


Abstract
Genome annotation in differently evolved organisms presents challenges because the lack of sequence-based homology limits the ability to determine the function of putative coding regions. To provide an alternative to annotation by sequence homology, we developed a method that takes advantage of unusual trypanosomatid biology and skews in nucleotide composition between coding regions and upstream regions to rank putative open reading frames based on the likelihood of coding. The method is 93% accurate when tested on known genes. We have applied our method to the full complement of open reading frames on Chromosome I of Trypanosoma brucei, and we can predict with high confidence that 226 putative coding regions are likely to be functional. Methods such as the one described here for discriminating true coding regions are critical for genome annotation when other sources of evidence for function are limited.
INTRODUCTION
Characterization of putative coding regions is a prerequisite for converting raw genomic sequence data into biologically relevant information. Standard approaches to this task provide less assistance, however, when the organism is evolutionarily distant from well-characterized counterparts and the process is frustrated by the lack of sequence homology based suggestions of function. This is the case for Trypanosoma brucei, a protist with many unusual biological features, limited experimental characterization, and estimated to be over 800 million years diverged from its nearest well-studied neighbor, Saccharomyces cerevisiae (1).
This paper presents a novel approach to the challenge of annotating unusual organisms. We utilize unique aspects of trypanosome genes to better distinguish true coding regions from computationally valid but non-functional predictions. The approach detailed here suggests ways in which utilizing unusual features of a particular organism can significantly enhance the annotation.
T.brucei is just one representative of the broad family of trypanosomatids, some members of which are significant pathogens of humans and livestock. T.brucei and Trypanosoma cruzi cause African sleeping sickness and Chagas’ disease, respectively, and a related species, Leishmania major, causes cutaneous leishmaniasis. They share complex life cycles with transmittal to humans and livestock effected through insect vectors, and all are adept at host immune evasion. As such, these are some of the most insidious parasites known, often with fatal consequences for their hosts (2,3). The genomes of T.brucei, T.cruzi and L.major are currently being sequenced.
Unlike most other eukaryotes, the majority of coding regions of trypanosomes are not interrupted by introns. More surprising, perhaps, is that no Pol II promoter for any protein-coding gene has been identified to date. Gene regulation appears to occur mainly after the initiation of RNA transcription, but the mechanisms have yet to be characterized in detail (4,5).
In part because of these unusual features and the large evolutionary distance to well-studied model organisms, the analysis of the T.brucei genome has yielded many more putative coding regions than can be assigned functions. Over 500 coding regions have been noted on Chromosome I of T.brucei, but just 26% of these have a function assignment (as reported in the EMBL database, see Data for details). This is summarized in Table 1, which shows the results of using a standard prokaryotic open reading frame (ORF) finder (Glimmer 2.0) followed by sequence homology searches of the major databases for Chromosome I of T.brucei. Glimmer 2.0 is reported to be up to 98% accurate at identifying prokaryotic coding regions and is based in part on a probabilistic analysis of codon usage (6). For comparison, the current annotation of the same chromosome by the Sanger Institute is also presented. As can be seen in this table, Glimmer not only finds a large number of ORFs; less than one-quarter can be assigned a function based on sequence homology evidence. The paucity of annotations for T.brucei was the motivation for the development of the organism-specific method described here.
Table 1. Results of ORF finding and annotation on Chromosome I of T.brucei using two approaches.
| Total number of ORFs predicted | Total with assigned function | |
|---|---|---|
| Glimmer 2.0 | 428 | 91 |
| (0.21) | ||
| Sanger Institute | 509 | 133 |
| (0.26) |
Glimmer 2.0 is a prokaryotic ORF finder and was trained on the data described in Data and run on the entire Chromosome I sequence. The results from the Sanger Institute’s analysis are a summary of their entries in the EMBL database; see Data for more information.
The crucial point is that over three-quarters of the putative coding regions identified to date on T.brucei chromosome I have no assigned function. At least some of these putative coding regions are likely to be computational artifacts, but distinguishing between true coding regions and such artifacts is difficult in the absence of evidence for function. Experimental demonstration of a functional role for a given ORF is the most conclusive evidence. However, in organisms such as T.brucei, the set of experimentally characterized genes is small compared to the number of possible coding regions. To experimentally characterize each computationally identified coding region would require painstaking effort. This effort could be better directed if ORFs were first ranked by the likelihood that they are functional genes. Such a ranking would facilitate the experimental characterization of the most likely coding regions first, with subsequent efforts clarifying the status of less likely coding regions. We therefore sought a computational approach that could assign a probability that a particular coding region might represent a real gene.
The method we present here is able to rank ORFs because it takes account of skews in nucleotide composition between coding regions and the regions immediately upstream of the translation start. These regions contain important signals involved in mRNA maturation. In trypanosomatids, most protein-coding genes are arrayed in cassettes that are co-transcribed into poly-cistronic mRNAs (7,8) that are cleaved into individual, mature mRNAs by a process known as trans-splicing, which adds a 39 nucleotide (nt) spliced leader (SL) sequence to the 5′ UTR of each transcript. As with other eukaryotes, the 3′ end of the mRNA is poly-adenylated to produce the complete, processed monocistronic mRNA for each gene in the array (9–14).
Figure 1 illustrates how trans-splicing might occur in a simplified polycistronic mRNA. trans-splicing is mechanistically similar to the more familiar intron-exon or cis-splicing and much of the spliceosomal machinery may be common to both processes (15). The trans-splicing signal appears to be a composite of several elements, but the key feature is a poly-pyrimidine (TC-rich) tract that precedes the AG used as the splice acceptor site (8,11,16–22). We can localize these tracts to the approximately 400 nt immediately upstream of known coding regions because 5′ untranslated regions (UTRs) are relatively short (50–250 nt) in T.brucei (based on a survey of GenBank entries). A clear over-representation of pyrimidines in upstream regions is visible when runs of pyrimidines are counted in upstream versus coding regions (Fig. 2).
Figure 1.
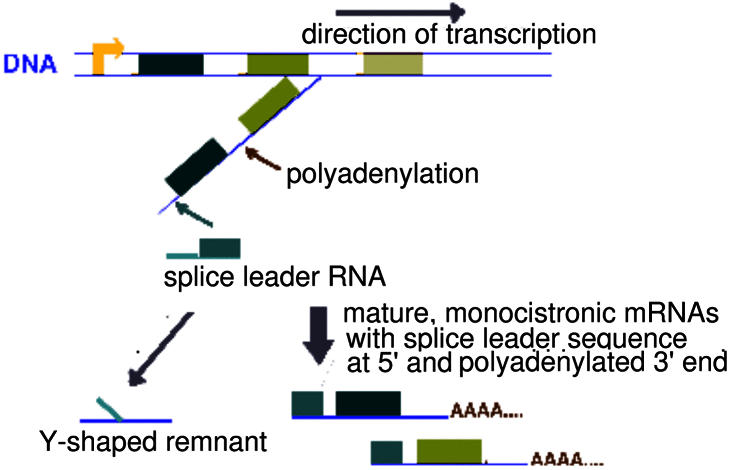
Schematic of trans-splicing in trypanosomatids. Genes are arrayed in polycistronic units, shown here as colored boxes (ORFs). Transcription yields a polycistronic precursor RNA with multiple transcripts strung together. The actual trans-splicing process adds a 39 nucleotide (nt) spliced leader sequence (shown in aqua) to the 5′ upstream sequence of each coding region to yield the monocistronic mRNAs [adapted from (16)]. As with cis-splicing, trans-splicing chemically bonds the guanine in the GT dinucleotide of the 5′ donor site to an adenine at what is known as the branch point. Following this bonding, the 3′ AG dinucleotide is used to demarcate the 3′ end of the region to be spliced. However, unlike in cis-splicing, two separate RNAs, the spliced leader RNA and the precursor RNA, are joined in the trans-splicing process. Therefore, the 5′ donor site is actually on the spliced leader sequence while the AG of the 3′ acceptor site is in the upstream region of the gene undergoing trans-splicing. The by-product of trans- splicing is therefore a Y-shaped RNA remnant, containing a portion of the spliced leader sequence and a segment of the upstream region of the gene. This Y-shaped RNA is the direct corollary of the lariat structure produced by cis-splicing during the removal of introns from precursor mRNA (9,12,14,16–18).
Figure 2.

An over-representation of pyrimidines can be seen when runs of T and C are compared between upstream flanking regions and known coding regions. In this figure, the x-axis indicates the number of pyrimidines in a row (i.e. TCTC would be four pyrimidines in a row). The y-axis indicates the number of such instances found in the entire set of sequences.
Although the trans-splicing signal is difficult to characterize computationally at this time, we have taken advantage of the dramatic skews in nucleotide composition associated with trans-splicing to identify likely coding regions. That is, we use the trans-splicing regions as a model of sequences that are likely to be non-coding, as it is highly unlikely that a trans-splicing signal will occur in the middle of a genuine coding region. We compared the dinucleotide frequency in these upstream regions with that of known coding regions to develop a predictive model of genuine coding regions. When the model was applied to an independent test set of known coding regions, it was 93% accurate at identifying true coding regions. The method has the advantage of being specific to T.brucei, enabling the identification of novel genes that share little or no sequence homology with previously characterized proteins in this or other organisms. This approach can be used to rank ORFs so that those most likely to be functional can be experimentally characterized first. Such efforts are critical to improve our understanding of this unusual organism.
DEFINITIONS
In this work, we will use certain common terms in ways that may require some explanation. We define an open reading frame (ORF) as a segment of sequence initiated by a start codon at the 5′ end of the sequence, a consecutive set of translatable codons and a stop codon that terminates the segment. There are no other features associated with an ORF; an ORF may or may not have any valid biological function.
We only consider protein-coding regions, so an ORF may be a coding ORF if it shares significant sequence-based homology to a known protein-coding gene at either the nucleotide or amino acid level. Other forms of evidence of functionality may also be used to declare an ORF as likely to be coding. In contrast, no-evidence ORFs (nORFs) are ORFs for which there is no evidence of functionality, or they lack all hallmarks of known coding regions. They may be computational artifacts.
Annotation is used in a very limited sense of the word: to refer to the individual descriptions of function assigned to a putative coding region. Each description of a coding region can be of two forms. A functional annotation is one giving a precise indication of a protein’s role in the biology of the organism (e.g. protein phosphatase). However, most annotation efforts label all identified ORFs with some kind of description. Therefore, there will also be putative coding regions with the annotation ‘hypothetical’ or ‘putative’, when these coding regions lack clear evidence of specific function. These are referred to as hypothetical annotations to distinguish them from functional annotations.
MATERIALS AND METHODS
We first explored features of the set of functionally annotated ORFs on Chromosome I against those with hypothetical annotations. The Wellcome Trust Sanger Institute has identified 509 ORFs in their submission to the EMBL database (see Data for accession numbers and other details). Of these, 91 have a functional annotation for a protein function. An additional 42 ORFs have annotations for DNA or RNA level features. The remainder is annotated with uninformative descriptors such as hypothetical. We could discern no significant differences in codon usage bias or other features of nucleotide composition between functionally annotated ORFs and those with hypothetical annotations (data not shown).
To validate true coding regions in the absence of other information, we wished to compare known coding regions to sequences we could be absolutely certain did not code for functional proteins. Although comparison to random sequence can be instructive in some instances, the objective here was to distinguish between classes of sequence from within the same organism. The poly-cistronic nature of transcription in T.brucei makes it difficult to identify truly inter-intergenic, non-coding sequences without first characterizing the poly-cistronic arrays. Identifying these arrays is difficult in the absence of complete knowledge of the genome. For a suitable model of non-coding regions, we therefore turned to the only sequences from T.brucei that were available in some abundance: sequences immediately upstream of known coding regions.
To facilitate comparisons of sequences of differing lengths and composition, we compared the upstream regions to coding regions at the dinucleotide level using transition probabilities. We applied maximum likelihood estimation (MLE) to estimate these probabilities since MLE allows for estimation from a relatively small sample size. MLE based transition probabilities are calculated by the formula:
![]()
where akl is the transition probability that the nucleotide l follows the nucleotide k. ckl is the number of times the dinucleotide combination kl occurs. In the denominator, we calculate the sum of the transition probabilities of all nucleotides that could follow k, represented by l′ as any of the four nucleotides (23).
We evaluated several standard methods for classification of sequences using the dinucleotide transition probabilities as variables (data not shown). For each classification method, we trained on a set of data and then evaluated the performance of each method on an independent dataset. The method that performed with the highest accuracy as a sequence classifier was linear discriminant analysis (LDA). LDA as implemented in the statistical package R was used for this analysis (24).
Data
For any classification method involving training and testing, several sets of data are required. A model is first developed using training data, which must be of the highest standard to ensure that the model accurately reflects known features. To test the actual performance of the model, other datasets are required. Where possible, evaluation on an independent dataset is desirable. It is the performance of a method on this test dataset that determines the overall accuracy of the method (25). Our datasets are described below and are included in the Supplementary Material.
Training set. Our training dataset was composed of proven coding and upstream regions. For coding regions, we selected T.brucei genes from GenBank where the entire coding sequence could be confidently identified. We used two approaches to collect data on upstream, non-coding regions. We generated some data specifically for this analysis through mRNA extraction, reverse transcription to cDNA and polymerase chain reaction (RT–PCR) amplification with subsequent sequencing. We supplemented this by mapping a limited set of previously characterized expressed sequence tags (ESTs) that contained the SL sequence to more recently determined genomic sequence.
Thirty-five cDNA sequences were selected from GenBank that contained at least 200 nt of upstream sequence and a complete coding region. For an additional 15 genes, we used sequences from genomic bacterial artificial chromosomes (BACs) sequenced by the Wellcome Trust Sanger Institute and The Institute for Genome Research (TIGR) that matched known genes in GenBank at the nucleotide level (at least 95% identity across 95% or more of the coding region).
For each of these genes, we designed a 3′ gene-specific primer. For the 5′ primer, we used a portion of the 39 nt leader sequence that occurs at the 5′ end of all T.brucei mRNAs. Primers were designed by the Primer3 program, and the optimal primer selected manually from the suggestions provided [S.Rozen and H.J.Skaletsky (1996,1997). Code available at http://www-genome.wi.mit.edu/genome_software/other/primer3.html. Sequences are included in the Supplementary Material].
RNA was extracted from cultures of the bloodstream and insect midgut stages of T.brucei Lister 427. Reverse transcription to cDNA was carried out using the Stratagene ProStar FirstStrand RT kit with random priming (with supplied random primers). Amplification by PCR was conducted with the following cycling parameters: 2:00 min at 94°C, then 30 cycles of 0:10 min at 94°C, 0:30 min at 60°C, 1:00 min at 72°C. A final extension period of 7:00 min at 72°C completed the PCR program. Amplifications used the Expand High Fidelity PCR polymerase from Roche Applied Science. The annealing temperature of 60°C was selected to minimize non-specific priming.
Ten microliters from each PCR reaction were used for gel electrophoresis on a 2% (w/v) agarose gel, and reactions with a clear single band were selected for sequencing. A total of 29 such reactions yielded clear sequence data. These were then mapped back to the original genomic sequence by BLAST comparisons using all default parameters (26). Each of the sequenced fragments was compared against the entire set of 50 genes. For each mapping, the best high-scoring pair (HSP) was used if the match length exceeded 50 nt. Mappings and extraction of upstream sequences were done in part by manual review of BLAST results and partially via ad hoc Perl scripts written for this purpose using the BLAST parser BPlite.pm [I.Korf (1999) BPlite—Lightweight BLAST parser. http://sapiens.wustl.edu/∼ikorf/BPlite.html].
We also extracted 146 5′ ESTs from dbEST (27) that contained the entire spliced leader sequence and mapped these to all the genomic data available from the Sanger Institute databases [genomic survey sequences (GSS), genomic contigs from Chromosomes IX and X and BACs from other chromosomes]. As with the experimentally generated data, we used BLAST and Perl scripts to map the ESTs and identify the upstream regions. This yielded an additional 77 upstream regions.
Our final set of confirmed upstream sequences had 106 sequences. To match this set of sequences, we collected an independent set of 106 coding sequences from GenBank. While upstream sequences for T.brucei are limited in GenBank, experimentally characterized coding regions are relatively more abundant. Therefore, some of the coding and upstream sequences were contiguous, but others were unrelated. The 212 sequences (106 upstream and 106 coding) yielded the transition probabilities used as the training data. The sequence data is included in the Supplementary Material.
Independent test dataset. To evaluate our method, we compiled an independent set of sequences composed of 103 coding regions with credible function assignments: 44 genes from Chromosome I and 59 genes from Chromosome II. We are reasonably confident that these are true coding regions based on their high percentage identity and good percent coverage to known genes. Four hundred nucleotides of sequence immediately upstream of the translation start were used as examples of non-coding regions. Since 5′ untranslated regions (UTRs) are relatively short (50–250 nt) in T.brucei (based on survey of GenBank entries), the 400 nt of non-coding sequence could be expected to contain sequence upstream of the UTR region. Transition probabilities for these sequences constituted the independent test dataset.
Other data. Finally, the method was applied to all 509 protein-coding ORFs identified by Sanger Institute on Chromosome I as reported in their submission to the European Molecular Biology Laboratory database on November 28, 2002 (EMBL accession numbers AL929603-AL929605 and AL929607). For each ORF, we used the entire coding region as documented by Sanger Institute. A LDA-based prediction for each of the 509 ORFs is included in the Supplementary Material and can be viewed interactively on our website at: http://bioinformatics.rit.edu/∼shuba/bin/motif-er.cgi
Genomic data for use in this web-based viewer were obtained from the Sanger Institute website at: http://www.sanger.ac.uk/Projects/T_brucei/.
Sequencing of the T.brucei genome was accomplished as part of the Trypanosome Genome Network, with support by The Wellcome Trust. Our use of these data conforms to the data release policy of the T.brucei genome project at the Sanger Institute.
Web interface
Users can access our method via a web interface at: http://bioinformatics.rit.edu/∼shuba/bin/orbit.cgi.
Users may submit up to 500 000 nucleotides of sequence data for analysis. Sequence data in the form of FASTA-formatted sequences can be submitted either as a contiguous sequence or individual sequences. However, the current web interface implementation makes no attempt to identify ORFs or other sequence structures. It is assumed that the users will have already identified likely ORFs and upstream regions prior to the use of our interface. For larger datasets or for processing raw genomic sequence into ORFs followed by predictions using our method, please contact the authors for a standalone version.
RESULTS
Our aim in the construction of the LDA model was to predict the likelihood that a given sequence is coding. To ensure that our model performs at a reasonable accuracy, we first consider the likelihood that any individual prediction will be correct. We consider two portions of the output from LDA analysis to determine the 95% confidence interval, or the region in which we can be 95% certain that an individual prediction is correct. LDA assigns a score to each sequence that is derived from the likelihood that the sequence was classified correctly. The LDA method selects a standard score cut-off for assigning samples into classes (28). In this case, a score of zero or less led to classification of a sequence as coding while a score greater than zero resulted in the sequence being labeled as non-coding.
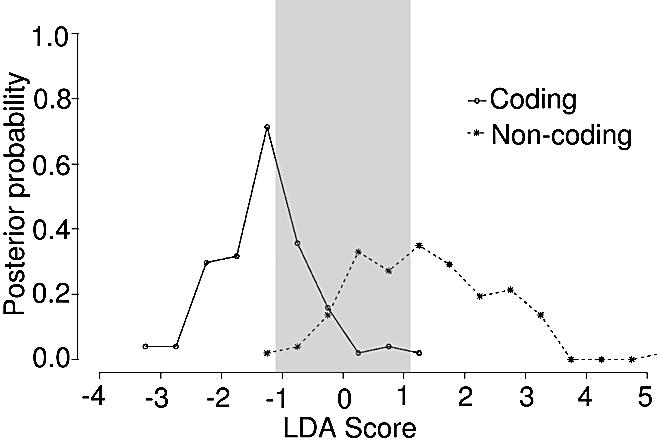
We consider this LDA assigned score in conjunction with a second measure, the actual probability of correct classification. Our aim is to identify the LDA scores associated with a probability of correct classification ≥0.95. Figure 3 shows the LDA assigned score versus the posterior probability (likelihood of correct classification) for the training data. Figure 3 suggests that a score less than –1 or greater than +1 indicates at least 95% confidence in the prediction. Some sequences with scores between –1 and +1, on the other hand, will be misclassified. This will be an important consideration when we evaluate the model performance on the independent test dataset.
Figure 3.

The posterior probability for the coding regions in the training data is shown in the left panel. The gray lines indicate the minimum score required for 95% confidence in an individual prediction. In this case, it corresponds to a LDA assigned score of –1 or lower. Similarly, the panel on the right shows the posterior probability distribution for non-coding sequences and that the 95% prediction interval corresponds to a LDA assigned score of +1 or higher.
Evaluation on the independent test set
Having established the score ranges for high confidence, we could evaluate the model’s overall accuracy on the independent test dataset. Figure 4 shows the distribution of LDA scores. We know from Figure 3 that there will be some misclassification when sequences score between –1 and +1. This is confirmed by the score distributions shown in Figure 4 where there is an overlap between the scores for known coding sequences and known non-coding sequences in the region between –1 and +1.
Figure 4.
The distribution of scores for the independent test dataset of 103 known coding regions and 103 known non-coding regions is shown here. The curves overlap in the score range from –1 to +1 as expected based on the results of Figure 3.
Despite some misclassification, the overall accuracy of this method is 93%. A more exact measure of the performance of this method is to consider the sensitivity (number of coding regions correctly identified from the total set of known coding regions) and the specificity (number of non-coding regions correctly identified from the set of all known non-coding regions). Sensitivity and specificity are calculated as follows: Sensitivity (Sn) = TP/(TP + FN), and Specificity (Sp) = TN/(TN + FP). Accuracy is defined as the average of sensitivity and specificity: Accuracy = TP + TN/(TP + FP + TN + FN) where TP is true positives, TN true negatives, FP false positives and FN false negatives. Throughout, we will highlight the sensitivity and specificity rather than the accuracy because these two measures of performance present a more nuanced understanding of predictive ability (29). While overall accuracy can often be quite high, by considering sensitivity and specificity we can ascertain where the method does well and where it is likely to fail.
Considering these measures of performance for our data, the method appears to be extremely sensitive and quite specific (Table 2). True coding regions (sensitivity) are correctly identified 96% of the time as shown. The ability to identify non-coding regions (specificity) is somewhat reduced but remains quite high: 90% of known non-coding regions are correctly identified.
Table 2. The results of evaluating our model on the independent test set of known coding regions and known non-coding regions.
| Known coding | Known non-coding | ||
|---|---|---|---|
| (total: 103) | (total: 103) | ||
| (A) | |||
| Predicted coding | True positives | False positives | |
| 99 | 10 | ||
| (0.96) | (0.1) | ||
| Predicted non-coding | False negatives | True negatives | |
| 4 | 93 | ||
| (0.04) | (0.9) | ||
| Sensitivity | Specificity | ||
| 0.96 | 0.9 | ||
| Accuracy: 0.93 | |||
| (B) | |||
| Score < –1 | True positives | False positives | |
| 73 | 1 | ||
| (0.71) | (0.01) | ||
| Score > +1 | False negatives | True negatives | |
| 1 | 62 | ||
| (0.01) | (0.6) | ||
| Sensitivity | Specificity | ||
| 0.99 | 0.98 | ||
| Accuracy: 0.99 | |||
(A) shows the overall proportion of sequences classified by the method as coding or non-coding. (B) shows the distribution of sequences with high confidence scores. Sensitivity and specificity are provided as measures of performance. In all cells, values in parentheses indicate the proportion out of the entire set of coding and non-coding sequences (103 each).
We can now dissect model performance in the regions where the score and associated prediction are of high confidence (less than –1 or greater than +1) as well as in the low confidence range between –1 and +1. The results indicate that for 71% (73/103) of the sequences classified as coding, we are ≥95% confident that the assignment is correct (Table 2B). For the remaining 29%, however, some misclassification is likely. Of the sequences in this region, 29 are predicted to be coding. Twenty-six of these are known coding regions corresponding to a sensitivity of 0.90. However, specificity is significantly lowered. Of the 40 ORFs predicted to be non-coding, only 31 are in fact non-coding sequences (specificity of 0.78). The reduced specificity and sensitivity are expected given that predictions in this region are unreliable.
Application to chromosome I
With this understanding, we analyzed the entire complement of 509 ORFs identified by the Sanger Institute. Our method predicts that 321 of the 509 ORFs are coding. Although we cannot validate every single ORF that our method predicts is likely to be coding, we can compare the distribution of functional annotations to hypothetical annotations across the four score levels of our method. This is shown in Figure 5.
Figure 5.

Breakdown of ORFs based on Sanger Institute annotation versus LDA based annotation. The Sanger Institute provides three sub-classifications of hypothetical annotations. Those ORFs that show sequence-based homology to hypothetical annotations in other organisms are termed ‘conserved hypothetical protein’ (denoted here as ‘Conserved’ or ‘C’). If an ORF is on the opposite strand from the predicted coding strand or has unusual GC composition, it is sometimes labeled ‘hypothetical protein, unlikely’ (based on details provided on the Sanger Institute web pages and EMBL entry). We have shown these here as ‘Unlikely’ or ‘U’. Finally, some ORFs are annotated merely as ‘hypothetical protein’ and we refer to these as ‘predicted’ or ‘P’. For those ORFs with a function assignment, we use the term ‘Assigned function’ or ‘A’. Plots on the left side of this figure show the distribution of annotations for ORFs that our method would label as likely coding, while plots on the right side if this figure show ORFs that our method would identify as unlikely to be true coding regions.
The Sanger Institute utilizes three forms of hypothetical annotations. Those ORFs that show clear sequence homology to hypothetical annotations in other organisms are denoted as ‘conserved hypothetical protein’ in the EMBL entries. These ORFs constitute the set of so-called orphan genes, a set of conserved ORFs found in many organisms but for which functions have yet to be determined. A second category of ORFs have no sequence homology and were determined to be unlikely to be true coding regions based on several heuristics, such as being on the opposite strand from the postulated coding strand (based on details provided on the Sanger Institute web pages and in the EMBL entries). These ORFs are marked ‘hypothetical protein, unlikely’ in the EMBL annotation. A third set of ORFs are marked only as ‘hypothetical’ and we refer to these as ‘predicted’ ORFs since their coding status is ambiguous.
What is striking from the results in Figure 5 is that 73% of those ORFs annotated as ‘hypothetical, unlikely’ score as non-coding regions based on the LDA analysis (55% score as highly unlikely and an additional 18% are in the ambiguous region). A small percentage of these ORFs (14%) do score as very likely coding regions, but this suggests that at least some of the heuristics used to mark an ORF as unlikely are perhaps not exact. Similarly, 78% of the ORFs annotated as ‘hypothetical, conserved’ are very likely to be true coding regions based on the LDA analysis. None of these ORFs score in the range where we could be reasonably confident that they are non-coding.
Of the ORFs with functional, protein annotations (91 in the EMBL entry), 86 are correctly identified as likely to be coding, and 58 score in the high confidence range. Only five ORFs are misclassified, yielding an overall true positive identification rate of 95%. These results reinforce the validity of this approach as a means to rank ORFs based on their likelihood of coding.
To further validate our method, we evaluated the mRNA expression of 47 of the identified ORFs on Chromosome I via RT–PCR. We were able to confirm expression from 76% of ORFs predicted to be coding by our method and validate the lack of expression from 52% of ORFs predicted to be non-coding (data not shown). Our computational evaluation and results are reinforced by these findings and provide preliminary proof of principle of the validity of our approach.
DISCUSSION
The method described here allows for annotation of likely coding regions based on sequence composition in the absence of other evidence for function. At the dinucleotide level there are significant differences between coding and non-coding regions in T.brucei, and these variations in dinucleotide profiles are the basis of the success of our approach. The result is a method that is 93% accurate when tested on functionally annotated genes and their corresponding upstream regions. The method is extremely sensitive, identifying 96% of true coding regions, and quite specific, finding 90% of true non-coding regions. In applying our method to Chromosome I, we correctly identify 66 out of 70 functionally annotated ORFs for a true positive identification rate of 95%.
With increasing understanding of the biology of trypanosomes, we may be able to extend our model to include other features of coding and non-coding regions. Currently, our ability to reliably identify non-coding regions is somewhat low, with a specificity of only 0.6 for high confidence predictions. This is a consequence of using sequences that were immediately upstream of coding regions as our model for non-coding regions. These regions were expected to contain signals for the unusual trans-splicing process. Obviously, not all non-coding regions will have these signals and we currently misclassify some non-coding regions because they lack the hallmark signals of trans-splicing. However, we were limited in terms of reliable sequence data that could be guaranteed not to contain a functional coding region (see Materials and Methods). At any rate, our focus has been on identifying likely functional coding regions, and this is amply addressed by the high sensitivity of the current model.
We may also be able to improve on our model, particularly for sequences that score in the ambiguous zone, as additional data from genome sequencing become available. Currently, the data sets described in Data represent the most complete set of known, non-coding regions and fully characterized coding regions available for this organism. This has limited the approaches that can be reasonably applied to the challenge of annotating ORFs in the absence of other evidence for function.
Compositional methods for the evaluation of coding regions have been explored in a number of organisms (23,30). These methods often use higher order nucleotide combinations than we present here. For example, one such method, Hexamer, evaluates hexa-nucleotide frequencies (23). Given the limited data available for T.brucei, however, such an approach would have been statistically unfeasible. In our data set, even tri-nucleotide combinations could not be satisfactorily determined for all sequences because of the length and compositional skew of the selected non-coding regions. In the absence of sufficient training data, therefore, such methods may yield dangerously inaccurate predictions. An alternative to direct compositional approaches are sequence log likelihood ratios (23), which are sometimes used to compare the overall composition of one region with that of another. However, this approach proved to be a very poor discriminator between coding and non-coding regions in our data set (data not shown).
With the accumulation of a larger set of confirmed coding and non-coding regions, we can apply more powerful non-linear classifiers such as neural nets (NNs) or probabilistic approaches such as hidden Markov models (HMMs). We were unable to apply these methods in the current situation because our training set is essentially the only such dataset available for T.brucei. It is too small a set for reliable estimation of all the parameters required by these methods (23). Indeed, we have separately established that this data set is a poor training set even for existing predictive tools that use probabilistic approaches, such as Glimmer 2.0 (data not shown).
One possible solution to expand our limited dataset of confirmed coding regions is by using syntenic comparisons across the members of the trypanosomatid family. That is, we could use functionally annotated ORFs from L.major or T.cruzi to identify likely functional coding regions in T.brucei. Unfortunately, such synteny-based analyses fail in this instance because L.major is believed to have diverged from T.brucei and T.cruzi about 340 million years ago. The divergence dates for T.brucei and T.cruzi are estimated to be greater than 110 million years ago (1). With such large evolutionary distances even amongst the closest relatives of T.brucei, syntenic comparisons cannot be made with any reasonable confidence.
While synteny cannot be used to improve on predictions in T.brucei, some of the biology of T.brucei is conserved amongst the other members of the family. trans-splicing appears to be common to all the trypanosomatids studied to date. It is quite likely that our method can be applied to other trypanosomatids, given suitable training datasets. With appropriate data, our method may even be applied to other classes of organisms such as Caenorhabditis elegans, where trans-splicing appears to occur along with cis-splicing of certain genes (31).
The key benefit of this approach is that it is derived from a model of true coding regions. Therefore, this approach can be used to validate ORFs that lack other evidence of function. By considering the LDA score assigned to a putative coding region, ORFs can be classified into one of four classes: high confidence coding, low confidence coding, possibly non-coding and likely non-coding. Refining the set of ORFs likely to be coding in this manner should help focus efforts to experimentally characterize and classify those coding regions most likely to be functional genes. Our approach can be used as a means of annotation in the absence of more substantial evidence from experimental characterization or sequence homology.
The results described here further demonstrate the importance of developing organism-specific approaches for gene verification in conjunction with more generalized approaches. While generalized tools can provide a first-pass evaluation of likely coding regions, the assumptions that underlie these methods may not necessarily hold true in the organism of interest. This is the case in T.brucei where it appears that not all ORFs are coding. It is in such cases that the application of methods derived from the biology of the organism can improve on and refine predictions so as to facilitate experimental characterization and investigation. Such work will be crucial to gaining a better understanding of unusual organisms, particularly those that are evolutionarily distant from their better-studied counterparts.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank Dr Raphael Clifford for suggesting the use of transition probabilities and linear discriminant analysis for this problem. Dr Mike Cummings, Dr Sridhar Sadasivan and Dr Eduardo Fajardo suggested several important additions and modifications to the analysis and presentation of the LDA results. Dr Joanna Lowell provided valuable assistance with the generation of the training dataset. T.brucei sequence data were obtained from the Sanger Institute website at: http://www.sanger.ac.uk/Projects/T_brucei/. Sequencing of the T.brucei genome was accomplished as part of the Trypanosome Genome Network with support by The Wellcome Trust. The authors would like to thank Dr Matthew Berriman and the members of the T.brucei sequencing group for updates on sequencing status and early access to assembled genomic data. The sequencing of Chromosome I by Sanger Institute has just been published (32). This work was supported in part by a grant to T.G. from the Burroughs Wellcome Fund New Investigator in Molecular Parasitology award (no. 1001530).
REFERENCES
- 1.Fernandes A.P., Nelson,K. and Beverley,S.M. (1993) Evolution of nuclear ribosomal RNAs in kinetoplastid protozoa: perspectives on the age and origins of parasitism. Proc. Natl Acad. Sci. USA, 90, 11608–11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pepin J. and Meda,H. (2001) The epidemiology and control of human African trypanosomiasis. Adv. Parasitol., 49, 71–132. [DOI] [PubMed] [Google Scholar]
- 3.Jernigan J. and Pearson,R. (1993) Chemotherapy of leishmaniasis, Chagas’ disease and African trypanosomiasis. Curr. Opin. Infect. Dis., 6, 794–802. [Google Scholar]
- 4.Clayton C. (2002) Life without transcriptional control? From fly to man and back again. EMBO J., 21, 1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanhamme L. and Pays,E. (1995) Control of gene expression in trypanosomes. Microbiol. Rev., 59, 223–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delcher A.L., Harmon,D., Kasif,S. and White,O. (1999) Improved microbial gene identification with GLIMMER. Nucleic Acids Res., 27, 4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myler P., Audelman,L., deVos,T., Hixon,G., Kiser,P., Lemley,C., Magness,C., Rickel,E., Sisk,E. and Sunkin,S. et al. (1999) Leishmania major Friedlin Chromosome I has an unusual distribution of protein-coding genes. Proc. Natl Acad. Sci. USA, 96, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vijayasarathy S., Ernest,I., Itzhaki,J., Sherman,D., Mowatt,M., Michels,P. and Clayton,C. (1990) The genes encoding fructose bisphosphate aldolase in Trypanosoma brucei are interspersed with unrelated genes. Nucleic Acids Res., 18, 2967–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clayton C.E., Fueri,J., Itzhaki,J., Bellofatto,V., Wisdom,G., Vijayasarathy,S. and Mowatt,M. (1990) Transcription of the procyclic acidic repetitive protein genes of Trypanosoma brucei. Mol. Cell. Biol., 10, 3036–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudenko G., Le Blanco,S., Smith,J., Lee,M., Rattray,A. and van derPloeg,L. (1990) Procyclic acidic repetitive protein (PARP) genes located in an unusually small a-amanitin-resistant transcription unit: PARP promoter activity assayed by transient DNA transfection of Trypanosoma brucei. Mol. Cell. Biol., 10, 3492–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ullu E., Matthews,K.R. and Tschudi,C. (1993) Temporal order of RNA-processing reactions in trypanosomes: rapid trans-splicing precedes polyadenylation of newly synthesized tubulin transcripts. Mol. Cell. Biol., 13, 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J. and van derPloeg,L.H. (1991) Maturation of polycistronic pre-mRNA in Trypanosoma brucei: Analysis of trans splicing and poly(A) addition at nascent RNA transcripts from hsp70 locus. Mol. Cell. Biol., 11, 3180–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Estrano C., Tschudi,C. and Ullu,E. (1998) Exonic sequences in the 5′ untranslated region of α-tubulin mRNA modulate trans splicing in Trypanosoma brucei. Mol. Cell. Biol., 18, 4620–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews K.R., Tschudi,C. and Ullu,E. (1994) A common pyrimidine-rich motif governs trans-splicing and polyadenylation of tubulin polycistronic pre-mRNA in trypanosomes. Genes Dev., 8, 491–501. [DOI] [PubMed] [Google Scholar]
- 15.Palfi Z., Lucke,S., Lahm,H., Lane,W., Kruft,V., Bragado-Nilsson,E., Seraphin,B. and Bindereif,A. (2000) The spliceosomal snRNP core complex of Trypanosoma brucei: cloning and functional analysis reveals seven Sm protein constituents. Proc. Natl Acad. Sci. USA, 16, 8967–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee M.G.-S. and van derPloeg,L.H. (1997) Transcription of protein-coding genes in trypanosomes by RNA polymerase I. Annu. Rev. Microbiol., 51, 463–489. [DOI] [PubMed] [Google Scholar]
- 17.Patzelt E., Perry,K. and Agabian,N. (1989) Mapping of branch sites in trans-spliced pre-mRNAs of Trypanosoma brucei. Mol. Cell. Biol., 9, 4291–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hug M., Hotz,H.R., Hartmann,C. and Clayton,C. (1994) Hierarchies of RNA-processing signals in a trypanosome surface antigen mRNA precursor. Mol. Cell. Biol., 14, 7428–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapotas N. and Bellofatto,V. (1993) Differential response to RNA trans-splicing signals within the phosphoglycerate kinase gene cluster in Trypanosoma brucei. Nucleic Acids Res., 21, 4067–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Revelard P., Lips,S. and Pays,E. (1993) Alternative splicing within and between alleles of the ATPase gene 1 locus of Trypanosoma brucei. Mol. Biochem. Parasitol., 62, 93–102. [DOI] [PubMed] [Google Scholar]
- 21.Sutton R.E. and Boothroyd,J. (1988) Trypanosome trans-splicing utilizes 2′-5′ branches and a corresponding debranching activity. EMBO J., 7, 1431–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metzenberg S. and Agabian,N. (1996) Human and fungal 3′ splice sites are used by Trypanosoma brucei for trans splicing. Mol. Biochem. Parasitol., 83, 11–23. [DOI] [PubMed] [Google Scholar]
- 23.Durbin R., Eddy,S., Krogh,A. and Mitchison,G. (1998) Biological Sequence Analysis. Cambridge University Press, Cambridge, UK. [Google Scholar]
- 24.Ihaka R. and Gentleman,R. (1996) R: A language for data analysis and graphics. J. Comput. Graph. Stat., 5, 299–314. [Google Scholar]
- 25.Mendenhall W. and Sincich,T. (1996) A Second Course in Statistics: Regression Analysis, chapter 8.9: Model Validation. Prentice Hall, Upper Saddle River, NJ, USA, pp. 489–492. [Google Scholar]
- 26.Altschul S., Gish,W., Miller,W., Myers,E. and Lipman,D. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 27.Boguski M., Lowe,T. and Tolstoshev,C. (1993) dbEST—database for ‘expressed sequence tags’. Nature Genet., 4, 332–333. [DOI] [PubMed] [Google Scholar]
- 28.Venables W. and Ripley,B. (1999) Modern Applied Statistics with S-Plus. 3rd edn. Springer Verlag, Heidelberg. [Google Scholar]
- 29.Sokal R. and Rohlf,J. (1995) Biometry. 3rd edn. W.H. Freeman and Company, New York, p. 69. [Google Scholar]
- 30.Burge C. and Karlin,S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol., 268, 78–94. [DOI] [PubMed] [Google Scholar]
- 31.Nilsen T. (1993) Trans-splicing of nematode premessenger RNA. Annu. Rev. Microbiol., 47, 413–440. [DOI] [PubMed] [Google Scholar]
- 32.Hall N., Berriman,M., Lennard,N.J., Harris,B.H., Hertz-Fowler,C., Bart-Delabesse,E.N., Gerrard,C.S., Atkin,R.J., Barron,A.J., Bowman,S. et al. (2003) The DNA sequence of chromosome I of an African trypanosome: gene content, chromosome organization, recombination and polymorphism. Nucleic Acids Res., 31, 4864–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.