was analyzed (left panels). The percentage of Gr-1hi cells with reduced SSC (“degranulation”) and increased FSC (“activation”) were gated in the bottom right quadrant. The data shown are shown from a representative experiment (right panel). (c) Generation of O2 − is exaggerated in HckF/F PMNs. PMNs from HckY/Y (white symbols) and HckF/F (black symbols) mice were seeded into fibrinogen-coated 96-well plates. The cells were stimulated with TNFα (20 ng/ml), fMLP (200 nM), or vehicle and incubated at 37°C for the indicated periods of time. O2 − production was then measured as described in Materials and Methods. The data represents means ± SD from quadruplicate cultures.
The pronounced endotoxemic response of HckF/F mice to LPS is a likely consequence of enhanced activation and degranulation of PMNs. We therefore undertook FACS® analysis of bone marrow cells to characterize the in vitro activation of PMNs from HckF/F and HckY/Y mice by TNFα. Cytokine-dependent PMN activation (and priming) typically results in an increase in cell size (as evidenced by an increase in “forward scatter”), whereas degranulation results in a decrease in “side scatter” (20). By arbitrarily setting the FACS® gates so that less than 10% of all unstimulated, Gr1hi cells (i.e., PMNs) of either genotype fell within the lower right quadrant, we were able to monitor TNFα activation of PMNs by quantifying the increase in the proportion of PMNs (Fig. 9 b, left panel). When preadhered to fibrinogen-coated plates, this response was markedly enhanced in PMNs isolated from HckF/F mice when compared with cells obtained from HckY/Y mice (Fig. 9 b, right panel). By contrast, the difference in the activation response was less pronounced between PMNs isolated from HckF/F and HckY/Y mice when the cells were kept in suspension during activation with TNFα. Similarly, PMN degranulation induced by PMA, which is independent of cell adhesion to extracellular matrix proteins (33), was indistinguishable between cells derived from HckF/F or HckY/Y mice (Fig. 9 b). Finally, the ability of PMNs isolated from HckF/F and wild-type mice to elicit cytokine-induced superoxide (O2 −) production, an adhesion-dependent process (34), was investigated. As revealed in Fig. 9 c, PMNs from HckF/F mice released approximately twofold more superoxide in response to TNFα or fMLP stimulation than did their wild-type counterparts when plated on fibrinogen-coated microtiter plates. By contrast, no significant difference in superoxide production was observed between PMNs of the two genotypes when plated on uncoated microtiter plates (unpublished data). Collectively, these data suggest latent priming of PMNs from HckF/F mice which results in their enhanced adhesion-dependent activation in response to inflammatory cytokines such as TNFα.
Discussion
To define the physiological function(s) of the Src family kinase Hck, we have used a “knock-in” gene targeting strategy to introduce a hypermorphic, “gain-of-function” mutation into the hck allele in the germline of mice. This approach ensured that all the cis-acting elements of the hck gene, which collectively serve to regulate its temporal and spatial expression patterns, remained intact. Thus, we can confidently attribute all the phenotypic alterations in the resulting HckF/F mice to deregulated Hck kinase activity rather than to the aberrant and/or ectopic expression of a transgene, which is a limitation of traditional transgenic “gain-of-function” mouse models. The latter point is significant in light of the finding that transgenic expression of constitutively active mutants of the SFK c-Src (35, 36), Lck (37), or Blk (38) result in tumorigenesis at the site of transgene expression. By contrast, we find no evidence for cellular transformation as a primary consequence of introducing a constitutively activating mutation into the endogenous loci of the murine hck (herein) or lyn genes (26), although we cannot exclude that Hck activity in the HckF/F knock-in mice may be below that required for tumor formation. Remarkably, deregulated Hck kinase activity resulted in a plethora of Hck-specific cellular activities and patho-physiological responses that were hitherto masked by functional redundancy among SFKs in Hck “loss-of-function” mutant mice.
Requirement for Tight Control of Hck Activity.
Our biochemical analysis of BMDMs and neutrophils isolated from HckF/F mice revealed only a small increase in net-tyrosine phosphorylation of endogenous proteins and a relatively modest increase in cellular activation of leukocytes following acute in vitro stimulation. However, the highly reproducible occurrence and progression of the pulmonary disease observed in HckF/F mice illustrates the severe patho-physiological consequences arising from the cumulative effects of apparently subtle alterations in the innate immune response. We therefore propose that the careful and tight control of the catalytic activity of Hck is a critical determinant in preventing the destruction of host organs (e.g., lung) by an inappropriate innate immune response (e.g., chronic inflammation). Unlike tyrosine phosphorylation-dependent signal transduction events emanating from growth factor receptors that are often terminated by internalization of the ligand-bound receptor complex, integrin-mediated “outside-in” signaling may be limited through the inactivation of integrin-associated tyrosine kinases (e.g., SFKs). The catalytic activity of the SFK Hck for example, is tightly regulated by the coordinated phosphorylation of the key regulatory Y388 and Y499 residues, which ensure that temporal changes in kinase activity remain within physiological levels. Our analysis of HckF-expressing cells suggests that the cellular machinery attempts to counteract the inappropriately activated tyrosine kinase by several mechanisms: (a) reducing the steady-state levels of hck F transcripts; (b) reducing the expression level of the HckF protein (possibly via an ubiquitin- and proteasome-dependent degradation pathway (39); and (c) by activating PTPs capable of inactivating Hck. The finding that HckF is not significantly tyrosine phosphorylated in BMDM derived from HckF/F mice and that treatment of these BMDMs with the PTP inhibitor pervanadate enhanced tyrosine phosphorylation of cellular proteins argues that PTPs play a significant role in suppressing the catalytic (signal transducing) activity of the mutant HckF. Intriguingly, mice homozygous for the mev hypomorphic allele of SHP-1 exhibit spontaneous consolidation of the lung with myeloid cell infiltrates independent of T and B lymphocytes (40, 41) that are remarkably similar in appearance to the leukocyte infiltrates observed in the lungs of HckF/F mice. In addition, BMDMs isolated from mev/v mice adhere and spread to a greater extent through β2-integrin mediated contacts than do their wild-type counterparts (42), and our preliminary observations suggest hyperphosphorylation (and activation) of SHP-1 in HckF/F cells (unpublished data). Similarly, the lack of CD45, which dephosphorylates the autocatalytic Y-residue of Hck and Lyn during integrin mediated-adhesion, results in enhanced adhesion of BMDMs in vitro (43).
Exaggerated Innate Immune Response in HckF/F Mice.
Many of the current gene knock-out models for asthma suggest that T cells are the main determining factor for establishing airway responsiveness and that alterations in the TH1/TH2 balance, irrespective of exogenous factors, may affect airway behavior. Here, we provide evidence that baseline abnormalities in lung function can also result as the cumulative consequence of a mild imbalance in the innate immune response. The abundance of eosinophils, mucus cell hyperplasia and elevated levels of IL-5 and eotaxin in the lungs of HckF/F mice are traits typically associated with atopic asthma in humans, a chronic inflammatory disorder of the airways that is associated with reversible airway obstruction and bronchial hyper-responsiveness. Macrophage activation is a frequent but underestimated feature of chronic asthma (44). As macrophages produce eotaxin (45) and through their TGFα release can induce cytokine and chemokine secretion by neighboring cells (e.g., epithelium), we propose that the underlying cause of the asthma-like pathology observed in HckF/F mice is most likely the consequence of the hyperresponsive nature of macrophages within the mice. Thus, the pulmonary response resulting from disregulated tyrosine phosphorylation in the myeloid cell compartment, and the associated exacerbation of the inflammatory response, is distinct from those observed in mouse models where airway hyperreactivity is due to genetic manipulation of the cytokine network, including mutants for IL-4 (46, 47), IL-5 (48), IL-9 (49), IL-13 (46, 50), eotaxin (51), as well as GM-CSF (52) and its downstream transcription factor PU.1 (53). In particular, the distinctive accumulation of eosinophilic material together with Charcot-Leyden crystals in the alveolar compartments is an unusual observation, even in mice that have intense eosinophilic inflammation induced by helminthic parasites or allergen inhalation.
An overwhelming systemic inflammatory response to endotoxin (i.e., LPS) is central to the pathogenesis (e.g., tissue damage, multi-organ failure, and death) of septic shock. Macrophage-derived pro-inflammatory cytokines (e.g., TNFα, IL1β, and GM-CSF) as well as nitric oxide, are critical mediators of endotoxic shock (54). Adhesion of PMNs to the vascular endothelium and their extravasation into the underlying tissue is necessary for endotoxic shock as mice deficient for ICAM-1 are resistant to LPS-induced toxic shock (55). Notably, Hck−/−Fgr−/− compound mutant mice are likewise resistant to LPS-induced toxic shock (10). Thus, the enhanced susceptibility of HckF/F mice to systemic administration of LPS is likely to be the consequence of constitutive priming of the leukocyte population.
Constitutive Priming of HckF/F Leukocytes?
Previous in vitro analysis of PEMs and BMDMs derived from Hck−/− Fgr−/− and Hck−/−Fgr−/−Lyn−/− compound mutant mice have functionally placed these SFKs downstream of leukocyte integrins (i.e., β1- and β2/β3-integrins in BMDMs and PEMs, respectively; see references 19 and 56), and upstream of the actin cytoskeleton-associated proteins paxillin, Pyk2, cortactin, and the signaling proteins Syk, c-Cbl, and PI 3-kinase (57). Accordingly, BMDMs derived from these compound mutant mice exhibit a defect in cell adhesion and migration (19, 57). Similarly, PEMs from Hck−/−Fgr−/− Lyn−/− compound mutant mice are defective in adhesion-dependent priming, culminating in impaired respiratory burst and degranulation responses to pro-inflammatory cytokines, such as TNFα (15, 56). Although it has been reported that Hck localizes to secretory granules in human PMNs and thus might potentially regulate the degranulation process (58), TNFα-induced degranulation of PMNs from HckF/F mice was still a cell adhesion–dependent process. Thus, we hypothesize that constitutive activation of Hck partially mimics cytokine (e.g., INF-γ) mediated leukocyte priming, characterized by integrin clustering (“inside-out” signaling) and increased avidity for ligands (e.g., intercellular adhesion molecule 1 [ICAM-1]) on endothelial cells. In vivo this is likely to result in enhanced tissue invasion by innate cells in response to an inflammatory stimulus (e.g., bacterial infection), as β2-integrin mediated adhesion of leukocytes to the endothelium is an important initiation signal for diapedesis. Indeed, mutations that impair the function of the β-subunit of the leukocyte integrins predispose individuals to life-threatening infections (59).
Although previous studies of “loss-of-function” models for SFKs identified circulating macrophages and PMNs as the major cell types susceptible to impairments in Hck-mediated signaling, our study suggests that this kinase also serves a major function in eosinophils. It is well established that Mac-1 (αmβ2; CD11b/CD18 integrin) mediated cell adhesion is important for eosinophil activation, degranulation, and the secretion (paracrine and autocrine) of eosinophilopoietic factors (60, 61). Hence, β2-integrin–mediated priming of eosinophils, either by IL-5–mediated cell adhesion, ligation of Mac-1 or addition of recombinant ICAM-1, triggered Y-phosphorylation of cbl, paxilin, enhanced cellular degranulation (61), and prolonged eosinophil survival in vitro (62). Thus, these observations imply that hyperactive HckF protein may also result in “constitutive” eosinophil priming, associated with enhanced cellular adhesion and degranulation.
Mutations in proteins involved in balancing (adhesion-dependent) cellular tyrosine phosphorylation in myeloid lineage cells (e.g., SHP1, CD45, SHIP, and CSK) often result in severe pathologies and associated reduction in the life spans of the corresponding mutant mice (40, 63–65). By contrast, dysregulated tyrosine phosphorylation in eosinophils, PMNs, and macrophages caused by the presence of the HckY499F mutation, appears not to affect the overall life span of HckF/F mice. Based on documented evidence for rarely occurring activating somatic nonsense mutations in the COOH-terminal end of human c-Src (66, 67), it is tempting to speculate that the hck gene might represent a potential candidate susceptibility locus in addition to those associated with allergy and bronchial hyperresponsiveness on chromosome 5q31-q33 (68).
Acknowledgments
We are grateful to all the former members of the Dunn laboratory for their valuable input into this work. Jessica Jones and Dianne Grail are thanked for expert technical assistance, Val Feakes for histological support, and Janna Stickland for photography.
Footnotes
Abbreviations used in this paper: BAL-F, bronchoalveloar lavage fluid; BMDM, bone marrow–derived macrophage; ES, embryonic stem; F, phenylalanine; fMLP, N-formyl-methionyl-leucyl-phenylalanine; MMP, metalloproteinase; NO, nitric oxide; PEEP, positive end expiratory pressure; PTP, protein tyrosine phosphatase; SFK, Src family kinases; SRBC, sheep red blood cell; Y, tyrosine.
References
- 1.Kefalas, P., T.R. Brown, and P.M. Bricknell. 1995. Signalling by the p60c-src family of protein-tyrosine kinases. Int. J. Biochem. Cell Biol. 27:551–563. [DOI] [PubMed] [Google Scholar]
- 2.Sicheri, F., and J. Kuriyan. 1997. Structures of Src-family tyrosine kinases. Curr. Opin. Struct. Biol. 7:777–785. [DOI] [PubMed] [Google Scholar]
- 3.Cartwright, C.A., W. Eckhart, S. Simon, and P.L. Kaplan. 1987. Cell transformation by pp60c-src mutated in the carboxy-terminal regulatory domain. Cell. 49:83–91. [DOI] [PubMed] [Google Scholar]
- 4.Kmiecik, T.T., and D. Shalloway. 1987. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 49:65–73. [DOI] [PubMed] [Google Scholar]
- 5.Piwnica-Worms, H., K.B. Saunders, T.M. Roberts, A.E. Smith, and S.H. Cheng. 1987. Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell. 49:75–82. [DOI] [PubMed] [Google Scholar]
- 6.Kawakami, T., Y. Kawakami, S.A. Aaronson, and K.C. Robbin. 1988. Acquisition of transforming properties by FYN, a normal SRC-related human gene. Proc. Natl. Acad. Sci. USA. 85:3870–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amrein, K.E., and B.M. Sefton. 1988. Mutation of a site of tyrosine phosphorylation in the lymphocyte-specific tyrosine protein kinase, p56lck, reveals its oncogenic potential in fibroblasts. Proc. Natl. Acad. Sci. USA. 85:4247-4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziegler, S.F., S.D. Levin, and R.M. Perlmutter. 1989. Transformation of NIH 3T3 fibroblasts by an activated form of p59hck. Mol. Cell. Biol. 9:2724–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowell, C.A., P. Soriano, and H.E. Varmus. 1994. Functional overlap in the src gene family: inactivation of hck and fgr impairs natural immunity. Genes Dev. 8:387–398. [DOI] [PubMed] [Google Scholar]
- 10.Lowell, C.A., and G. Berton. 1998. Resistance to endotoxic shock and reduced neutrophil migration in mice deficient for the Src-family kinases Hck and Fgr. Proc. Natl. Acad. Sci. USA. 95:7580–7584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ernst, M., D.P. Gearing, and A.R. Dunn. 1994. Functional and biochemical association of hck with the LIF/IL-6 receptor signal transducing subunit gp130 in embryonic stem cells. EMBO J. 7:1574–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coyle, A.J., G. Le Gros, C. Bertrand, S. Tsuyuki, C.H. Heusser, M. Kopf, and G.P. Anderson. 1995. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am. J. Respir. Cell Mol. Biol. 13:54–59. [DOI] [PubMed] [Google Scholar]
- 13.Both, N.J., E. Kwak, and E. Klootwijk-van Dijke. 1980. Erythrocyte production and survival in Rauscher murine leukemia virus-infected BALB/c mice. Cancer Res. 40:4270–4275. [PubMed] [Google Scholar]
- 14.Lowell, C.A., L. Fumagalli, and G. Berton. 1996. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J. Cell Biol. 133:895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinagawa, K., and G.P. Anderson. 2000. Rapid isolation of homogeneous murine bronchoalveolar lavage fluid eosinophils by differential lectin affinity interaction and negative selection. J. Immunol. Methods. 237:65–72. [DOI] [PubMed] [Google Scholar]
- 16.Wienands, J., O. Larbolette, and M. Reth. 1996. Evidence for a preformed transducer complex organized by the B cell antigen receptor. Proc. Natl. Acad. Sci. USA. 93:7865–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falk, W., R.H. Goodwin, and E.J. Leonard. 1980. A 48-well micro chemotaxis assembly for rapid and accurate measurement of leukocyte migration. J. Immunol. Methods. 33:239–247. [DOI] [PubMed] [Google Scholar]
- 18.Meng, F., and C.A. Lowell. 1997. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 185:1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tkalcevic, J., M. Novelli, M. Phylactides, J.P. Iredale, A.E. Segal, and J. Roes. 2000. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 12:201–210. [DOI] [PubMed] [Google Scholar]
- 20.Stuehr, D., and C.F. Nathan. 1989. Nitric oxide: A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med. 169:1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berton, G., C. Laudanna, C. Sorio, and F. Rossi. 1992. Generation of signals activating neutrophil functions by leukocyte integrins: LFA-1 and gp150/95, but not CR3, are able to stimulate the respiratory burst of human neutrophils. J. Cell Biol. 116:1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cartledge, K., A.R. Dunn, and G. Scholz. 2000. Generation and characterization of monoclonal antibodies to the Src-family kinase Hck. Hybridoma. 19:323–330. [DOI] [PubMed] [Google Scholar]
- 23.Ernst, M., A. Oates, and A.R. Dunn. 1996. gp130-mediated signal transduction in ES cells involves activation of Jak and ras/MAP kinase pathways. J. Biol. Chem. 271:30136–30143. [DOI] [PubMed] [Google Scholar]
- 24.Coleman, D.L., J.A. Chodakewitz, A.H. Bartiss, and J.W. Mellors. 1988. Granulocyte-macrophage colony-stimulating factor enhances selective effector functions of tissue-derived macrophages. Blood. 72:573–578. [PubMed] [Google Scholar]
- 25.Scholz, G., K. Kartledge, and A.R. Dunn. 2000. Hck enhances the adherence of lipopolysaccharide-stimulated macrophages via Cbl and phosphatidylinositol 3-kinase. J. Biol. Chem. 275:14615–14623. [DOI] [PubMed] [Google Scholar]
- 26.Harder, K.W., L.M. Parsons, J. Armes, N. Evans, N. Kountouri, R. Clark, C. Quilici, D. Grail, G.S. Hodgson, A.R. Dunn, and M.L. Hibbs. 2001. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 15:603–615. [DOI] [PubMed] [Google Scholar]
- 27.Clutterbuck, E., J.G. Shields, J. Gordon, S.H. Smith, A. Boyd, R.E. Callard, H.D. Campbell, I.G. Young, and C.J. Sanderson. 1987. Recombinant human interleukin 5 is an eosinophil differentiation factor but has no activity in standard human B cell growth factor assays. Eur. J. Immunol. 17:1743–1750. [DOI] [PubMed] [Google Scholar]
- 28.Rothenberg, M.E., J.A. MacLean, E. Pearlman, A.D. Luster, and P. Leder. 1997. Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J. Exp. Med. 185:785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hantos, Z., B. Daroczy, B. Suki, S. Nagy, and J.J. Fredberg. 1992. Input impedance and peripheral inhomogeneity of dog lungs. J. Appl. Physiol. 72:168–178. [DOI] [PubMed] [Google Scholar]
- 30.Sly, P.D., M.J. Hayden, F. Petak, and Z. Hantos. 1996. Measurement of low-frequency respiratory impedance in infants. Am. J. Respir. Crit. Care Med. 154:161–166. [DOI] [PubMed] [Google Scholar]
- 31.Xing, Z., J. Gauldie, G. Cox, H. Baumann, M. Jordana, X.F. Lei, and M.K. Achong. 1998. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Invest. 101:311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo, L., R.S. Johnson, and J.C. Schuh. 2000. Biochemical characterization of endogenously formed eosinophilic crystals in the lungs of mice. J. Biol. Chem. 275:8032–8037. [DOI] [PubMed] [Google Scholar]
- 33.Fuortes, M., W.W. Jin, and C. Nathan. 1994. Beta 2 integrin-dependent tyrosine phosphorylation of paxillin in human neutrophils treated with tumor necrosis factor. J. Cell Biol. 127:1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sacks, T., C.F. Moldow, P.R. Craddock, T.K. Bowers, and H.S. Jacob. 1978. Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. An in vitro model of immune vascular damage. J. Clin. Invest. 61:1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weissenberger, J., J.P. Steinbach, G. Malin, S. Spada, T. Rulicke, and A. Aguzzi. 1997. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene. 14:2005–2013. [DOI] [PubMed] [Google Scholar]
- 36.Webster, M.A., R.D. Cardiff, and W.J. Muller. 1995. Induction of mammary epithelial hyperplasias and mammary tumors in transgenic mice expressing a murine mammary tumor virus/activated c-src fusion gene. Proc. Natl. Acad. Sci. USA. 92:7849–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abraham, K.M., S.D. Levin, J.D. Marth, K.A. Forbush, and R.M. Perlmutter. 1991. Thymic tumorigenesis induced by overexpression of p56lck. Proc. Natl. Acad. Sci. USA. 88:3977–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malek, S.N., D.I. Dordai, J. Reim, H. Dintzis, and S. Desiderio. 1998. Malignant transformation of early lymphoid progenitors in mice expressing an activated Blk tyrosine kinase. Proc. Natl. Acad. Sci. USA. 95:7351–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hakak, Y., and G.S. Martin. 1999. Ubiquitin-dependent degradation of active Src. Curr. Biol. 9:1039–1042. [DOI] [PubMed] [Google Scholar]
- 40.Shultz, L.D., D.R. Coman, C.L. Bailey, W.G. Beamer, and C.L. Sidman. 1984. “Viable motheaten,” a new allele at the motheaten locus. I. Pathology. Am. J. Pathol. 116:179–192. [PMC free article] [PubMed] [Google Scholar]
- 41.Yu, C.C., H.W. Tsui, B.Y. Ngan, M.J. Shulman, G.E. Wu, and F.W. Tsui. 1996. B and T cells are not required for the viable motheaten phenotype. J. Exp. Med. 183:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roach, T.I., S.E. Slater, L.S. White, X. Zhang, P.W. Majerus, E.J. Brown, and M.L. Thomas. 1998. The protein tyrosine phosphatase SHP-1 regulates integrin-mediated adhesion of macrophages. Curr. Biol. 8:1035–1038. [DOI] [PubMed] [Google Scholar]
- 43.Roach, T., S. Slater, M. Koval, L. White, E.C. McFarland, M. Okumura, M. Thomas, and E. Brown. 1997. CD45 regulates Src family member kinase activity associated with macrophage integrin-mediated adhesion. Curr. Biol. 7:408–417. [DOI] [PubMed] [Google Scholar]
- 44.Hallsworth, M.P., C.P. Soh, S.J. Lane, J.P. Arm, and T.H. Lee. 1994. Selective enhancement of GM-CSF, TNF-alpha, IL-1 beta and IL-8 production by monocytes and macrophages of asthmatic subjects. Eur. Respir. J. 7:1096–1102. [PubMed] [Google Scholar]
- 45.Watanabe, K., P.J. Jose, and S.M. Rankin. 2002. Eotaxin-2 generation is differentially regulated by lipopolysaccharide and IL-4 in monocytes and macrophages. J. Immunol. 168:1911–1918. [DOI] [PubMed] [Google Scholar]
- 46.Grunig, G., M. Warnock, A.E. Wakil, R. Venkayya, F. Brombacher, D.M. Rennick, D. Sheppard, M. Mohrs, D.D. Donaldson, R.M. Locksley, and D.B. Corry. 1998. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 282:2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gavett, S.H., D.J. O'Hearn, C.L. Karp, E.A. Patel, B.H. Schofield, F.D. Finkelman, and M. Wills-Karp. 1997. Interleukin-4 receptor blockade prevents airway responses induced by antigen challenge in mice. Am. J. Physiol. 272:L253–L261. [DOI] [PubMed] [Google Scholar]
- 48.Foster, P.S., S.P. Hogan, A.J. Ramsay, K.I. Matthaei, and I.G. Young. 1996. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 183:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Temann, U., G.P. Geba, J.A. Rankin, and R.A. Flavell. 1998. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J. Exp. Med. 188:1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng, T., Z. Zhu, Z. Wang, R.J. Homer, B. Ma, R.J. Riese, H.A. Chapman, S.D. Shapiro, and J.A. Elias. 2000. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J. Clin. Invest. 106:1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothenberg, M.E., A.S.D. Luster, and P. Leder. 1995. Murine eotaxin: an eosinophil chemoattractant inducible in endothelial cells and in interleukin 4-induced tumor suppression. Proc. Natl. Acad. Sci. USA. 92:8960–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanley, E., G.J. Lieschke, D. Grail, D. Metcalf, G. Hodgson, J.A. Gall, D.W. Maher, J. Cebon, V. Sinickas, and A.R. Dunn. 1994. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc. Natl. Acad. Sci. USA. 91:5592–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shibata, Y., P.Y. Berclaz, Z.C. Chroneos, M. Yoshida, J.A. Whitsett, and B.C. Trapnell. 2001. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 15:557–567. [DOI] [PubMed] [Google Scholar]
- 54.Correll, P.H., A. Iwama, S. Tondat, G. Mayrhofer, T. Suda, and A. Bernstein. 1997. Deregulated inflammatory response in mice lacking the STK/RON receptor tyrosine kinase. Genes Funct. 1:69–83. [DOI] [PubMed] [Google Scholar]
- 55.Xu, H., J.A. Gonzalo, Y. St Pierre, I.R. Williams, T.S. Kupper, R.S. Cotran, T.A. Springer, and J.C. Gutierrez-Ramos. 1994. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J. Exp. Med. 180:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mocsai, A., E. Ligeti, C.A. Lowell, and G. Berton. 1999. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J. Immunol. 162:1120–1126. [PubMed] [Google Scholar]
- 57.Suen, P.W., D. Ilic, E. Caveggion, G. Berton, C.H. Damsky, and C.A. Lowell. 1999. Impaired integrin-mediated signal transduction, altered cytoskeletal structure and reduced motility in Hck/Fgr deficient macrophages. J. Cell Sci. 112:4067–4078. [DOI] [PubMed] [Google Scholar]
- 58.Möhn, H., V. Le Cabec, S. Fischer, and I. Maridonneau-Parini. 1995. The src-family protein-tyrosine kinase p59hck is located on the secretory granules in human neutrophils and translocates towards the phagosome during cell activation. Biochem. J. 309:657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson, D.C., and T.A. Springer. 1987. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu. Rev. Med. 38:175–194. [DOI] [PubMed] [Google Scholar]
- 60.Kato, M., R.T. Abraham, S. Okada, and H. Kita. 1998. Ligation of the beta2 integrin triggers activation and degranulation of human eosinophils. Am. J. Respir. Cell Mol. Biol. 18:675–686. [DOI] [PubMed] [Google Scholar]
- 61.Kato, M., H. Kita, H. Kimura, A. Tachibana, Y. Motegi, K. Tokuyama, and A. Morikawa. 2000. Stimulation of the beta(2) integrin, alpha(M)beta(2), triggers tyrosine phosphorylation and cellular degranulation on human eosinophils. Int. Arch. Allergy Immunol. 122(Suppl. 1):33–35. [DOI] [PubMed] [Google Scholar]
- 62.Chihara, J., T. Kakazu, I. Higashimoto, N. Saito, K. Honda, S. Sannohe, H. Kayaba, and O. Urayama. 2000. Signaling through the beta2 integrin prolongs eosinophil survival. J. Allergy Clin. Immunol. 106:S99–S103. [DOI] [PubMed] [Google Scholar]
- 63.Byth, K.F., L.A. Conroy, S. Howlett, A.J. Smith, J. May, D.R. Alexander, and N. Holmes. 1996. CD45-null transgenic mice reveal a positive regulatory role for CD45 in early thymocyte development, in the selection of CD4+CD8+ thymocytes, and B cell maturation. J. Exp. Med. 183:1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Helgason, C.D., J.E. Damen, P. Rosten, R. Grewal, P. Sorensen, S.M. Chappel, A. Borowski, F. Jirik, G. Krystal, and R.K. Humphries. 1998. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 12:1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Imamoto, A., and P. Soriano. 1993. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 73:1117–1124. [DOI] [PubMed] [Google Scholar]
- 66.Irby, R.B., W. Mao, D. Coppola, J. Kang, J.M. Loubeau, W. Trudeau, R. Karl, D.J. Fujita, R. Jove, and T.J. Yeatman. 1999. Activating src mutation in a subset of advanced human colon cancers. Nat. Genet. 21:187–190. [DOI] [PubMed] [Google Scholar]
- 67.Sugimura, M., K. Kobayashi, S. Sagae, Y. Nishioka, S. Ishioka, K. Terasawa, T. Tokino, and R. Kudo. 2000. Mutation of the SRC gene in endometrial carcinoma. Jpn. J. Cancer Res. 91:395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Los, H., G.H. Koppelman, and D.S. Postma. 1999. The importance of genetic influences in asthma. Eur. Respir. J. 14:1210–1227. [DOI] [PubMed] [Google Scholar]