

Abstract
Limitation of clonal expansion of activated T cells is necessary for immune homeostasis, and is achieved by growth arrest and apoptosis. Growth arrest and apoptosis can occur passively secondary to cytokine withdrawal, or can be actively induced by religation of the T cell receptor (TCR) in previously activated proliferating T cells. TCR-induced apoptosis appears to require prior growth arrest, and is mediated by death receptors such as Fas. We tested whether TCR religation affects T cell responses to interleukin (IL)-2, a major T cell growth and survival factor. TCR ligation in activated primary human T cells blocked IL-2 induction of signal transducer and activator of transcription (STAT)5 DNA binding, phosphorylation of STAT5, Janus kinase (Jak)1, Jak3, and Akt, and kinase activity of Jak1 and Jak3. Inhibition was mediated by the mitogen-activated protein kinase kinase (MEK)–extracellular stimulus–regulated kinase (ERK) signaling pathway, similar to the mechanism of inhibition of IL-6 signaling we have described previously. TCR ligation blocked IL-2 activation of genes and cell cycle regulatory proteins, and suppressed cell proliferation and expansion. These results identify TCR-induced inhibition of IL-2 signaling as a novel mechanism that underlies antigen-mediated feedback limitation of T cell expansion, and suggest that modulation of cytokine activity by antigen receptor signals plays an important role in the regulation of lymphocyte function.
Keywords: cytokines, signal transduction, inhibition, mitogen-activated protein kinases, gene activation
Uncontrolled exuberance of an immune response leads to direct toxicity to the host and may predispose to autoimmunity. Prominent examples include mortality in human patients unable to limit responses to superantigens produced by Staphylococcus aureus 1, in mice that are unable to restrain CD4+ T cell responses to lymphocytic choriomeningitis virus 2, and autoimmune syndromes in mice and humans (for a review, see reference 3). Several mechanisms of limiting T cell responses have been described. Negative signaling by molecules such as CTL-associated molecule 4 (CTLA4), and by cytokines such as IL-10 and TGF-β, can suppress T cell proliferation 3. Growth arrest and apoptosis secondary to low levels of proliferative or antiapoptotic cytokines such as IL-2 and type I IFNs have been implicated in the limitation and termination of responses to several pathogens and antigens, including viruses and Staphylococcus enterotoxins A (SEA)1 and SEB 3 4 5 6 7. Apparently paradoxical feedback inhibition by recurrent stimulation with the antigen that initiated the response is an important mechanism of restraining ongoing T cell responses 3. TCR-induced feedback inhibition occurs by a combination of growth arrest 2 8 9 10 11 12 13 and induced apoptotic cell death 14, which appear to be linked, as growth arrest precedes, and may be required for, apoptosis 15. Antigen-mediated feedback inhibition is especially effective when persistent or high-affinity antigens interact with TCRs on activated and rapidly cycling cells 16.
TCR-induced apoptosis is mediated by specific death receptors such as Fas and TNF receptor 1, and by modulation of signaling by these receptors 14 17 18 19 20 21. TCR-induced growth arrest in T cell hybridomas and lines that do not regulate growth in a normal, physiological fashion has been linked to downregulation of cell cycle regulatory proteins such as cyclin D3 and cyclin-dependent kinase 2 (cdk2) 10 15. Mechanisms underlying TCR-induced growth arrest of primary T cells, which are dependent on growth factors and cytokines for proliferation and survival 5 6 7 22, are not known. We considered the hypothesis that TCR ligation in cycling cytokine-dependent T cells may block cytokine activity by inhibiting signal transduction. Such inhibition of signaling would limit T cell responses by several mechanisms, including growth arrest and apoptosis secondary to decreased survival signals, and would sensitize cells for apoptosis mediated by death receptors, which appears to depend on prior growth arrest 15 23.
One important component of TCR signaling is the activation of a kinase cascade involving protein kinase C (PKC)→ Raf→mitogen-activated protein kinase kinase (MEK)1, MEK2→extracellular stimulus–regulated kinase (ERK)1, ERK2 24. The ERKs constitute one family of mitogen-activated protein kinases (MAPKs) that are downstream effector kinases in a signaling pathway activated by a large number of extracellular ligands. Interestingly, cellular proliferative responses to ERKs are highly dependent on cell type and activation status, and ERKs can induce either proliferation or growth arrest and senescence 25 26 27. Furthermore, we have previously shown that activation of the MEK-ERK pathway blocks signaling and activation of signal transducer and activator of transcription (STAT)3 by IL-6 28 29, and a PKC-dependent pathway has been shown to enhance susceptibility to Fas-mediated apoptosis 30. Therefore, we tested whether activation of the MEK-ERK pathway by religation of the TCR in previously activated T cells inhibits IL-2 signaling, thus inducing a functional withdrawal of cells from the effects of IL-2. Activation of IL-2 signaling pathways and target genes was inhibited by TCR ligation, and inhibition of signaling correlated with decreased proliferation and subsequent cell death. These results demonstrate that cross-talk between two major signaling pathways in T cells has important functional consequences for modulating the extent of cell expansion.
Materials and Methods
Cell Culture.
Mononuclear cells (MNCs) were obtained from whole blood from disease-free volunteers by density gradient centrifugation using Ficoll metrizoate (Lymphoprep; GIBCO BRL). MNCs were depleted of CD56+ NK cells using the MACS separation system (Miltenyi Biotec) and CD56-specific, paramagnetic beads. In pilot experiments, CD8+ or CD4+ cells were also depleted, and in all cases depletion was verified using flow cytometry (see below). In experiments with superantigens, cells were activated with SEA (Sigma Chemical Co.) in complete media (CM) containing RPMI (GIBCO BRL) supplemented with 10% fetal bovine serum (Hyclone) and 2 mM l-glutamine for 3 d, and SEA-reactive T cells were expanded in IL-2 and added to adherence-purified monocytes. Otherwise, purified OKT3 antibody (Coulter) was added to NK cell–depleted MNCs at a 1:1,000 dilution that strongly activated T cells in initial titration experiments, and cells were harvested after 3 d, washed, and used for experiments or expanded in IL-2 as noted in the figure legends. An aliquot was saved for flow cytometric analysis of cell surface phenotype, and remaining cells (>90% CD3+) were plated in wells that had been coated with 10 μg/ml of rabbit anti–mouse Ig and 3 μg/ml of either anti-CD3 (clone UCHT-1; PharMingen) or an isotype-matched irrelevant control antibody. Initial dose–response experiments demonstrated that the effects of anti-CD3 saturated at 1–2 μg/ml. Additional anti-CD3 antibodies that were used with similar results were 4B5 (Boehringer Mannheim) and HIT3a (Immunotech), and anti-CD4 antibodies (RPA-T4; PharMingen) were also used as an additional control.
Flow Cytometry.
Cells were analyzed using flow cytometry as described previously 31 using the following antibodies: T cell and activation markers, UCHT-1 and 4B5 (anti-CD3), RPA-T4 (anti-CD4), HIT8a (anti-CD8), M-A251 (anti-CD25), CF1 and Mik-β3 (anti-CD122), AG184 (anti-γc); NK cell marker, 3G8 (anti-CD16); monocyte marker, IV.3 (anti-CD32 monocyte-specific epitope); and 48607 (anti–CC chemokine receptor 2 [CCR2]). AG184, M-A251, HIT8a, and Mik-β3 were obtained from PharMingen, 4B5 and 3G8 from Boehringer Mannheim, CF1 from Immunotech, IV.3 from Medarex, and 48607 from R&D Systems. Analyses were done using a FACScan™ flow cytometer with CELLQuest software™ (Becton Dickinson).
Cell Lysis, Electrophoretic Mobility Shift Assay, Immunoprecipitation, and Immunoblotting.
Cell extracts were obtained as described previously 29. Extracts corresponding to 3.3 × 105 cells (∼12 μg of protein) were incubated for 15 min at room temperature with 0.5 ng of 32P-labeled double stranded oligonucleotide containing the gamma-activated sequence (GAS) STAT-binding site from the IFN regulatory factor (IRF) promoter in a 15-μl binding reaction containing 40 mM NaCl and 2 μg of poly-dI-dC (Amersham Pharmacia Biotech), as described previously 29, and complexes were resolved on nondenaturing 4.5% polyacrylamide gels. For immunoblotting, cell lysates were fractionated on 7.5 or 10% polyacrylamide gels using SDS-PAGE, transferred to polyvinylidene fluoride membranes (Millipore), and incubated with specific antibodies, and enhanced chemiluminescence was used for detection. mAbs against Janus kinase (Jak)1 (clone 73) and STAT5 (clone 89) were obtained from Transduction Laboratories, anti-cyclin D3 (D-7) from Santa Cruz Biotechnology, anti-underphosphorylated retinoblastoma protein (Rb, G99-549) from PharMingen, and antiphosphotyrosine (4G10) and anti-tyrosine phosphorylated STAT5 from Upstate Biotechnology. Polyclonal antibodies against Jak1 (Q-19), STAT5 (C-17), and cyclin D3 (C-16) were from Santa Cruz Biotechnology, and anti-tyrosine phosphorylated Jak1 was from Quality Control Biochemicals. In most cases, results were confirmed using two different antibodies. Immunoprecipitations were performed by adjusting cell extracts obtained from 20 × 106 cells to a volume of 0.5 ml and incubating with 2–4 μg of specific antibody for 4 h at 4°C. Immunoprecipitates were collected using protein A and protein G agarose beads (Pierce Chemical Co.), washed twice in lysis buffer and once in PBS, resolved by SDS-PAGE, and analyzed by immunoblotting.
In Vitro Kinase Assays.
Jak1 and Jak3 immunoprecipitates were obtained from 40 × 106 cells that had been lysed in buffer containing 0.5% NP-40, 10% glycerol, 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 0.1 mM EDTA, 1 mM dithiothreitol, 1 mM Na3VO4, and protease inhibitors, as described previously 29, using Jak1 (Q-19) or Jak3 antibodies (provided by J. O'Shea, National Institutes of Health, Bethesda, MD). After washes in lysis buffer and kinase buffer containing 20 mM Hepes (pH 7.4), 50 mM NaCl, 5 mM MgCl2, 5 mM MnCl2, and 0.1 mM Na3VO4, 25% of immunoprecipitates were saved for immunoblotting, and the remainder was incubated for 30 min at room temperature in 40 μl of kinase buffer with 1 μCi of [γ-32P]ATP. 1 μM unlabeled ATP was added to Jak3 kinase reactions. 32P-labeled proteins were washed in kinase buffer and PBS, and analyzed using SDS-PAGE and autoradiography.
Transfections and Reporter Gene Assays.
Kit225 cells, a human IL-2–dependent cell line 32, were maintained in CM containing 20 U/ml of IL-2, and deprived of IL-2 for 24 h before transfection. 107 cells were transfected using electroporation and the Bio-Rad Gene Pulser (320 V, 960 μF) with 20 μg of total DNA (10 μg of 5× gamma response region [GRR] GAS-luciferase reporter [32]), 2 μg of lacZ encoding plasmid, and 8 μg of empty control vector or vector encoding constitutively active MEK1 (containing the S218E, S222D mutations and an NH2-terminal deletion of residues 30–49) or kinase-inactive MEK1 29. Cells were stimulated with 100 U/ml of IL-2 for 6 h, and luciferase activity was measured as described previously 29, and normalized for β-galactosidase activity encoded by the cotransfected control plasmid.
Analysis of mRNA Levels.
Total cellular RNA was isolated using Trizol (GIBCO BRL) according to the instructions of the manufacturer. For reverse transcription (RT)-PCR, RNA was treated with RNase-free DNase, and cDNA was obtained using Moloney murine leukemia virus reverse transcriptase (GIBCO BRL). 2.5% of each cDNA was subjected to 25 cycles of PCR using conditions that result in a single specific amplification product of the correct size as described previously 33: 30 s denaturation at 94°C, 1 min annealing at 55°C, and 30 s extension at 72°C in a GeneAmp 9600 thermal cycler (Perkin-Elmer Corp.). dNTPs were used at 100 μM, and 1 μCi of [α-32P]dATP was added to each reaction. No amplification products were obtained when reverse transcriptase was omitted, indicating the absence of contaminating genomic DNA. Amplification was empirically determined to be in the linear range. Oligonucleotide primers for the cytokine-inducible SH2 protein (CIS) and PGE2R genes were provided by C. Beadling and K. Smith (Weill Medical College of Cornell University [34]), and additional primer sequences are Fos: CCG AGA TTG CCA ACC TGC TGA A and CAC TGG GCC TGG ATG ATG C; suppressor of cytokine signaling (SOCS)1 (three sets): GGA CGC CTG CGG ATT CTA CTG G and TCG CGG AGG ACG GGG TTG AG, CGA CGG CGG CCA GAA CCT TC and GTG AAA GCG GCC GGC CTG AAA GT, and CTC CGG CTG GCC CCT TCT GTA G and TGA AAG CGG CCG GCC TGA AAG TG; SOCS3: CAC TAC ATG CCG CCC CCT GGA G and TCG CCC CCG GAG TAG ATG TAA T; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH): GTG AAG GTC GGA GTC AAC and TGG AAT TTG CCA TGG GTG.
Proliferation Analysis.
105 cells in 100 μl of medium were seeded in triplicate in 96-well tissue culture plates that had been coated with 10 μg/ml of rabbit anti–mouse Ig and 3 μg/ml of anti-CD3 or control antibody. Cells were pulsed for 4 h with 10 μCi/ml of [3H]thymidine and harvested using an automated cell harvester (Harvester 96; Tomtec), and [3H]thymidine incorporation was quantitated using a Microbeta Trilux Scintillation Counter (Wallac). Cell counts were performed in duplicate, and cell viability was determined using trypan blue and propidium iodide exclusion.
Results
TCR Ligation Blocks Activation of STAT5 by IL-2.
IL-2 is a major growth factor for activated T cells and plays an important role in lymphocyte expansion and differentiation during an immune response (for reviews, see references 35 36 37 38). Activation of STAT5 is an important component of IL-2 signaling 38, and the effect of TCR cross-linking on IL-2 induction of STAT5 DNA binding activity was analyzed. Since staphylococcal superantigens are high-affinity naturally occurring TCR ligands that initially lead to vigorous immune responses, which subsequently are strongly limited 1, the effect of SEA on IL-2 signaling was determined. Freshly isolated MNCs from normal donors were depleted of NK cells, activated with SEA, and SEA-reactive T cells were expanded in IL-2. T cells were then added to tissue culture wells containing APCs in the presence or absence of SEA for 1 h, stimulated with IL-2 for 10 min, and cell extracts were assayed for STAT DNA-binding activity by electrophoretic mobility shift assays (EMSAs; Fig. 1 A). Treatment with IL-2 resulted in the rapid induction of a DNA–protein complex (lane 3) that bound the IRF GAS oligonucleotide specifically and corresponded to STAT5 (33; and data not shown), and treatment with SEA resulted in significant inhibition of IL-2 activation of STAT5 DNA binding activity (lane 4); similar results were obtained with SEB (data not shown). Immunoblotting demonstrated comparable levels of STAT5 in all extracts (Fig. 1 A, lower panel), indicating that SEA treatment suppressed activation of DNA binding and did not decrease STAT5 protein levels.
Figure 1.

TCR ligation blocks IL-2 activation of STAT5 DNA binding. Preactivated T cells (>90% CD3+) were added to wells containing purified monocytes, and SEA (2 μg/ml) was added (A); or to wells coated with isotype-matched control or anti-CD3 antibodies (B), for 1 h, followed by a 10-min stimulation with IL-2 (100 U/ml). Cell extracts were analyzed using EMSA with the IRF oligonucleotide and immunoblotting (IB) as described previously (reference 33).
To determine whether APCs or APC products were required for inhibition, TCRs were cross-linked using plate-bound anti-CD3 antibodies in the absence of APCs. Activation of STAT5 DNA binding was suppressed by TCR ligation (Fig. 1 B). Suppression of STAT5 activation by TCR ligation (40–90% inhibition relative to control) was consistently detected in >40 independent experiments using different blood donors, and was observed with a broad range of IL-2 doses (between 2 and 100 U/ml), with cross-linking times of 0.5–4 h, with purified CD4+ or CD8+ cells, and with several antibodies that ligate the TCR, but not with irrelevant or anti-CD4 antibodies (data not shown). Consistent with previous reports that TCR signaling is less dependent on costimulation in activated T cells 39, co-cross-linking of CD28 had no additional effect, but pharmacological mimicking of maximal TCR stimulation using PMA plus ionomycin more strongly inhibited STAT5 activation (data not shown).
Tyrosine phosphorylation of STAT5 is required for DNA binding 38, and the effect of SEA or TCR cross-linking on IL-2–induced tyrosine phosphorylation was examined. IL-2–induced tyrosine phosphorylation of STAT5 was suppressed by SEA treatment (Fig. 2 A, lanes 3 and 4) or TCR cross-linking (Fig. 2 B, lanes 2 and 4). Immunoblotting demonstrated comparable levels of STAT5 in all extracts (Fig. 2). Rapid and transient activation of STAT5 tyrosine phosphorylation, which has been reported by one group after TCR cross-linking (detected after 7 min, and absent after 15 min [40]), was not detected in the time frame of these experiments. The results showing inhibition of STAT5 tyrosine phosphorylation confirm the DNA binding results (Fig. 1), and suggest that TCR ligation blocks STAT5 activation. The mechanism of inhibition of IL-2 signaling by TCR cross-linking was further explored using the more simple system with plate-bound antibodies, which eliminated potential contributions from APCs or ligation of additional T cell surface proteins.
Figure 2.

TCR ligation blocks STAT5 tyrosine phosphorylation. After 1 h exposure to SEA (2 μg/ml; A) or plate-bound control or anti-CD3 antibodies (B), T cells were stimulated for 10 min with IL-2 (100 U/ml) and cell extracts were prepared. Cell extracts were analyzed by immunoblotting with antiphosphotyrosine (P-tyr)–STAT5 antibodies (A), or STAT5 immunoprecipitates were blotted with 4G10 antiphosphotyrosine antibodies (B). Filters were stripped and reprobed with anti-STAT5 antibodies.
Inhibition of IL-2 Signaling Occurs Upstream of STAT5 Activation, and Akt Activation is Also Blocked.
Activation of STAT5 by IL-2 depends on membrane-proximal events including the phosphorylation and activation of Jak1 and Jak3 kinases 38. IL-2–induced tyrosine phosphorylation of Jak1 and Jak3 was suppressed by TCR cross-linking (Fig. 3A and Fig. B), and Jak kinase activity was suppressed in parallel (Fig. 3A and Fig. B). Similar results were obtained using an antibody specific for tyrosine-phosphorylated Jak1 (data not shown). These results indicate that TCR cross-linking suppressed the activation of IL-2 signaling upstream of STAT5 activation, and suggest that additional IL-2 signaling pathways downstream of the Jaks may be affected. Activation of the Akt kinase (also termed protein kinase B) by IL-2 is important for proliferation and suppression of apoptosis 32 41, and the effect of TCR cross-linking on activation of Akt was investigated using immunoblotting with antibodies specific for the activated, phosphorylated form of Akt (Fig. 3 C). TCR cross-linking effectively suppressed IL-2 activation of Akt (lane 4). These results indicate that TCR cross-linking in activated T cells blocks IL-2 signal transduction at a proximal step, and suppresses the activation of several signaling pathways important for the proliferative response to IL-2.
Figure 3.
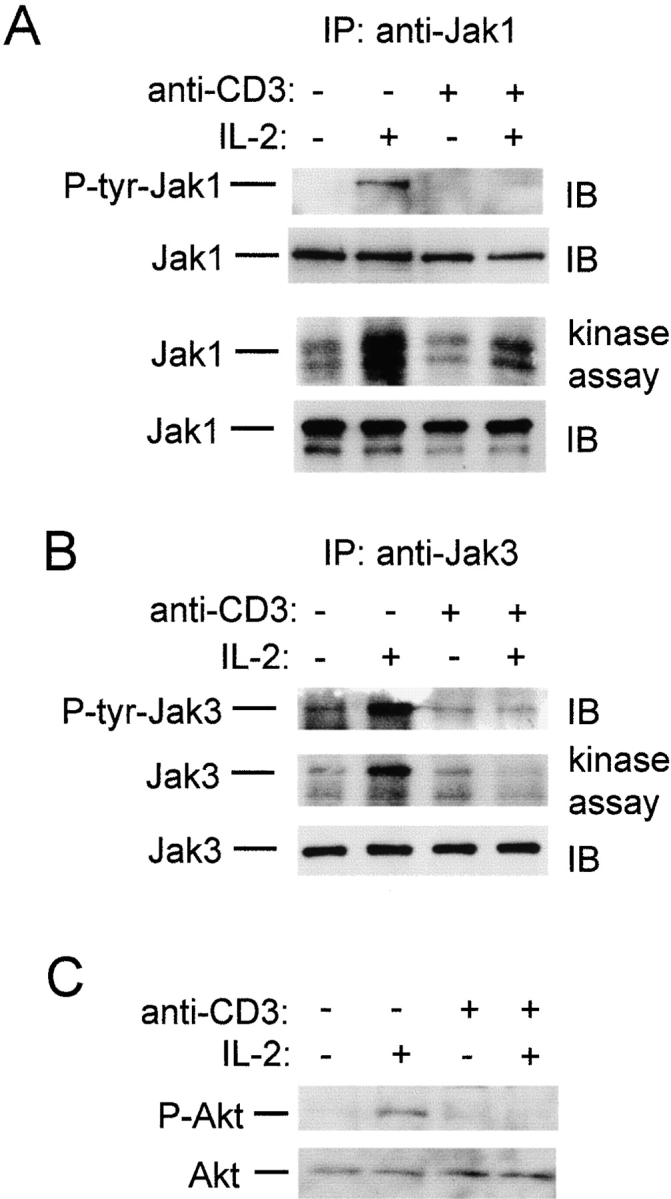
TCR ligation blocks activation of Jak1, Jak3, and Akt. Preactivated T cells were subjected to TCR ligation for 1 h, followed by a 10-min stimulation with IL-2 (100 U/ml). (A) Jak1 immunoprecipitates (IP) were analyzed using immunoblotting (IB) with 4G10 (top panel), and the same filter was reprobed with anti-Jak1 antibodies (second panel). Alternatively, Jak1 immunoprecipitates were used in in vitro kinase assays (75% of each immunoprecipitate, third panel) and analyzed using immunoblotting (25% of each immunoprecipitate, bottom panel). (B) Jak3 immunoprecipitates were obtained, 75% of each immunoprecipitate was used in in vitro kinase assays (middle panel), and 25% of each immunoprecipitate was analyzed using immunoblotting with 4G10 and Jak3 antibodies (top and bottom panels). (C) Cell extracts were analyzed by immunoblotting with antibodies specific for serine 473–phosphorylated Akt (upper panel) and against Akt (lower panel).
TCR ligation could block STAT5 activation by inhibiting the expression or the function of components of the IL-2R–Jak-STAT signaling pathway. Expression levels of the IL-2R and the associated Jak1 and Jak3 kinases were investigated (Fig. 4). Flow cytometry experiments demonstrated that, as expected, TCR ligation resulted in decreased cell surface expression of CD3 (Fig. 4 A), thus confirming the efficacy of TCR engagement 39. In contrast, TCR cross-linking did not have a significant effect on cell surface expression of the IL-2R α (CD25), β (CD122), or γc chains (Fig. 4 A). TCR cross-linking also had no effect on cellular levels of Jak1 or Jak3 (Fig. 4 B). The results demonstrating no changes in IL-2R, Jak1, Jak3, and STAT5 levels after TCR cross-linking indicate that cross-linking does not modulate the expression of the components of the IL-2R–Jak-STAT pathway that are required for STAT5 activation, and suggest instead that TCR cross-linking modulates their function.
Figure 4.
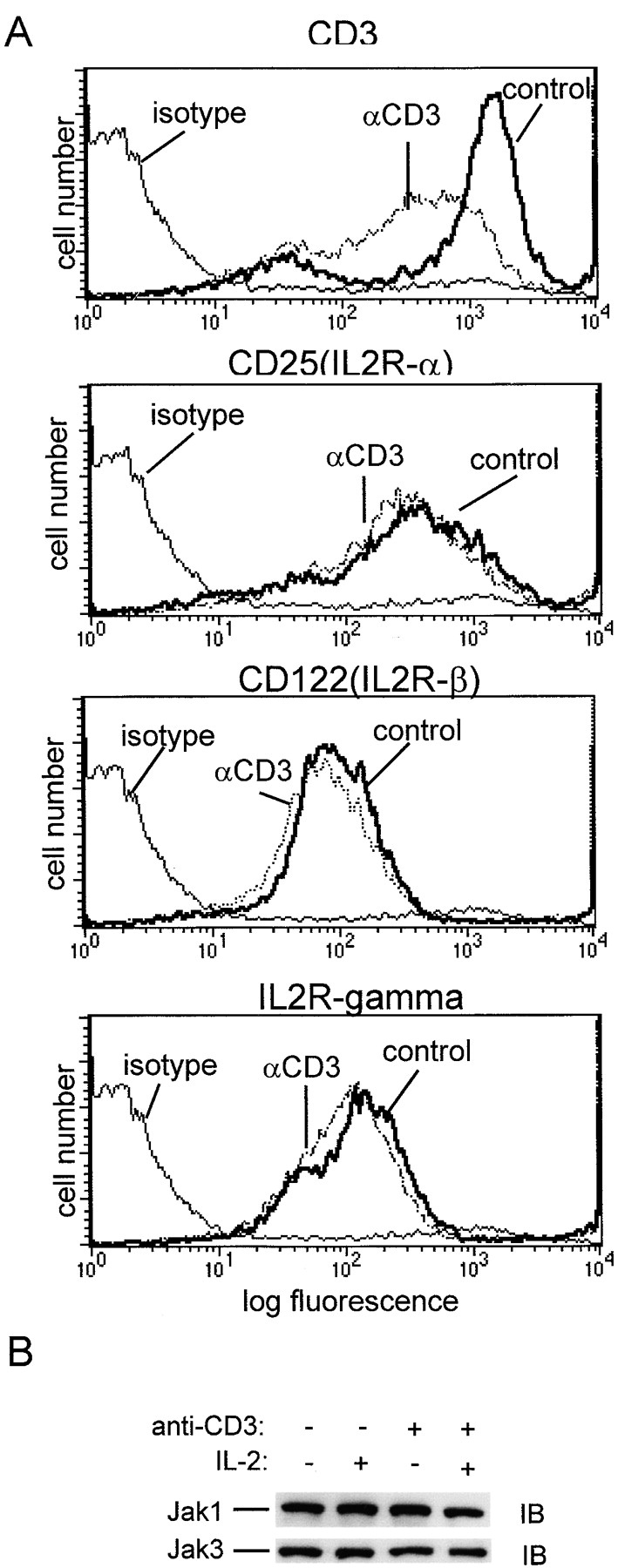
TCR ligation does not affect IL-2R, Jak1, or Jak3 expression. Preactivated T cells were subjected to TCR ligation for 1 h and analyzed by flow cytometry (A), or cell extracts were analyzed by immunoblotting (IB; B).
Role of the MEK-ERK Signaling Pathway in Inhibition of IL-2 Signaling.
We have previously demonstrated that triggering of the kinase cascade leading to MEK and ERK activation blocks IL-6 activation of Jaks upstream of STAT3 activation without altering expression of IL-6Rs or Jaks 28 29, and others have shown that activation of the MEK-ERK pathway can lead to suppression of proliferation 26 27. Since cross-linking of the TCR activates a kinase cascade involving PKC→Raf→MEK1, MEK2→ERK1, ERK2, we investigated whether these kinases play a role in inhibition of IL-2 signaling (Fig. 5). The specific PKC inhibitor GF109203X, but not the calcineurin inhibitor cyclosporin A, reversed TCR inhibition of STAT5 activation (Fig. 5 A), and inhibition was also reversed by the MEK inhibitor PD98059 (Fig. 5 B). These results suggest that the PKC-dependent activation of the MEK-ERK pathway plays an important role in inhibition of IL-2 signaling. The role of this pathway was further investigated using PMA, a direct activator of PKC and a strong activator of downstream MEKs and ERKs. Preincubation for 15 min with PMA resulted in a strong suppression of STAT5 activation (Fig. 5 C), consistent with the rapid inhibition of IL-6 signaling by this pathway 29. PMA treatment did not alter IL-2R expression (data not shown), and cytokine signaling was not globally inhibited by PMA, since IFN-α signaling was not inhibited in T cells (Fig. 5 C, lanes 5 and 6) or in myeloid cells 29. The role of MEKs and the functional significance of inhibition of IL-2 signaling mediated by these kinases was investigated using transient transfections of the IL-2–responsive Kit225 cell line and reporter gene assays (Fig. 5 D). IL-2 treatment induced a sevenfold increase in activity of the 5× GRR GAS-luciferase reporter gene, which contains five copies of the GAS site from the FcγRI promoter, and was previously shown to be induced in these cells in an IL-2– and STAT5-dependent manner 32. This induction was nearly completely blocked when cells were cotransfected with a plasmid encoding constitutively active MEK1, but not kinase-inactive MEK1 (Fig. 5 D). These results, taken together, indicate that activation of the PKC→ Raf→MEK→ERK pathway plays an important role in inhibition of IL-2 signaling by TCR ligation in activated T cells, consistent with previously reported MEK-dependent inhibition of proliferation in PC12 cells and primary fibroblasts 26 27.
Figure 5.
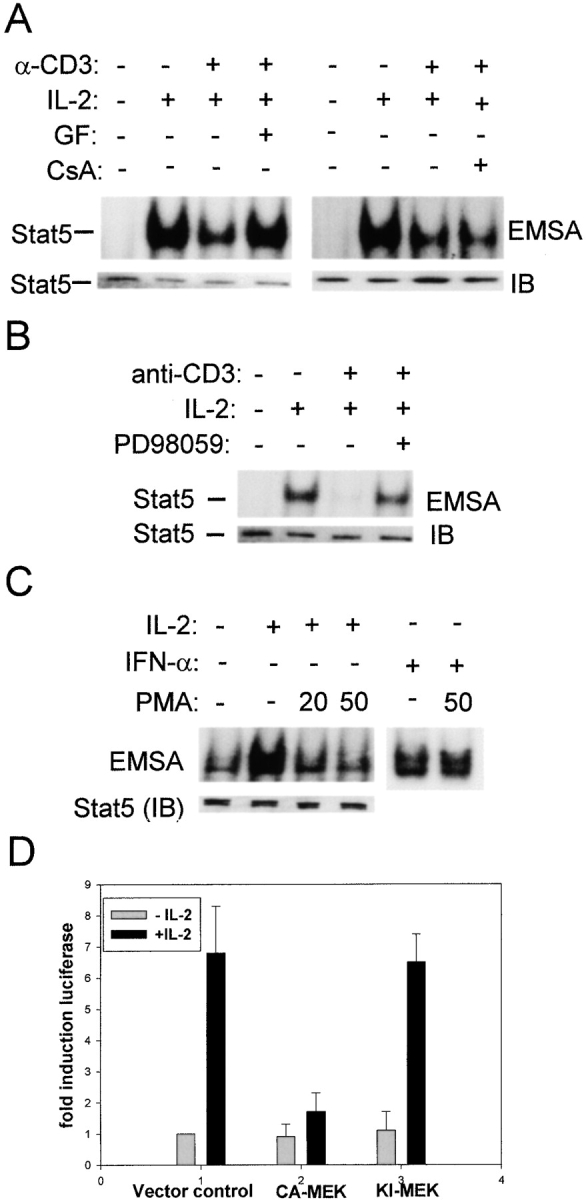
TCR inhibition of IL-2 signaling is mediated by the MEK-ERK signaling pathway. (A and B) Preactivated T cells were treated for 30 min with 1 μM GF109203X (GF), 0.5 μg/ml cyclosporin A (CsA), or 50 μM PD98059, followed by TCR ligation for 1 h and IL-2 (100 U/ml) stimulation for 10 min. (C) Cells were treated with PMA (20 or 50 ng/ml) for 15 min before a 10-min stimulation with IL-2 (100 U/ml) or IFN-γ (100 U/ml). (D) Kit225 cells were cotransfected in duplicate with 5× GRR GAS-luciferase, a β-galactosidase encoding plasmid, and either a control empty vector, or, as indicated, the same vector containing a constitutively active (CA) or kinase inactive (KI) MEK1 cDNA. Cells were split and stimulated with IL-2 (100 U/ml) for 6 h. Luciferase activity was normalized according to β-galactosidase activity, and a representative experiment out of five performed is shown.
We have previously shown that rapid MEK-ERK–dependent inhibition of IL-6 signaling occurs independently of new protein or RNA synthesis 29, presumably through phosphorylation of preexisting signaling components. Inhibition of IL-2 signaling by PMA occurred within 15 min (Fig. 5); inhibition by TCR ligation occurred reproducibly within 30 min, and was less consistent with shorter incubation periods, possibly secondary to the time required for cells to settle from suspension onto the bottom of antibody-coated wells (data not shown). The requirement for new protein and RNA synthesis for inhibition of IL-2 signaling by TCR ligation was investigated using cycloheximide and actinomycin D under conditions where these agents effectively blocked new protein and RNA synthesis, respectively (28 29 31; and data not shown). TCR ligation blocked IL-2 activation of STAT5 in the presence of cycloheximide and actinomycin D (Fig. 6), indicating that inhibition can occur in the absence of new synthesis of inhibitory molecules. These results do not exclude the possibility that TCR ligation may induce expression of inhibitory molecules that contribute to inhibition of IL-2 signaling, especially at later time points. SOCS1 and SOCS3 are the two members of the SOCS family of proteins that are currently known to inhibit Jak-STAT signal transduction, and therefore the expression of these genes after TCR ligation was examined (Fig. 7). Semiquantitative RT-PCR that had been validated using Northern hybridization was used as described previously 33. Positive control panels show that Fos and IFN-γ mRNA levels were dramatically induced by TCR ligation with the expected kinetics (third and fourth panels). SOCS3 mRNA was modestly and transiently increased after TCR ligation (top panel), suggesting that SOCS3 may contribute to inhibition of IL-2 signaling. Activation of the MEK-ERK pathway by PMA did not increase SOCS3 mRNA levels (top panel, lane 6), indicating that inhibition of IL-2 signaling by the MEK-ERK pathway is not mediated by SOCS3. SOCS1 mRNA levels were regulated in a complex fashion by TCR ligation (second panel); this result was obtained in multiple independent experiments using three different sets of SOCS1 primers. Although we have not yet resolved whether the regulation of SOCS1 may involve opposing effects on transcription and RNA stability, it is clear that neither TCR ligation nor PMA treatment resulted in increased SOCS1 levels. Taken together, the results indicate that one rapid, MEK-dependent component of inhibition of IL-2 signaling is independent of the synthesis of new inhibitory proteins, and suggest that inhibition of IL-2 signaling by the MEK-ERK pathway is not dependent on induction of SOCS1 or SOCS3 proteins. We have not excluded the possibility that induction of inhibitory proteins, such as SOCS3, may contribute to inhibition of IL-2 signaling, possibly at later time points.
Figure 6.

Inhibition of IL-2 signaling after TCR ligation does not require new RNA or protein synthesis. Preactivated T cells were preincubated for 30 min with cycloheximide (CHX, 40 μg/ml) or actinomycin D (Act D, 5 μg/ml), then subjected to TCR ligation for 1 h followed by 10 min of stimulation with IL-2.
Figure 7.

Regulation of SOCS mRNA levels after TCR ligation. Preactivated T cells were added to wells containing anti-CD3 antibodies, and cells were harvested at the indicated time points. mRNA levels were determined using semiquantitative RT-PCR, as described previously (reference 33).
TCR Ligation Suppresses IL-2 Induction of Genes and Cell Cycle Regulators, and IL-2–dependent Cell Expansion.
The functional consequences of TCR-induced inhibition of IL-2 signaling were investigated by analyzing expression of CIS, PGE2R, and CCR2, which are induced by IL-2, but not by the TCR 34 42 43. Ligation of the TCR before addition of IL-2 effectively blocked the induction of CIS and PGE2R mRNA (Fig. 8 A), and suppressed expression of CIS and PGE2R genes even when cells were exposed to high doses of IL-2 before TCR ligation (Fig. 8 B). PMA plus ionomycin more strongly inhibited expression of IL-2–dependent genes (Fig. 8 B), which correlated with stronger inhibition of STAT5 (data not shown). CIS transcription is STAT5 dependent 43, and the CIS and PGE2R genes are regulated by IL-2 at the transcriptional level, indicating that TCR ligation likely inhibited gene expression by blocking IL-2–induced transcription. TCR-mediated suppression of IL-2 induction of CCR2 42 was confirmed in our system using flow cytometry (Fig. 8 C). These results, together with the suppression of IL-2 activation of reporter gene activity (Fig. 5 D), demonstrate that inhibition of IL-2 signaling by TCR ligation in activated T cells has important functional consequences for modulation of gene expression.
Figure 8.
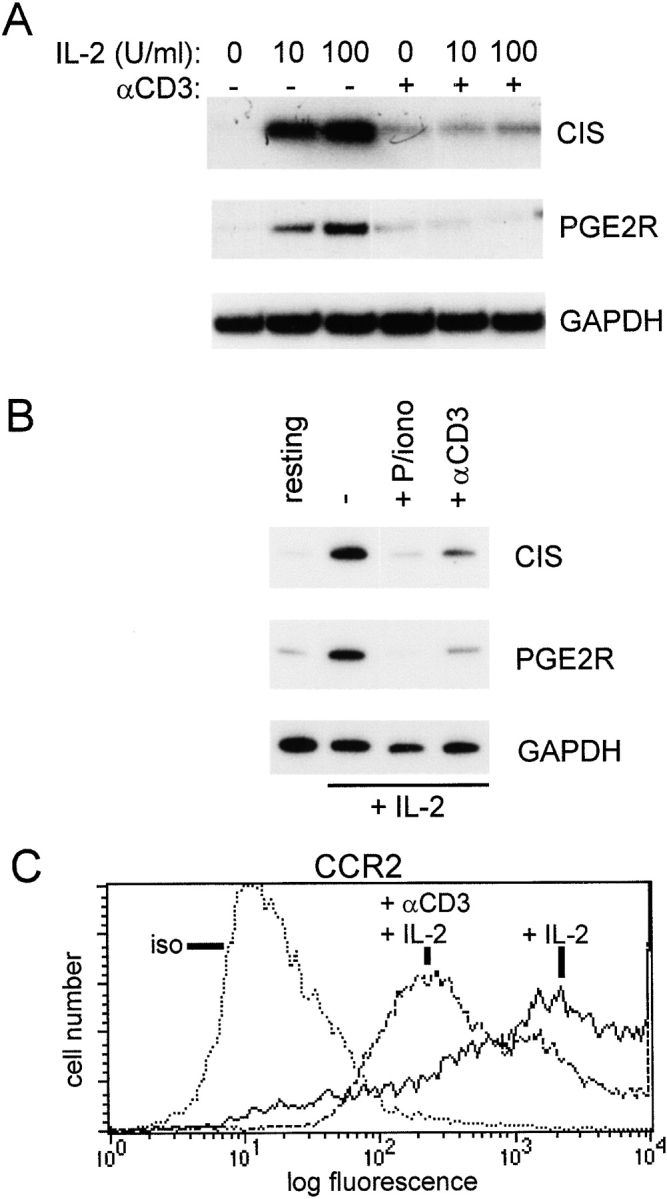
TCR inhibition of IL-2–induced gene expression. (A) Preactivated T cells were subjected to TCR ligation for 1 h followed by 1 h of stimulation with IL-2, and mRNA levels were determined using semiquantitative RT-PCR, as described previously (reference 33). (B) T cells activated with OKT3 for 3 d were cultured for an additional 2 d in IL-2 (100 U/ml), and then added to wells containing control or anti-CD3 antibodies for 5 h. resting, cells not exposed to IL-2. P/iono, PMA and ionomycin. (C) Cell surface CCR2 expression after 24 h of TCR ligation was determined using flow cytometry. iso, isotype control.
Important events for IL-2–induced progression into S phase of the cell cycle include upregulation of cyclin D3 and cdk2 levels 32 44 45, leading to increased cdk activity and phosphorylation of Rb. TCR ligation in rapidly proliferating, IL-2–dependent T cells resulted in downregulation of cyclin D3 and cdk2 levels (Fig. 9). Downregulation of cyclin D3 and cdk2 levels would be predicted to result in increased levels of underphosphorylated Rb, and suppression of proliferation. As predicted, TCR cross-linking resulted in increased levels of underphosphorylated Rb (Fig. 9). The effects of PMA or PMA plus ionomycin were comparable to, or greater than, the effects of TCR ligation (Fig. 9), and PMA plus ionomycin increased underphosphorylated Rb levels more dramatically in other experiments (data not shown). These results show that TCR ligation, or pharmacologic mimicking of TCR ligation, blocks IL-2 signals that are important for cell cycle progression.
Figure 9.

TCR regulation of cell cycle regulatory proteins. T cells activated with OKT3 for 3 d were cultured for an additional 2 d in IL-2 (100 U/ml), and then added to wells containing control or anti-CD3 antibodies. Cell extracts were prepared after 5 h of TCR ligation or PMA and ionomycin (P/iono) treatment, and analyzed using immunoblotting. The same filter was sequentially probed with the indicated antibodies. under-P-Rb, underphosphorylated Rb.
Proliferation of activated T cells in these cultures was highly dependent on exogenous IL-2 (data not shown), consistent with previous reports 22. TCR ligation in rapidly proliferating, IL-2–dependent T cells led to a 65% decrease in T cell proliferation as assessed by [3H]thymidine incorporation (Fig. 10 A; a representative experiment out of four performed is shown). Cell counts performed on parallel wells at the same time point (after 4 h of TCR ligation) demonstrated comparable numbers of viable cells (data not shown), indicating that decreased [3H]thymidine incorporation reflected inhibition of proliferation rather than cell death. PMA inhibited proliferation comparably to TCR cross-linking, and PMA plus ionomycin was more effective (Fig. 10 A). The effect of TCR ligation on T cell expansion was determined. 24 h after plating of proliferating IL-2–dependent T cells, control cultures demonstrated a 90% increase in the number of viable cells, whereas cultures subjected to TCR ligation demonstrated only a 10% increase in viable cells (Fig. 10 B; a representative experiment out of five performed is shown). The decreased cell numbers in TCR-stimulated cultures were primarily secondary to suppression of growth, although a small increase in cell death was detected 24 h after TCR ligation (Fig. 10 B). Direct cross-linking of Fas receptors using agonist antibodies at this early point in culture (after 2 d of culture in exogenous high dose IL-2) had minimal effect on cell numbers, indicating that the decrease in T cell numbers detected (Fig. 10 B) was not secondary to a Fas-mediated mechanism. As predicted, TCR signaling affected the distribution of T cells in different phases of the cell cycle, and Fig. 8 C shows a decrease in actively cycling T cells in S and G2/M phases of the cell cycle after SEA treatment. SEA treatment induced an increase in apoptosis, as assessed by numbers of cells containing subdiploid levels of DNA, from 4 to 12% of cells (Fig. 10 C), and the possible role of a Fas-mediated mechanism in apoptosis has not yet been investigated. TCR ligation resulted in decreased cell numbers throughout the subsequent 5 d of culture, with a resulting fourfold lower yield of viable cells in TCR-stimulated as opposed to control cultures (Fig. 10 D; a representative experiment out of three performed is shown). The effect of PMA was comparable to, and the effect of PMA plus ionomycin greater than, the effect of TCR ligation (Fig. 10 D). Increased cell death contributed more to the lower cell recovery 5 d after TCR cross-linking (Fig. 10 E) than 1 d after TCR ligation (Fig. 10 B). Cells became sensitive to apoptosis triggered by cross-linking cell surface Fas receptors during the final 2–3 d of these cultures (data not shown), and the relative contributions of cytokine withdrawal and Fas-dependent mechanisms to this increased cell death have not yet been resolved. These results demonstrate that TCR-induced inhibition of IL-2 signaling leads to growth arrest, and suggest that inhibition is linked to subsequent cell death.
Figure 10.

Inhibition of IL-2–dependent T cell expansion by TCR ligation. T cells activated with OKT3 or SEA for 3 d and cultured in IL-2 for 2 d to obtain rapidly proliferating cells were used. (A) [3H]Thymidine incorporation. [3H]Thymidine was added to triplicate wells 2 h after cells were plated, and cells were harvested and [3H]thymidine incorporation measured after an additional 4 h. A representative experiment out of four performed is shown. (B) Cell counts from duplicate wells were obtained 24 h after cells were plated. Cell viability was determined using trypan blue and propidium iodide exclusion. A representative experiment out of five performed is shown. (C) Cell cycle analysis was performed on T cells 24 h after addition of APCs and SEA, as described previously (reference 33). (D and E) Cell counts were obtained 3 and 5 d after cells were plated. D shows viable cell numbers, and E shows relative proportions of live and dead cells on the fifth day of culture. A representative experiment out of three performed is shown.
Discussion
In T cell activation, initial encounter with antigen and TCR ligation induces quiescent cells to enter the G1 phase of the cell cycle and to express cytokine receptors, and renders them competent to progress through the cell cycle in response to cytokines and growth factors such as IL-2 22. In this initial phase, the cells are not cytokine dependent, and MEK-ERK signaling downstream of TCR ligation plays a role in activation and priming for expansion 25. Upon subsequent exposure to cytokines, T cells enter a proliferative phase during which they are cytokine dependent for growth and survival 22. At this point, persistence of antigen leads to negative feedback and limitation of clonal expansion. The best-characterized mechanism of negative feedback is TCR-triggered apoptosis mediated by death receptors such as Fas and TNF receptor 1 14, and defects in apoptosis have been implicated in autoimmunity in mice and human patients 14 46 47 48. The results described herein identify TCR-induced, rapidly acting, and MEK-dependent inhibition of IL-2 signaling as a novel physiological mechanism that underlies growth arrest of primary proliferating T cells. Given the recent evidence that TCR-induced apoptosis is dependent on prior growth arrest 15, it is possible that TCR-induced blockade of IL-2 signaling and subsequent growth arrest may represent an important first step that, along with additional factors, makes cells susceptible to undergo apoptosis at a later time point. Apoptosis may be secondary to decreased survival signals or may occur by a Fas-dependent mechanism, and the relationship between inhibition of IL-2 signaling and apoptosis will be further investigated in future work. Growth arrest was observed before the onset of significant levels of apoptosis (Fig. 10), and may correspond to an additional level of limiting T cell responses that functions in addition to apoptosis. This growth arrest may contribute to the limitation in T cell expansion in the absence of cell death that has been described under certain circumstances, such as high dose paralysis occurring in the presence of high doses of antigens, and in persistent infections 2 9 11 12 23. IL-2–dependent T cell expansion was limited but not blocked, suggesting that this mechanism does not completely prevent the normal progression of an immune response, but that interplay between TCR and cytokine signals of varying intensities and at different time points in T cell activation determines the magnitude of the response.
At first glance, inhibition of IL-2 signaling and proliferation by an MEK-dependent mechanism may appear paradoxical, since activation of the MEK-ERK pathway by the TCR in resting T cells primes the cells for proliferation 22 25 49, and activation of the MEK-ERK pathway by Ras has been linked to proliferation in transformed cells 50. However, it is becoming increasingly clear that the effects of MEK-ERK signaling on cell growth depend on the physiological state of the cell, and either proliferation or growth arrest and senescence may occur 25 26 27. During activation, T cells progress from a quiescent, cytokine-independent state to a proliferative state where growth and survival are exquisitely cytokine dependent. It is only during this cytokine-dependent state that the antiproliferative aspects of TCR-triggered MEK-ERK signaling become apparent, through inhibition of cytokine signal transduction.
TCR ligation and MEK-ERK signaling blocked IL-2 induction of STAT5, activation of a STAT5-dependent reporter gene (Fig. 5), and activation of expression of the IL-2–inducible CIS, PGE2R, and CCR2 genes (Fig. 8). The CIS gene is activated primarily by STAT5 43, and thus TCR-mediated inhibition of CIS expression provides a functional correlate to inhibition of STAT5 activation. It is not yet clear if inhibition of PGE2R and CCR2 expression is secondary to blocking STAT5 or other IL-2 signaling pathways, but inhibition of these genes clearly demonstrates that TCR inhibition of IL-2 signaling has important functional consequences for T cell phenotype. Inhibition of IL-2 signaling also resulted in inhibition of IL-2–dependent proliferation. Suppression of proliferation may be mediated, at least in part, through inhibition of STAT5, which has been shown to play an important role in proliferation of primary T cells 51 52 53. Another important mechanism by which IL-2 stimulates T cell proliferation is through induction of cdk2 54 and cyclin D3 expression 32 45, with concomitant phosphorylation of Rb and progression from G1 into S phase of the cell cycle. IL-2 regulation of cyclin D3 and Rb has been shown to be mediated by Akt 32, and activation of Akt was blocked by TCR cross-linking (Fig. 3). As predicted, this resulted in decreased levels of IL-2–induced cyclin D3 and cdk2 expression, and decreased phosphorylation of Rb (Fig. 9). Thus, our results demonstrate that TCR ligation blocked IL-2–dependent proliferation of T cells by blocking at least two signaling pathways important for proliferation.
A recent report by Welte et al. 40 described rapid activation of STAT5 tyrosine phosphorylation that was detected 7 min after TCR ligation in the murine D10 Th2 clone, and was very transient, such that tyrosine phosphorylation was not detected 15 min after TCR ligation. TCR activation of STAT5 tyrosine phosphorylation was significantly weaker than activation by IL-2, and activation of STAT5 DNA binding after TCR ligation was detected only in transfected 293 cells that overexpressed the relevant signaling components 40. In our system, STAT5 activity was examined 0.5–4 h after TCR ligation, and thus we would not have detected very rapid and transient activation of STAT5. Our results are consistent with a previous report that TCR ligation did not induce STAT5 activation 55. Welte et al. suggested that TCR-mediated STAT5 activation played a role in TCR-induced proliferation based on results obtained using overexpression of a dominant negative STAT5-Y694F, which contains a mutation in the STAT5 tyrosine residue that becomes phosphorylated. In contrast to Welte et al. 40, we show that TCR ligation blocks subsequent activation of STAT5 by IL-2, and blocks IL-2–dependent proliferation (Fig. 10). The most likely explanation for these different findings is that they represent differences between quiescent T cells, where TCR ligation induces proliferation 40, and actively cycling T cells, where TCR ligation induces growth arrest (Fig. 10). Alternatively, although D10 cells do not produce IL-2, they produce proliferative cytokines such as IL-4, and high levels of overexpressed STAT5-Y694 may have resulted in loss of specificity in receptor docking and blocked proliferation 40 by blocking STAT signaling by cytokines such as IL-4.
Inhibition of cytokine signaling is a rapidly emerging area, and several constitutive mechanisms of downmodulating STAT activity have been described, including dephosphorylation, proteolytic degradation, or association with inhibitory molecules 56 57 58 59 60 61. We have previously described a rapidly inducible mechanism of inhibition of IL-6 signaling 28 29. Similarities between the mechanisms of inhibition of IL-6 signaling in myeloid cells and of IL-2 signaling in T cells include rapid induction within 15–60 min of addition of inhibitory factors, independence of new protein or RNA synthesis (at early time points), blockade of signaling at a proximal step in signal transduction without any changes in expression of receptors, Jaks, or STATs, and dependence on the MEK-ERK kinase cascade. These results suggest that inhibition of IL-2 and IL-6 signaling is achieved through MEK-dependent modification of IL-2 and IL-6 signaling components. Another previously described inducible mechanism for inhibiting the Jak-STAT pathway is cytokine-mediated induction of expression of SOCS/Jak binding (JAB)/STAT-inducible STAT inhibitor (SSI) proteins, which interact with and inhibit Jaks 62 63 64. Induction of SOCS3 may contribute to the inhibition of IL-2 signaling (Fig. 7), and inhibition of IL-6 signaling by GM-CSF at later time points also depended on new protein synthesis, likely of SOCS proteins 28. Thus, two distinct inhibitory mechanisms, mediated by MAPKs and SOCS proteins, may function in tandem to achieve effective inhibition of signaling at both early and later time points after addition of an inhibitory factor. Inhibition of IL-2 and IL-6 signaling in T cells, myeloid cells, and several cell lines by the MEK-ERK pathway indicates that this inhibitory mechanism functions in a number of cell types and physiological settings. Interestingly, additional important immune receptors that activate the MEK-ERK pathway, FcRs and CR3, have also been shown to inhibit cytokine and Jak-STAT signaling, although the mechanisms of inhibition and the role of the MEK-ERK pathway were not resolved 65 66. Thus, since the major receptors that mediate interactions of immune cells with antigenic stimuli, antigen, Fc, and complement receptors, activate the MEK-ERK pathway, cross-talk between the MEK-ERK and Jak-STAT pathways may play a particularly important role in modulation of cytokine signaling in immune cells.
Acknowledgments
We thank Carol Beadling, Constance Hsia, Kendall Smith, and Edward Yang for helpful discussions, and Selina Chen-Kiang, Keith Elkon, and Philip King for reviewing the manuscript. We are grateful to Carol Beadling and Kendall Smith for providing cells, plasmids, and oligonucleotides, and to J. O'Shea for providing Jak3 antibodies.
This work was supported by grants from the American Cancer Society, the National Institutes of Health, and the SLE Foundation (to L.B. Ivashkiv). K.B. Hisert was supported by National Institutes of Health Medical Scientist Training Program grant GM07739.
Footnotes
1used in this paper: CCR2, CC chemokine receptor 2; cdk, cyclin-dependent kinase; CIS, cytokine-inducible SH2 protein; EMSA, electrophoretic mobility shift assay; ERK, extracellular stimulus–regulated kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GAS, gamma-activated sequence; GRR, gamma response region; IRF, IFN regulatory factor; Jak, Janus kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase; MNC, mononuclear cell; PKC, protein kinase C; Rb, retinoblastoma protein; RT, reverse transcription; SEA, Staphylococcus enterotoxin A; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription
I.-H. Lee's present address is Hospital for Rheumatic Diseases, Hanyang University, Seoul 133-792, Korea.
References
- Kotzin B.L., Leung D.Y., Kappler J., Marrack P. Superantigens and their potential role in human disease. Adv. Immunol. 1993;54:99–166. doi: 10.1016/s0065-2776(08)60534-9. [DOI] [PubMed] [Google Scholar]
- Oxenius A., Zinkernagel R.M., Hengartner H. Comparison of activation versus induction of unresponsiveness of virus-specific CD4+ and CD8+ T cells upon acute versus persistent viral infection. Immunity. 1998;9:449–457. doi: 10.1016/s1074-7613(00)80628-7. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Abbas A.K. Homeostasis and self-tolerance in the immune systemturning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- Kuroda K., Yagi J., Imanishi K., Yan X.J., Li X.Y., Fujimaki W., Kato H., Miyoshi-Akiyama T., Kumazawa Y., Abe H., Uchiyama T. Implantation of IL-2-containing osmotic pump prolongs the survival of superantigen-reactive T cells expanded in mice injected with bacterial superantigen. J. Immunol. 1996;157:1422–1431. [PubMed] [Google Scholar]
- Vella A.T., Dow S., Potter T.A., Kappler J., Marrack P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 1998;95:3810–3815. doi: 10.1073/pnas.95.7.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack P., Kappler J., Mitchell T. Type I interferons keep activated T cells alive. J. Exp. Med. 1999;189:521–530. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar A.N., Salmon M. Cellular environments and apoptosistissue microenvironments control activated T-cell death. Immunol. Today. 1997;18:72–76. doi: 10.1016/s0167-5699(97)01003-7. [DOI] [PubMed] [Google Scholar]
- Ashwell J.D., Cunningham R.E., Noguchi P.D., Hernandez D. Cell growth cycle block of T cell hybridomas upon activation with antigen. J. Exp. Med. 1987;165:173–184. doi: 10.1084/jem.165.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matis L.A., Glimcher L.H., Paul W.E., Schwartz R.H. Magnitude of response of histocompatibility-restricted T-cell clones is a function of the product of the concentrations of antigen and Ia molecules. Proc. Natl. Acad. Sci. USA. 1983;80:6019–6023. doi: 10.1073/pnas.80.19.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake S., Nakano H., Park S.Y., Yamazaki T., Takase K., Matsushime H., Kato A., Saito T. Induction of G1 arrest by down-regulation of cyclin D3 in T cell hybridomas. J. Exp. Med. 1995;182:401–408. doi: 10.1084/jem.182.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau G.J., Moldwin R.L., Lancki D.W., Kim D.K., Fitch F.W. Inhibition of IL-2 driven proliferation of murine T lymphocyte clones by supraoptimal levels of immobilized anti-T cell receptor monoclonal antibody. J. Immunol. 1987;139:114–122. [PubMed] [Google Scholar]
- Suzuki G., Kawase Y., Koyasu S., Yahara I., Kobayashi Y., Schwartz R.H. Antigen-induced suppression of the proliferative response of T cell clones. J. Immunol. 1988;140:1359–1365. [PubMed] [Google Scholar]
- Russell J.H., White C.L., Loh D.Y., Meleedy-Rey P. Receptor-stimulated death pathway is opened by antigen in mature T cells. Proc. Natl. Acad. Sci. USA. 1991;88:2151–2155. doi: 10.1073/pnas.88.6.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Lenardo M.J. Molecules involved in cell death and peripheral tolerance. Curr. Opin. Immunol. 1997;9:818–825. doi: 10.1016/s0952-7915(97)80184-7. [DOI] [PubMed] [Google Scholar]
- Lissy N.A., Van Dyk L.F., Becker-Hapak M., Vocero-Akbani A., Mendler J.H., Dowdy S.F. TCR antigen-induced cell death occurs from a late G1 phase cell cycle check point. Immunity. 1998;8:57–65. doi: 10.1016/s1074-7613(00)80458-6. [DOI] [PubMed] [Google Scholar]
- Boehme S.A., Lenardo M.J. Propriocidal apoptosis of mature T lymphocytes occurs at S phase of the cell cycle. Eur. J. Immunol. 1993;23:1552–1560. doi: 10.1002/eji.1830230724. [DOI] [PubMed] [Google Scholar]
- Brunner T., Mogil R.J., LaFace D., Yoo N.J., Mahboubi A., Echeverri F., Martin S.J., Force W.R., Lynch D.H., Ware C.F. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- Dhein J., Walczak H., Baumler C., Debatin K.M., Krammer P.H. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- Ju S.T., Panka D.J., Cui H., Ettinger R., el-Khatib M., Sherr D.H., Stanger B.Z., Marshak-Rothstein A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- Refaeli Y., Van Parijs L., London C.A., Tschopp J., Abbas A.K. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- Zheng L., Trageser C.L., Willerford D.M., Lenardo M.J. T cell growth cytokines cause the superinduction of molecules mediating antigen-induced T lymphocyte death. J. Immunol. 1998;160:763–769. [PubMed] [Google Scholar]
- Smith K.A. Interleukin-2inception, impact, and implications. Science. 1988;240:1169–1176. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- Radvanyi L.G., Mills G.B., Miller R.G. Religation of the T cell receptor after primary activation of mature T cells inhibits proliferation and induces apoptotic cell death. J. Immunol. 1993;150:5704–5715. [PubMed] [Google Scholar]
- Su B., Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- Qian D., Weiss A. T cell antigen receptor signal transduction. Curr. Opin. Cell Biol. 1997;9:205–212. doi: 10.1016/s0955-0674(97)80064-6. [DOI] [PubMed] [Google Scholar]
- Lin A.W., Barradas M., Stone J.C., van Aelst L., Serrano M., Lowe S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qui M.S., Green S.H. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705–717. doi: 10.1016/0896-6273(92)90033-a. [DOI] [PubMed] [Google Scholar]
- Sengupta T.K., Schmitt E.M., Ivashkiv L.B. Inhibition of cytokines and JAK-STAT activation by distinct signaling pathways. Proc. Natl. Acad. Sci. USA. 1996;93:9499–9504. doi: 10.1073/pnas.93.18.9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta T.K., Talbot E.S., Scherle P.A., Ivashkiv L.B. Rapid inhibition of interleukin-6 signaling and stat3 activation mediated by mitogen-activated protein kinases. Proc. Natl. Acad. Sci. USA. 1998;95:11107–11112. doi: 10.1073/pnas.95.19.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B., Arron J., Choi Y. T cell receptor signals enhance susceptibility to Fas-mediated apoptosis. J. Exp. Med. 1997;186:1939–1944. doi: 10.1084/jem.186.11.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv L.B., Schmitt E.M., Castro A. Inhibition of transcription factor Stat1 activity in mononuclear cell cultures and T cells by the cyclic AMP signaling pathway. J. Immunol. 1996;157:1415–1421. [PubMed] [Google Scholar]
- Brennan P., Babbage J.W., Burgering B.M., Groner B., Reif K., Cantrell D.A. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity. 1997;7:679–689. doi: 10.1016/s1074-7613(00)80388-x. [DOI] [PubMed] [Google Scholar]
- Castro A., Sengupta T.K., Ruiz D.C., Yang E., Ivashkiv L.B. IL-4 selectively inhibits IL-2-triggered stat5 activation, but not proliferation, in human T cells. J. Immunol. 1999;162:1261–1269. [PubMed] [Google Scholar]
- Beadling C., Johnson K.W., Smith K.A. Isolation of interleukin 2-induced immediate-early genes. Proc. Natl. Acad. Sci. USA. 1993;90:2719–2723. doi: 10.1073/pnas.90.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnitz L.M., Abraham R.T. Interleukin-2 receptor signaling mechanisms. Adv. Immunol. 1996;61:147–199. [PubMed] [Google Scholar]
- Taniguchi T., Minami Y. The IL-2/IL-2 receptor systema current overview. Cell. 1993;73:5–8. doi: 10.1016/0092-8674(93)90152-g. [DOI] [PubMed] [Google Scholar]
- Gaffen, S.L., M.A. Goldsmith, and W.C. Greene. 1998. Interleukin-2 and the interleukin-2 receptor. The Cytokine Handbook. Academic Press, Inc., San Diego. 73–103.
- Leonard W.J., O'Shea J.J. Jaks and STATsbiological implications. Annu. Rev. Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Iezzi G., Karjalainen K., Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8:89–95. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- Welte T., Leitenberg D., Dittel B.N., al-Ramadi B.K., Xie B., Chin Y.E., Janeway C.A., Jr., Bothwell A.L.M., Bottomly K., Fu X.Y. STAT5 interaction with the T cell receptor complex and stimulation of T cell proliferation. Science. 1999;283:222–225. doi: 10.1126/science.283.5399.222. [DOI] [PubMed] [Google Scholar]
- Ahmed N.N., Grimes H.L., Bellacosa A., Chan T.O., Tsichlis P.N. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher P., Seitz M., Baggiolini M., Moser B. Interleukin-2 regulates CC chemokine receptor expression and chemotactic responsiveness in T lymphocytes. J. Exp. Med. 1996;184:569–577. doi: 10.1084/jem.184.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A., Masuhara M., Mitsui K., Yokouchi M., Ohtsubo M., Misawa H., Miyajima A., Yoshimura A. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood. 1997;89:3148–3154. [PubMed] [Google Scholar]
- Firpo E.J., Koff A., Solomon M.J., Roberts J.M. Inactivation of a Cdk2 inhibitor during interleukin 2-induced proliferation of human T lymphocytes. Mol. Cell Biol. 1994;14:4889–4901. doi: 10.1128/mcb.14.7.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J.M. IL-2-dependent induction of G1 cyclins in primary T cells is not blocked by rapamycin or cyclosporin A. Int. Immunol. 1993;5:1199–1209. doi: 10.1093/intimm/5.10.1199. [DOI] [PubMed] [Google Scholar]
- Drappa J., Vaishnaw A.K., Sullivan K.E., Chu J.L., Elkon K.B. Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N. Engl. J. Med. 1996;335:1643–1649. doi: 10.1056/NEJM199611283352204. [DOI] [PubMed] [Google Scholar]
- Fisher G.H., Rosenberg F.J., Straus S.E., Dale J.K., Middleton L.A., Lin A.Y., Strober W., Lenardo M.J., Puck J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- Rieux-Laucat F., Le Deist F., Hivroz C., Roberts I.A., Debatin K.M., Fischer A., de Villartay J.P. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- DeSilva D.R., Jones E.A., Favata M.F., Jaffee B.D., Magolda R.L., Trzaskos J.M., Scherle P.A. Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J. Immunol. 1998;160:4175–4181. [PubMed] [Google Scholar]
- Weinberg R.A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Moriggl R., Topham D.J., Teglund S., Sexl V., McKay C., Wang D., Hoffmeyer A., van Deursen J., Sangster M.Y., Bunting K.D. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- Teglund S., McKay C., Schuetz E., van Deursen J.M., Stravopodis D., Wang D., Brown M., Bodner S., Grosveld G., Ihle J.N. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Nakajima H., Liu X.W., Wynshaw-Boris A., Rosenthal L.A., Imada K., Finbloom D.S., Hennighausen L., Leonard W.J. An indirect effect of Stat5a in IL-2-induced proliferationa critical role for Stat5a in IL-2-mediated IL-2 receptor alpha chain induction. Immunity. 1997;7:691–701. doi: 10.1016/s1074-7613(00)80389-1. [DOI] [PubMed] [Google Scholar]
- Modiano J.F., Domenico J., Szepesi A., Lucas J.J., Gelfand E.W. Differential requirements for interleukin-2 distinguish the expression and activity of the cyclin-dependent kinases Cdk4 and Cdk2 in human T cells. J. Biol. Chem. 1994;269:32972–32978. [PubMed] [Google Scholar]
- Beadling C., Guschin D., Witthuhn B.A., Ziemiecki A., Ihle J.N., Kerr I.M., Cantrell D.A. Activation of JAK kinases and STAT proteins by interleukin-2 and interferon alpha, but not the T cell antigen receptor, in human T lymphocytes. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:5605–5615. doi: 10.1002/j.1460-2075.1994.tb06898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M., Lee C., Strehlow I., Schindler C. Functionally distinct isoforms of STAT5 are generated by protein processing. Immunity. 1997;6:691–701. doi: 10.1016/s1074-7613(00)80445-8. [DOI] [PubMed] [Google Scholar]
- Kim T.K., Maniatis T. Regulation of interferon-gamma-activated STAT1 by the ubiquitin-proteasome pathway. Science. 1996;273:1717–1719. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- Haspel R.L., Salditt-Georgieff M., Darnell J.E., Jr. The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends upon a protein tyrosine phosphatase. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:6262–6268. [PMC free article] [PubMed] [Google Scholar]
- Shuai K., Liao J., Song M.M. Enhancement of antiproliferative activity of gamma interferon by the specific inhibition of tyrosine dephosphorylation of Stat1. Mol. Cell Biol. 1996;16:4932–4941. doi: 10.1128/mcb.16.9.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragane Y., Kulms D., Luger T.A., Schwarz T. Down-regulation of interferon gamma-activated STAT1 by UV light. Proc. Natl. Acad. Sci. USA. 1997;94:11490–11495. doi: 10.1073/pnas.94.21.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.D., Liao J., Liu B., Rao X., Jay P., Berta P., Shuai K. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- Endo T.A., Masuhara M., Yokouchi M., Suzuki R., Sakamoto H., Mitsui K., Matsumoto A., Tanimura S., Ohtsubo M., Misawa H. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Naka T., Narazaki M., Hirata M., Matsumoto T., Minamoto S., Aono A., Nishimoto N., Kajita T., Taga T., Yoshizaki K. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Starr R., Willson T.A., Viney E.M., Murray L.J., Rayner J.R., Jenkins B.J., Gonda T.J., Alexander W.S., Metcalf D., Nicola N.A., Hilton D.J. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Feldman G.M., Chuang E.J., Finbloom D.S. IgG immune complexes inhibit IFN-gamma-induced transcription of the Fc gamma RI gene in human monocytes by preventing the tyrosine phosphorylation of the p91 (Stat1) transcription factor. J. Immunol. 1995;154:318–325. [PubMed] [Google Scholar]
- Marth T., Kelsall B.L. Regulation of interleukin-12 by complement receptor 3 signaling. J. Exp. Med. 1997;185:1987–1995. doi: 10.1084/jem.185.11.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]