

Abstract
Although elevated levels of IgE in asthmatic patients are strongly associated with lung infiltration by activated T helper (Th) 2 cells, the physiological role of immunoglobulin E (IgE) in the airway remains largely undefined. Lymphotoxin-deficient α (LTα−/−) mice exhibit increased airway inflammation, paradoxically accompanied by diminished levels of IgE and reduced airway hyperresponsiveness in response to both environmental and induced antigen challenge. The severe lung inflammation in LTα−/− mice is Th1 in nature and can be alleviated by IgE reconstitution. Conversely, depletion of IgE in wild-type mice recapitulates the lung pathologies of LTα−/− mice. Therefore, this work has revealed that lymphotoxin is essential for IgE production, and a physiological role of IgE in the airway may consist of maintaining the balance of Th1 and Th2 responses to prevent aberrant inflammation.
Keywords: bronchial hyperresponsiveness, cytokines, T helper cells, infiltration, allergen
Introduction
Several papers have shown that the development of bronchial asthma is associated with elevated IgE levels, the predominance of Th2 cytokine production, and eosinophilic inflammation of the airways (1–4). In addition to clinical correlations of asthma with high IgE levels, genetic analyses of families with asthma have also revealed a link between bronchial hyperresponsiveness (BHR) and high IgE levels (5, 6). Allergen-linked IgE signaling participates in the initiation of immediate hypersensitivity reactions and BHR by triggering mast cell degranulation (3, 7, 8). Allergens trigger mast cells by cross-linking surface FcɛRI-bound IgE, resulting in the release of IL-4, IL-13, and other factors, and ultimately promoting a Th2 response and allergic airway inflammation (9–13). Moreover, reduced expression of T-bet, a transcription factor for Th1 cytokines, induces a phenotype of murine asthma analogous to both acute and chronic human asthma, thus providing further support for the necessity of an optimal balance between Th1 and Th2 to maintain lung homeostasis and prevent aberrant inflammation. (10). In contrast to the data that Th2-mediated inflammation is dominant in allergen-induced asthma, several papers have shown surprisingly that the addition of allergen-specific Th1 cells can exacerbate airway inflammation (11–13). To control moderate-to-severe allergic asthma, humanized monoclonal antibodies directed against IgE have been used for treatment, but their efficacy is rather limited (14, 15). Surprisingly, IgE may not be essential for allergen-induced BHR and airway inflammation (16); thus far, the physiological role of IgE in the airway has not been demonstrated.
Lymphotoxin (LT) α has been identified in two molecular forms as follows: a secreted form consisting of an LTα3 homotrimer that binds to TNF receptors and a membrane-associated heterotrimeric form with LTαβ2 that binds to LTβR. LTα-deficient (LTα−/−) and LTβR−/− mice lack LNs but show a spontaneous accumulation of a diverse repertoire of immune cells in the lungs. The mechanisms underlying this spontaneous accumulation have not been adequately addressed (17–19). It is curious that LTα−/− mice have a strong inflammatory response to antigen challenge in the airway, even in the absence of draining LNs (20, 21). Here, we report that the infiltrating cells are actively participating in airway inflammation, and their accumulation in the lung can be paradoxically attributed to diminished serum IgE levels in LTα−/− mice. Therefore, our results uncover a hitherto unappreciated physiological role of IgE in the protection of the airway from inappropriate inflammation and BHR.
Materials and Methods
Mice.
LTα−/− and LTβR−/− mice were backcrossed to C57BL/6 mice and maintained under specific pathogen-free conditions as described previously (18, 22). TNF−/− and C57BL/6 mice were purchased from the Jackson Laboratory. LTβR−/− mice were obtained from Taconic Farms. Animal care and use were in accordance with institutional and National Institutes of Health guidelines.
Histology.
Lung tissues for histological examination were fixed in 10% buffered formalin. Lung tissues of IgE−/− mice were provided by H. Oettgen (Boston Children's Hospital, Boston, MA). 3-μm sections were obtained from the paraffin blocks and stained using both the hematoxylin and eosin and Masson's trichrome staining methods. All sections were examined qualitatively by an experienced pathologist in a blind fashion.
Flow Cytometric Analysis.
All antibodies for flow cytometric analysis were purchased from BD Biosciences. Bronchoalveolar lavage (BAL) and lung cells were incubated in Fc-block, 2.4G2, for 20 min, stained in PBS containing 1% FBS (GIBCO BRL) plus 0.01% NaN3 for 30 min on ice, and analyzed by flow cytometry on a FACScan™ (BD Biosciences). For statistical analysis, p-values were determined by two-tailed Student's t test. For intracellular cytokine staining, lung cells were obtained by collagenase digestion and stimulated with 50 ng/ml PMA, 500 ng/ml ionomycin, and 10 μg/ml Brefelden A in complete RPMI 1640 media with 10% FCS for 6 h at 37°C, 5% CO2. Extracellular staining was performed using anti–CD4-PE for 30 min on ice. The cells were fixed with 2% paraformaldehyde. For intracellular staining, cells were permeabilized with 0.5% saponin in PBS, stained with either IL-4–allophycocyanin or IFNγ-allophycocyanin in permeabilization buffer, and analyzed by FACS®.
Isolation of Lung Leukocytes and BAL Cells.
Lung tissues were digested three times by shaking (175 revolutions/min) for 30 min at 37°C in RPMI 1640 medium containing 1.5 mg/ml collagenase VIII and 2% FBS. Lung cells were passed through a nytex filter. Bronchial lavage was performed by delivering 1 ml of cold RPMI 1640 containing 2% FBS into the trachea and gently aspirating the fluid. The first lavage was centrifuged, and supernatant was stored at −20°C for cytokine analysis by ELISA. Lavage was repeated three times, and cells collected from each wash were pooled for FACS® analysis.
Cytokine and IgE ELISA.
The lung tissue was homogenized in 500 μl PBS containing protease inhibitors. The lysate was collected by centrifugation at 12,000 revolutions/min for 15 min. Cytokine production from the lung lysate and BAL fluid was measured by ELISA. The total IgE concentration from the serum (n = 9) was measured by ELISA according to the manufacturer's protocol (BD Biosciences).
Respiratory Mechanics.
Assessment of cholinergic airway constrictor responsiveness was done with a computer-controlled small-animal ventilator (Flexivent; SCIREQ). In brief, the mice were anesthetized with 0.1 ml per 10 g body weight of a mixture containing 2 mg/ml xylazine and 40 mg/ml ketamine hydrochloride given i.p. Anesthesia was maintained by supplemental administration of 30% of the initial dose at ∼25-min intervals, as required. Heart rate was monitored by EKG with needle electrodes. After tracheostomy, the trachea was cannulated with a blunted 18-gauge metal needle. The mouse was quasi-sinusoidally ventilated with a nominal tidal volume of 10 ml/kg at a frequency of 150 breaths/min and a positive end-expiratory pressure of 2 cm H2O. To determine the differences in airway response to methacholine between WT and LTα−/− mice, each mouse was challenged with seven doses of methacholine aerosol (0, 0.1, 1, 5, 10, 20, and 40 mg/ml in saline) for 12 s. Before each aerosol challenge, the animal was given two deep inspirations to standardize volume history. After each methacholine challenge, respiratory system resistance was recorded during tidal breathing every 10 s for 2 min, and the peak resistance was selected as the bronchoconstrictor response to that methacholine dose. Analysis of variance is used to analyze the differences in airway response to methacholine between WT and LTα−/− mice.
Schistosoma mansoni Egg Antigen (SEA) Sensitization and Challenge.
Mice were sensitized i.p. on day 0 with 2.5 × 103 inactivated S. mansoni eggs. On day 7, the mice were challenged intratracheally (i.t.) with 50 μg of soluble SEA. 4 d after challenge, mice were killed, BAL was collected, and lungs were dissected for digestion. Strong Th2-dominant airway inflammation with 70–90% eosinophils in BAL could be detected after challenge (23, 24). In IgE reconstitution experiments, LTα−/− mice were injected i.p. on days −21, −14, and −7 with either mouse Ig or polyclonal IgE purified from WT mice. S. mansoni sensitization on day 0, SEA challenge on day 7, and harvest on day 11 were performed as described in Fig. 4. In IgE depletion experiments, B6 mice were injected i.p. on days −21, −14, and −7 with 30 μg of either rat Ig or anti-IgE antibodies as follows: EM95 or R1E4 (8, 25). All reagents were free of endotoxin (EU < 0.25).
Figure 4.

IgE reconstitution of LTα−/− mice results in the reduction of the total cell number and a shift from a Th1 to Th2 cytokine profile in the lung and BAL cells. (A) LTα−/− mice (n = 3) were treated i.p. with 500 ng of mouse Ig or purified IgE once per week for 3 wk. 1 wk after the last treatment, the total number of cells in the lung and BAL of B6, mIg-, or IgE-treated LTα−/− mice were analyzed by trypan blue staining. IgE-reconstituted LTα−/− mice have similar total cell numbers compared with control mice (P < 0.01). (B) Reconstituted LTα−/− have reduced leukocytes in the BAL and lung compared with control treated mice (P < 0.01). The cell number was determined by calculating the total number of cells and the absolute number of each cell type from the FACS® profile. IFN-γ and IL-5 levels were determined by ELISA from BAL fluids of mIg- or purified IgE-treated LTα−/− mice. (P < 0.05; IgE vs. mIg). (C) LTα−/− mice (n = 3) were injected i.p. with either 500 ng mouse Ig or purified IgE on day −21, −14, and −7. On day 0, mice were injected with either mIg or IgE and sensitized i.p. with 2.5 × 103 inactivated S. mansoni eggs. On day 7, the mice were challenged i.t. with 50 μg SEA. On day 11, mice were killed and analyzed for the total number of cells in BAL and digested lung (P < 0.01; IgE vs. mIg). (D) The type of cells present in the BAL of SEA-challenged and mIg- or IgE-reconstituted LTα−/− mice was determined by calculating the total number of cells and the absolute number of each cell type from the FACS® profiles. IFN-γ and IL-5 levels were measured from BAL fluids of SEA-challenged and mIg- or IgE-reconstituted LTα−/− mice (P < 0.001 for IFN-γ; P < 0.05 for IL-5; IgE vs. mIg). Data represent the mean ± SD from a representative experiment. Experiments were repeated four times by different individuals with similar results.
Results
Increased Airway Inflammation and Remodeling in LTα−/− Mice.
To carefully determine the nature of the previously reported accumulation of leukocytes in the lungs of LTα−/− mice (17–19), detailed histopathological analysis was performed. The lungs of LTα−/− mice do not merely harbor leukocytes but gradually develop a chronic airway inflammation that worsens with age (Fig. 1 A). The infiltration consists of mainly mononuclear cells predominantly in the peribronchial and perivascular areas. Histopathological analysis of lung tissues in LTα−/− mice also revealed significant airway remodeling. These changes include bronchial wall thickening, subepithelial fibrosis, increased goblet cell mass, myoblast–myocyte hyperplasia and hypertrophy, epithelial cell hypertrophy, and increased deposition of collagen below the basement membrane; these are all clear indicators of chronic airway inflammation. Furthermore, the thickness of this subepithelial collagen layer was visualized by Masson's trichrome staining, which stains collagen. In naive LTα−/− mice, the collagen layer under the basement membrane was much thicker than that of WT mice (Fig. 1 A). To further determine the type and number of infiltrating cells in the lung tissue, lungs from WT and LTα−/− mice were digested with collagenase. The suspension of cells was analyzed by flow cytometry and a three- to fivefold increase in total cell number was found in LTα−/− mice (Fig. 1 B) with an increased proportion of activated T cells, as measured by CD69 expression, in LTα−/− mice compared with WT mice (Fig. 1 C). Thus, LTα−/− mice, without deliberate challenge, appear to develop a spontaneous, cumulative airway inflammation with active remodeling not seen in WT mice housed in the same specific pathogen-free facility.
Figure 1.
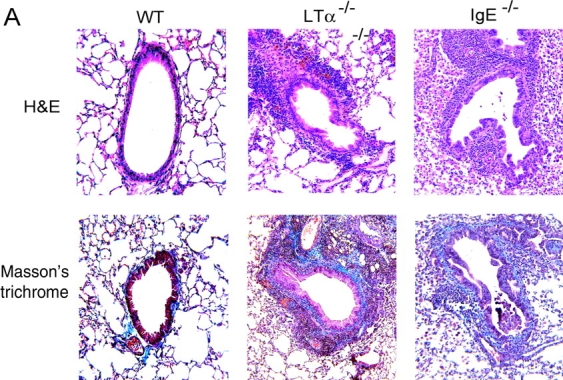


Increased inflammation and airway remodeling in LTα−/− and IgE−/− mice. (A) Lung tissues from WT, LTα−/−, and IgE−/− mice (3 mo old) were fixed in 10% buffered formalin and embedded in paraffin. Hematoxylin and eosin staining (top, H&E) and Masson's trichrome staining (bottom) of B6, LTα−/−, and IgE−/− mice are presented. (B) The lung (left) and BAL cells (right) were isolated by collagenase digestion and lavage, respectively, from 3-mo-old B6 (white bars) and LTα−/− (black bars) mice (n = 5 per group). The total number of cells was analyzed with trypan blue staining. The number of leukocytes in the lung of LTα−/− mice is significantly higher than that of the WT (P < 0.001). BAL cells from five mice were pooled for further analysis by FACS®. (C) Lung cells were dually stained with FITC-conjugated anti-CD69 and PE-conjugated anti-CD3. The fluorescence intensity was analyzed from the gated lymphocyte population of B6 (left) and LTα−/− (right) mice. The percentage of CD69+ cells among CD3+ cells is presented. (D) The forward/side scatter dot-plot profiles were analyzed from BAL cells of B6 (left) and LTα−/− (right) mice; (a), (b), and (c) represent the lymphocyte, granulocyte, and macrophage population, respectively. (E) The gated lymphocyte population (a) from the forward/side scatter dot-plot profiles of BAL cells (D) was analyzed for CD4 and B220 expression by FACS® analysis. (F) CCR3 and CD3 expression was analyzed from both the gated lymphocyte (a) and granulocyte (b) populations. (G) The type of BAL cells was determined by calculating the absolute number of each cell type from the FACS® profiles and by the total number of cells (P < 0.0001; WT vs. LTα−/−).
(H) B6 and LTα−/− mice were analyzed by three-color staining with FITC–anti-B220, cychrome–anti-CD4, and PE–anti-CD8 or PE–anti-NK1.1. The gated lymphocyte population (a) from the forward/side scatter dot-plot profiles of the lung cells was analyzed for CD4 and B220 (P < 0.01, WT vs. LTα−/− mice). The percentage of CD8+ or NK1.1+ cells gated from B220−CD4− was evaluated. (I) CCR3 and CD3 expression was analyzed from both the gated lymphocyte (a) and granulocyte (b) population. (J) The total number of cells was calculated. The type of lung cells was also determined by calculating the absolute number of each cell type from the FACS® profiles. The results are representative of five independent experiments.
To more closely define the inflammation in the airway, BAL cells were collected from WT and LTα−/− mice and analyzed. An increase in total BAL cells was also found in LTα−/− mice (Fig. 1 B), corresponding to escalating infiltration in the lung tissue. More impressively, LTα−/− mice had a much higher percentage of BAL lymphocytes than WT mice as determined by flow cytometry (Fig. 1 D, area a, 6.7 ± 0.8% in WT mice, 70.2 ± 1.6% in LTα−/− mice). These results were consistent with differential cell counts from cytospin preparations of BAL (unpublished data). CD4+ T and B cells were the major subsets of the increased lymphocyte population in LTα−/− BAL (Fig. 1, E and G). The percentage of CD8+ T cells was also increased, and NK1.1+ cells were reduced (Fig. 1 H). The forward/side scatter dot-plot profile of WT and LTα−/− BAL cells also showed a reduction in the granulocyte population in LTα−/− mice (Fig. 1 D, area b, 8.3% in WT mice, 2.8% in LTα−/− mice). CCR3 is a β chemokine receptor found predominantly, but not exclusively, on eosinophils (26, 27). To determine whether LTα−/− mice present with eosinophilia, cells from the BAL were stained with an anti-CCR3 antibody, and the number of CCR3 positive cells was reduced in airways of LTα−/− (Fig. 1 F, 22 ± 1.3% in WT mice, 7 ± 0.4% in LTα−/− mice). This result was consistent with the reduced percentage of eosinophils in LTα−/− mice determined by cytospin; however, the total number of eosinophils was not reduced (unpublished data). Although the absolute cell numbers of CD4- and B220-expressing cells were markedly increased in the BAL of LTα−/− mice compared with WT mice (∼30- and 80-fold, respectively), the number of macrophages remained unperturbed (Fig. 1 G). Similarly to the BAL, the percentages of CD4-, CD8-, and B220-expressing cells in the lungs of LTα−/− mice were increased (Fig. 1 H). Because the total number of leukocytes in the lung increased significantly (P > 0.0001), the actual total number of CD4, CD8, and B220+ cells increased an impressive 8–14 fold. The percentage of NK cells and CCR3+ cells was greatly reduced, and the absolute number of these cells was also slightly reduced in the lung of LTα−/− mice (Fig. 1, I and J). Together, these observations implied that the profound increase of total cell numbers in the airways of LTα−/− mice was largely due to the increased infiltration of lymphocytes, a feature of chronic inflammation.
Interaction between LT and Its Receptor Is Required for IgE Production.
Increased airway inflammation is often associated with elevated serum IgE concentrations. Despite the increased airway inflammation in naive LTα−/− mice, unexpectedly we found IgE concentrations reduced to nearly undetectable levels (Fig. 2) . LTα−/− mice lack both soluble and membrane LT. Either membrane or soluble LT could be required for IgE production; therefore, the sera from LTβ−/− mice were collected. These mice, which lack only the membrane form of LT, also have low IgE levels. To better define the receptor involvement, serum from LTβR−/− mice was collected and found to share the IgE defect observed in LTα−/− mice, thus identifying a new ligand and receptor pair required for IgE production. Interestingly, TNF, another closely related cytokine that shares the same receptors as soluble LT, is dispensable for IgE production (Fig. 2). These data revealed the presence of membrane LT and its receptor, LTβR−/−, are critical for IgE production.
Figure 2.

LT is required for IgE production. The sera from various 12–16-wk-old LTα−/− mice (n = 9) were collected, and total IgE concentration was measured by ELISA. Data represent the mean ± SE.
Mice Deficient in IgE Also Develop Airway Inflammation.
Antigen-induced bronchial hyperreactivity and eosinophilic inflammation can occur in the absence of IgE in a mouse model of asthma, but whether there is an increase of airway inflammation without deliberate challenge had not been shown (16). Fixed lung tissues of IgE−/− mice were obtained and showed a chronic airway inflammation by 3–4 mo of age (Fig. 1 A). The infiltration mainly consisted of mononuclear cells, predominantly in the peribronchial and perivascular areas in a pattern similar to LTα−/− mice. Further histopathological analysis of IgE−/− lung tissues revealed bronchial wall thickening, subepithelial fibrosis, increased goblet cell mass, myoblast–myocyte hyperplasia and hypertrophy, epithelial cell hypertrophy, and increased deposition of collagen below the basement membrane. In fact, 6-mo-old BCR−/− mice have also increased infiltration in both the perivascular and peribronchial areas (unpublished data). Together, these data show that naive IgE−/− mice, similar to the LTα−/− mice, had increased airway inflammation compared with WT controls, which supports the importance of maintaining appropriate IgE levels for airway homeostasis.
The Airway Inflammation of LTα−/− Mice Is Characterized by an Increased Th1 Response.
To study the cytokine profile of the lung and BAL of LTα−/− mice, first we measured the levels of IFN-γ, a prototypic cytokine for a Th1 response. The levels of IFN-γ, in both the lung and BAL of LTα−/− mice were markedly higher than those of WT mice in the absence of active challenge (Fig. 3 A). Despite the increased T cell infiltration in their lungs, LTα−/− mice have reduced Th2 cytokines, represented by a dramatic reduction of IL-5 on a per–T cell basis (Fig. 3 B). IL-4 levels in the BAL of LTα−/− mice were also reduced profoundly (2.5 ± 0.8 pg/ml vs. 11 ± 1.8 pg/ml of WT mice; P < 0.001). Together, these data suggest a deliberate Th1-type inflammation.
Figure 3.

LTα−/− airways display a Th1 phenotype even after a strong Th2 antigen challenge. The lung tissues from B6 and LTα−/− mice (n = 3–5 per group) were homogenized in PBS containing proteinase inhibitors, and the supernatants were collected by centrifugation. Cytokines from the BAL were measured after a 1-ml flushing of the airways through the trachea. IFN-γ (A) and IL-5 (B) levels were measured by ELISA from the lung lysates (left) and BAL fluids (right). (C and D) B6 and LTα−/− mice were sensitized i.p. on day 0 with 5 × 103 inactivated soluble S. mansoni egg antigen (SEA). On day 7, the mice were challenged i.t. with SEA. On day 11, mice were killed, and cytokines from the lung and BAL were analyzed. The lung lysates and BAL fluids of those mice were subjected to ELISA. IFN-γ (C) and IL-5 (D) levels are shown (P < 0.05). Data represent the mean ± SD from a representative experiment. Experiments were repeated four times by different individuals with similar results. For intracellular staining, lung cells (E) and splenocytes (F) were collected and stimulated as described in Materials and Methods. Extracellular staining was performed using anti–CD4-PE, and intracellular staining was performed using either IL-4–allophycocyanin or IFNγ-allophycocyanin.
Stimulation of the airway with allergen may stimulate IgE-bound cells to release an initial burst of cytokines, such as IL-4, which bias the initial response to Th2 (9, 28). Intratracheal challenge of WT mice with SEA, subsequent to priming with S. mansoni eggs, results in a robust Th2-mediated lung inflammation (23, 24). To determine whether this Th2-skewing model of airway inflammation could deviate the T cell response in LTα−/− mice, LTα−/− mice were primed and challenged with SEA. LTα−/− mice develop a much more severe airway inflammation in response to SEA challenge than WT mice (4.6 ± 0.4 × 106 cells in BAL and 52 ± 4.3 × 106 cells in the lung of LTα−/− mice vs. 1.8 ± 0.2 × 106 in BAL and 16 ± 3 × 106 in the lung of WT mice; P < 0.001). This response remains characterized by Th1 polarization including increased IFN-γ and reduced IL-4 or IL-5 in response to SEA (Fig. 3, C and D). To determine whether the altered cytokine patterns are associated with an increased Th1/Th2 ratio in the lung of naive LTα−/− mice, intracellular staining of anti–IFN-γ and IL-4 in combination with T cell markers was performed (Fig. 3, E and F). Clearly, increased IFN-γ and reduced IL-4 from CD4+ T cells in LTα−/− mice were also demonstrated. Therefore, the preexisting milieu in the lungs of LTα−/− mice, conditioned by reduced IgE levels, not only resists deviation to a Th2-mediated response, but also exacerbates the overall Th1 inflammatory reaction. To study whether such altered cytokine patterns were specific to the lung, intracellular cytokines of lymphocytes from the spleen of WT mice and LTα−/− mice were studied. Interestingly, cytokine profiles in the spleen were unchanged (Fig. 3 F). These data suggest that the altered cytokine patterns might not be systemic but limited to the lung microenvironment.
IgE Reconstitution Reverses Th1-dominant Airway Inflammation in LTα−/− Mice.
LTα−/− mice exhibit multiple defects in addition to IgE deficiency; for example, their absence of LNs, which may contribute to the increase of infiltrates in the lungs (17, 18, 29). To circumvent the numerous defects associated with LTα−/− and define the relevant contribution of IgE, 12-wk-old LTα−/− mice with spontaneous airway inflammation were reconstituted with polyclonal purified murine IgE. After three infusions (once a week for 3 wk), serum IgE concentrations in reconstituted LTα−/− mice peaked at 30–50% of WT levels. When compared with control murine Ig treatments or no treatment, this short-term IgE reconstitution resulted in a remarkable reduction of cell infiltration in the lung and BAL of LTα−/− mice, predominantly in the CD4+ and macrophage population (Fig. 4 A). To further test whether reconstitution of IgE in LTα−/− mice depolarizes the established Th1 microenvironment in addition to alleviating the cellular infiltration, Th1/Th2 cytokine profiles were analyzed. IFN-γ levels in the BAL fluid of IgE-reconstituted LTα−/− mice were reduced to undetectable levels, whereas IL-5 increased dramatically (Fig. 4 B). Consistently, IL-4 levels in BAL fluid in IgE-reconstituted LTα−/− mice were also restored to the level of WT mice (8.6 ± 1.3 pg/ml in reconstituted group; 2.2 ± 0.8 pg/ml in control-treated group). After SEA challenge, the total cell number and cytokine profiles of IgE-reconstituted mice matched that of WT mice, in both the lung and the BAL (Fig. 4, C and D). IgE reconstitution in LTα−/− mice reduced their airway inflammation and restored the WT Th2 response, implicating IgE deficiency in LTα−/− mice as the central defect responsible for the Th1 airway inflammation in the lungs.
Recapitulation of Th1-dominant Airway Inflammation in WT Mice by Depletion of IgE.
To further define the importance of IgE in the airway inflammation, we determined whether reduced IgE in WT mice can sufficiently recapitulate the airway inflammation seen in LTα−/− mice. To deplete IgE, WT mice were treated with mAb against mouse IgE (EM95), once a week for 3 wk. The serum, lung, and BAL cells of the IgE-depleted mice were collected and analyzed 1 wk after the last treatment, when IgE concentrations were undetectable. The total number of leukocytes in the lung and BAL of EM95-treated WT mice increased to the level of LTα−/− mice (Fig. 5 A). A severalfold increase of immune cells in both BAL and the lung was observed in IgE-depleted mice. Additionally, cytokine expression shifted from the prototypic Th2 to Th1 profile, with profoundly increased IFN-γ levels and reduced IL-4 in the BAL fluid (Fig. 5 B). The percentage of lymphocyte populations in IgE-depleted WT mice (19%) increased compared with mice treated with the control rat Ig (Fig. 5 C, area a, 2.8%). After treatment, the absolute number of CD4+, CD8+, and B220+ cells increased >20-fold, resulting in a profile similar to LTα−/− mice (unpublished data). The granulocyte populations also increased after the anti-IgE treatment, whereas macrophage populations were reduced as shown (Fig. 5 C, areas b and c). Interestingly, the percentage of CCR3+ cells was reduced in the BAL of WT mice treated with EM95 (Fig. 5 D). After depletion of serum IgE (day −21, −14, and −7), mice were sensitized and challenged with SEA to test if antigens that favor a Th2 response can reverse the Th1 bias resulting from the lack of IgE. After SEA challenge, IgE-depleted mice had three- to fivefold more leukocytes in both the lung and BAL than WT mice or control Ig-treated mice recapitulating the inflammation in LTα−/− mice (5.8 ± 1.8 × 106 in BAL and 74 ± 6.3 × 106 in the lung of anti-IgE–treated mice vs. 1.8 ± 0.2 × 106 in BAL and 16 ± 3 × 106 in the lung of WT mice; P < 0.001). Furthermore, the elevated Th1 cytokine profile in IgE-depleted WT mice after SEA challenge reiterates the role of IgE in minimizing Th1-mediated inflammation (Fig. 5, E and F).
Figure 5.
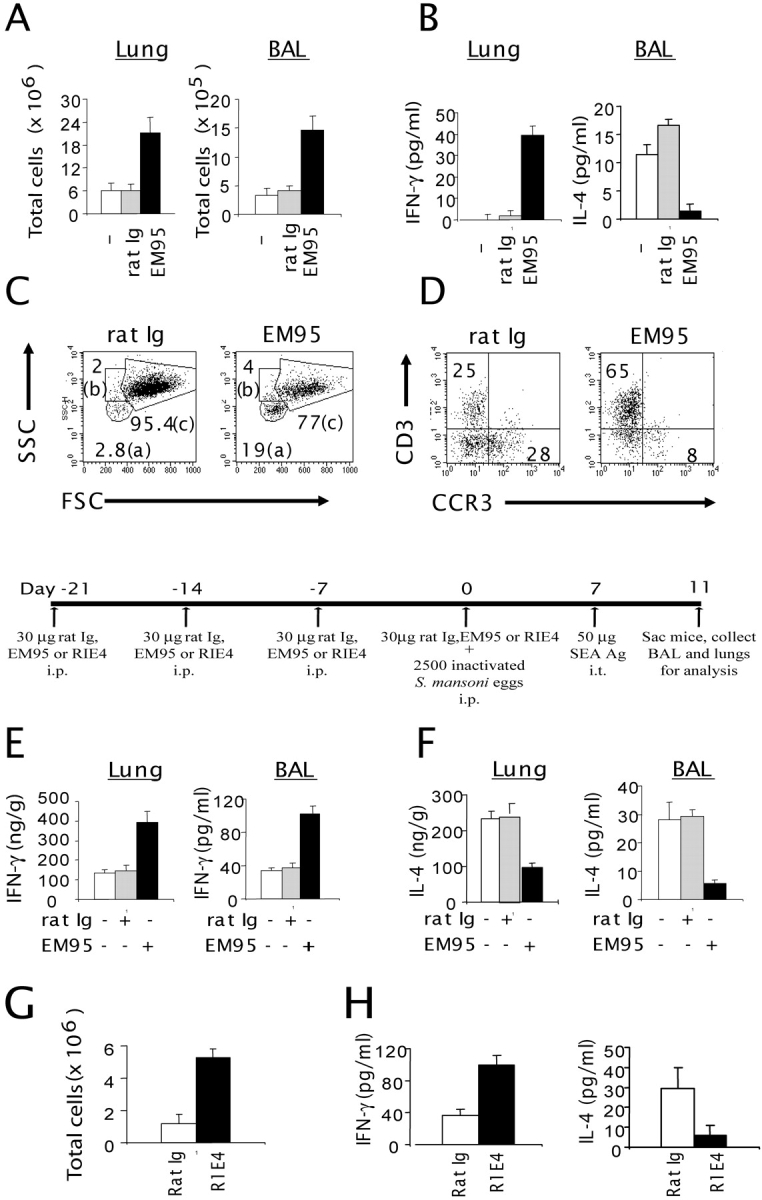
Increased cell number and altered cytokine production in the lung and BAL cells of IgE-depleted WT mice. (A) B6 mice (n = 3) were treated i.p. with 50 μg of rat Ig or anti-IgE (EM95) once per week for 3 wk. 1 wk after the last treatment, the total number of cells in the lung and BAL was analyzed (P < 0.01; EM95 vs. control group). (B) IFN-γ and IL-4 levels were determined by ELISA in BAL fluids from B6 mice treated with either rat Ig or anti-IgE (P < 0.05; EM95 vs. control group). (C) The forward/side scatter dot-plot profiles were analyzed from B6 mice treated with either rat Ig (left) or EM95 (right). (D) The CCR3 and CD3 expression was analyzed from both the gated lymphocyte (a) and granulocyte population (b). (E) B6 mice were injected i.p. on day −21, −14, and −7 with 30 μg rat Ig or EM95. On day 0, the mice were injected i.p. with 30 μg rat Ig or anti-IgE sensitized i.p. with 2.5 × 103 inactivated S. mansoni eggs. On day 7, the mice were challenged i.t. with SEA. On day 11, mice were killed and analyzed. IFN-γ (E) and IL-4 levels (F) were measured from the lung lysates (left) and BAL fluids (right) by ELISA. Similar experiments were performed using a noncross-linking anti-IgE antibody, R1E4 (G and H). The number of BAL cells (G) and cytokine profiles (H) were determined as described in A and B (P < 0.01; R1E4 vs. control group). Data represent the mean ± SD from a representative experiment. Experiments were repeated four times by different individuals with similar results.
EM95 binds both soluble IgE and surface-bound IgE captured by FcɛRI on mast cells. Cross-linking of the latter in vitro by anti-IgE antibody has been shown to induce mast cell degranulation and release of Th2 cytokines (8). This side effect of EM95 is unlikely to be a confounding factor in our system, as the treated mice show a Th1-skewed profile, including lower IL-4 levels, despite potential anti-IgE–triggered Th2 cytokines from mast cells or other FcɛRI+ cells. To avoid mast cell degranulation during anti-IgE treatment, WT mice were treated with a different anti-IgE (R1E4) mAb. R1E4, which binds only serum IgE via the Fc portion, is incapable of causing mast cell degranulation via IgE cross-linking, so that after sufficient receptor turnover and serum depletion, mast cells are incapable of eliciting IgE-mediated IL-4 production (25). The result from a short-term treatment of WT mice with R1E4 was consistent with our findings with the EM95 treatment as follows: an increase of Th1-skewed airway inflammation even with SEA challenge (Fig. 5, G and H). Consistently, the total number of leukocytes increased three- to fivefold (Fig. 5 H). T and B cells increased two- to threefold, and macrophages increased three- to fourfold, whereas NK cells remained unchanged (unpublished data). The data clearly underscore the importance of IgE in balancing Th1/Th2 responses and controlling airway inflammation in response to both environmental antigens and induced allergen.
Failure to Increase BHR to Nonspecific Challenge in LTα−/− Mice.
Increased spontaneous airway inflammation is often associated with increased BHR (3, 7, 8). To evaluate the severity of BHR in LTα−/− mice, we used intravenous delivery of methacholine to assess airway constrictor responsiveness in anesthetized, tracheostomized, mechanically ventilated mice, using the Flexivent computerized mouse ventilator, which calculates respiratory system resistance automatically. Unexpectedly, LTα−/− mice with severe airway inflammation failed to respond to methacholine challenge. LTα−/− mice showed inert response to even a high dose of mechacholine when compared with B6 mice (Fig. 6 A). The peak resistance in the bronchoconstrictor response to various methacholine doses from all WT mice and LTα−/− mice are summarized in Fig. 6 B. The overall response in resting LTα−/− mice is lower than that of WT mice (Fig. 6 B, P < 0.001 as determined by analysis of variance). In contrast to Th2-dominant airway inflammation with increased BHR seen in WT mice, LTα−/− mice showed a rather distinct Th1 airway inflammation with reduced BHR.
Figure 6.

Methacholine dose–response curves for respiratory system resistance in a WT mouse versus an LTα−/− mouse. Invasive measurement was used to determine respiratory system resistance in anesthetized, tracheostomized, mechanically ventilated mice. (A) Dose-dependent bronchoconstriction was induced by i.v. methacholine administration to WT mice versus LTα−/− mice. (B) The peak resistance from each dose from pooled data from three experiments was analyzed to compare both groups (n = 4; P < 0.005).
Discussion
In this paper, we have demonstrated that LT is critical for IgE production, and that a physiological role of IgE in the airway is to minimize Th1-dominant airway inflammation. Remarkably, the spontaneous Th1-mediated airway inflammation phenotype of LTα−/− mice, with or without challenge, can be rescued by short-term IgE reconstitution, and is recapitulated in both IgE−/− mice and IgE-depleted WT mice. These findings demonstrate the indispensable role of physiological levels of serum IgE in maintaining lung homeostasis and BHR as well as modulating a Th1/Th2 balance, thus providing a novel model by which to study how IgE affects airway inflammation.
There may be multiple mechanisms responsible for the control of the Th1/Th2 balance and airway inflammation exerted by IgE. The ability of IgE to enhance mast cell survival and FcɛRI expression in vitro has been described previously (30, 31). It is possible that reduced IgE levels in LTα−/− mice impair antigen-mediated mast cell degranulation and cytokine production, such as IL-4, that are required for a Th2 response. The resulting reduction in IL-4 may lead to a Th1-dominated inflammatory response. Whether the defect is due to mast cell maturation defects, reduced IgE avidity on the mast cell surface, or perhaps defects in other IgE binding cells remains to be investigated.
In this paper, we have demonstrated clearly that impaired IgE levels and increased Th1 responses occur in LTα−/− mice with or without antigen challenge. However, recent works evaluated the role of LNs in the development of airway inflammation using LTα−/− mice and found the generation of Th2-mediated airway inflammation could still occur in LTα−/− mice (20, 21). Although baseline IgE levels have not been determined in these analyses, antigen-specific IgE responses can be detectable in OVA–GM-CSF–challenged LTα−/− mice, and both Th1-associated IFNγ and Th2-associated IL-5/IL-13 were higher in OVA–GM-CSF–challenged LTα−/− mice, suggesting that LNs are not essential for Th2-mediated airway inflammation (20). Several differences may contribute to the discrepancies between the analyses as follows. First, OVA–GM-CSF very strongly activates the Th2 pathway and may overcome the defect in LTα−/− mice after repeated challenge. Second, it is possible that the relative increase in IFN-γ may be accountable for the dominating Th1 response in LTα−/− mice. In fact, all three models have shown increased IFN-γ levels. Third, the age of the mice may also provide an explanation for the discrepancies between the two analyses. 5–7-wk-old mice were used in the previous two studies, whereas 12–16-wk-old mice were used in our study. We have found the level of baseline IgE is undetectable in 5–7-wk-old B6 mice but gradually elevates after 8 wk. We have also noticed that it is difficult to induce Th1-dominant airway inflammation in 5–6-wk-old LTα−/− mice (unpublished data). Fourth, our work has used freshly isolated cells from the lung, whereas other works used restimulated splenocytes in vitro. Furthermore, different antigens/adjuvant, challenge route, and the assays for cytokine analysis may also account for potential discrepancies.
Our paper implies that a physiological role of IgE in the lung is to minimize airway inflammation via balancing Th1/Th2 responses. We hypothesize a possible mechanism in which physiological levels of IgE allow controlled degranulation and cytokine production of mast cells or other cells at levels required to routinely clear airway antigens without provoking excessive inflammatory infiltration. When IgE concentrations are excessively low, IgE-mediated mast cell responses could be impaired, resulting in a poor ability to clear the inhaled environmental antigen. The accumulated antigens lead to the increase of infiltrates in the lung responding to retained antigen may actually cause tissue damage. Our paper has an impact on the recent administration of anti-IgE in the clinic. High IgE levels during inflammation or other pathological conditions that favor Th2 responses or asthmatic patients with pathologically high IgE concentrations may benefit from anti-IgE therapy in some cases. However, depletion of IgE resulting in levels below normal may trigger a destructive Th1 inflammation. Conversely, there may be a subset of yet unrecognized patients for whom lower IgE concentrations may cause a severe Th1-mediated airway inflammation. It will be interesting to determine whether such patients have low IgE levels and reduced BHR. Thus, a physiological role of IgE in the airway is required for balancing both Th1 and Th2 responses to prevent immoderate inflammation dominated by either.
Acknowledgments
The authors gratefully thank Dr. H. Oettgen for providing lung tissue blocks of IgE−/− mice, as well as Y. Wang and A. Tesciuba for technical assistance.
These analyses were supported in part by National Institutes of Health grants to Y.X. Fu and A.I. Sperling.
Abbreviations used in this paper: BAL, bronchoalveolar lavage; BHR, bronchial hyperresponsiveness; i.t., intratracheally; LT, lymphotoxin; SEA, Schistosoma mansoni egg antigen.
References
- 1.Yazdanbakhsh, M., P.G. Kremsner, and R. van Ree. 2002. Allergy, parasites, and the hygiene hypothesis. Science. 296:490–494. [DOI] [PubMed] [Google Scholar]
- 2.Robinson, D.S., Q. Hamid, S. Ying, A. Tsicopoulos, J. Barkans, A.M. Bentley, C. Corrigan, S.R. Durham, and A.B. Kay. 1992. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N. Engl. J. Med. 326:298–304. [DOI] [PubMed] [Google Scholar]
- 3.Maddox, L., and D.A. Schwartz. 2002. The pathophysiology of asthma. Annu. Rev. Med. 53:477–498. [DOI] [PubMed] [Google Scholar]
- 4.Lewis, D.B. 2002. Allergy immunotherapy and inhibition of Th2 immune responses: a sufficient strategy? Curr. Opin. Immunol. 14:644–651. [DOI] [PubMed] [Google Scholar]
- 5.Burrows, B., F.D. Martinez, M. Halonen, R.A. Barbee, and M.G. Cline. 1989. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N. Engl. J. Med. 320:271–277. [DOI] [PubMed] [Google Scholar]
- 6.Postma, D.S., E.R. Bleecker, P.J. Amelung, K.J. Holroyd, J. Xu, C.I. Panhuysen, D.A. Meyers, and R.C. Levitt. 1995. Genetic susceptibility to asthma–bronchial hyperresponsiveness coinherited with a major gene for atopy. N. Engl. J. Med. 333:894–900. [DOI] [PubMed] [Google Scholar]
- 7.Sears, M.R., B. Burrows, E.M. Flannery, G.P. Herbison, C.J. Hewitt, and M.D. Holdaway. 1991. Relation between airway responsiveness and serum IgE in children with asthma and in apparently normal children. N. Engl. J. Med. 325:1067–1071. [DOI] [PubMed] [Google Scholar]
- 8.Coyle, A.J., K. Wagner, C. Bertrand, S. Tsuyuki, J. Bews, and C. Heusser. 1996. Central role of immunoglobulin (Ig) E in the induction of lung eosinophil infiltration and T helper 2 cell cytokine production: inhibition by a nonanaphylactogenic anti-IgE antibody. J. Exp. Med. 183:1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss, D.L., and M.A. Brown. 2001. Regulation of IL-4 production in mast cells: a paradigm for cell-type-specific gene expression. Immunol. Rev. 179:35–47. [DOI] [PubMed] [Google Scholar]
- 10.Finotto, S., M.F. Neurath, J.N. Glickman, S. Qin, H.A. Lehr, F.H. Green, K. Ackerman, K. Haley, P.R. Galle, S.J. Szabo, et al. 2002. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 295:336–338. [DOI] [PubMed] [Google Scholar]
- 11.Hansen, G., G. Berry, R.H. DeKruyff, and D.T. Umetsu. 1999. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J. Clin. Invest. 103:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Randolph, D.A., C.J. Carruthers, S.J. Szabo, K.M. Murphy, and D.D. Chaplin. 1999. Modulation of airway inflammation by passive transfer of allergen-specific Th1 and Th2 cells in a mouse model of asthma. J. Immunol. 162:2375–2383. [PubMed] [Google Scholar]
- 13.Randolph, D.A., R. Stephens, C.J. Carruthers, and D.D. Chaplin. 1999. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J. Clin. Invest. 104:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milgrom, H., R.B. Fick, Jr., J.Q. Su, J.D. Reimann, R.K. Bush, M.L. Watrous, and W.J. Metzger. 1999. Treatment of allergic asthma with monoclonal anti-IgE antibody. rhuMAb-E25 study group. N. Engl. J. Med. 341:1966–1973. [DOI] [PubMed] [Google Scholar]
- 15.Barnes, P.J. 2000. Anti-IgE therapy in asthma: rationale and therapeutic potential. Int. Arch. Allergy Immunol. 123:196–204. [DOI] [PubMed] [Google Scholar]
- 16.Mehlhop, P.D., M. van de Rijn, A.B. Goldberg, J.P. Brewer, V.P. Kurup, T.R. Martin, and H.C. Oettgen. 1997. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc. Natl. Acad. Sci. USA. 94:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banks, T.A., B.T. Rouse, M.K. Kerley, P.J. Blair, V.L. Godfrey, N.A. Kuklin, D.M. Bouley, J. Thomas, S. Kanangat, and M.L. Mucenski. 1995. Lymphotoxin-alpha-deficient mice: effects on secondary lymphoid organ development and humoral immune responsiveness. J. Immunol. 155:1685–1693. [PubMed] [Google Scholar]
- 18.Futterer, A., K. Mink, A. Luz, M.H. Kosco-Vilbois, and K. Pfeffer. 1998. The lymphotoxin b receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 9:59–70. [DOI] [PubMed] [Google Scholar]
- 19.Alimzhanov, M.B., D.V. Kuprash, M.H. Koscovilbois, A. Luz, R.L. Turetskaya, A. Tarakhovsky, K. Rajewsky, S.A. Nedospasov, and K. Pfeffer, K. 1997. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc. Natl. Acad. Sci. USA. 94:9302–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gajewska, B.U., D. Alvarez, M. Vidric, S. Goncharova, M.R. Stampfli, A.J. Coyle, J.C. Gutierrez-Ramos, and M. Jordana. 2001. Generation of experimental allergic airways inflammation in the absence of draining lymph nodes. J. Clin. Invest. 108:577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Constant, S.L., J.L. Brogdon, D.A. Piggott, C.A. Herrick, I. Visintin, N.H. Ruddle, and K. Bottomly. 2002. Resident lung antigen-presenting cells have the capacity to promote Th2 T cell differentiation in situ. J. Clin. Invest. 110:1441–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Togni, P., J. Goellner, N.H. Ruddle, P.R. Streeter, A. Fick, S. Mariathasan, S.C. Smith, R. Carlson, L.P. Shornick, S.J. Strauss, et al. 1994. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 264:703–707. [DOI] [PubMed] [Google Scholar]
- 23.Tesciuba, A.G., S. Subudhi, R.P. Rother, S.J. Faas, A.M. Frantz, D. Elliot, J. Weinstock, L.A. Matis, J.A. Bluestone, and A.I. Sperling. 2001. Inducible costimulator regulates Th2-mediated inflammation, but not Th2 differentiation, in a model of allergic airway disease. J. Immunol. 167:1996–2003. [DOI] [PubMed] [Google Scholar]
- 24.King, C.L., J. Xianli, I. Malhotra, S. Liu, A.A. Mahmoud, and H.C. Oettgen. 1997. Mice with a targeted deletion of the IgE gene have increased worm burdens and reduced granulomatous inflammation following primary infection with Schistosoma mansoni. J. Immunol. 158:294–300. [PubMed] [Google Scholar]
- 25.Keegan, A.D., C. Fratazzi, B. Shopes, B. Baird, and D.H. Conrad. 1991. Characterization of new rat anti-mouse IgE monoclonals and their use along with chimeric IgE to further define the site that interacts with Fc epsilon RII and Fc epsilon RI. Mol. Immunol. 28:1149–1154. [DOI] [PubMed] [Google Scholar]
- 26.Humbles, A.A., B. Lu, D.S. Friend, S. Okinaga, J. Lora, A. Al-Garawi, T.R. Martin, N.P. Gerard, and C. Gerard. 2002. The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness. Proc. Natl. Acad. Sci. USA. 99:1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heath, H., S. Qin, P. Rao, L. Wu, G. LaRosa, N. Kassam, P.D. Ponath, and C.R. Mackay. 1997. Chemokine receptor usage by human eosinophils. The importance of CCR3 demonstrated using an antagonistic monoclonal antibody. J. Clin. Invest. 99:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Umetsu, D.T., J.J. McIntire, O. Akbari, C. Macaubas, and R.H. DeKruyff. 2002. Asthma: an epidemic of dysregulated immunity. Nat. Immunol. 3:715–720. [DOI] [PubMed] [Google Scholar]
- 29.Fu, Y.X., and D.D. Chaplin. 1999. Development and maturation of secondary lymphoid tissues. Annu. Rev. Immunol. 17:399–433. [DOI] [PubMed] [Google Scholar]
- 30.Asai, K., J. Kitaura, Y. Kawakami, N. Yamagata, M. Tsai, D.P. Carbone, F.T. Liu, S.J. Galli, and T. Kawakami. 2001. Regulation of mast cell survival by IgE. Immunity. 14:791–800. [DOI] [PubMed] [Google Scholar]
- 31.Kalesnikoff, J., M. Huber, V. Lam, J.E. Damen, J. Zhang, R.P. Siraganian, and G. Krystal. 2001. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 14:801–811. [DOI] [PubMed] [Google Scholar]